


Abstract
Acquiring foreign spacer DNA into the CRISPR locus is an essential primary step of the CRISPR–Cas pathway in prokaryotes for developing host immunity to mobile genetic elements. Here, we investigate spacer integration in vitro using proteins from Pyrococcus furiosus and demonstrate that Cas1 and Cas2 are sufficient to accurately integrate spacers into a minimal CRISPR locus. Using high-throughput sequencing, we identified high frequency spacer integration occurring at the same CRISPR repeat border sites utilized in vivo, as well as at several non-CRISPR plasmid sequences which share features with repeats. Analysis of non-CRISPR integration sites revealed that Cas1 and Cas2 are directed to catalyze full-site spacer integration at specific DNA stretches where guanines and/or cytosines are 30 base pairs apart and the intervening sequence harbors several positionally conserved bases. Moreover, assaying a series of CRISPR repeat mutations, followed by sequencing of the integration products, revealed that the specificity of integration is primarily directed by sequences at the leader-repeat junction as well as an adenine-rich sequence block in the mid-repeat. Together, our results indicate that P. furiosus Cas1 and Cas2 recognize multiple sequence features distributed over a 30 base pair DNA region for accurate spacer integration at the CRISPR repeat.
INTRODUCTION
CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) arrays, found on the genomes of roughly half of bacteria and the majority of archaea, harbor a genetic memory of previously encountered nucleic acid invaders (1,2). Cas (CRISPR-associated) proteins recurrently add short, ∼30–40 bp DNA sequences (called spacers) to CRISPR arrays, and coordinate the defense against the nucleic acid invaders upon subsequent infections (1,3,4). CRISPR uptake of spacer DNA fragments occurs through the process of adaptation and leads to heritable genetic memories of the infecting virus or other mobile genetic element (5–8). During adaptation, segments of the invader are cleaved and typically processed at protospacer adjacent motifs (PAMs) before being directionally integrated into the CRISPR genetic memory bank (9–12). The CRISPR locus includes a leader sequence followed by an array of short repeating DNA sequences, separated by unique spacers derived from past infections (13). In order to defend against the invading mobile genetic element, the CRISPR locus is transcribed and processed into CRISPR RNAs (crRNAs) that guide effector Cas proteins to the invading nucleic acid through base complementarity (3,14,15). When bound to a complementary target nucleic acid, crRNA-associated Cas nucleases degrade the invader and the threat is silenced (16–19).
Diverse CRISPR–Cas systems have evolved that each employ distinct Cas protein family members and mechanisms for defense and have been categorized into six types and >30 subtypes (20,21). In contrast, the proteins that perform the integration of new spacers into CRISPR arrays, Cas1 and Cas2, are common components found in the various types of active CRISPR–Cas systems, and sequences of CRISPR leaders and repeats tend to coevolve with Cas1 sequences (20,22).
The model organism Pyrococcus furiosus, an extreme hyperthermophile, has proven to be useful for investigating the understudied archaeal adaptation mechanisms (12,23). The P. furiosus genome contains type I-A, I-G and III-B CRISPR–Cas systems that use crRNA guides generated from seven active CRISPR arrays (4,24–28). New spacers are added to each of these CRISPR arrays by a single, shared set of Cas1 and Cas2 proteins (23), after proper processing of the protospacer by Cas4–1 and Cas4–2 (12). Cas1 has been shown to functionally and physically interact with Cas4 proteins in several studied systems (12,29–31). P. furiosus Cas4–1 and Cas4–2 nucleases are necessary for PAM recognition and removal during spacer generation, for setting spacer length, and for ensuring that new spacers are efficiently integrated in the correct orientation that supports immunity (12). However, in the absence of both Cas4–1 and Cas4–2 proteins, Cas1 and Cas2 proteins retain the ability to accurately integrate spacers at CRISPR repeats in vivo (12) raising the key question of what specific cis-acting determinants guide Cas1 and Cas2 proteins to localize at CRISPR loci.
The P. furiosus cas1 and cas2 genes are encoded immediately adjacent to genes of the type I-G Cas proteins (Cst module) (23), suggesting that they co-evolved to function as the adaptation module associated with the type I-G system (rather than III-B or I-A system). Supplementary Figure S1 displays a model of P. furiosus adaptation based on in vivo experiments conducted with P. furiosus (12,23) and in vitro experiments with type I-E and I-A systems (11,30,32–36). The spacer integration step of this process is achieved through a two-step transesterification reaction, where the 3′ hydroxyl groups of the incoming, predominately double-stranded DNA spacer attack both borders of the first repeat sequence, on opposite strands (34,37). The majority of studies suggest that type I and II systems first attack the top strand at the leader-repeat junction (LR), followed by a second attack of the repeat-spacer junction (RS) on the bottom strand (8,38). Direct, in vitro kinetic tests on the order of attacks have only been done for I-E and II-A systems (6,32,35), and these tests should be expanded to include more systems, given their diversity and ability to achieve immunity through unique mechanisms.
Details of the CRISPR spacer integration reaction have begun to be elucidated through in vivo and in vitro studies across a variety of CRISPR–Cas systems. In both type I and II systems, a Cas14–Cas22 complex has been found consisting of two Cas1 dimers bridged by a central Cas2 dimer, with Cas1 serving as the integrase and Cas2 likely playing a structural role (11,31,34,35,39,40). The Cas1–Cas2 integrase complex is directed to integrate new spacers at the leader-proximal repeat (1,6,7,39,41,42), but the mechanisms guiding this targeted integration are proving to be quite diverse across different subtypes. Some systems require non-Cas host factors, such as the leader-binding integration host factor (IHF), to guide Cas1–Cas2 to the first repeat in the array (32,35,43,44). In contrast, the type II-A systems lack a requirement for IHF and instead have an intrinsic ability to direct spacer integration, likely due to protein-DNA interactions between Cas1 and sequences surrounding the leader-repeat junction (6,40,41). It remains to be determined how spacer integration is directed in adaptation systems of archaea, which lack IHF, but preliminary findings implicate both sequence and protein components. The large (531 bp) archaeal leader in Sulfolobus solfataricus is important for type I-A adaptation, and in vitro spacer integration required unidentified host factor(s) from cell lysate and ATP hydrolysis (36). As this has been the only report on in vitro spacer integration in archaeal systems, there is still much unknown about how spacers are directed for integration at CRISPR repeats in archaeal organisms.
Sequences in the leader and repeat have been shown to influence proper spacer integration. Two in vivo studies investigating I-E and I-B spacer addition and repeat duplication identified mid-repeat motifs that orchestrate accurate spacer integration at LR and RS sites (45,46). In the I-E study, the essential motifs were situated in palindromic inverted repeats (IRs) within the CRISPR repeat (45), while in the I-B study, accurate integration sites relied on a motif between the IRs. While IRs are a common feature among many groups of CRISPR repeats, both studies found that the ability for IRs to form cruciform structures was not necessary for adaptation (45,46). Recent high resolution cryo-EM and X-ray crystallography structures showed Cas1–Cas2 bound to pre-spacers being integrated into minimal CRISPR loci (11,35,40), and suggested that these important mid-repeat motifs likely serve as binding sites for the integrase machinery. Several in vitro studies have investigated cis-acting elements of the repeat and leader through a variety of sequence mutations (6,8,32,35,36,40). However, results have varied depending on the system, again highlighting the mechanistic diversity of CRISPR systems. It is important to note that these in vitro mutational studies have been limited by drawing conclusions solely on apparent integration efficiency rather than determining the sites of integration by direct sequencing methods.
