

Abstract
Reactive oxygen species (ROS) contribute to sensitization of pain pathways during neuropathic pain, but little is known about the primary sources of ROS production and how ROS mediate pain sensitization. Here, we show that the NADPH oxidase isoform Nox4, a major ROS source in somatic cells, is expressed in a subset of nonpeptidergic nociceptors and myelinated dorsal root ganglia neurons. Mice lacking Nox4 demonstrated a substantially reduced late-phase neuropathic pain behavior after peripheral nerve injury. The loss of Nox4 markedly attenuated injury-induced ROS production and dysmyelination processes of peripheral nerves. Moreover, persisting neuropathic pain behavior was inhibited after tamoxifen-induced deletion of Nox4 in adult transgenic mice. Our results suggest that Nox4 essentially contributes to nociceptive processing in neuropathic pain states. Accordingly, inhibition of Nox4 may provide a novel therapeutic modality for the treatment of neuropathic pain.
Introduction
Pain in response to tissue damage (inflammatory pain) or to lesions to the nervous system (neuropathic pain) is characterized by a sensitization of the nociceptive system. This sensitization clinically manifests as pain in response to normally innocuous stimuli (allodynia), increased response to noxious stimuli (hyperalgesia), or spontaneous pain in the absence of any stimulus. Because pain can persist long after the initial injury has been resolved, it causes major health problems (Basbaum et al., 2009; Schmidtko et al., 2010). Accumulating evidence suggests that reactive oxygen species (ROS), such as hydrogen peroxide (H2O2) and superoxide (O2−), and the active superoxide byproduct peroxynitrite essentially contribute to the sensitization during persistent pain (Salvemini et al., 2011). Accordingly, the nociceptive behavior was inhibited in animal models of pain after systemic or intrathecal administration of antioxidants, free radical scavengers, and peroxynitrite-decomposition catalysts (Kim et al., 2004; Wang et al., 2004; Lu et al., 2011; Yowtak et al., 2011; Janes et al., 2012). However, the sources of ROS production and how ROS contribute to pain sensitization remain poorly understood.
NADPH oxidases of the Nox family are a group of enzymes whose sole known function is the production of ROS by catalyzing electron transfer from NADPH to molecular O2. Four rodent genes of the catalytic subunit Nox (Nox1–Nox4) have been identified, each with tissue-specific expression and different functions in intracellular signaling (Sorce and Krause, 2009). Recent studies suggest that Nox enzymes are important ROS sources during pain sensitization. For example, administration of Nox inhibitors blocked the development of morphine-induced hyperalgesia and antinociceptive tolerance (Doyle et al., 2010). Nox1 mRNA was detected at high levels in the mouse paw, and mice lacking Nox1 demonstrated a reduced nociceptive behavior in inflammatory pain models (Ibi et al., 2008). Nox2 is induced in spinal cord microglia cells after peripheral nerve injury and contributes to neuropathic pain hypersensitivity (Kim et al., 2010).
Nox4 differs from other Nox enzymes in that it generates predominantly H2O2 rather than O2− (Takac et al., 2011). Nox4 mRNA has been detected in dorsal root ganglia (Ibi et al., 2008), but its cellular distribution and its function in the peripheral nervous system remain elusive. Moreover, the mechanisms by which Nox4 mediates pain sensitization have not been clarified yet. In the present study, we therefore investigated the distribution of Nox4 in the peripheral nervous system, and we analyzed the nociceptive behavior of Nox4-deficient mice in models of acute, inflammatory, and neuropathic pain. We demonstrate for the first time that Nox4-derived ROS in peripheral nerves essentially contribute to the processing of nerve dysmyelination and neuropathic pain hypersensitivity after nerve injury.
Materials and Methods
Animals
The generation of mice lacking Nox4 (Nox4−/− mice) has been described previously (Zhang et al., 2010). Experiments were performed in 6- to 12-week-old mice of either sex backcrossed onto C57BL/6 background. Tamoxifen-inducible Nox4 knock-out mice (Nox4–CreERT2 mice) were produced by crossing homozygous Nox4-floxed mice and heterozygous tamoxifen-inducible CreERT2 mice in which the Cre recombinase, fused to a mutated estrogen ligand-binding domain (ERT2) that requires the presence of tamoxifen for activity (Indra et al., 1999), is under the control of the ubiquitous ROSA26 promoter. C57BL/6N mice (Harlan) were additionally used for RT-PCR analyses. Animals were housed on a 12 h light/dark cycle with standard rodent chow and water available ad libitum. All experiments were performed in accordance with the International Association for the Study of Pain and were approved by the local Ethics Committee for Animal Research.
Behavioral testing
Littermate mice were used in all behavioral studies. All investigations were done by a blinded observer.
Rotarod test.
Motor coordination was assessed with a rotarod treadmill for mice (Ugo Basile) at a constant rotating speed of 32 rpm. All mice had five training sessions before the day of the experiment. The fall-off latency was averaged from five tests, and the cutoff time was 120 s.
Cold-plate and hot-plate test.
Mice were placed on a cooled or heated metal plate surrounded by a Plexiglas cylinder (Hot/Cold Plate; Ugo Basile). The time between placement and shaking or licking of a hindpaw was recorded. Temperatures of 5, 48, 50, and 52°C were tested with cutoff times of 90, 80, 60, and 40 s, respectively, to prevent tissue damage. Only one test per animal was performed because repeated measures might cause profound latency changes (Mogil et al., 1999).
Formalin test.
A 5% formaldehyde solution (15 μl; Formalin) was injected subcutaneously into the dorsal site of one hindpaw (Hunskaar et al., 1985). The time spent licking the Formalin-injected paw was recorded in 5 min intervals up to 50 min after Formalin injection.
Mechanical paw sensitivity.
Paw-withdrawal latency after mechanical stimulation was measured using a Dynamic Plantar Aesthesiometer (Ugo Basile). This device pushes a thin probe (0.5 mm diameter) with increasing force through a wire-grated floor against the plantar surface of the paw from beneath, and it automatically stops and records the latency time at which the animal withdraws the paw. The force increased from 0 to 5 g in 10 s (0.5 g/s ramp) and was then held at 5 g for an additional 10 s (Schmidtko et al., 2008b; Heine et al., 2011). The latency time was calculated as the average of four to six consecutive exposures with at least 20 s in between.
Zymosan- or complete Freund's adjuvant-induced hyperalgesia.
Fifteen microliters of a zymosan A suspension (5 mg/ml in 0.1 m PBS, pH 7.4; Sigma-Aldrich) or 20 μl of complete Freund's adjuvant (CFA; containing 1 mg/ml heat-killed Mycobacterium tuberculosis in 85% paraffin oil and 15% mannide monooleate; Sigma-Aldrich) was injected into the plantar subcutaneous space of a hindpaw (Meller and Gebhart, 1997).
Neuropathic pain.
The “spared nerve injury” (SNI) model (Decosterd and Woolf, 2000) and a modified version of the “chronic constriction injury” (CCI) model (Bennett and Xie, 1988) were used to investigate neuropathic pain behavior. Surgery was performed under isoflurane anesthesia. For SNI, two branches of the sciatic nerve were ligated and cut distally, leaving the sural nerve intact (Decosterd and Woolf, 2000). SNI-induced hypersensitivity was assessed by stimulation of the lateral surface of the hindpaw (sural nerve skin area). For CCI, the common sciatic nerve was exposed unilaterally at the mid-thigh level by blunt dissection. Proximal to the trifurcation of the sciatic nerve, ∼5 mm of nerve was freed of adhering tissue, and three silk ligatures (6-0) that constricted the nerve by ∼30–50% were tied around it with ∼1 mm spacing (Tegeder et al., 2006; Schmidtko et al., 2008a).
Tamoxifen induction.
Tamoxifen (Sigma-Aldrich) was prepared by dissolving in ethanol (10 mg/100 μl) and mixing this solution with 900 μl of corn oil for a final concentration of 10 mg/ml. Eight- to 10-week-old Nox4–CreERT2 mice were injected intraperitoneally with 100 μl of tamoxifen solution (1 mg, corresponding to ∼40 mg/kg tamoxifen) once a day at 10, 11, and 12 d after SNI.
