

Abstract
Müller cell gliosis, which is characterized by upregulated expression of glial fibrillary acidic protein (GFAP), is a universal response in many retinal pathological conditions. Whether down-regulation of inward rectifying K+ (Kir) channels, which commonly accompanies the enhanced GFAP expression, could contribute to Müller cell gliosis is poorly understood. We investigated changes of Kir currents, GFAP and Kir4.1 protein expression in Müller cells in a rat chronic ocular hypertension (COH) model, and explored the mechanisms underlying Müller cell gliosis. We show that Kir currents and Kir4.1 protein expression in Müller cells were reduced significantly, while GFAP expression was increased in COH rats, and these changes were eliminated by MPEP, a group I metabotropic glutamate receptors (mGluR I) subtype mGluR5 antagonist. In normal isolated Müller cells, the mGluR I agonist (S)-3,5-dihydroxyphenylglycine (DHPG) suppressed the Kir currents and the suppression was blocked by MPEP. The DHPG effect was mediated by the intracellular Ca2+-dependent PLC/IP3-ryanodine/PKC signaling pathway, but the cAMP-PKA pathway was not involved. Moreover, intravitreal injection of DHPG in normal rats induced changes in Müller cells, similar to those observed in COH rats. The DHPG-induced increase of GFAP expression in Müller cells was obstructed by Ba2+, suggesting the involvement of Kir channels. We conclude that overactivation of mGluR5 by excessive extracellular glutamate in COH rats could contribute to Müller cell gliosis by suppressing Kir channels.
Introduction
Reactivation of glial cells (gliosis) occurs in almost all forms of CNS disorders (Sofroniew, 2005). Specifically, gliosis of Müller cells, a major type of glial cells in the retina, is a common response in a variety of retinal pathological alternations (Bringmann et al., 2006; 2009), which contributes to neurodegeneration (Goureau et al., 1999; Kashiwagi et al., 2001; Tezel et al., 2001; Tezel and Wax, 2003; Bringmann et al., 2009). Müller cell gliosis is characterized by upregulated expression of glial cytoskeletal proteins, glial fibrillary acidic protein (GFAP) and vimentin, and downregulated membrane K+ conductance, especially inward rectifying K+ channel (Kir) 4.1-mediated currents (Francke et al., 1997, 2001; Bringmann et al., 2000; Pannicke et al., 2006). It is well established that inhibition of Müller cell K+ channels leads to depolarization of the cell membrane (Pannicke et al., 2000, Bringmann et al., 2002a), which disturbs the characteristic hyperpolarized resting potential of the cell and could result in a loss of its neuron-supportive functions. In experimental glaucoma models and DBA/2J mice, which develop ocular hypertension, GFAP expression in Müller cells was enhanced, suggesting reactivation of the cells (Wang et al., 2000; Woldemussie et al., 2004; Xue et al., 2006; Inman and Horner, 2007; Bolz et al., 2008; Zhang et al., 2009). However, the evidence now available concerning changes in K+ channels, following the onset of glaucoma, seems inconsistent. It was reported that Kir currents were downregulated in glial cells isolated from diseased human retinas, including those from patients with glaucoma (Francke et al., 1997). However, no significant alterations of membrane K+ channels were detected in DBA/2J mice (Bolz et al., 2008). The first purpose of this investigation was to explore whether Müller cell gliosis is indeed related to Kir channels in experimental glaucoma. We here show that the increased GFAP expression in Müller cells in a rat chronic ocular hypertension (COH) model is accompanied by a progressive, significant reduction of Kir currents and Kir proteins in these cells. We further provide evidence that the increased GFAP expression may be a consequence of reduced activity of Kir channels.
A second question we wanted to address was what caused the changes in Kir channels in experimental glaucoma. Considerable evidence suggests that excess extracellular glutamate, a major excitatory neurotransmitter in the mammalian retina, may be an important risk factor for retinal injury in glaucoma due to overactivation of both ionotropic and metabotropic glutamate receptors (mGluRs) (Salt and Cordeiro, 2006; Seki and Lipton, 2008). Overactivation of mGluRs has been shown to contribute to pathogenesis of various CNS disorders (Nicoletti et al., 1999; Lau and Tymianski, 2010; Caraci et al., 2012). Our data show that overactivation of group I mGluR (mGluR I) may be involved in Müller cell gliosis in the rat COH model via inhibiting Kir channels, a condition that could be ameliorated by suppressing these receptors.