Here, we established an in vitro system capable of accurately integrating spacer DNA at the proper junctions of P. furiosus CRISPR repeats. Interestingly, spacers also became integrated into plasmid DNA at a subset of non-CRISPR (‘off-target’) DNA regions that share specific sequence features with CRISPR repeats. We found P. furiosus Cas1 and Cas2 to be necessary and sufficient for complete (full-site) spacer integration. Specific DNA determinants important for efficient and accurate integration of spacer DNA were identified both by evaluating reaction efficiency and monitoring the precise location of spacer integration by sequencing for an extensive panel of CRISPR repeat and leader mutant target DNAs. Together, our results indicate that P. furiosus Cas1 and Cas2 recognize multiple sequence features distributed over an ∼30 bp DNA stretch to enable accurate integration of spacer DNA into the repeats of target CRISPR loci. Our results implicate specific sequence elements and their relative spacing to each other in properly positioning the P. furiosus Cas1 and Cas2 integrase proteins for accurate spacer integration at the CRISPR repeat.
MATERIALS AND METHODS
Cloning, expression and purification
The cas1 (PF1117) and cas2 (PF1118) genes were amplified from Pyrococcus furiosus COM1 genomic DNA, individually cloned into pET21d expression vectors, and transformed into Escherichia coli BL21 RIPL strain. Cultures were grown at 37°C in 200 ml of Luria broth to an OD600 of 0.4–0.6, and expression of the C-terminal 6× histidine tagged proteins was induced with 1 mM IPTG during overnight growth at room temperature. Harvested cells were lysed in 40 mM Tris, 500 mM KCl, 10% glycerol, 5 mM imidazole, pH 7.5. Following thermal precipitation at 70°C for 30 min, the cell lysate was centrifuged at 14 000 rpm at 4°C for 30 min and the soluble fraction was collected and filtered (0.8 μm filter pore size Millex filter unit, Millipore). His-tagged proteins were purified by Ni2+ affinity column chromatography, using a stepwise increase of imidazole (10, 20, 50, 100, 250 and 500 mM) in 40 mM Tris, 500 mM KCl, 10% glycerol, pH 7.5. Peak elution fractions were dialyzed using Slide-a-lyzer mini dialysis cassettes (Thermo Fisher) into 40 mM Tris, 200 mM KCl, 10% glycerol, pH 7.5, and stored at 4°C prior to use for functional assays. C-terminal 6× histidine tagged proteins were confirmed to be functional for adaptation in vivo (data not shown), ruling out potential detrimental effects of the tag.
DNA substrate preparation
DNA oligonucleotides were from Eurofins Genomics (for minimal CRISPR substrates and PCR primers) and Integrated DNA Technologies (for pre-spacer DNA and hairpin CRISPR substrates). Oligonucleotides used to make pre-spacers and CRISPR substrates were separated by 15% denaturing polyacrylamide gel electrophoresis in 1× TBE, detected by ethidium bromide staining, and the band of the expected oligonucleotide size was excised. Oligonucleotides were eluted from the gel slices overnight at 4°C in 500 μl of elution buffer (0.5 M ammonium acetate, 1 mM EDTA (pH 8.0), and 0.1% SDS), extracted with phenol/chloroform/isoamyl alcohol (pH 8.0), ethanol precipitated, and resuspended in 20 mM Tris, 100 mM KCl, 5% glycerol, pH 7.5. Corresponding oligonucleotides were annealed by incubating at 95°C for 5 min followed by slow cooling until 23°C. Annealing was confirmed by 10% non-denaturing polyacrylamide gel electrophoresis in 1× TBE. Pre-spacers and half-site CRISPR substrates were 5′ radiolabeled with T4 polynucleotide kinase (PNK) and [γ-32P] ATP. In the case of the half-site substrates, a second gel extraction and precipitation was performed after annealing. Oligonucleotide sequences can be found in Supplementary Table S1.
Spacer integration assay
Unless stated otherwise, a final concentration of 1 μM Cas1, 1 μM Cas2, 1 mM DTT and 10 mM MgCl2 was incubated in reaction buffer (20 mM Tris, 100 mM KCl, 5% glycerol, pH 7.5) at 4°C for 1 h. 20 nM of radiolabed pre-spacer or 100 nM of unlabeled pre-spacer was added to the reactions and incubated at 4°C for an additional 15 min. Finally, plasmid or linear DNA was added to a final concentration of 5 nM for pCR7, or 100 nM for minimal CRISPR substrate, and the reaction was incubated at 70°C for 1 h. Reactions with pCR7 were quenched with EDTA and proteinase K (Life Technologies). Products were mixed with gel loading dye (purple, NEB) and separated on 1% agarose gels in 1× TAE. Reactions with minimal CRISPR substrates were quenched with an equal volume of Gel Loading Buffer II (Thermofisher) and 25 mM EDTA. Samples were boiled for 5 min before separation by 12% denaturing 7 M urea-containing polyacrylamide gel electrophoresis in 1× TBE. Gels were dried and radioactivity was detected with phosphorimaging (Storm 840 Scanner GE Healthcare).
Analysis of integration by high-throughput sequencing
Library preparation
To analyze integrations by high-throughput sequencing, the spacer integration assay was performed as described above using unlabeled pre-spacer. Following incubation, DNA was isolated using the DNA Clean and Concentrator Kit (Zymo Research, Irvine, CA, USA). For the plasmid integration samples, excess un-integrated pre-spacer was removed using Agencourt AMPure XP beads (Beckman Coulter, Indianapolis, IN, USA). Next, Illumina adapter sequence with an N10 random primer was annealed to the plasmid DNA and extended (thermocycler conditions: 98°C for 30 s, 25°C for 30 s, 35°C for 30 s, 45°C for 30 s, and 72°C for 5 min). Following extension, excess adapter was removed using AMPure beads, and PCR was done to amplify plasmid DNA that contained integrated pre-spacer: forward primers were specific for the pre-spacer, while reverse primers targeted the Illumina adapter introduced with the random anneal and extension step. This amplified both full-site and half-site integration events with no discrimination. Illumina barcodes and additional adapter sequences were added with a final PCR and the resulting library was separated on a 1% agarose gel to select for DNA in a 400–700 bp size range. DNA was isolated using the Zymo Gel DNA Recovery Kit (Zymo Research, Irvine, CA, USA) and sequenced on an Illumina MiSeq in a 100 by 50 cycle run. Only the 100 bp Read 1 data was used in this analysis.
For the minimal linear CRISPR substrate products, 1 μl of eluted DNA was used as a PCR template. Primers to add Illumina adaptor sequences were annealed to the newly integrated spacer and the 3′ end of either the plus or minus strand of the CRISPR substrate. DNA Clean and Concentrator Kit (Zymo Research, Irvine, CA) was used to isolate the PCR product, and 1 μl of this product was used as the template for a second PCR using primers to add Illumina barcodes. These products were purified on a 1% agarose gel and extracted with a Gel Purification Kit (Zymo Research, Irvine, CA).
Mapping integration events
After sequencing, samples were de-multiplexed by barcode and analyzed to determine sites of integration. For plasmid data, the complete pre-spacer sequence was located in each read and 50 bp of sequence immediately downstream from the end of the pre-spacer was extracted. These 50 bp sequences were aligned to the appropriate plasmid reference using Bowtie (47). To visualize the distribution of integration events, alignment output files were converted into coverage files using bedtools (48) and displayed on a custom UCSC genome browser track hub (https://www.genome.ucsc.edu). An initial inspection of the integration tracks revealed that large peaks occurred outside of the CRISPR arrays and data suggested that these peaks were due to particular sequence and spatial features. These trends were both analyzed in an unbiased plasmid-wide manner. To determine sequence preferences at the sites of integration, the base at the integration point, along with upstream and downstream context sequence, was extracted from the reference sequence (bedtools) and used to make sequence logos (49). For spacing trends, we took the browser track files and assessed the distances between two large peaks occurring on the same strand or on opposite strands. To do this, 500 random 50 bp intervals (‘windows’) were selected for each plasmid. Within each of these windows, the two highest peaks on the plus strand and the minus strand were identified and the bp distance between these peaks was determined (highest to second highest on the plus strand, highest to second highest on the minus strand, highest plus strand to highest minus strand). Distance values from all 500 windows were then binned and counted. For the minimal linear CRISPR integration data, the spacer-target junction was determined from each read and counts for each potential integration point were totaled.