Immunostaining
Mice were intracardially perfused with 0.9% saline, followed by 4% paraformaldehyde in PBS, pH 7.4, under deep isoflurane anesthesia. The lumbar spinal cord (L4–L5), dorsal root ganglia (DRGs) (L4–L5), and the proximal sciatic nerve were dissected, postfixed for 10 min in the same fixative, and cryoprotected in 20% sucrose overnight. Tissues were frozen in tissue freezing medium (Leica) on dry ice, cryostat sectioned at a thickness of 14–16 μm, and stored at −80°C. For immunofluorescence, sections were permeabilized for 5 min in PBST (0.1% Triton X-100 in PBS), blocked for 1 h using 10% normal goat or donkey serum and 3% bovine serum albumin (BSA) in PBS, and incubated with primary antibodies diluted in 3% BSA in PBST overnight at 4°C or for 2 h at room temperature. The following antibodies were used: rabbit anti-Nox4 (1:800; Anilkumar et al., 2008), rat anti-substance P (1:200; BD Biosciences), mouse anti-NF200 (clone N52; 1:1000; Sigma-Aldrich), rabbit anti-Iba1 (1:500; Wako), and chicken anti-MPZ (1:500; Neuromics). Sections were then washed in PBS and stained with secondary antibodies conjugated with Alexa Fluor 488 (Invitrogen) or Cy3 (Sigma-Aldrich). For staining with Griffonia simplicifolia isolectin B4 (IB4), sections were incubated with Alexa Fluor 488-conjugated IB4 (10 μg/ml in PBS; Invitrogen) for 2 h at room temperature. After immunostaining, slides were immersed for 5 min in 0.06% Sudan black B (in 70% ethanol) to reduce lipofuscin-like autofluorescence (Schnell et al., 1999; Schmidtko et al., 2008c), washed in PBS, and coverslipped. In double-labeling experiments, primary antibodies were consecutively incubated. Images were taken using an Axio Observer.Z1 microscope (Carl Zeiss) equipped with a monochrome CCD camera (AxioCam Mrm; Carl Zeiss). Images were taken with different filters, pseudocolored, and superimposed using the Carl Zeiss AxioVision 4.7.2 software. Adjustment of brightness and contrast was done using Adobe Photoshop CS software (Adobe Systems). Controls were performed by omitting the first and/or the second primary antibodies and by incubating tissues of Nox4−/− mice.
Cell counting
Serial sections of entire L4–L5 DRGs from wild-type (WT) and Nox4−/− mice (three to four mice per genotype) were cut (16 μm), and three sections, at least 100 μm apart, per DRG per animal were counted. Only cells containing nuclei and showing staining clearly above background were included. Specificity of Nox4 immunoreactivity was confirmed by simultaneous staining of coembedded tissues of WT and Nox4−/− mice. The percentage of Nox4-positive cells expressing marker was calculated by dividing the number of Nox4-positive cells colocalized with marker by the total number of Nox4-positive cells. The percentage of marker-positive DRG neurons in WT and Nox4−/− mice is expressed as a proportion of marker-positive cells per total number of DRG neurons.
Microglia quantification
Microglia activation after SNI was investigated by an observer blinded to the animal genotype using Iba1 immunostaining on sections of 16 μm thickness as described above. To find the precise localization of the area affected by the SNI surgery, Alexa Fluor 488-conjugated IB4 was additionally incubated on the slides together with the Cy3-conjugated secondary antibody. In the area affected by SNI surgery, the IB4 staining pattern in lamina II fades (Casals-Díaz et al., 2009). Spinal cord sections at a distance of ∼300 μm (seven to nine sections per mouse, four mice per genotype) were stained, and images of the dorsal horn (laminae I–IV) were captured under a 10× objective. Only slides in which the IB4 staining was interrupted were analyzed. Using NIH ImageJ software equipped with the MacBiophotonics MBF plugin (McMaster Biophotonics Facility, McMaster University, Hamilton, Ontario, Canada), Iba1 image contrast was adjusted such that the background level just disappeared, and the same cutoff level was used for all images (command: image > adjust > color threshold). Images were then converted into 8-bit (command: image > type > 8-bit), the cell bodies were selected according to their size using nucleus counter plugin (command: plugins > particle analysis > nucleus counter; smallest particle size, 50; threshold method, otsu), and Iba1-positive cell bodies were counted. Similar results were obtained in control experiments using manual counting of Iba1-positive cell bodies.
Morphological analysis of sciatic nerves
Under deep isoflurane anesthesia, mice were perfused with 0.9% saline, followed by a solution containing 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 m cacodylate buffer, pH 7.4. Proximal sciatic nerves were dissected, postfixed in the same fixative overnight, and embedded in Epon. Control and SNI-operated nerves of Nox4−/− and WT mice were cross-sectioned at a thickness of 1 μm and stained with toluidine blue using standard procedures. The morphology of injured nerves was analyzed at a distance of 1 mm proximal to the lesion. The number of myelinated axons as well as the number of infoldings and outfoldings of the myelin sheath were counted, and the percentage of infoldings and outfoldings per total number of myelinated axons was calculated. An infolding and outfolding was defined as a fiber containing one or more myelin infolds (into the axon) or one containing one or more redundant loop(s) of myelin flanking a primary myelinated axon (Robinson et al., 2008). Axonal diameter was measured using NIH ImageJ software equipped with the MacBiophotonics MBF plugin. Images were converted into 8-bit (command: image > type > 8-bit), and the image threshold was adjusted so that the background level (nonmyelinated axons and other cells) disappeared. The diameter of the areas surrounded by the myelin sheath was measured (command: analyze > set measurements > feret's diameter; command: analyze > analyze particles; size, 50 to infinity; circularity, 0.0–1.0; Show, Overlay Masks; Settings, Display Results; Clear results, Summarize; Exclude on edges, In situ Show), and the mean value of the smallest (MiniFeret) and longest diameter (Feret) of the area surrounded by the myelin was calculated and taken as the axon diameter. Myelin thickness was measured using the line selection tool at three individual points of each axon, and the mean value was taken for additional analysis. Using a scale bar, measured values were converted into micrometers, and the g ratio was calculated. A total number of 857 myelinated axons from three mice per genotype were analyzed.
H2O2 detection
H2O2 was detected with Amplex Red (Invitrogen), which in the presence of horseradish peroxidase reacts stoichiometrically with H2O2 to generate the red fluorescent oxidation product resorufin (Rhee et al., 2010). Lumbar spinal cord (L4–L5), lumbar DRGs (L4–L5), and sciatic nerves (10-mm-long segments proximal to the lesion) were rapidly dissected and homogenized in ice-cold 500 mm phosphate buffer containing 10 mm sodium azide, incubated for 5 min at room temperature, and centrifuged at 10,000 × rpm for 10 min at 4°C. The supernatant was transferred into a 96-well plate (μCLEAR, BLACK; Greiner Bio One), and samples and H2O2 standards were incubated with Amplex Red (100 μm) and horseradish peroxidase (0.2 U/ml) for 30 min at room temperature. Fluorescence intensities were acquired using a plate reader at an excitation of 540 nm and emission of 595 nm. Experiments were performed in the presence and absence of catalase (1000 U/ml). Data are presented as the catalase-sensitive part of the Amplex Red oxidation (Schröder et al., 2009) and were normalized to H2O2 levels in the spinal cord of naive WT mice.
Western blot
Sciatic nerves (10-mm-long segments proximal to the lesion) were rapidly dissected, frozen in liquid nitrogen, and stored at −80°C until use. Samples were homogenized in Phosphosafe buffer (Novagen) combined with a protease inhibitor mixture (Complete Mini; Roche Diagnostics) and centrifuged at 14,000 × g for 1 h. Extracted proteins (20 μg/lane) were separated by SDS-PAGE and blotted onto a nitrocellulose membrane. Membranes were blocked in blocking buffer (diluted 1:1 in PBS; Odyssey blocking buffer; LI-COR Bioscience) for 1 h at room temperature and then incubated with chicken anti-MPZ (1:500; Neuromics), rabbit anti-PMP22 (1:500; Abcam), or mouse anti-GAPDH (1:2000; Ambion) dissolved in blocking buffer containing 0.1% Tween 20 overnight at 4°C. After incubation with secondary antibodies for 1 h at room temperature, proteins were detected using an Odyssey Infrared Imaging System (LI-COR Bioscience). Quantification of band densities was done using NIH ImageJ software.
Real-time RT-PCR
Mice were exsanguinated under deep isoflurane anesthesia, and tissues were dissected, snap frozen in liquid nitrogen, and stored at −80°C. Total RNA from lumbar DRGs (L4–L5), lumbar spinal cord (L4–L5), and brain was extracted under RNase-free conditions using a RNA isolation kit (for spinal cord and brain: RNeasy Lipid Tissue Mini kit, Qiagen; for DRGs: RNAqueous Micro kit, Ambion) according to the instructions of the manufacturer, DNase treated for 15 min to minimize genomic DNA contamination, and quantified with a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies). cDNA was synthesized using 200 ng of RNA, random hexamer primers, RT-Enhancer, and the Verso-enzyme of the Verso kit (Thermo Fisher Scientific). Real-time RT-PCR was performed with an ABI Prism 7500 Sequence Detection System (Applied Biosystems) using TaqMan Gene Expression Assays for murine Nox4 (Mm01317086_m1), Nox1 (Mm00549170_m1), Nox2 (Mm01287742_m1), Nox3 (Mm01339132_m1), GAPDH (Mm99999915_g1), and β-actin (Mm00607939_s1), purchased from Applied Biosystems. Reactions (total volume, 10 μl) were performed in duplicate or triplicate by incubating at 95°C for 10 min, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. Water controls were included to ensure specificity. Relative expression of target gene levels was determined using the 2−ΔΔCt method, with Ct indicating the cycle number at which the signal of the PCR product crosses an arbitrary threshold set within the exponential phase of the PCR. The amount of sample RNA was normalized to GAPDH. Control experiments revealed similar results if β-actin was used for normalization.