Materials and Methods
Animals and rat COH model.
All experimental procedures described here were in accordance with the National Institutes of Health (NIH) guidelines for the Care and Use of Laboratory Animals and the guidelines of Fudan University on the ethical use of animals. During this study, all possible efforts were made to minimize the number of animals used and their suffering. Male Sprague Dawley rats, weighing 100–300 g and obtained from SLAC Laboratory Animal Company, were housed under conditions of a 12 h light/dark cycle. The rat COH model was reproduced following a procedure described previously in detail (Naka et al., 2010; Wu et al., 2010; Chen et al., 2011). Briefly, rats were anesthetized with an intramuscular injection of a mixture of ketamine (25 mg/kg) and xylazine (10 mg/kg), and the eyes were further anesthetized locally with a topical application of 0.4% oxybuprocaine hydrochloride eye drops (Benoxil, Santen Pharmaceutical). Two or three episcleral veins in the left eye were carefully separated and cauterized under an OPMI VISU 140 microscope (Carl Zeiss). In sham-operated control eyes, the surgery was done using a similar procedure, except for not occluding the vines. As a convention, sham-operation was done on the eyes of other rats, but not on the contralateral eye of the operated animal. After surgery, the eyes were flushed with antibiotic eye drops and covered with antibiotic ointment. Intraocular pressure (IOP) was measured using a handheld digital tonometer (Tonopen XL; Mentor O&O), under general and local anesthesia as described above. The average value of five consecutive acceptable measurements with a deviation <5% was recorded. All measurements were performed in the morning to avoid possible circadian differences. The IOPs of both eyes were measured before surgery as a baseline, immediately after surgery (day 0), the first day after surgery (G1d), and weekly thereafter.
Preparation of isolated Müller cells.
Animals were deeply anesthetized with 4% sodium pentobarbital and killed by decapitation. Retinas were removed quickly and incubated in oxygenated Hank's solution containing the following (in mm): NaCl 137, NaHCO3 0.5, NaH2PO4 1, KCl 3, CaCl2 2, MgSO4 1, HEPES 20, sodium pyruvate 1, and glucose 16, adjusted to pH 7.4 with NaOH. The retinas were then digested with 1.6 U/ml papain (Worthington Biochemical) in Hank's solution supplemented with 0.2 mg/ml l-cysteine and 0.2 mg/ml bovine serum albumin, for 35 min at 35°C. The solution was bubbled continuously with 100% O2 and adjusted to pH 7.4 with NaOH. After several rinses with Hank's solution, the retinas were mechanically dissociated with fire-polished Pasteur pipettes, and the cell suspension was plated onto a culture dish mounted on an inverted microscope (IX 70; Olympus Optical). Müller cells with typical morphological features (Ishii et al., 1997; Wurm et al., 2006) were chosen for whole-cell patch-clamp recording within 2–3 h after dissociation.
Whole-cell patch-clamp recordings.
Dissociated Müller cells were continuously perfused with an external solution containing the following (in mm): NaCl 135, KCl 3, CaCl2 2, MgCl2 1, HEPES 10, glucose 11, pyruvate sodium 1, TTX 0.0005, and sucrose 10, adjusted to pH 7.4 with NaOH. Patch pipettes were made by pulling BF150–86-10 glass (Sutter Instrument Co.) on a P-97 flaming/brown micropipette puller (Sutter Instrument Co.) and fire polished (Model MF-830; Narishige) for recording. The pipette resistance was typically 8–10 MΩ after filling with an internal solution consisting of (in mm): NaCl 20, K-gluconate 130, CaCl2 1, MgCl2 2, EGTA 1, HEPES 10, GTP-Na 0.1, and ATP-Mg 2, adjusted to pH 7.2 with KOH and to 290–300 mOsm/L. Whole-cell membrane currents were recorded from Müller cells using an Axopatch 200B amplifier, Digidata 1322A data acquisition board, and Clampex 8.0 software (Molecular Devices). Fast capacitance was fully cancelled and cell capacitance was partially cancelled as much as possible by the amplifier circuits. Seventy percent of the series resistance of the recording electrode was compensated. Analog signals were sampled at 5 kHz, filtered at 1 kHz, and stored for further analysis. All recordings were made at room temperature (20−25°C).