RESULTS
P. furiosus Cas1 and Cas2 facilitate spacer integration in vitro
To determine the requirements for the integration of new spacers into the CRISPR array, Cas1 and Cas2 recombinant proteins cloned from P. furiosus were expressed and purified from E. coli and tested in spacer integration assays with a synthetic pre-spacer and a plasmid containing a minimal CRISPR array (pCR7) (Figure 1). The CRISPR array had a complete 508 bp leader followed by three 30 bp repeats, separated by two 37 bp spacers derived from P. furiosus CRISPR locus 7 (Figure 1A). The 37 bp sequence of the pre-spacer DNA was selected due to its high frequency of acquisition from a plasmid during transformation into P. furiosus (23). When pCR7 was incubated in vitro with Cas1, Cas2, and radiolabeled pre-spacers in the presence of MgCl2, full-site or half-site integration products (which cause plasmid nicking detectable by agarose gel electrophoresis and DNA staining) were detected by autoradiography (Figure 1B, compare lanes 1–5). Spacer integration was also observed using plasmids that lacked the leader sequence (pCR7 no leader) or control plasmids devoid of a CRISPR locus (pControl) (Figure 1B). Radiolabeled integration products were detected in the nicked and linear conformation of the plasmid, and required the presence of Cas1, Cas2 and MgCl2 (Figure 1B). Cas1 alone was able to produce extremely low levels of integration, while Cas2 alone could not (Supplementary Figure S2A) indicating the importance of both Cas1 and Cas2 in catalyzing spacer integration into the target DNA molecules. Pre-spacers with blunt-ends or 3′ overhangs (five nucleotides in length) were both integrated into pCR7 (Supplementary Figure S2B), but subsequent experiments were carried out using pre-spacers with 3′ overhangs. Moreover, integration occurred efficiently into linear plasmid DNA, indicating that integration by Cas1 and Cas2 is not strictly dependent on the supercoiled topology of circular plasmid DNA (Supplementary Figure S2B).
Figure 1.

P. furiosus Cas1 and Cas2 facilitate spacer integration in vitro. (A) Schematic of the in vitro spacer integration reaction. pCR7 containing a minimal CRISPR array was incubated with Pyrococcus furiosus (Pfu) Cas1 and Cas2 purified from E. coli and radiolabeled pre-spacers (PS). Integration products include half-site or full-site integration (nicked form of the plasmid). (B) Integration assay comparing pCR7, pCR7 with a deletion of the leader, and pControl lacking the entire leader and CRISPR array. Products separated by agarose gel electrophoresis and detected by EtBr followed by autoradiography. Noncontiguous portions of the same gel are indicated by dashed lines. (C–E) Sites of spacer integration were identified by high-throughput sequencing; peak heights in tracks C–G directly reflect the number of reads that showed an integration event at that position in the plasmid. Integration sites along pCR7, pCR7 no leader, and pControl, respectively. Brackets above the tracks highlight sites of integration at the CRISPR and a highly preferred non-CRISPR integration site, which was investigated further in later experiments. (F, G) Increased resolution of integration at CRISPR repeats of pCR7 and pCR7 no leader.
We next mapped the precise sites of spacer integration in each of the tested plasmids by high-throughput sequencing of the integration products (Figure 1C–E). Integration into pCR7 occurred at very low levels at sites distributed throughout the entire plasmid with a marked increased preference at each of the three CRISPR repeats (occurring precisely at the same top strand and bottom strand repeat junctions as is observed in vivo) (Figure 1C and F and (23)), as well as several non-CRISPR locations (Figure 1C). The high frequency of integration at the first repeat was lost when the adjacent leader was deleted, but the levels of integration at the second and third repeat remained high (Figure 1D and G). The non-CRISPR integration sites remained when the leader or entire CRISPR locus was absent from the plasmid (Figure 1D and E). One such highly preferred non-CRISPR site of integration (bracketed in Figure 1C) was investigated further in subsequent experiments. This exact site was also highly preferred in an alternative plasmid backbone containing the same non-CRISPR sequence (pControl2) (Supplementary Figure S3).
Non-CRISPR integration sites resemble CRISPR repeats
A closer look at the most highly targeted non-CRISPR plasmid DNA integration sites revealed similarities between these sites and the CRISPR repeat, both in size and sequence identity (Figure 2). Pairs of high frequency integration sites on opposite strands, similar to the pattern observed at the repeats, were observed scattered along the plasmid and the distances between the paired non-CRISPR integration sites were consistent with the size of a CRISPR repeat (30 bp), as shown with the non-CRISPR example (Figure 2A). We quantified this spacing trend by randomly selecting windows across the plasmid and determining the distance between the largest peaks on opposite strands. There was a significant trend wherein the highest plus strand peak was 30–31 bp from the highest minus strand peak for both plasmid DNA that contained (pCR7) or lacked (pControl) the CRISPR array (Figure 2B and C). In contrast, when the same analysis was performed for peaks that were both on the plus strand or both on the minus strand, no such 30 bp spacing preference was observed, suggesting the paired peaks on opposite strands are due to the Cas1 and Cas2-catalyzed, full-site integration (two-step transesterification reaction) (Supplementary Figure S4).
Figure 2.

Non-CRISPR integration sites resemble CRISPR repeats. (A) Spacing between maximum integration peaks on plasmid tracks shown in Figure 1 were measured within 500 random 50 bp windows. (B, C) The number of times each bp spacing between maximum peaks was counted on pCR7 (B) and pControl (C). (D, E) Percent of total integration sites located at guanine, cytosine, adenine, and thymine on pCR7 (D) and pControl (E). (F) The top ten 30 bp non-CRISPR sites on pCR7. List includes any sequences that had more than 40 reads at either the ‘LR’ or ‘RS’ location of the sequence. Homology is highlighted for each nucleotide that matches the CRISPR repeat. (G) WebLogo of non-CRISPR sequences listed in F. (H, I) WebLogo of all integration events on pCR7 (H) and pControl (I).
In addition to trends in peak spacing, we also noted that certain bases were conserved in the highly integrated non-CRISPR sites. We therefore analyzed the sequences surrounding these non-CRISPR integration sites to determine if there was any similarity to the leader-repeat elements of the CRISPR locus. The nucleotide identity at the site of integration across the entire plasmid exhibited a strong preference for guanine, with the second most preferred base being cytosine, which is in agreement with the nucleotide identity of the natural P. furiosus CRISPR repeat borders (Figure 2D and E). Sequences for the top ten most highly integrated non-CRISPR sites of 30 bp spacing are listed in Figure 2F, and their locations are indicated on the plasmid map in Supplementary Figure S5. A WebLogo created from these top ten sequences revealed similarities to the repeat, particularly at the borders and the center of the repeat (Figure 2G). Specifically, nucleotides G1, T2, A6, A12, T14, A17, A18, A20, A23, T24, T25, A28, and A29 of the repeat, and C-3, C-4, G-7, A-8, A-9 and A-10 of the leader were often enriched in the non-CRISPR sites (Figure 2G). When all integration sites were considered (not just ones with a paired peak pattern), repeat-like features were conserved; most notably, G1, T2, T3, A6, T14, A17, A18, A29, as well as C-3 in the leader (Figure 2H and I). The same spacing and sequence trends are seen when analyzing plasmids lacking the leader (pCR7 no leader) and missing the entire leader-CRISPR array (pControl), demonstrating that this phenomenon is not due to the presence of the CRISPR locus but rather reflects an intrinsic integration site preference of Cas1 and Cas2 (Supplementary Figures S4 and S6). Taken together, this analysis of integration sites across the plasmid suggests that spacer integration by P. furiosus Cas1 and Cas2 proteins is being directed by specific sequences organized in a defined spatial configuration.