Statistics
Statistical analysis was performed with SPSS software using the Student's t test for paired comparisons or one-way ANOVA for multiple comparisons, followed by a Fisher's post hoc test. When mice were tested at different time points, we used a repeated-measures ANOVA, and differences between groups at each time point were analyzed with a Fisher's post hoc test. Rotarod fall-off latencies were analyzed with Mann–Whitney U test and are expressed as median and interquartile range. All other data are presented as the mean ± SEM. For all tests, a probability value p < 0.05 was considered as statistically significant.
Results
Nox4 is expressed in DRG neurons
We first examined the Nox4 distribution in DRGs by immunostaining (Fig. 1A) and found that 13% of sensory neurons expressed Nox4. Specificity of the Nox4 antibody (Anilkumar et al., 2008) was confirmed by the absence of immunoreactivity in DRGs of Nox4−/− mice (Fig. 1A). The detailed distribution of Nox4 in DRGs was investigated by double-labeling immunostaining experiments with established markers. Interestingly, 68% of Nox4-positive cells bound the lectin IB4, a marker of the nonpeptidergic population of unmyelinated nociceptors (Fig. 1B,E), whereas there was virtually no overlap of Nox4-positive cells with substance P, a marker of peptidergic unmyelinated nociceptors (Fig. 1C,E). In addition, 15% of Nox4-positive cells expressed NF200, a neurofilament marker of neurons with myelinated axons (Fig. 1D,E). Hence, our results indicate that Nox4 is predominately expressed in IB4-binding nociceptors. Because this subset of nonpeptidergic primary afferent neurons plays a particular role in the sensitization of pain pathways (Basbaum et al., 2009), we hypothesized that Nox4 might contribute to nociceptive processing.
Figure 1.
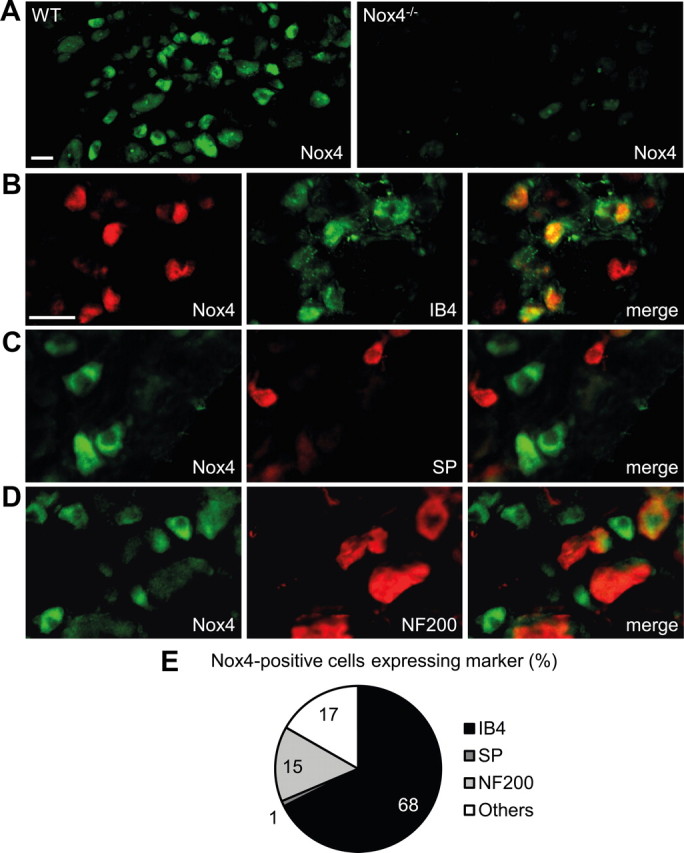
Expression of Nox4 in DRGs. A, Immunofluorescence of Nox4 in lumbar DRGs of WT mice and Nox4−/− mice reveals specific Nox4 expression in 12.8 ± 0.8% of WT DRG neurons (4456 cells counted, n = 4). B–D, Typical examples of Nox4 immunoreactivity in subpopulations of DRG neurons labeled using IB4 binding or using antibodies to substance P (SP) or NF200, respectively. E, Quantitative summary of DRG cell populations expressing Nox4 protein from experiments represented in B–D. Most Nox4-expressing DRG neurons bind IB4 (68%) and are therefore unmyelinated and nonpeptidergic, whereas Nox4 almost never colocalizes with the substance P-containing peptidergic subpopulation of unmyelinated neurons. A few Nox4-expressing cells coexpress NF200 (15%), indicating that they are myelinated DRG neurons. Scale bars, 25 μm.
Basal sensitivity and inflammatory pain behavior are normal in Nox4−/− mice
To assess the role of Nox4 in nociceptive processing in vivo, we compared the nociceptive behavior of Nox4−/− mice with that of littermate WT mice in various animal models of pain. We tested both male and female mice (Mogil and Chanda, 2005; Greenspan et al., 2007), but no significant main effects of sex were detected in any assay. Nox4−/− mice are viable and fertile, normal in size, and do not display any gross physical or behavioral abnormalities (Zhang et al., 2010). The overall frequencies of DRG neuron populations positive for substance P, IB4, or NF200 were similar between WT and Nox4−/− mice (Fig. 2A). The macroscopic morphology of DRGs and the spinal cord, and the distribution of terminals of nociceptive and thermoreceptive primary afferents in the superficial dorsal horn (assessed by lectin IB4 binding and substance P immunoreactivity) appeared normal in Nox4−/− mice (data not shown), suggesting that the lack of Nox4 did not affect the morphology or general structural properties of sensory neurons. Quantitative real-time RT-PCR analyses revealed that the expression of other Nox enzymes in DRGs, the spinal cord, and brain are not compensatory upregulated in Nox4−/− mice (Nox1 and Nox3 mRNA was not detected, whereas Nox2 mRNA levels were similar in both genotypes; Fig. 2B). Furthermore, the motor coordination and balance is not impaired in Nox4−/− mice, as analyzed in the rotarod test [median fall-off latencies: Nox4−/− mice, 103.4 s (interquartile range, 73.1–120.0 s); WT mice, 108.8 s (interquartile range, 78.2–120.0 s); p = 0.801; n = 11–12 per group].
Figure 2.

Basal characteristics, acute pain behavior, and inflammatory pain behavior are not impaired in Nox4−/− mice. A, Percentages of DRG neurons binding IB4 or immunoreactive for substance P (SP) or NF200 were similar in WT and Nox4−/− mice (3340 cells counted, n = 3–4 mice per genotype). B, Expression of Nox mRNA in DRGs, spinal cord and brain of WT and Nox4−/− mice assessed by quantitative real-time RT-PCR. Nox4 mRNA was only detected in tissues from WT mice, whereas Nox2 mRNA levels were similar in tissues from WT and Nox4−/− mice. Nox1 or Nox3 mRNA were not reliably detected (Ct values > 35) in both genotypes (n = 3–4). C, Cold-plate and hot-plate tests. The latency of Nox4−/− mice to exhibit nocifensive behaviors was similar to that of WT littermates at cold (5°C) and hot (48–52°C) temperatures (n = 12). D, Formalin test. Both genotypes showed a similar biphasic response to 5% Formalin injected into a hindpaw (n = 8). E, F, Paw-withdrawal latency times after mechanical stimulation after injection of zymosan (E) or CFA (F) into a hindpaw. Mechanical hypersensitivity did not differ between genotypes at all times tested (n = 7–9). Data are presented as mean ± SEM.
To determine acute nociception in Nox4−/− mice, we measured the latency times to acute thermal stimuli using the cold-plate (5°C) and hot-plate (48, 50, and 52°C) test. No significant differences in latency times were found between Nox4−/− and WT mice (Fig. 2C), indicating that the lack of Nox4 does not affect the immediate response to acute noxious thermal stimulation. To test whether Nox4 deficiency affects the rapid sensitization in pain pathways, we performed the 5% Formalin test. However, we found no significant differences in Formalin-induced pain sensitivity between Nox4−/− mice and their control littermates (Fig. 2D). We then tested the extent of mechanical hyperalgesia evoked by injection of zymosan or CFA into a hindpaw, two models of inflammatory pain. As shown in Figure 2, E and F, Nox4−/− mice developed mechanical hypersensitivity in the injected hindpaw that was indistinguishable from that of WT mice in both models. Together, these data suggest that Nox4, despite its distinctive localization in a subset of nociceptive primary afferent neurons, is not critically involved in the development of exaggerated pain sensitivity during inflammatory pain.