Intravitreal injection.
Rats were anesthetized with an intramuscular injection of a mixture of ketamine and xylazine. The pupil was dilated with tropicamide drops, and 10 μm (S)-3,5-dihydroxyphenylglycine (DHPG), 10 μm MPEP, 200 μm BaCl2, or 50 μm CoCl2 dispersed in 2 μl of 0.9% saline were injected into the vitreous space through a postlimbus spot using Hamilton microinjector (Hamilton) under a stereoscopic microscope (Carl Zeiss). A 30-gauge needle was inserted 2 mm behind the temporal limbus and directed toward the optic nerve. Eyes that received only an injection of saline in the same manner served as a vehicle control.
Western blot analysis.
Western blot analysis was conducted as previously described with some modifications (Zhao et al., 2010; Chen et al., 2011). Rat retinas were lysed in lysis buffer (50 mm Tris-Cl, 150 mm NaCl, 1% Triton X-100, 0.1% aprotinin, 1 mm phenylmethylsulfonyl fluoride, 1 mm sodium orthovanadate, and 25 mm sodium fluoride, pH 7.4), and protein concentrations were determined by the BCA method (Pierce). Protein samples (1.0 μg/μl, 15 μl) were subjected to 10% SDS-PAGE using a Mini-Protean 3 electrophoresis system (Bio-Rad) and electrotransferred to polyvinylidene fluoride membranes using a Mini Trans-Blot electrophoretic transfer system (Bio-Rad). The membranes were blocked with 5% skimmed milk at room temperature for 1 h, and then incubated with mouse monoclonal antibody against GFAP (1:1000 dilution; Sigma), rabbit polyclonal antibody against Kir4.1 (1:400 dilution; Alomone) or mouse monoclonal antibody against β-actin (1:2000, Sigma) overnight at 4°C. The blots were washed with TBST and incubated with HRP-conjugated goat anti-mouse or anti-rabbit IgG (1:4000; Jackson ImmunoResearch Laboratories) for 1 h at room temperature and visualized with enhanced chemofluorescence reagent (Pierce). For sequential immunoblotting, the blots were reblocked, tested for residual signal, and then stripped with restore Western blot stripping buffer (Pierce) if necessary. The protein bands were quantitatively analyzed with NIH image analysis software.
Immunohistochemistry.
Immunofluorescent staining was performed following the procedure described in detail previously (Zhao et al., 2010; Yang et al., 2011). After rats were anesthetized and perfused with 4% paraformaldehyde (PFA, in 0.1 m PB, pH 7.4), the left eyeballs were removed and post-fixed in 4% PFA for 2–4 h, followed by dehydration with graded sucrose solutions at 4°C (4 h in 20% and overnight in 30%). The retinas were vertically sectioned at 14 μm thickness on a freezing microtome (Leica). The slices were collected and mounted on chrome-alum-gelatin-coated slides. After washing with 0.01 m PBS, pH 7.4, the sections were blocked in 6% bovine serum albumin (Sigma) in PBS plus 0.1% Triton X-100 at room temperature for 2 h, and then incubated with mouse monoclonal anti-GFAP (1:400 dilution; Sigma), polyclonal rabbit anti-Kir4.1 (1:400 dilution; Alomone) primary antibodies at 4°C for 48 h. Immunoreactive proteins were visualized by incubating with FITC-conjugated goat anti-mouse IgG (1:200 dilution; Jackson ImmunoResearch Laboratories) and cy3-conjugated goat anti-rabbit IgG (1:200 dilution; Jackson ImmunoResearch Laboratories). The samples were mounted with anti-fade mounting medium with DAPI (Vector Laboratories) and the immunofluorescence images were visualized with an Olympus FV1000 confocal laser-scanning microscope using a 40× oil-immersion objective lens. To avoid any possible reconstruction stacking artifact, the double labeling was precisely evaluated by sequential scanning of single-layer optical sections at intervals of 1.0 μm.