Full-site integration
To further elucidate the mechanism of integration at the repeat, an assay using a minimal linear CRISPR array was established (Figure 3A and B). Unlike plasmid integration data, the results of these assays allowed us to distinguish half-site and full-site spacer DNA integrations. When using a linear CRISPR array with only 10 bp of the leader, a single repeat, and a single spacer (Figure 3A), products representing integration at both leader-repeat (LR) and repeat-spacer (RS) borders of the repeat were observed (Figure 3C), and confirmed through high-throughput sequencing (Figure 3D). The same high degree of specificity was observed when using the sequence of the highly preferred non-CRISPR site described above (Figures 1, 2, Supplementary Figure S3), even though the sequence is only 43% identical to the wild type (WT) repeat (Figure 3A, C, and D). This orientation was selected because of the higher repeat sequence conservation, but it is important to note that the alternative orientation also has conserved elements identified to influence integration location (G1, T3, A6, T14, A18, A20, T24 and A29 as well as the C-3). When the sequences of the non-CRISPR substrate that are conserved with the WT repeat were mutated (NC Mut), efficiency was reduced (Figure 3C) and the specificity of integration at the sites 30 bp apart was abolished to <1% of the total reads (Figure 3D).
Figure 3.

Integration into a minimal CRISPR reveals full-site integration. (A) Annotated sequence of minimal linear CRISPR. LR marks leader-repeat integration site. RS marks repeat-spacer integration site. IR1 and IR2 mark inverted repeats. Below the WT sequence, non-CRISPR (NC) and non-CRISPR mutant (NC Mut) sequences are displayed. Dots above NC sequence indicate homology with WT repeat. (B) Schematic of in vitro spacer integration assay with a minimal linear CRISPR containing a single repeat. Potential reaction products include full-site or half-site integration at either the leader-repeat junction (LR) or the repeat-spacer junction (RS). (C) Integration products separated by denaturing PAGE and detected with autoradiography. Noncontiguous lanes from the same gel are indicated by dashed lines. (D) Sequenced integration products displayed as % of reads mapped to sites of integration. (E, F) Continuation of LR half-site (E) or RS half-site (F) substrates to full-site integration detected with radiolabel on bottom strand of the spacer (E) or top strand of leader (F). (G, H) Spacer disintegration from LR (G) or RS (H) half-site substrates detected with radiolabel on spacer.
Next, to determine if an integrated spacer at one of the two repeat junctions would progress to a full-site integration, CRISPR DNA substrates modeled to be half-site DNA intermediates were assayed (Figure 3E–H). Half-site substrates were rapidly transformed into full-site products regardless of which junction of the repeat the spacer started at (Figure 3E and F). Disintegration of the half-site spacer was also separately examined since it is expected to reflect the reverse reaction of spacer integration that should be occurring at some level in the reactions (50). Half-site spacer disintegration was observed at lower levels (Figure 3G and H) than full-site conversion (Figure 3E and F) showing that most, if not all, products observed with the half-site substrates resulted in full-site integration rather than disintegration followed by reintegration at the second site. Additionally, disintegration did not require the presence of Cas2 (Supplementary Figure S7C and D), which was needed for the progression to full-site (Supplementary Figure S7A and S7B). The results show that P. furiosus Cas1 and Cas2 execute accurate second-site integrations by attacking the proper top or bottom strand leader-repeat or repeat-spacer junctions.
We next addressed if P. furiosus Cas1 and Cas2 catalyze sequential, two-step transesterification reactions and if there is a preference for first-site integration at the leader-repeat junction vs. the repeat-spacer junction (Figure 4). This was investigated using CRISPR substrates with a DNA hairpin structure at either the leader end or the spacer end, which allowed full-site products to be distinguished from the two possible half-site products on the basis of the size of fragments separated by gel electrophoresis. Both hairpin substrates resulted in rapid, full-site integration but half-site products at both the leader-repeat and repeat-spacer junctions were also observed as expected (Figure 4A and B). In order to confirm that the full-site product was indeed a result of the two-step integration event from a single spacer rather than two separate half-site integration events by two independent spacers, a dideoxy group was substituted for the 3′ hydroxyl on one strand or the other of the pre-spacer. With the lack of second 3′ hydroxyl group required for the sequential second-site integration, reactions were halted after the first-site integration event, verifying that the full-site product was not produced by independent half-site integrations by two separate spacers. Additionally, with the dideoxy pre-spacers, the LR and RS half-site products were observed at approximately equal levels, suggesting no preference for one border of the repeat over the other for the first transesterification attack (Figure 4C). When the dideoxy was present on both strands (no 3′ hydroxyl groups), no integration was observed, as expected. Furthermore, full-site integration was also observed with the highly preferred, non-CRISPR plasmid sequence (Figure 4D) described above (Figures 1–3, Supplementary Figure S3). In addition to versatility for the first-site of integration, no preference for orientation was observed for the newly integrated spacer, suggesting that pre-spacer internal sequences do not guide orientation to ensure functional crRNA-mediated invader defense (Supplementary Figure S8 and (12)).
Figure 4.
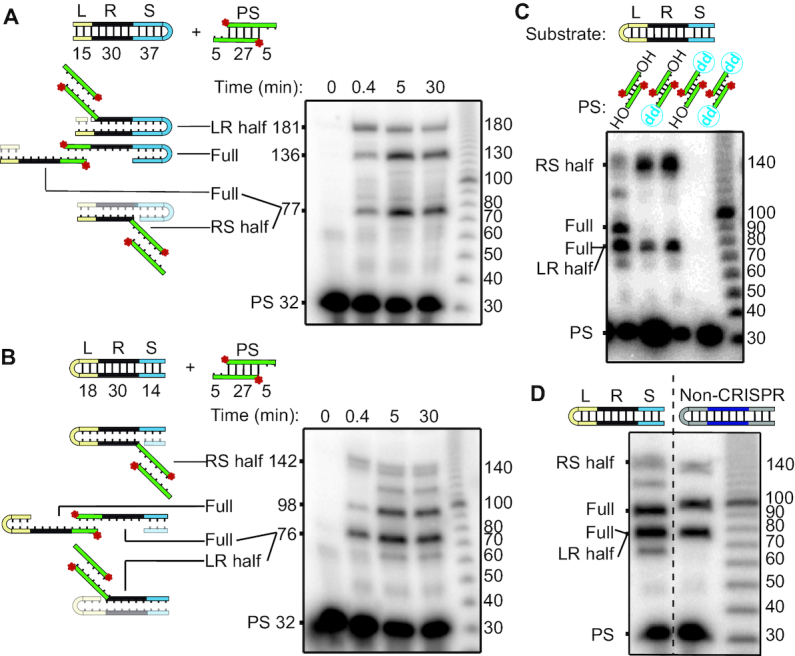
Integration into hairpin CRISPRs suggests no preferred order to the two-step transesterification attacks. (A, B) Full-site integration detected using a hairpin turn in the (A) spacer or (B) leader ends of the minimal CRISPR substrates. (C) Site of first transesterification attacks identified with pre-spacers with 3′ dideoxy groups on top, bottom, or both strands. (D) Full-site integration into hairpin substrate containing the non-CRISPR sequence. Noncontiguous lanes from the same gel are indicated by dashed lines.