Neuropathic pain behavior is attenuated in Nox4−/− mice
We then examined the behavior of Nox4−/− mice and littermate WT mice after SNI, an animal model of persistent peripheral neuropathic pain. Mechanical hypersensitivity of the affected hindpaw developed similarly in Nox4−/− and WT mice during the first 7 d after nerve injury (Fig. 3A). Interestingly however, at later time points (i.e., between 10 d after nerve injury and the end of the 35 d observation period), the extent of mechanical hypersensitivity was significantly reduced in Nox4−/− mice compared with WT mice (Fig. 3A). Notably, equivalent results were observed in another model of peripheral nerve injury, the CCI model. During the first 7 d after CCI surgery, mechanical hypersensitivity was indistinguishable in both groups. However, at later stages, the hypersensitivity was considerably reduced in Nox4−/− mice compared with WT mice (Fig. 3B). Together, these data suggest that Nox4 essentially contributes to the maintenance of neuropathic pain after peripheral traumatic axonal injury, but it seems not to be critically involved in the induction of neuropathic pain during the first days after injury.
Figure 3.
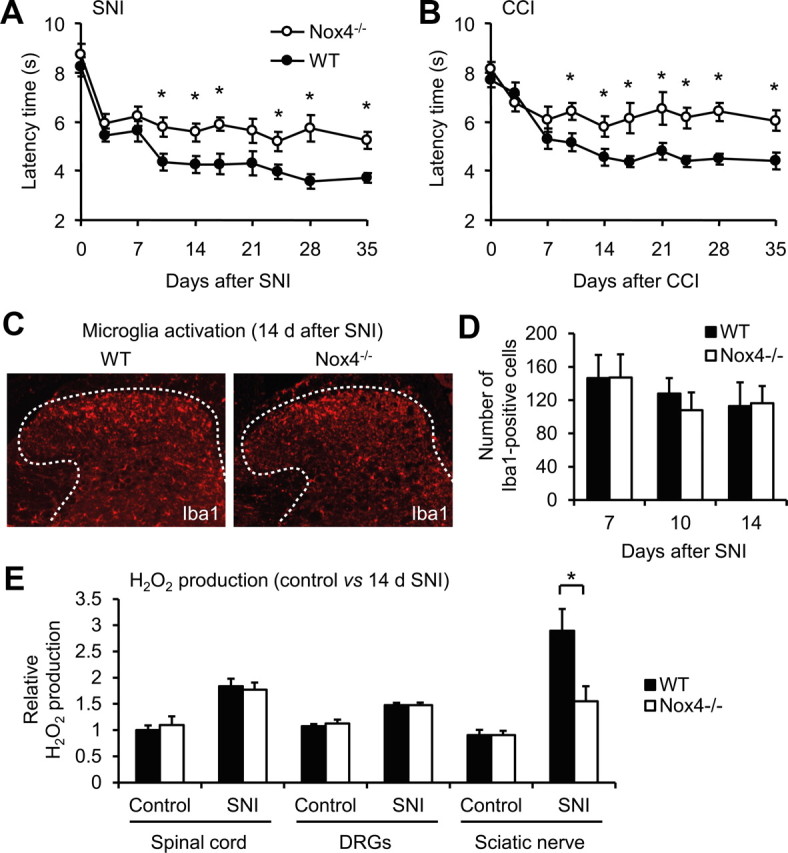
Reduced neuropathic pain behavior and injury-induced ROS production in Nox4−/− mice. A, B, Paw-withdrawal latency times of Nox4−/− and WT mice after mechanical stimulation in the SNI (A) and CCI (B) models of neuropathic pain. Nox4−/− mice demonstrated reduced mechanical hypersensitivity compared with WT littermates during 10–35 d after SNI or CCI (n = 12–14). C, D, Peripheral nerve injury induced comparable activation of microglia in the spinal cords of Nox4−/− and WT mice, as visualized using an antibody against Iba1. Representative examples of Iba1 immunofluorescence (red) in the spinal cord 14 d after SNI are presented in C; dotted lines delineate gray matter. Quantitative analysis of Iba1-positive cells in the dorsal horn 7–14 d after SNI are shown in D (n = 4 mice per group). E, H2O2 production in the spinal cord, DRGs, and the sciatic nerve of control animals and day 14 SNI animals. The SNI-induced H2O2 production was significantly lower in the sciatic nerve of Nox4−/− mice compared with WT mice. Data are presented as mean ± SEM. *p < 0.05, comparing Nox4−/− and WT mice.
Previous studies demonstrated that spinal cord microglia activation contributes to the pathological hypersensitivity after peripheral nerve injury (Costigan et al., 2009). Because this microglia reaction was markedly reduced in Nox2-deficient mice (Kim et al., 2010), we assessed the SNI-induced microglia reaction in Nox4−/− mice. However, we did not detect any differences in expression of the microglia marker Iba1 in the spinal cord at 7, 10, and 14 d after SNI (Fig. 3C,D). These data point to a limited role of Nox4, in contrast to Nox2, in peripheral nerve injury-induced microglia activation in the spinal cord. Furthermore, the data also indicate that Nox4 and Nox2 affect peripheral nerve injury-induced neuropathic pain signaling by different mechanisms.
SNI-induced ROS production in peripheral nerves is reduced Nox4−/− mice
To determine the sites of Nox4-induced ROS production after peripheral nerve injury, we measured H2O2 levels in the spinal cord, DRGs, and the sciatic nerve (proximal nerve stump) of WT and Nox4−/− mice using the Amplex Red assay. We compared H2O2 levels in naive control animals and in animals 14 d after SNI (i.e., at a time point of reduced hypersensitivity in Nox4−/− mice). In control animals, H2O2 was detected to a similar extent in WT and Nox4−/− mice, indicating a limited contribution of Nox4 to basal H2O2 production in the investigated tissues (Fig. 3E). After SNI, H2O2 levels were increased in all investigated tissues of both genotypes. Notably, in the sciatic nerve, the SNI-induced H2O2 production was significantly lower in Nox4−/− mice compared with WT mice, whereas in the spinal cord and in DRGs, it was comparable between genotypes (Fig. 3E). These data demonstrate that H2O2 production is considerably increased in peripheral nerves after injury and that injury-induced H2O2 production in peripheral nerves mostly depends on Nox4. Furthermore, the data point to a limited contribution of Nox4 to SNI-induced H2O2 production in DRGs and the spinal cord.
SNI-induced dysmyelination of peripheral nerves is reduced in Nox4−/− mice
The fact that Nox4 essentially contributes to injury-induced H2O2 production in peripheral nerves points to a peripheral mechanism underlying the attenuated neuropathic pain behavior in Nox4−/− mice. We speculated that a disruption of myelin sheath integrity of peripheral nerves might be involved, because previous work demonstrated that (1) the myelin sheath is highly susceptible to degeneration caused by H2O2 (Konat and Wiggins, 1985; Richter-Landsberg and Vollgraf, 1998; Laszkiewicz et al., 1999; Mronga et al., 2004; Perfeito et al., 2007), (2) the myelin sheath is disrupted and the proximal nerve stump seals off after peripheral nerve injury (Inoue et al., 2004; Devor, 2006a; Nagai et al., 2010), and (3) the dysmyelination of peripheral nerves induced by nerve injury or by delivery of bioactive lipids, such as lysophosphatidic acid, is accompanied by spontaneous action potentials in primary afferent nerves and sensitization of sensory processing, thereby contributing to neuropathic pain hypersensitivity (Devor et al., 1989; Wallace et al., 2003; Devor, 2006b; Ueda, 2008; Zhu et al., 2012). To monitor the time course of injury-induced dysmyelination of peripheral nerves in the SNI model, we first analyzed the protein levels of MPZ, the main peripheral myelin protein, in C57BL/6 mice during 21 d after SNI. As shown in Figure 4, SNI induced a significant decrease of MPZ expression in the proximal sciatic nerve stump during the entire observation period. These data indicate that a persistent dysmyelination occurs in the proximal sciatic nerve stump after SNI.
Figure 4.

MPZ expression in the sciatic nerve after SNI. Western blot analysis of the myelin-specific protein MPZ in the proximal sciatic nerve stump of C57BL/6 mice revealed that MPZ protein expression is significantly decreased from 1 to 21 d after SNI. GAPDH was used as loading control. n = 3 mice per group. Data are presented as mean ± SEM (*p < 0.05).
We then explored the myelination status in the proximal sciatic nerve stump of WT and Nox4−/− mice 14 d after SNI. In WT mice, the protein levels of MPZ were significantly reduced in the day 14 SNI sciatic nerve compared with the uninjured sciatic nerve, as expected (Fig. 5A). Similarly, we observed reduced protein levels of PMP22, another major myelin component in the peripheral nervous system, in the day 14 SNI sciatic nerve of WT mice (Fig. 5A). In Nox4−/− mice, basal MPZ and PMP22 expression in uninjured sciatic nerves was slightly, but not significantly, decreased compared with WT mice. However, a decrease in MPZ or PMP22 protein expression in injured sciatic nerves did not occur in Nox4−/− mice (Fig. 5A), indicating that Nox4-dependent H2O2 production essentially contributes to SNI-induced dysmyelination.
Figure 5.