Reagents and drug application.
KN-62, KN-93, U73122, MPMQ, and MPEP were obtained from Tocris Bioscience. All the other chemicals were from Sigma-Aldrich. U73122, chelerythrine chloride, Bis IV, MPMQ, MPEP, and forskolin were first dissolved in dimethyl sulfoxide (DMSO) and then added to the extracellular or internal solution, with the final concentration of DMSO being <0.1%. The other chemicals were freshly dissolved in the extracellular or internal solution. Drugs were delivered by a superfusion drug application system (DAD-8VCSP, ALA Scientific Instruments), which has eight pressurized 5 ml reservoirs, each with its own control valve to feed fluid through an eight-to-one tubing manifold (500 μm inner diameter; ALA Scientific Instruments). The open/close switch of each valve was manually controlled. Once the valve was open, the solution in the corresponding reservoir was pressure ejected by nitrogen gas along the pipes. With the large flow pipes, 10–90% whole-cell solution exchanges were achieved in <2.5 ms.
Data analysis.
The data analysis was performed using Clampfit 8.0 (Molecular Devices) and Igor 4.0 (WaveMetrics). Data are presented as means ± SEM. A one-way ANOVA with Bonferroni's post hoc test (multiple comparisons), Mann–Whitney test (comparisons between two groups), and t test (paired data) were used as appropriate. A value of p < 0.05 was considered significant.
Results
Suppression of Kir currents in the rat COH model
Kir currents were first identified in isolated rat Müller cells (Fig. 1A). The currents, induced in a Müller cell by a series of hyperpolarized voltage pulses from a holding potential of −80 mV in increments of −20 mV at different concentrations of extracellular potassium ([K+]o), are shown in Figure 1B. The channels opened rapidly and did not inactivate when hyperpolarization was maintained. The currents were increased in amplitudes with increasing [K+]o. Figure 1C shows that bath application of Ba2+ (1 mm) significantly suppressed the currents from a Müller cell, induced by stepping to test potentials between −160 to +20 mV from a holding potential of −80 mV, in a reversible manner, and the average data (n = 10) are plotted in Figure 1D. Consistently, bath application of Ba2+ yielded similar effects on ramp currents recorded from another Müller cell (Fig. 1E), and addition of Cs+ (1 mm) did not further suppress the current in the presence of Ba2+ (data not shown). These properties of the currents are characteristic of K+-selective Kir currents (Newman, 1993; Iandiev et al., 2006; Wurm et al., 2006). Moreover, the I–V relationships of the currents, shown in Figure 1, D and E, exhibited a weakly inward rectifying feature, suggesting that these currents may be largely mediated by Kir4.1 channels (Tada et al., 1998; Kofuji et al., 2002; Butt and Kalsi, 2006).
Figure 1.
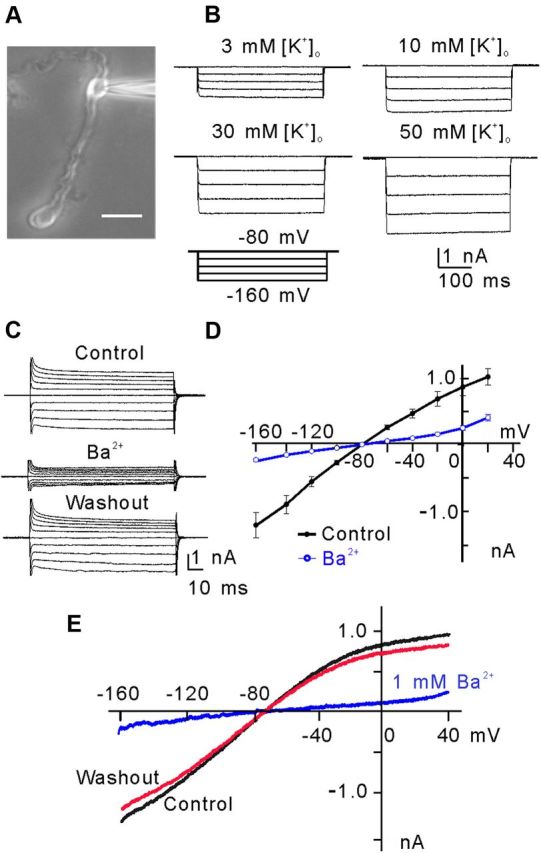
Inwardly rectifying K+-selective current (Kir) in rat retinal Müller cells. A, Micrograph showing a typical isolated rat Müller cell. Scale bar, 10 μm. B, [K+]o dependence of hyperpolarization-activated Kir currents of Müller cells. The currents were evoked by a series of hyperpolarized voltage pulses from a holding potential of −80 mV in increments of −20 mV. Note that the current amplitudes were increased with an increase in [K+]o. C, Ba2+ (1 mm) blocked the membrane currents of a Müller cell, voltage clamped at −80 mV and stepped to −160 mV in −10 mV increments and then to +20 mV in 10 mV increments. D, Membrane currents plotted against the step potentials. Note the K+-selective weakly rectifying Kir4.1-like I–V relationship (n = 10). The Kir currents were reduced significantly by Ba2+ (1 mm). E, Currents elicited by a 1 s voltage ramp from −160 mV to +40 mV before (control), after the application of Ba2+ (1 mm) and washout of Ba2+.