Together these results indicate that full-site spacer integration reactions facilitated by P. furiosus Cas1 and Cas2 occur approximately equally well from either direction. Additionally, the abundant full-site integration products observed with the CRISPR and non-CRISPR sequences (Figure 4) suggests that many or all of the paired integration sites on the plasmids (Figure 1C–E) are full-site integration events. Finally, the large leader and region upstream of the non-CRISPR sequence can be shortened to 10 bp while maintaining accuracy of spacer integration (Figure 3A–D). However, a mutation to repeat-conserved nucleotides of the non-CRISPR substrate abolished this specificity (Figure 3A–D), supporting the notion that Cas1 and Cas2 are guided by specific sequence elements described above (Figure 2).
Sequences within the repeat guide integration to occur at a distance of 30 bp
The observation that spacers were selectively integrated at CRISPR repeat and repeat-like sequences alike led us to further investigate the characteristics of the repeat sequence that are directing Cas1 and Cas2-facilitated spacer integration. We introduced a series of mutations to the minimal CRISPR DNA substrates (Figure 5A and B) and examined the effects of each mutation on spacer integration efficiency (level of integration observed on a gel with autoradiography relative to the WT repeat) and specificity (location of integration determined and quantified through sequencing). The first (G1 of the top strand) and last (C30 of the bottom strand) nucleotides of the WT repeat are the sites for transesterification attacks during integration, and when both of these nucleotides were mutated to adenine or thymine (G1A, C30T and G1T, C30A mutants), the efficiency and specificity were both reduced severely, while a mutation to cytosine and guanine (G1C, C30G mutant) did not significantly alter the reaction (Figure 5C and F). Individual mutations of the first or last nucleotide did not have the same severe effect as the simultaneous mutations, except in the case of G1T mutation which was detrimental to both integration efficiency and specificity (Supplementary Figure S9). Thus, the identity of the base at the sites of transesterification attack on the CRISPR repeat is an important component for specifying integration at a CRISPR repeat by the Cas1 and Cas2 proteins.
Figure 5.

Integration efficiency and specificity is altered by mutations to the CRISPR repeat. (A) Annotated sequence of minimal linear CRISPR. LR marks leader-repeat integration site. RS marks repeat-spacer integration site. IR1 and IR2 mark inverted repeats. (B) List of substitution, insertion, and deletion mutations made to the minimal CRISPR. (C, D, E) Integration assays comparing mutations to the first and last nucleotide of the repeat (C), block mutations of the leader and repeat (D), and 3 bp insertions or deletions at the center of the repeat (E). (F–H) Integration sites along the minimal CRISPRs. Integration sites mapping to the nucleotide level resolution for all mutations can be found in Supplementary Figure S11, and a summary of integration specificity at LR and RS junctions of all mutants can be found in Supplementary Table S2.
DNA base substitution block mutations throughout the minimal CRISPR DNA substrates were used to detect additional elements that coordinate accurate and specific spacer integration (Figure 5). Mutation of the 10 bp leader (B1 mutant) resulted in a moderate reduction in integration efficiency and specificity, while a mutation to the eight nucleotides spanning the leader-repeat junction (B2 mutant) had a detrimental effect on both efficiency and specificity of integration (Figure 5D and G). Even with an intact leader sequence, a mutation to the first four nucleotides of the repeat (B3 mutant) still caused major disruption, especially to the leader-repeat integration site which was reduced to <2% of total integration reads (Figure 5G). In contrast, if the last four nucleotides of the repeat were mutated (B8 mutant), there was no significant effect on either the efficiency or specificity of integration relative to WT (Figure 5D and G). However, if both the first and last four nucleotides were mutated simultaneously (B10 mutant), the specificity at both junctions was drastically impacted, more so than B3 mutation alone (Figure 5D and G). Mutations to the inverted repeats (B4 and B7 mutants) did not have a significant effect, and if they were mutated together (B9 mutant), efficiency was somewhat reduced but specificity remained largely intact excluding a critical role for any potential DNA cruciform structure in directing integration (Figure 5D and G). In the case of a mid-repeat mutation (B5 mutant), integration at the repeat-spacer junction was inefficient but remained highly specific to the correct sites of integration. However, a more 3′ mid-repeat mutation (B6 mutant), which appeared to have integration efficiency similar to WT levels, severely disrupted the specificity of integration (Figure 5D and G). This demonstrated that integration products that appear to be the expected length on the denaturing polyacrylamide gel could actually be off-target integration products. To confirm this, we changed the length of the minimal CRISPR DNA substrate so that off-target products would be a length that was distinct from the leader-repeat and repeat-spacer products (Supplementary Figure S10). We noticed that the sequence substituted in the B6 mutant (5′-ACCCCTC-3′) had partial sequence overlap with a sequence (5′-CCCCT-3′) found in the leader immediately upstream (−1 to −5) of the first repeat that we considered might be responsible for the altered integration specificity. However, this possibility was ruled out when the same dramatic loss of spacer integration fidelity with a B6 mutant was observed by replacement with a different sequence with no similarity to sequences within the leader (B6b; 5′-GTTTTCT-3′) (Supplementary Figure S10). It is clear that this adenine-rich section of the internal CRISPR repeat (5′-CAAAAGA-3′ at position 16–22 of the repeat) is contributing significantly to proper spacer integration.
The finding that the vast majority of integration events on pCR7 were at a distance of 30 bases (Figure 2B) from one another suggested that the size of the repeat is a consequence of the proper spacing of the important sequence elements specifying integration sites. To test this, three nucleotides were either inserted or deleted from the center of the repeat in a region that can withstand base substitutions (B5 mutant). Expanding the spacing between sequence elements within the repeat through base insertion (Ins mutant) caused an apparent reduction in efficiency with a clear disruption in integration specificity at both borders of the repeat (Figure 5E and H). Similarly, reducing the spacing between sequence elements within the repeat by base deletion (Del mutant) did not dramatically reduce overall efficiency of integration but did significantly impair the specificity of integration at both borders of the repeat (Figure 5E and H). The drastic effects of these distance-altering mutations on integration fidelity suggest that proper spacing of the important repeat elements is crucial for accurate spacer integration.
Taken together, the results of the block mutations and insertions and deletions (Figure 5D, E, G, H) revealed that sequences immediately adjacent to the leader-repeat junction (5′-CCCT|GTTA-3′ at positions −4 through +4) as well as in an adenine-rich internal-repeat region (5′-CAAAAGA-3′ at positions 16–22 of the repeat) are particularly critical elements for accurate spacer integration at the LR and RS borders of the repeat. Paired integration sites on opposite strands of the CRISPR DNA, at a set distance of 30 bp, relies heavily on these two motifs. These results are in agreement with the size and sequence preferences of the non-CRISPR plasmid integration sites (Figures 2 and 3).