MPZ and PMP22 expression in the sciatic nerve of WT and Nox4−/− mice after SNI. A, Western blot analysis of the myelin-specific proteins MPZ and PMP22 in the day 14 SNI sciatic nerve (proximal nerve stump) and the uninjured control sciatic nerve. GAPDH was used as loading control. Note that MPZ and PMP22 protein expression is significantly decreased after SNI in WT mice but not in Nox4−/− mice. n = 3 mice per group. Data are presented as mean ± SEM (*p < 0.05). B, Immunostaining of the day 14 SNI sciatic nerve shows increased MPZ immunoreactivity in Nox4−/− mice compared with WT mice, whereas immunoreactivity of the neuronal marker NF200 is similar in both genotypes. Scale bar, 10 μm.
We further assessed the MPZ localization in the day 14 proximal sciatic nerve stump by immunostaining on longitudinal sections. As shown in Figure 5B, in both genotypes, most MPZ immunoreactivity was colocalized with the neurofilament marker NF200, which reflects the presence of MPZ in myelin sheaths surrounding axons. However, the MPZ immunofluorescence intensity was considerably reduced in nerves of WT mice compared with those of Nox4−/− mice (Fig. 5B), confirming the differences in MPZ protein levels detected by Western blot analyses. Similar differences in immunofluorescence intensities between WT and Nox4−/− mice were observed in immunostainings using the PMP22 antibody (data not shown). Together, these results indicate that peripheral nerve injury causes a drop of MPZ and PMP22 protein levels in the myelin sheaths surrounding the axons of injured nerves proximal to the site of injury and that Nox4-dependent H2O2 production is essential for this effect.
We then performed histological examinations of sciatic nerves on semithin cross sections stained with toluidine blue. Figure 6A depicts a normal appearance of the sciatic nerve from naive WT and Nox4−/− mice, with small- and large-diameter myelinated fibers regularly distributed in both genotypes. Two weeks after SNI, the proximal segments of injured nerves displayed severe fiber dystrophy and a decrease of axon density (number of axons per square millimeter) in all animals (Fig. 6B), consistent with previous reports (Inoue et al., 2004; Nagai et al., 2010). To estimate the myelination status, we measured the axon (inner myelin sheath circle) and myelin diameters in the day 14 SNI sciatic nerve using NIH ImageJ software and calculated the g ratio, i.e., the numerical ratio between axon and fiber diameter. In both genotypes, lowest g ratios were detected in small-diameter myelinated fibers (Fig. 6C). This observation suggests that 14 d after SNI small-diameter axons were encapsulated by myelin sheaths with a higher relative thickness (indicated by a lower g ratio), which was similar between WT and Nox4−/− mice. In contrast, a decrease of relative myelin thickness (indicated by a higher g ratio) occurred with increasing axon diameter (Fig. 6C). Interestingly, the proportion of these enlarged axons with relatively thin myelin sheaths was decreased in Nox4−/− mice compared with WT mice (Fig. 6C), suggesting that Nox4 contributes to the injury-induced processing of dysmyelination of large-diameter fibers. Finally, we assessed the injury-induced myelin infoldings and outfoldings (Fig. 6B), which predominately affect large-diameter fibers and are indicative of dysmyelination (Tersar et al., 2007). As shown in Figure 6D, the percentage of myelin infoldings and outfoldings in the day 14 SNI sciatic nerve was significantly reduced in Nox4−/− mice compared with WT mice, suggesting that injury-induced dysmyelination depends on Nox4. Altogether, our data show that Nox4-dependent H2O2 production essentially contributes to the changes in myelination that occur in the proximal stump of peripheral nerves as a response to nerve injury.
Figure 6.

Structural alterations in the sciatic nerve of WT and Nox4−/− mice after SNI. A, Toluidine blue-stained semithin cross sections of sciatic nerves of naive mice demonstrate normal appearance of myelinated fibers in WT and Nox4−/− mice. B, Toluidine blue stainings in proximal segments 1 mm from the injured site 14 d after SNI show marked alterations in the sciatic nerve of WT and Nox4−/− mice, including reduced axonal density, enlarged axons, and myelin infoldings and outfoldings. C, Scatter plots display g ratios of individual fibers as a function of the respective axon diameter (n = 348 and 243 fibers for WT and Nox4−/− mice, respectively). Note that the proportion of enlarged axons with thin myelin sheaths (expressed by a higher g ratio) is reduced in Nox4−/− mice. D, The percentage of myelin infoldings and outfoldings in the day 14 SNI sciatic nerve is significantly reduced in Nox4−/− mice. Data are presented as mean ± SEM (*p < 0.05). n = 3 animals per genotype. Scale bar, 5 μm.
Inducible deletion of Nox4 attenuates neuropathic pain behavior
Global gene ablation may cause developmental compensatory adaptations that might affect phenotypic changes in the genetically manipulated adult animal. To circumvent these issues, we crossed mice carrying a conditional null allele of Nox4 (Nox4fl/fl) with tamoxifen-inducible ERT2–Cre0/+ transgenic mice, with the resulting homozygous inducible conditional Nox4 knock-out mice (Nox4fl/fl;ERT2–Cre0/+) referred to as Nox4–CreERT2 mice. The Nox4fl/fl littermates were referred to as control mice. Animals were subjected to SNI to induce mechanical hypersensitivity. Ten days after SNI, mice were injected intraperitoneally with tamoxifen (1 mg/d for 3 d) to activate Cre recombinase and knockdown Nox4, and paw-withdrawal latency times were measured up to 21 d after the first injection. As shown in Figure 7A, mechanical hypersensitivity was not affected during the first 10 d after tamoxifen treatment. However, at later stages, the mechanical hypersensitivity was gradually reduced in tamoxifen-treated Nox4–CreERT2 mice, whereas in control mice, which also received tamoxifen, as well as in Nox4–CreERT2 mice without tamoxifen treatment, the hypersensitivity remained nearly constant until the end of the 21 d observation period (Fig. 7A). Investigation of Nox4 expression levels in Nox4–CreERT2 mice 21 d after tamoxifen treatment revealed a decrease of Nox4 mRNA to 83% (p = 0.116) in the spinal cord and to 54% (p = 0.008) in DRGs compared with vehicle-treated Nox4–CreERT2 mice (Fig. 7B). These data imply that our tamoxifen treatment protocol was sufficient for significant knockdown of Nox4 in DRGs. In conclusion, persisting neuropathic pain behavior is attenuated by temporal somatic knockdown of Nox4 in adult mice, suggesting that Nox4 could be a therapeutic target for the treatment of neuropathic pain.
Figure 7.

Reduced neuropathic pain behavior after inducible deletion of Nox4. A, Paw-withdrawal latency times after mechanical stimulation in tamoxifen-inducible Nox4–CreERT2 mice and littermate control mice during SNI-induced neuropathic pain. Tamoxifen (1 mg/d) or vehicle was injected intraperitoneally on 3 consecutive days starting 10 d after SNI surgery (indicated by arrows). Mechanical hypersensitivity was gradually reduced in tamoxifen-treated Nox4–CreERT2 mice in contrast to tamoxifen-treated control mice or vehicle-treated Nox4–CreERT2 mice (n = 8–10). B, Nox4 mRNA expression in DRGs and the spinal cord at the end of the observation period (i.e., 21 d after the first tamoxifen injection) assessed by quantitative real-time RT-PCR. Tamoxifen treatment significantly reduced Nox4 mRNA levels in DRGs of Nox4–CreERT2 mice as evaluated by quantitative real-time RT-PCR. Data are presented as mean ± SEM. *p < 0.05, comparing vehicle- and tamoxifen-treated Nox4–CreERT2 mice.
Discussion
In this study, we demonstrate that the H2O2-producing enzyme Nox4 is expressed in a population of nociceptive primary afferent neurons. Analyzing Nox4-deficient mice points to an essential function of Nox4 in injury-induced H2O2 production, dysmyelination, and the maintenance of neuropathic pain after peripheral nerve injury, whereas Nox4 does not essentially contribute to the processing of acute or inflammatory pain.
This study highlights the specific roles of different Nox enzymes for nociceptive signaling. Nox enzymes differ from most other ROS sources in that ROS generation is their primary function and not a byproduct of other biological reactions (Sorce and Krause, 2009). The best understood molecular mechanism by which ROS (in particular, H2O2) regulate cell functions occurs through redox-sensitive protein cysteine residues in local environments that lower the pKa of the cysteine such that the thiolate anion is the dominate form, thereby increasing the susceptibility of cysteine to oxidation (for review, see Forman et al., 2010). These specific actions mediated by ROS derived from different Nox enzymes located in different cells of the nociceptive system might be a plausible explanation for a selective contribution of Nox4 to neuropathic pain signaling [similar to Nox2 (Kim et al., 2010)] but not to inflammatory pain signaling [in contrast to Nox1 (Ibi et al., 2008)] and for the antinociceptive effects that have been observed in various animal models of neuropathic and inflammatory pain after administration of ROS scavengers and antioxidants (Kim et al., 2004; Wang et al., 2004; Yowtak et al., 2011). One has to consider, however, that the ROS scavengers that inhibited the pain behavior very effectively, such as phenyl-N-tert-butylnitrone and 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxy, are nonspecific in that they scavenge a wide range of ROS indiscriminately (Goldstein and Lestage, 2000; Wilcox and Pearlman, 2008). It is therefore likely that the antinociceptive effects of these compounds are based on scavenging ROS derived from Nox enzymes and other ROS sources.