We then examined whether Kir currents of Müller cells were changed in the COH model (Naka et al., 2010; Chen et al., 2011; Wu et al., 2010). In this model, the average IOP of the operated eyes was sharply increased to 30.0 ± 0.3 mmHg on G1d (n = 39, p < 0.001) from 19.2 ± 0.2 mmHg before surgery (n = 39). The IOP was declined to 27.5 ± 0.4 mmHg on the first weak after surgery (G1w, n = 39, p < 0.001) and then maintained at higher levels during the next 5 weeks (27.0 ± 0.4 mmHg on G2w, n = 32; 26.8 ± 0.3 mmHg on G3w, n = 27; 26.6 ± 0.3 mmHg on G4w, n = 23; 26.7 ± 0.4 mmHg on G5w, n = 18; 26.2 ± 0.5 mmHg on G6w, n = 10; p < 0.001). Meanwhile, the average IOPs in the sham-operated eyes kept unchanged during 6 weeks after surgery, ranging from 19.3 ± 0.2 mmHg to 20.0 ± 0.2 mmHg (n = 10∼39), which were not significantly different from that before surgery (19.4 ± 0.2 mmHg, n = 39) (p > 0.05). Kir currents of Müller cells isolated from the operated eyes and the sham-operated eyes were recorded at different time points, using stepping voltages. Figure 2, A and B, show the currents recorded from representative cells and the averaged I–V relationships, respectively. The averaged current amplitudes by voltage steps of −160 mV are represented as a function of time after the operation in Figure 2C. It is evident that the currents were reduced in amplitudes at all time points. The peak current amplitudes were decreased to approximately 47% of control even on G1d (47.8 ± 4.5% control, n = 8, p = 0.018), then further to 36.3 ± 7.4% of control on G1w (n = 8, p = 0.008), and remained at this level for next 2 weeks (G2w: 33.4 ± 7.9%, n = 12, p < 0.001; G3w: 28.5 ± 16.5% of control, n = 8, p = 0.003). Although the reduction of the current amplitudes became less significant on G4w (63.7 ± 10.4% of the control, n = 8, p = 0.024) and on G6w (51.9 ± 14.1% of control, n = 11, p = 0.004), they were still lower than control (Fig. 2C).
Figure 2.