DISCUSSION
The ability to accurately integrate new spacer DNA sequences into a CRISPR repeat in a site-specific manner is critical for generating functional CRISPR RNAs that endow prokaryotic organisms with heritable immunity against viruses and other potentially detrimental mobile genetic elements. Our work provides the first in vitro characterization of spacer integration for the hyperthermophilic archaeon P. furiosus, and importantly sheds light on the mechanism by which Cas1 and Cas2 are directed to integrate spacers into CRISPR repeats. Our results are consistent with a model (Figure 6) in which P. furiosus Cas1 and Cas2 are guided to perform properly spaced two-site integration at the leader-proximal repeat by certain sequences, most notably motifs at the leader-repeat junction and central region of the repeat. Proper ∼30 bp spacing of integration sites (which spans three helical turns of double-stranded DNA) is likely maintained by the architecture of the presumed P. furiosus Cas1–Cas2 integrase complex, while the fidelity of integration is governed by sequence preferences and ultimately, by the overall structural properties of the repeat. Furthermore, integration at both junctions of the CRISPR repeat relies on the proper spacing of sequences throughout the DNA, allowing the two-site, full spacer integration to occur without a strict order, potentially even simultaneously.
Figure 6.
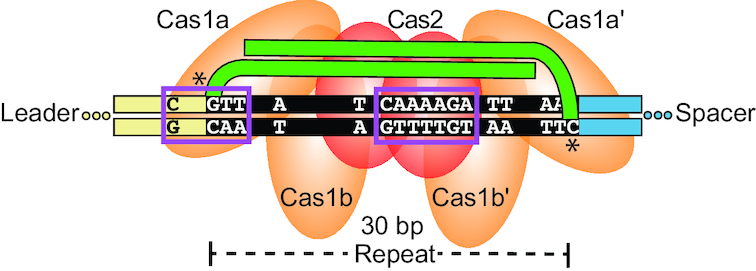
P. furiosus spacer integration model. Cas1 (orange) and Cas2 (red) have an affinity for several elements within the repeat (black) and leader (tan) in order to guide accurate spacer (green) integration. The architecture of the protein-spacer-CRISPR ternary complex positions the two-step transesterification attacks 30 bp apart at the preferred G and C bases of the repeat borders. Boxed in purple are the most essential motifs including the sequence spanning the leader-repeat junction and the internal adenine-rich region of nucleotides 16–22 (Block 6). Other nucleotides highlighted throughout the entire repeat collectively contribute to the additive specificity.
Our study yielded valuable insight into CRISPR elements governing spacer integration by analyzing effects on spacer integration efficiency and specificity for a wide array of leader and repeat mutations (Figure 5) as well as through careful inspection of off-target DNA integration sites (Figure 2). By sequencing integration products to confirm spacer positioning, we were able to more fully interpret the effects of DNA mutations. Some products that were the expected size actually represented off-target integration sites (Blocks 2, 3 and 6 of Figure 5), and some mutations that caused reduced efficiency still maintained a preference for LR and RS sites (G1A-C30T, Blocks 1, 5, and 9 of Figure 5). Thus, analyzing in vitro integration products through gel electrophoresis to address efficiency, combined with sequencing to address specificity, provided a more comprehensive approach to studying integration than gel analysis alone. In addition, given the rich information gleaned from inspection of off-target DNA integration sites, we conclude that off-target events can provide insight into key, context-independent elements that are recognized by proteins during adaptation. Although off-target integration poses potential deleterious effects in vivo (6), evidence suggests that non-canonical spacer integration might serve in the spontaneous generation of CRISPR loci (51). The specific DNA sequences we identified from these approaches (Figures 2 and 5; represented in our model, Figure 6), suggest that points distributed throughout the repeat collectively contribute to accurate spacer integration. Block DNA mutations (Figure 5) determined that some of these elements hold a greater importance than others, including two essential motifs at the leader-repeat junction and central region of the repeat, which are discussed in greater detail below.
Leader-repeat motif
Based on our findings (Figures 2 and 5), the motif at the leader-repeat junction is defined by GTT at positions 1–3 of the repeat and a C at the −3 position of the leader (Figure 6). The preference for guanine at sites of integration has been previously observed for Cas1 and Cas2 in I-A (36,50), I-B (46), I-E (34,50), II-A (6,40) and V-C systems (52), further implicating G1 as a key feature for directing integration to the leader-repeat junction. We predict that Cas1 directly interacts with this motif to guide integration at G1, which agrees with X-ray crystallographic evidence revealing base-specific DNA contacts of Cas1 at positions T1 and T2 of the repeat and −1 through −4 of the leader in a type II-A system (40). The importance of the C at position −3 is further supported by its conservation across all group 1 leader sequences of the Pyrococcales, with the exception of CRISPR locus 8 of P. furiosus, which has low activity compared to the other active loci (23,53). Similarly, a WebLogo of integration sites for the archaeal type I-A system of S. solfataricus revealed a preference for a C at the −2 position (36). Our identification of an important leader-repeat motif for the P.furiosus CRISPR–Cas system is in agreement with several studies that report the sequence spanning the LR junction to be important for spacer integration examined in vivo (7,37,41,42,46,54) and in vitro (32,36,40). This important motif is unique to the first repeat, and it may play a role in making the leader-proximal repeat the preferred repeat in the CRISPR array in vivo.
Adenine-rich central motif
The centrally located repeat motif identified in this study is defined as an adenine-rich sequence located at positions 16–22 of the repeat (Figure 6). The adenine bases at positions 17–18 likely contribute significantly to this motif, as they were particularly conserved in the non-CRISPR sites of integration (Figure 2). We predict this motif plays an upstream binding role to orient Cas1 and Cas2 in the correct position for integration at the two borders of the repeat, since we found that mutations to this motif (Block 6) significantly disrupted both LR and RS integration sites, as opposed to just one or the other (Figure 5 and Supplementary Figure S10). Our model is supported by other in vitro studies, which demonstrated motifs in the mid-repeat to be important for Cas1 and Cas2 binding to the repeat (33) and completion from half-site to full-site integration (35). In vivo studies have also implicated internal motifs as Cas1 and Cas2 docking sites that coordinate the proper spacing of the paired integration sites (45,46). While the internal motifs identified across different systems vary in location and attributes, they could all be playing a common role to recruit and properly orient Cas1 and Cas2 for accurately spaced integration.
Unique properties of P. furiosus Cas1 and Cas2
Prior work characterizing the integration properties of Cas1 and Cas2 from different organisms and distinct CRISPR types, have revealed variability in mechanisms governing: (i) the order of spacer integration at LR and RS sites and (ii) how integration is directed to the leader-proximal repeat vs. downstream repeats. There is in vitro evidence suggesting that Cas1 and Cas2 from type I-A (36,50), type I-E (32) and type II-A (6,40) systems attack at the leader-repeat junction first followed by integration at the repeat-spacer junction. The affinity of Cas1 and Cas2 for the sequence spanning the leader-repeat junction reported in vitro (32,36,40,50) is presumably what causes this to be the first integration site. In contrast, our results with P. furiosus Cas1 and Cas2 show that full-site integration occurs by an unordered or simultaneous attack at both borders of the repeat (Figures 3 and 4). This difference could be explained by the relative importance of different motifs in the repeat: in systems where Cas1 and Cas2 primarily recognize the leader-repeat sequence, there is sequential integration. In P. furiosus, Cas1 and Cas2 have a much wider recognition zone, spanning sequence elements spread throughout the entire repeat (Figure 2), leading to simultaneous or unordered integration events. Additionally, some systems that do demonstrate sequential integration also report a stable Cas1–Cas2 complex (6,32). While our work shows a clear requirement for both Cas1 and Cas2 for efficient, full-site integration (Figures 3, 4, Supplementary Figure S7), P. furiosus Cas1 and Cas2 do not form a stable complex in vitro (our unpublished results) and instead could be brought together by interacting with the spacer and/or CRISPR repeat DNA elements.