The neuropathic pain behavior was reduced not only in Nox4−/− mice but also in tamoxifen-inducible Nox4–CreERT2 mice that were treated with tamoxifen after the SNI-induced mechanical allodynia was fully developed. This observation rules out several potentially confounding factors, such as developmental defects or compensatory mechanisms, that could have complicated the interpretation of the reduced neuropathic pain behavior in Nox4−/− mice. In addition, these data suggest that reducing Nox4 expression, or presumably selective inhibition of Nox4 activity, may be beneficial in neuropathic pain states that already persist for a longer time. We used a mild tamoxifen treatment protocol (1 mg/d, i.p., for 3 d) to avoid possible undesired tamoxifen-induced side effects that might impair the analysis of nociceptive behavior. In Nox4–CreERT2 mice, the tamoxifen treatment led to a reduction of Nox4 expression in DRGs but not in the spinal cord and to a reduction of mechanical hypersensitivity. The long interval between tamoxifen treatment and the occurrence of significant behavioral effects (18–21 d after the first tamoxifen injection) might be attributable to a long half-life of Nox4 mRNA and protein, because the half-life of individual mRNAs and proteins can vary greatly so that it can take weeks after tamoxifen injection until the target protein is lost (Feil et al., 2009). The selective reduction of Nox4 expression in DRGs but not in the spinal cord confirms previous reports that tamoxifen and its metabolite 4-hydroxytamoxifen, which induces the Cre-dependent recombination in vivo, do not easily cross the blood–brain barrier, thus limiting recombination in the spinal cord (Feil et al., 2009). However, the fact that the neuropathic pain behavior was significantly attenuated in these mice suggests that Nox4 expressed in primary afferent neurons rather than in the spinal cord exerts the “pain-relevant ” effects after peripheral nerve injury and that inhibition of Nox4 in primary afferent neurons might have therapeutic potentials for the treatment of neuropathic pain. Given the crucial role of Nox2-derived ROS in the innate immune response to invading pathogens and the possibility that pharmacological Nox2 inhibition could severely compromise a patient's immunological function (Drummond et al., 2011), selective Nox4 inhibitors might have therapeutic advantages over Nox2 inhibitors or unspecific ROS scavengers that also scavenge ROS derived from Nox2. However, because Nox4 is expressed in many cells throughout the body (Bedard and Krause, 2007), treatment with selective Nox4 inhibitors might also be associated with side effects.
We detected Nox4 immunoreactivity only in somata of DRG neurons but not in the sciatic nerve (data not shown). In addition to the Nox4 antibody used in this study, we tested several commercially available Nox4 antibodies that, however, turned out to be unspecific. The most likely explanation for the lack of Nox4 protein detection in the sciatic nerve are steric effects that impair the antibody binding. However, our H2O2 measurements clearly demonstrated Nox4-dependent H2O2 production in the proximal nerve stump in response to injury, strongly suggesting that Nox4 exerts its pain-relevant effects in the axons of peripheral nerves.
We show here that Nox4 plays an important role in dysmyelination after peripheral nerve injury. In response to peripheral nerve injury, myelinating Schwann cells are activated and their myelin properties are modified, resulting in altered conduction properties of nociceptive fibers (Devor, 2006b; Thacker et al., 2007; Nagai et al., 2010). In particular, the myelin degradation of Aβ afferents promotes susceptibility of their axonal plasma membrane to pronociceptive stimuli, leading to ectopic depolarization and mechanical allodynia (Kobayashi et al., 2008). Accordingly, neuropathic pain behaviors occurred after experimental demyelination of peripheral or central neurons (Wallace et al., 2003; Inoue et al., 2004; Moalem-Taylor et al., 2007; Zhu et al., 2012) and in mutant mice with aberrant myelination or loss of myelin (Gillespie et al., 2000; Chen et al., 2006). Moreover, neuropathic pain accompanies many demyelinating human diseases, such as Guillain-Barré syndrome, Charcot-Marie-Tooth type I disease, and multiple sclerosis (Ueda, 2008). We here provide several lines of evidence that Nox4 orchestrates the dysmyelination after peripheral nerve injury. First, in the injured proximal nerve stump, we observed a marked, Nox4-dependent production of H2O2, which can induce rapid degradation of myelin-specific proteins in vitro, thereby affecting myelin sheath homeostasis (Konat and Wiggins, 1985; Richter-Landsberg and Vollgraf, 1998; Laszkiewicz et al., 1999; Mronga et al., 2004; Perfeito et al., 2007). Second, our Western blot and immunostaining analyses showed that the nerve injury-induced degradation of myelin-specific proteins was abolished in Nox4−/− mice. Finally, our morphological investigations demonstrated a decreased number of enlarged fibers with thin myelin sheaths and of myelin infoldings/outfoldings in injured sciatic nerves of Nox4−/− mice. Therefore, it is tempting to speculate that H2O2 derived from Nox4 mediates the dysmyelination, which in turn contributes to injury-induced neuropathic pain.
A key question posed by this study is how H2O2 derived from Nox4, which is mostly expressed in nonmyelinated DRG neurons, could mediate the dysmyelination in a different population of fibers. In addition to intracellular signaling, H2O2 can diffuse across biological membranes and regulate functions of neighboring cells (Bienert et al., 2006). This paracrine action of H2O2 is seemingly facilitated by aquaporin channels (Bienert et al., 2007; Miller et al., 2010), suggesting that a regulated entry contributes to overall signaling specificity. About the possible molecular mechanism(s) of H2O2-induced myelin degradation in Schwann cells we can only speculate. Myelin is a highly specialized plasma membrane that consists mostly of lipids and contains a specific set of >500 proteins (Patzig et al., 2011). MPZ and PMP22 form complexes in the myelin membrane, and alterations in either of MPZ or PMP22 are sufficient to destabilize the entire myelin structure (D'Urso et al., 1999). Interestingly, MPZ contains an extracellular disulfide bond that strongly affects MPZ adhesion properties and stability (Pfend et al., 2001). H2O2 could therefore theoretically change the MPZ conformation by direct reaction with MPZ protein thiols and thereby perturb the myelin structure. Another possible mechanism might involve glutamine synthetase (GS), which is expressed in myelinating Schwann cells and whose expression is controlled posttranscriptionally by proteasomal degradation. After peripheral nerve injury, GS is degraded in the distal nerve stump in an ROS-dependent manner, and the resulting increased levels of glutamate (a GS substrate) seem to induce dysmyelination (Saitoh and Araki, 2010). Moreover, the Nox4-mediated dysmyelination might include apoptosis of myelinating Schwann cells, as has been observed after H2O2 exposure to Schwann cells in vitro and after experimental diabetic neuropathy (Wang et al., 2005; Luo et al., 2010). Conversely, we cannot exclude the possibility that Nox4-derived H2O2 induces dysmyelination by an indirect mechanism, e.g., by another signaling molecule that is released from primary afferent neurons in response to high intracellular H2O2 levels. Thus, the precise mechanism of Nox4-mediated dysmyelination after nerve injury is still unclear and awaits additional investigation. However, our data point to a so far unrecognized signaling mechanism between nociceptive primary afferent neurons and myelinating Schwann cells.
In conclusion, our results show that Nox4 is particularly important for the maintenance of neuropathic pain after peripheral nerve injury. Hence, inhibition of Nox4 activity might have therapeutic potentials for an effective and persistent treatment of neuropathic pain.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (Sonderforschungsbereich 815-A14) and a grant from the Goethe University Frankfurt, Germany (Nachlass Held/Hecker). We thank Karin Schilling, Christine Manderscheid, Judith Fuchs, Annett Häussler, Heike Korff, Anke Biczysko, and Susanne Schütz for excellent technical assistance.
The authors declare no competing financial interests.