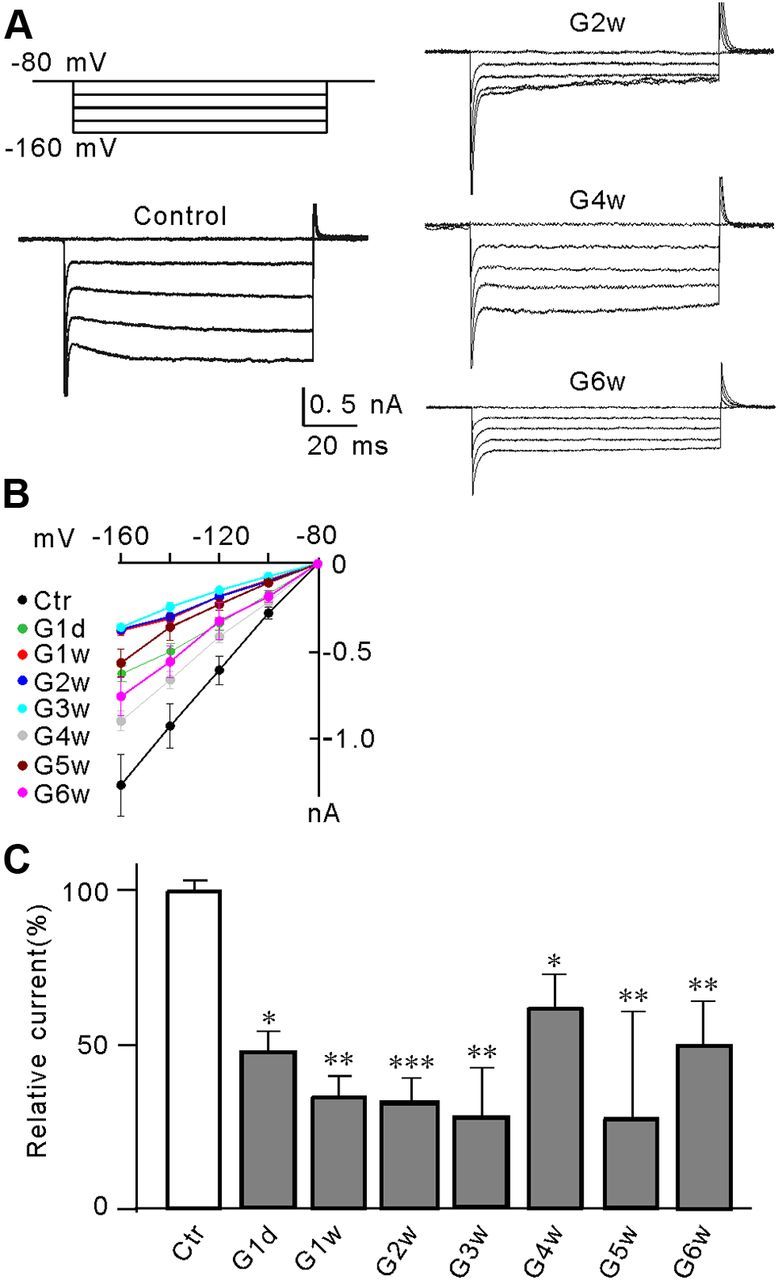
Suppression of Kir currents in Müller cells in a rat COH model. A, Voltage-clamp analysis of the Kir current changes at negative membrane potentials. Cells were clamped at −80 mV and stepped to −160 mV in −20 mV increments (top left). Whole-cell membrane currents were recorded in Müller cells isolated from sham-operated control (Ctr) and COH rats on the second (G2w), fourth (G4w), and sixth week (G6w) after the operation. B, The I–V relationships, showing voltage-dependent suppression of Kir current amplitudes in Müller cells obtained from Ctr and COH rats at the first day after surgery (G1d), and the first to sixth week (G1w–G6w) after the operation C, Summarized data showing that the average Kir current peak amplitudes decreased when the IOP was elevated. All data are normalized to control. Error bars represent SEM. n = 8∼15, *p < 0.05, **p < 0.01, and ***p < 0.001 versus control (Ctr).