In vivo, new spacers are almost exclusively integrated at the leader-proximal (first) repeat as opposed to downstream repeats of a CRISPR array (1,41,42). Two distinct mechanisms (factor-dependent and factor-independent) have been proposed to explain how various Cas1 and Cas2 proteins are normally guided to integrate selectively at the leader-proximal (first) repeat (6,32,35,36,40,43). For type I-E and I-F systems, this is only achieved in the presence of Integration Host Factor (IHF), which binds to specific elements in the leader and interacts with Cas1 and Cas2 to recruit the proteins to the first repeat (32,35,43). Similarly, the I-A system in S. sulfataricus required the presence of an unidentified host factor from the cell lysate to achieve specific integration at the first repeat (36). In contrast, Cas1 and Cas2 of II-A systems display an intrinsic preference for the first repeat (without additional factors) due to strong affinity for, and reliance on, the sequence surrounding the LR junction (6,40). We found that P. furiosus Cas1 and Cas2 integrate at each repeat of a CRISPR array in vitro (Figure 1), similar to what is observed for I-E and I-F systems, which integrate at every repeat in the absence of IHF (32,34,43). This is an indication that a yet unidentified factor in P. furiosus cells (there are no known close IHF homologs) may be responsible for directing the proteins to the leader-proximal repeat.
Functional integration
In vivo, P. furiosus integrates new spacers at the leader-proximal repeat with an orientation that results in crRNA that is complementary to the PAM-containing strand of the invading DNA and is functional for immunity (12). Since we did not observe orientation biases in our in vitro results, we predict that additional factors beyond Cas1 and Cas2 are involved in regulating a strict order to integration and in recognizing the leader to direct spacer integration to the first repeat. It is likely that a larger portion of the leader is important for maintaining specific integration, as was the case for the large leader of the archaeal thermophile, S. sulfataricus in vitro (36). Given that Cas4–1 and Cas4–2 are required for proper spacer orientation in vivo (12), it is possible that these two proteins play a role in directing the order of Cas1-directed transesterifications, perhaps via interactions between Cas4 proteins and Cas1 and/ or the leader-repeat motif. Considerable effort was directed at testing these hypotheses in vitro, but we were unable to identify the proper conditions for Cas4–1 and Cas4–2 activity (our unpublished data). Further genetic and biochemical analysis will be required to elucidate the factor(s) involved in targeting integration to the first repeat and to determine if the order of integration sites is regulated.
We have established the importance of distinct elements within the CRISPR DNA array for spacer integration, but how do these elements give rise to function? We suggest in our model that the properties of the sequence elements and their relative spacing collectively influence direct interaction with the Cas1–Cas2 proteins, as well as enable the flexibility of the repeat. Together these factors complement the architecture of the bound Cas1–Cas2 proteins to achieve properly spaced integration sites. This model is supported by the apparently functionally important bending of the repeat observed in the structures of Cas1–Cas2 in half-site and full-site integration conformations for I-E (35) and II-A systems (40). Adenine-tracts are known to confer intrinsic DNA bending properties (55,56). The adenine-richness of the identified key functional elements within the P. furiosus repeat (summarized in Figure 6), may provide the needed repeat DNA malleability for serving as target DNA for spacer integration. In conclusion, we demonstrated that P. furiosus Cas1 and Cas2 have an intrinsic specificity for the sequences and relative spacing of the CRISPR repeats in order to facilitate accurate and full-site integration. Future studies will address how these intrinsic activities of Cas1 and Cas2 are refined to ensure that integration is correctly oriented and localized to the leader-proximal repeat, as is observed in vivo.
DATA AVAILABILITY
Sequence data were deposited in the NCBI Sequence Read Archive under the BioProject ID PRJNA540982.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Terns and Graveley laboratories for helpful discussions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R35GM118160 to M.P.T., R35GM118140 to B.R.G.]. Funding for open access charge: National Institutes of Health [R35GM118160 to M.P.T., R35GM118140 to B.R.G.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D.A., Horvath P.. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007; 315:1709–1712. [DOI] [PubMed] [Google Scholar]
- 2. Mojica F.J., Diez-Villasenor C., Garcia-Martinez J., Soria E.. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005; 60:174–182. [DOI] [PubMed] [Google Scholar]
- 3. Brouns S.J., Jore M.M., Lundgren M., Westra E.R., Slijkhuis R.J., Snijders A.P., Dickman M.J., Makarova K.S., Koonin E.V., van der Oost J.. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008; 321:960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hale C.R., Zhao P., Olson S., Duff M.O., Graveley B.R., Wells L., Terns R.M., Terns M.P.. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009; 139:945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sternberg S.H., Richter H., Charpentier E., Qimron U.. Adaptation in CRISPR–Cas Systems. Mol. Cell. 2016; 61:797–808. [DOI] [PubMed] [Google Scholar]
- 6. Wright A.V., Doudna J.A.. Protecting genome integrity during CRISPR immune adaptation. Nat. Struct. Mol. Biol. 2016; 23:876–883. [DOI] [PubMed] [Google Scholar]
- 7. Yosef I., Goren M.G., Qimron U.. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012; 40:5569–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McGinn J., Marraffini L.A.. Molecular mechanisms of CRISPR–Cas spacer acquisition. Nat. Rev. Microbiol. 2019; 17:7–12. [DOI] [PubMed] [Google Scholar]
- 9. Deveau H., Barrangou R., Garneau J.E., Labonte J., Fremaux C., Boyaval P., Romero D.A., Horvath P., Moineau S.. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol. 2008; 190:1390–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shah S.A., Erdmann S., Mojica F.J., Garrett R.A.. Protospacer recognition motifs: mixed identities and functional diversity. RNA Biol. 2013; 10:891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang J., Li J., Zhao H., Sheng G., Wang M., Yin M., Wang Y.. Structural and mechanistic basis of PAM-dependent spacer acquisition in CRISPR–Cas systems. Cell. 2015; 163:840–853. [DOI] [PubMed] [Google Scholar]
- 12. Shiimori M., Garrett S.C., Graveley B.R., Terns M.P.. Cas4 nucleases define the PAM, length, and orientation of DNA fragments integrated at CRISPR loci. Mol. Cell. 2018; 70:814–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grissa I., Vergnaud G., Pourcel C.. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics. 2007; 8:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carte J., Wang R., Li H., Terns R.M., Terns M.P.. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 2008; 22:3489–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Charpentier E., Richter H., van der Oost J., White M.F.. Biogenesis pathways of RNA guides in archaeal and bacterial CRISPR–Cas adaptive immunity. FEMS Microbiol. Rev. 2015; 39:428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jackson R.N., van Erp P.B., Sternberg S.H., Wiedenheft B.. Conformational regulation of CRISPR-associated nucleases. Curr. Opin. Microbiol. 2017; 37:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marraffini L.A. CRISPR–Cas immunity in prokaryotes. Nature. 2015; 526:55–61. [DOI] [PubMed] [Google Scholar]
- 18. van der Oost J., Westra E.R., Jackson R.N., Wiedenheft B.. Unravelling the structural and mechanistic basis of CRISPR–Cas systems. Nat. Rev. Microbiol. 2014; 12:479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hille F., Richter H., Wong S.P., Bratovic M., Ressel S., Charpentier E.. The Biology of CRISPR–Cas: Backward and Forward. Cell. 2018; 172:1239–1259. [DOI] [PubMed] [Google Scholar]