References
- Anilkumar N, Weber R, Zhang M, Brewer A, Shah AM. Nox4 and nox2 NADPH oxidases mediate distinct cellular redox signaling responses to agonist stimulation. Arterioscler Thromb Vasc Biol. 2008;28:1347–1354. doi: 10.1161/ATVBAHA.108.164277. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- Bienert GP, Schjoerring JK, Jahn TP. Membrane transport of hydrogen peroxide. Biochim Biophys Acta. 2006;1758:994–1003. doi: 10.1016/j.bbamem.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Bienert GP, Møller AL, Kristiansen KA, Schulz A, Møller IM, Schjoerring JK, Jahn TP. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J Biol Chem. 2007;282:1183–1192. doi: 10.1074/jbc.M603761200. [DOI] [PubMed] [Google Scholar]
- Casals-Díaz L, Viv ó M, Navarro X. Nociceptive responses and spinal plastic changes of afferent C-fibers in three neuropathic pain models induced by sciatic nerve injury in the rat. Exp Neurol. 2009;217:84–95. doi: 10.1016/j.expneurol.2009.01.014. [DOI] [PubMed] [Google Scholar]
- Chen S, Velardez MO, Warot X, Yu ZX, Miller SJ, Cros D, Corfas G. Neuregulin 1-erbB signaling is necessary for normal myelination and sensory function. J Neurosci. 2006;26:3079–3086. doi: 10.1523/JNEUROSCI.3785-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci. 2009;32:1–32. doi: 10.1146/annurev.neuro.051508.135531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–158. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- Devor M. Response of nerves to injury in relation to neuropathic pain. In: McMahon SB, Koltzenburg M, editors. Wall and Melzack's textbook of pain. Philadelphia: Elsevier; 2006a. pp. 905–927. [Google Scholar]
- Devor M. Sodium channels and mechanisms of neuropathic pain. J Pain. 2006b;7:S3–S12. doi: 10.1016/j.jpain.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Devor M, Keller CH, Deerinck TJ, Levinson SR, Ellisman MH. Na+ channel accumulation on axolemma of afferent endings in nerve end neuromas in Apteronotus. Neurosci Lett. 1989;102:149–154. doi: 10.1016/0304-3940(89)90070-0. [DOI] [PubMed] [Google Scholar]
- Doyle T, Bryant L, Muscoli C, Cuzzocrea S, Esposito E, Chen Z, Salvemini D. Spinal NADPH oxidase is a source of superoxide in the development of morphine-induced hyperalgesia and antinociceptive tolerance. Neurosci Lett. 2010;483:85–89. doi: 10.1016/j.neulet.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10:453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Urso D, Ehrhardt P, Müller HW. Peripheral myelin protein 22 and protein zero: a novel association in peripheral nervous system myelin. J Neurosci. 1999;19:3396–3403. doi: 10.1523/JNEUROSCI.19-09-03396.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil S, Valtcheva N, Feil R. Inducible Cre mice. Methods Mol Biol. 2009;530:343–363. doi: 10.1007/978-1-59745-471-1_18. [DOI] [PubMed] [Google Scholar]
- Forman HJ, Maiorino M, Ursini F. Signaling functions of reactive oxygen species. Biochemistry. 2010;49:835–842. doi: 10.1021/bi9020378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie CS, Sherman DL, Fleetwood-Walker SM, Cottrell DF, Tait S, Garry EM, Wallace VC, Ure J, Griffiths IR, Smith A, Brophy PJ. Peripheral demyelination and neuropathic pain behavior in periaxin-deficient mice. Neuron. 2000;26:523–531. doi: 10.1016/s0896-6273(00)81184-8. [DOI] [PubMed] [Google Scholar]
- Goldstein S, Lestage P. Chemical and pharmacological aspects of heteroaryl-nitrones. Curr Med Chem. 2000;7:1255–1267. doi: 10.2174/0929867003374183. [DOI] [PubMed] [Google Scholar]
- Greenspan JD, Craft RM, LeResche L, Arendt-Nielsen L, Berkley KJ, Fillingim RB, Gold MS, Holdcroft A, Lautenbacher S, Mayer EA, Mogil JS, Murphy AZ, Traub RJ. Studying sex and gender differences in pain and analgesia: a consensus report. Pain. 2007;132(Suppl 1):S26–S45. doi: 10.1016/j.pain.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine S, Michalakis S, Kallenborn-Gerhardt W, Lu R, Lim HY, Weiland J, Del Turco D, Deller T, Tegeder I, Biel M, Geisslinger G, Schmidtko A. CNGA3: A target of spinal NO/cGMP signaling and modulator of inflammatory pain hypersensitivity. J Neurosci. 2011;31:11184–11192. doi: 10.1523/JNEUROSCI.6159-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunskaar S, Fasmer OB, Hole K. Formalin test in mice, a useful technique for evaluating mild analgesics. J Neurosci Methods. 1985;14:69–76. doi: 10.1016/0165-0270(85)90116-5. [DOI] [PubMed] [Google Scholar]
- Ibi M, Matsuno K, Shiba D, Katsuyama M, Iwata K, Kakehi T, Nakagawa T, Sango K, Shirai Y, Yokoyama T, Kaneko S, Saito N, Yabe-Nishimura C. Reactive oxygen species derived from NOX1/NADPH oxidase enhance inflammatory pain. J Neurosci. 2008;28:9486–9494. doi: 10.1523/JNEUROSCI.1857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indra AK, Warot X, Brocard J, Bornert JM, Xiao JH, Chambon P, Metzger D. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acid Res. 1999;27:4324–4327. doi: 10.1093/nar/27.22.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Rashid MH, Fujita R, Contos JJ, Chun J, Ueda H. Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nat Med. 2004;10:712–718. doi: 10.1038/nm1060. [DOI] [PubMed] [Google Scholar]
- Janes K, Neumann WL, Salvemini D. Anti-superoxide and anti-peroxynitrite strategies in pain suppression. Biochim Biophys Acta. 2012;1822:815–821. doi: 10.1016/j.bbadis.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, You B, Jo EK, Han SK, Simon MI, Lee SJ. NADPH oxidase 2-derived reactive oxygen species in spinal cord microglia contribute to peripheral nerve injury-induced neuropathic pain. Proc Natl Acad Sci U S A. 2010;107:14851–14856. doi: 10.1073/pnas.1009926107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HK, Park SK, Zhou JL, Taglialatela G, Chung K, Coggeshall RE, Chung JM. Reactive oxygen species (ROS) play an important role in a rat model of neuropathic pain. Pain. 2004;111:116–124. doi: 10.1016/j.pain.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Chattopadhyay S, Kato K, Dolkas J, Kikuchi S, Myers RR, Shubayev VI. MMPs initiate Schwann cell-mediated MBP degradation and mechanical nociception after nerve damage. Mol Cell Neurosci. 2008;39:619–627. doi: 10.1016/j.mcn.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konat GW, Wiggins RC. Effect of reactive oxygen species on myelin membrane proteins. J Neurochem. 1985;45:1113–1118. doi: 10.1111/j.1471-4159.1985.tb05530.x. [DOI] [PubMed] [Google Scholar]
- Laszkiewicz I, Mouzannar R, Wiggins RC, Konat GW. Delayed oligodendrocyte degeneration induced by brief exposure to hydrogen peroxide. J Neurosci Res. 1999;55:303–310. doi: 10.1002/(SICI)1097-4547(19990201)55:3<303::AID-JNR5>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Lu R, Kallenborn-Gerhardt W, Geisslinger G, Schmidtko A. Additive antinociceptive effects of a combination of vitamin C and vitamin E after peripheral nerve injury. PLoS One. 2011;6:e29240. doi: 10.1371/journal.pone.0029240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Chen B, Zheng R, Lin P, Li J, Chen H. Hydrogen peroxide induces apoptosis through the mitochondrial pathway in rat Schwann cells. Neurosci Lett. 2010;485:60–64. doi: 10.1016/j.neulet.2010.08.063. [DOI] [PubMed] [Google Scholar]
- Meller ST, Gebhart GF. Intraplantar zymosan as a reliable, quantifiable model of thermal and mechanical hyperalgesia in the rat. Eur J Pain. 1997;1:43–52. doi: 10.1016/s1090-3801(97)90052-5. [DOI] [PubMed] [Google Scholar]
- Miller EW, Dickinson BC, Chang CJ. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proc Natl Acad Sci U S A. 2010;107:15681–15686. doi: 10.1073/pnas.1005776107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moalem-Taylor G, Allbutt HN, Iordanova MD, Tracey DJ. Pain hypersensitivity in rats with experimental autoimmune neuritis, an animal model of human inflammatory demyelinating neuropathy. Brain Behav Immun. 2007;21:699–710. doi: 10.1016/j.bbi.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Mogil JS, Chanda ML. The case for the inclusion of female subjects in basic science studies of pain. Pain. 2005;117:1–5. doi: 10.1016/j.pain.2005.06.020. [DOI] [PubMed] [Google Scholar]