Kir4.1 and GFAP expression in the COH model
We further evaluated changes in Kir4.1 protein and GFAP protein expression in our COH model by immunohistochemistry and Western blotting. Figure 3A1 shows that in sham-operated retinal section (Control), Kir4.1 proteins were extensively expressed in Müller cells, predominantly at the endfeet, in cells of the ganglion cell layer (GCL) and the outer plexiform layer (OPL), and in the perivascular membranes. GFAP proteins were seen mainly at the endfeet of Müller cells (Fig. 3A2,3), which is consistent with previous reports (Kofuji et al., 2002; Iandiev et al., 2006; Wurm et al., 2006). Kir4.1 expression in Müller cells, especially at the endfeet, showed a remarkable reduction in retinal sections obtained at different times (G1w–G6w) from the COH rats (Fig. 3B1,C1,D1,E1). Consistently, total Kir4.1 proteins extracted from the operated eyes in the COH retinas detected by Western blot were significantly decreased, as illustrated in the representative results in Figure 3F. The protein level of Kir4.1 was greatly reduced to 22.6 ± 10.6% of control on G1w (n = 6, p < 0.001 vs values obtained from sham-operated control eyes and unoperated contralateral eyes of the same rats), and it remained at this low level until G6w (24.6 ± 9.3% on G2w, n = 6; 27.1 ± 11.4% on G4w, n = 6; 35.5 ± 16.1% of control on G6w, n = 6; p < 0.001 vs values obtained from sham-operated control eyes and unoperated contralateral eyes of the same rats, respectively) (Fig. 3G). Meanwhile, immunocytochemical experiments showed that GFAP expression in Müller cells was progressively enhanced in association with the elevated IOP (Fig. 3B2–E2), suggesting Müller cells gliosis. Consistent with this, Western blot experiments (Fig. 3F) showed that the average densities of GFAP proteins were considerably increased to 196.0 ± 21.5% (n = 6), 219.4 ± 24.1% (n = 6), 260.1 ± 23.3% (n = 6), and 400.3 ± 36.6% of control (n = 6) (p < 0.001 vs the values of sham-operated control and unoperated contralateral eyes of the same rats) on G1w, G2w, G4w, and G6w, respectively (Fig. 3H).
Figure 3.

Changes in Kir4.1 and GFAP expression in Müller cells of COH rats. A1–E1, Immunofluorescence labeling showing the changes in Kir4.1 protein expression in rat retinal vertical slices taken from sham-operated retina (Control), and those obtained at the first (G1w), second (G2w), fourth (G4w), and sixth week (G6w) after the operation. Note that Kir4.1 expression in the endfeet of Müller cells was reduced in the COH retinal sections (B1, C1, D1, E1). A2–E2, Immunohistochemical staining showing DAPI and GFAP protein expression in the same slices as shown in A1, B1, C1, D1, and E1, respectively. C3–E3, Merged images of A1–E1 and A2–E2. Scale bar, (for all the images) 20 μm. IPL, Inner plexiform layer; INL, inner nuclear layer; ONL, outer nuclear layer. F, Representative immunoblots showing the changes in Kir4.1 (top) and GFAP (middle) protein expression in sham-operated (Ctr) and COH retinal extracts obtained from both operated eyes and their unoperated contralateral eyes in the same rats at different time points (G1w, G2w, G4w, and G6w) after the operation. G, H, Bar charts summarizing the average densitometric quantification of immunoreactive bands of Kir4.1 (G) and GFAP (H) protein expression at different time points after the operation. All data are normalized to control (Ctr). n = 6 all groups. ***p < 0.001 versus the unoperated contralateral eyes in the same rats, and ###p < 0.001 versus Ctr.
Activation of mGluR I inhibits Kir currents of isolated Müller cells
What caused the reduction of Kir currents in the COH model? It was previously demonstrated that l-glutamate acted at mGluRs to inhibit outward K+ currents in isolated salamander Müller cells (Schwartz, 1993). Moreover, as the glutamate transporter levels were reduced significantly in glaucoma models, it has been suggested that the glutamate concentrations in the retina might have increased (Dreyer et al., 1996; Vorwerk et al., 2000; Martin et al., 2002; Guo et al., 2006; Salt and Cordeiro, 2006; Harada et al., 2007; Seki and Lipton, 2008). We therefore proposed a working hypothesis that increased extracellular glutamate in experimental glaucoma may overactivate mGluR I, thereby inhibiting Kir currents. This hypothesis was tested by examining effects of mGluR I agonist and antagonist on Kir currents. As shown in Figure 4A, the selective mGluR I agonist DHPG (10 μm) significantly and voltage-dependently inhibited Kir currents in isolated rat retinal Müller cells in a reversible manner. Figure 4B shows the DHPG action as a function of time. Bath application of DHPG (10 μm) rapidly decreased the Kir current peak amplitudes to 39.1 ± 5.9% of pretreatment levels in 200 s (n = 6, p < 0.001), and they then recovered gradually with normal bath solution washout. The DHPG effect was blocked by an intracellular dialysis of GDP-β-S (3 mm), a G protein inhibitor, through recording pipettes. During GDP-β-S dialysis, as shown in Figure 4C, bath application of 10 μm DHPG for 200 s had no effect on the Kir currents. The average peak current amplitudes in the presence of DHPG for 200 s were 98.2 ± 4.7% of the control levels (n = 5, p = 0.98). As mGluR I consists of two receptor subtypes, mGluR1 and mGluR5, to explore which one or whether both were involved in the DHPG effect, we used selective antagonists for these two subtypes. When isolated Müller cells were preincubated with MPMQ, a selective mGluR1 antagonist, 10 μm DHPG persisted to inhibit Kir currents in the presence of MPMQ. Bath application of DHPG for 200 s reduced the current amplitudes to 43.0 ± 2.8% of that before DHPG application (n = 6, p < 0.001) (Fig. 4D). In contrast, preincubation with 10 μm MPEP, a highly selective mGluR5 antagonist, almost completely eliminated the DHPG effect. In the presence of MPEP, Kir current amplitudes were hardly changed by 10 μm DHPG (94.5 ± 2.6% of control, n = 5, p = 0.97) (Fig. 4E). These results suggest that it was mGluR5, but not mGluR1, that was involved in the DHPG effect on Kir currents.