- 20. Makarova K.S., Wolf Y.I., Alkhnbashi O.S., Costa F., Shah S.A., Saunders S.J., Barrangou R., Brouns S.J., Charpentier E., Haft D.H. et al.. An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Microbiol. 2015; 13:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koonin E.V., Makarova K.S., Zhang F.. Diversity, classification and evolution of CRISPR–Cas systems. Curr. Opin. Microbiol. 2017; 37:67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alkhnbashi O.S., Shah S.A., Garrett R.A., Saunders S.J., Costa F., Backofen R.. Characterizing leader sequences of CRISPR loci. Bioinformatics. 2016; 32:i576–i585. [DOI] [PubMed] [Google Scholar]
- 23. Shiimori M., Garrett S.C., Chambers D.P., Glover C.V.C. 3rd, Graveley B.R., Terns M.P.. Role of free DNA ends and protospacer adjacent motifs for CRISPR DNA uptake in Pyrococcus furiosus. Nucleic Acids Res. 2017; 45:11281–11294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Majumdar S., Ligon M., Skinner W.C., Terns R.M., Terns M.P.. Target DNA recognition and cleavage by a reconstituted Type I-G CRISPR–Cas immune effector complex. Extremophiles. 2017; 21:95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Elmore J.R., Sheppard N.F., Ramia N., Deighan T., Li H., Terns R.M., Terns M.P.. Bipartite recognition of target RNAs activates DNA cleavage by the Type III-B CRISPR–Cas system. Genes Dev. 2016; 30:447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hale C.R., Cocozaki A., Li H., Terns R.M., Terns M.P.. Target RNA capture and cleavage by the Cmr type III-B CRISPR–Cas effector complex. Genes Dev. 2014; 28:2432–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Elmore J., Deighan T., Westpheling J., Terns R.M., Terns M.P.. DNA targeting by the type I-G and type I-A CRISPR–Cas systems of Pyrococcus furiosus. Nucleic. Acids. Res. 2015; 43:10353–10363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Majumdar S., Terns M.P.. CRISPR RNA-guided DNA cleavage by reconstituted Type I-A immune effector complexes. Extremophiles. 2019; 23:19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Plagens A., Tjaden B., Hagemann A., Randau L., Hensel R.. Characterization of the CRISPR/Cas subtype I-A system of the hyperthermophilic crenarchaeon Thermoproteus tenax. J. Bacteriol. 2012; 194:2491–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee H., Zhou Y., Taylor D.W., Sashital D.G.. Cas4-dependent prespacer processing ensures high-fidelity programming of CRISPR arrays. Mol. Cell. 2018; 70:48–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee H., Dhingra Y., Sashital D.G.. The Cas4-Cas1–Cas2 complex mediates precise prespacer processing during CRISPR adaptation. eLife. 2019; 8:e44248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nunez J.K., Bai L., Harrington L.B., Hinder T.L., Doudna J.A.. CRISPR immunological memory requires a host factor for specificity. Mol. Cell. 2016; 62:824–833. [DOI] [PubMed] [Google Scholar]
- 33. Moch C., Fromant M., Blanquet S., Plateau P.. DNA binding specificities of Escherichia coli Cas1–Cas2 integrase drive its recruitment at the CRISPR locus. Nucleic Acids Res. 2017; 45:2714–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nunez J.K., Lee A.S., Engelman A., Doudna J.A.. Integrase-mediated spacer acquisition during CRISPR–Cas adaptive immunity. Nature. 2015; 519:193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wright A.V., Liu J.J., Knott G.J., Doxzen K.W., Nogales E., Doudna J.A.. Structures of the CRISPR genome integration complex. Science. 2017; 357:1113–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rollie C., Graham S., Rouillon C., White M.F.. Prespacer processing and specific integration in a Type I-A CRISPR system. Nucleic Acids Res. 2018; 46:1007–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arslan Z., Hermanns V., Wurm R., Wagner R., Pul U.. Detection and characterization of spacer integration intermediates in type I-E CRISPR–Cas system. Nucleic Acids Res. 2014; 42:7884–7893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jackson S.A., McKenzie R.E., Fagerlund R.D., Kieper S.N., Fineran P.C., Brouns S.J.. CRISPR–Cas: adapting to change. Science. 2017; 356:eaal5056. [DOI] [PubMed] [Google Scholar]
- 39. Nunez J.K., Kranzusch P.J., Noeske J., Wright A.V., Davies C.W., Doudna J.A.. Cas1–Cas2 complex formation mediates spacer acquisition during CRISPR–Cas adaptive immunity. Nat. Struct. Mol. Biol. 2014; 21:528–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xiao Y., Ng S., Nam K.H., Ke A.. How type II CRISPR–Cas establish immunity through Cas1–Cas2-mediated spacer integration. Nature. 2017; 550:137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wei Y., Chesne M.T., Terns R.M., Terns M.P.. Sequences spanning the leader-repeat junction mediate CRISPR adaptation to phage in Streptococcus thermophilus. Nucleic Acids Res. 2015; 43:1749–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McGinn J., Marraffini L.A.. CRISPR–Cas systems optimize their immune response by specifying the site of spacer integration. Mol. Cell. 2016; 64:616–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fagerlund R.D., Wilkinson M.E., Klykov O., Barendregt A., Pearce F.G., Kieper S.N., Maxwell H.W.R., Capolupo A., Heck A.J.R., Krause K.L. et al.. Spacer capture and integration by a type I-F Cas1–Cas2-3 CRISPR adaptation complex. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E5122–E5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yoganand K.N., Sivathanu R., Nimkar S., Anand B.. Asymmetric positioning of Cas1-2 complex and Integration Host Factor induced DNA bending guide the unidirectional homing of protospacer in CRISPR–Cas type I-E system. Nucleic Acids Res. 2017; 45:367–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Goren M.G., Doron S., Globus R., Amitai G., Sorek R., Qimron U.. Repeat size determination by two molecular rulers in the type I-E CRISPR array. Cell Rep. 2016; 16:2811–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang R., Li M., Gong L., Hu S., Xiang H.. DNA motifs determining the accuracy of repeat duplication during CRISPR adaptation in Haloarcula hispanica. Nucleic Acids Res. 2016; 44:4266–4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Langmead B., Trapnell C., Pop M., Salzberg S.L.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Crooks G.E., Hon G., Chandonia J.M., Brenner S.E.. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rollie C., Schneider S., Brinkmann A.S., Bolt E.L., White M.F.. Intrinsic sequence specificity of the Cas1 integrase directs new spacer acquisition. eLife. 2015; 4:e08716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nivala J., Shipman S.L., Church G.M.. Spontaneous CRISPR loci generation in vivo by non-canonical spacer integration. Nat. Microbiol. 2018; 3:310–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wright A.V., Wang J.Y., Burstein D., Harrington L.B., Paez-Espino D., Kyrpides N.C., Iavarone A.T., Banfield J.F., Doudna J.A.. A functional mini-integrase in a two-protein-type V-C CRISPR system. Mol. Cell. 2019; 73:727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Norais C., Moisan A., Gaspin C., Clouet-d’Orval B.. Diversity of CRISPR systems in the euryarchaeal Pyrococcales. RNA Biol. 2013; 10:659–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Diez-Villasenor C., Guzman N.M., Almendros C., Garcia-Martinez J., Mojica F.J.M.. CRISPR-spacer integration reporter plasmids reveal distinct genuine acquisition specificities among CRISPR–Cas I-E variants of Escherichia coli. RNA Biol. 2013; 10:792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Haran T.E., Mohanty U.. The unique structure of A-tracts and intrinsic DNA bending. Q. Rev. Biophys. 2009; 42:41–81. [DOI] [PubMed] [Google Scholar]
- 56. Rettig M., Germann M.W., Wang S., Wilson W.D.. Molecular basis for sequence-dependent induced DNA bending. ChemBioChem. 2013; 14:323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data were deposited in the NCBI Sequence Read Archive under the BioProject ID PRJNA540982.