- Mogil JS, Wilson SG, Bon K, Lee SE, Chung K, Raber P, Pieper JO, Hain HS, Belknap JK, Hubert L, Elmer GI, Chung JM, Devor M. Heritability of nociception I: responses of 11 inbred mouse strains on 12 measures of nociception. Pain. 1999;80:67–82. doi: 10.1016/s0304-3959(98)00197-3. [DOI] [PubMed] [Google Scholar]
- Mronga T, Stahnke T, Goldbaum O, Richter-Landsberg C. Mitochondrial pathway is involved in hydrogen-peroxide-induced apoptotic cell death of oligodendrocytes. Glia. 2004;46:446–455. doi: 10.1002/glia.20022. [DOI] [PubMed] [Google Scholar]
- Nagai J, Uchida H, Matsushita Y, Yano R, Ueda M, Niwa M, Aoki J, Chun J, Ueda H. Autotaxin and lysophosphatidic acid1 receptor-mediated demyelination of dorsal root fibers by sciatic nerve injury and intrathecal lysophosphatidylcholine. Mol Pain. 2010;6:78. doi: 10.1186/1744-8069-6-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patzig J, Jahn O, Tenzer S, Wichert SP, de Monasterio-Schrader P, Rosfa S, Kuharev J, Yan K, Bormuth I, Bremer J, Aguzzi A, Orfaniotou F, Hesse D, Schwab MH, Möbius W, Nave KA, Werner HB. Quantitative and integrative proteome analysis of peripheral nerve myelin identifies novel myelin proteins and candidate neuropathy loci. J Neurosci. 2011;31:16369–16386. doi: 10.1523/JNEUROSCI.4016-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfeito R, Pereira J, Oliveira CR, Bettencourt-Relvas J, Rego AC. Trolox protection of myelin membrane in hydrogen peroxide-treated mature oligodendrocytes. Free Radic Res. 2007;41:444–451. doi: 10.1080/10715760601134442. [DOI] [PubMed] [Google Scholar]
- Pfend G, Matthieu JM, Garin N, Tosic M. Implication of the extracellular disulfide bond on myelin protein zero expression. Neurochem Res. 2001;26:503–510. doi: 10.1023/a:1010908828134. [DOI] [PubMed] [Google Scholar]
- Rhee SG, Chang TS, Jeong W, Kang D. Methods for detection and measurement of hydrogen peroxide inside and outside of cells. Mol Cells. 2010;29:539–549. doi: 10.1007/s10059-010-0082-3. [DOI] [PubMed] [Google Scholar]
- Richter-Landsberg C, Vollgraf U. Mode of cell injury and death after hydrogen peroxide exposure in cultured oligodendroglia cells. Exp Cell Res. 1998;244:218–229. doi: 10.1006/excr.1998.4188. [DOI] [PubMed] [Google Scholar]
- Robinson FL, Niesman IR, Beiswenger KK, Dixon JE. Loss of the inactive myotubularin-related phosphatase Mtmr13 leads to a Charcot-Marie-Tooth 4B2-like peripheral neuropathy in mice. Proc Natl Acad Sci U S A. 2008;105:4916–4921. doi: 10.1073/pnas.0800742105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh F, Araki T. Proteasomal degradation of glutamine synthetase regulates Schwann cell differentiation. J Neurosci. 2010;30:1204–1212. doi: 10.1523/JNEUROSCI.3591-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvemini D, Little JW, Doyle T, Neumann WL. Roles of reactive oxygen and nitrogen species in pain. Free Radic Biol Med. 2011;51:951–966. doi: 10.1016/j.freeradbiomed.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtko A, Luo C, Gao W, Geisslinger G, Kuner R, Tegeder I. Genetic deletion of synapsin II reduces neuropathic pain due to reduced glutamate but increased GABA in the spinal cord dorsal horn. Pain. 2008a;139:632–643. doi: 10.1016/j.pain.2008.06.018. [DOI] [PubMed] [Google Scholar]
- Schmidtko A, Gao W, König P, Heine S, Motterlini R, Ruth P, Schlossmann J, Koesling D, Niederberger E, Tegeder I, Friebe A, Geisslinger G. cGMP produced by NO-sensitive guanylyl cyclase essentially contributes to inflammatory and neuropathic pain by using targets different from cGMP-dependent protein kinase I. J Neurosci. 2008b;28:8568–8576. doi: 10.1523/JNEUROSCI.2128-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtko A, Gao W, Sausbier M, Rauhmeier I, Sausbier U, Niederberger E, Scholich K, Huber A, Neuhuber W, Allescher HD, Hofmann F, Tegeder I, Ruth P, Geisslinger G. Cysteine-rich protein 2, a novel downstream effector of cGMP/cGMP-dependent protein kinase I-mediated persistent inflammatory pain. J Neurosci. 2008c;28:1320–1330. doi: 10.1523/JNEUROSCI.5037-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtko A, Lötsch J, Freynhagen R, Geisslinger G. Ziconotide for treatment of severe chronic pain. Lancet. 2010;375:1569–1577. doi: 10.1016/S0140-6736(10)60354-6. [DOI] [PubMed] [Google Scholar]
- Schnell SA, Staines WA, Wessendorf MW. Reduction of lipofuscin-like autofluorescence in fluorescently labeled tissue. J Histochem Cytochem. 1999;47:719–730. doi: 10.1177/002215549904700601. [DOI] [PubMed] [Google Scholar]
- Schröder K, Kohnen A, Aicher A, Liehn EA, Büchse T, Stein S, Weber C, Dimmeler S, Brandes RP. NADPH oxidase Nox2 is required for hypoxia-induced mobilization of endothelial progenitor cells. Circ Res. 2009;105:537–544. doi: 10.1161/CIRCRESAHA.109.205138. [DOI] [PubMed] [Google Scholar]
- Sorce S, Krause KH. NOX enzymes in the central nervous system: from signaling to disease. Antioxid Redox Signal. 2009;11:2481–2504. doi: 10.1089/ars.2009.2578. [DOI] [PubMed] [Google Scholar]
- Takac I, Schröder K, Zhang L, Lardy B, Anilkumar N, Lambeth JD, Shah AM, Morel F, Brandes RP. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J Biol Chem. 2011;286:13304–13313. doi: 10.1074/jbc.M110.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegeder I, Costigan M, Griffin RS, Abele A, Belfer I, Schmidt H, Ehnert C, Nejim J, Marian C, Scholz J, Wu T, Allchorne A, Diatchenko L, Binshtok AM, Goldman D, Adolph J, Sama S, Atlas SJ, Carlezon WA, Parsegian A, Lötsch J, Fillingim RB, Maixner W, Geisslinger G, Max MB, Woolf CJ. GTP cyclohydrolase and tetrahydrobiopterin regulate pain sensitivity and persistence. Nat Med. 2006;12:1269–1277. doi: 10.1038/nm1490. [DOI] [PubMed] [Google Scholar]
- Tersar K, Boentert M, Berger P, Bonneick S, Wessig C, Toyka KV, Young P, Suter U. Mtmr13/Sbf2-deficient mice: an animal model for CMT4B2. Hum Mol Genet. 2007;16:2991–3001. doi: 10.1093/hmg/ddm257. [DOI] [PubMed] [Google Scholar]
- Thacker MA, Clark AK, Marchand F, McMahon SB. Pathophysiology of peripheral neuropathic pain: immune cells and molecules. Anesth Analg. 2007;105:838–847. doi: 10.1213/01.ane.0000275190.42912.37. [DOI] [PubMed] [Google Scholar]
- Ueda H. Peripheral mechanisms of neuropathic pain - involvement of lysophosphatidic acid receptor-mediated demyelination. Mol Pain. 2008;4:11. doi: 10.1186/1744-8069-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace VC, Cottrell DF, Brophy PJ, Fleetwood-Walker SM. Focal lysolecithin-induced demyelination of peripheral afferents results in neuropathic pain behavior that is attenuated by cannabinoids. J Neurosci. 2003;23:3221–3233. doi: 10.1523/JNEUROSCI.23-08-03221.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Schmeichel AM, Iida H, Schmelzer JD, Low PA. Ischemia-reperfusion injury causes oxidative stress and apoptosis of Schwann cell in acute and chronic experimental diabetic neuropathy. Antioxid Redox Signal. 2005;7:1513–1520. doi: 10.1089/ars.2005.7.1513. [DOI] [PubMed] [Google Scholar]
- Wang ZQ, Porreca F, Cuzzocrea S, Galen K, Lightfoot R, Masini E, Muscoli C, Mollace V, Ndengele M, Ischiropoulos H, Salvemini D. A newly identified role for superoxide in inflammatory pain. J Pharmacol Exp Ther. 2004;309:869–878. doi: 10.1124/jpet.103.064154. [DOI] [PubMed] [Google Scholar]
- Wilcox CS, Pearlman A. Chemistry and antihypertensive effects of tempol and other nitroxides. Pharmacol Rev. 2008;60:418–469. doi: 10.1124/pr.108.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yowtak J, Lee KY, Kim HY, Wang J, Kim HK, Chung K, Chung JM. Reactive oxygen species contribute to neuropathic pain by reducing spinal GABA release. Pain. 2011;152:844–852. doi: 10.1016/j.pain.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Brewer AC, Schröder K, Santos CX, Grieve DJ, Wang M, Anilkumar N, Yu B, Dong X, Walker SJ, Brandes RP, Shah AM. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci U S A. 2010;107:18121–18126. doi: 10.1073/pnas.1009700107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YL, Xie ZL, Wu YW, Duan WR, Xie YK. Early demyelination of primary A-fibers induces a rapid-onset of neuropathic pain in rat. Neuroscience. 2012;200:186–198. doi: 10.1016/j.neuroscience.2011.10.037. [DOI] [PubMed] [Google Scholar]