Figure 4.

Activation of mGluR I suppresses Kir currents in Müller cells. A, DHPG (10 μm), a selective mGluR I receptor agonist, inhibited remarkably and reversibly Kir currents. The currents were evoked by a series of pulses from a holding potential of −80 mV to −160 mV in −20 mV increments. The I–V relationships show voltage-dependent reduction in Kir current amplitudes. n = 10. B, Plot of Kir inward currents evoked by a voltage step from a holding potential of −80 mV to −160 mV as a function of time, showing that DHPG (10 μm) significantly suppressed the Kir current amplitudes. n = 6. The representative current traces (top) are taken from the time points as indicated. C, Internal dialysis of GDP-β-S (3 mm) prevented the DHPG-induced suppression of Kir currents. The representative current traces (top) are used to generate the plot. n = 5. D, Plot of Kir inward currents as a function of time, showing that preperfusion of MPMQ (10 μm) failed to block DHPG-induced reduction in Kir current amplitudes. n = 6. The representative current traces (top) are taken from the time points as indicated. E, Plot of Kir inward currents as a function of time, showing that preperfusion of MPEP (10 μm) blocked DHPG-induced reduction in Kir current amplitudes. n = 5. The representative current traces (top) are taken from the time points as indicated. All the current amplitudes are normalized to the amplitude of the first evoked event.
Changes in Kir and GFAP expression by activation of mGluR I receptor
How activation of mGluR I change the expression of Kir and GFAP was studied by immunohistochemistry and Western blotting. DHPG of 10 μm (2 μl) was first intravitreally injected into left eyes, and after 2 weeks retinal slices and extracts were made for immunohistochemistry and Western blot analysis. As shown in Figure 5, A2 and C2, Kir4.1 expression in Müller cells was remarkably decreased, as compared with those of normal saline (NS)-injected retinas (Fig. 5A1,C1). Similar observations were obtained in other five rats. Western blotting revealed that total Kir4.1 protein in DHPG-injected retinas was profoundly decreased to 10.8 ± 9.7% of NS-injected group (n = 6, p < 0.001; Fig. 5D). Meanwhile, DHPG injection induced a much stronger GFAP expression in Müller cells (Fig. 5B2,C2), as compared with that observed in normal ones (Fig. 5B1,C1). Western blotting indicated that total GFAP protein was increased to 172.4 ± 15.0% of NS-injected retinas (n = 6, p < 0.001; Fig. 5E), following DHPG injection. These changes in Kir and GFAP expression caused by activation of mGluR I were reminiscent of those observed in the COH model.
Figure 5.

DHPG decreases Kir4.1 protein expression and induces GFAP expression in Müller cells. DHPG (10 μm) or vehicle (0.9% saline, NS) were injected into the vitreous space in a volume of 2 μl. The retinas were sectioned 2 weeks after the injection. A1, A2, Immunofluorescence labeling showing the Kir4.1 protein expression in retinal vertical slices taken from the NS- (A1) or the DHPG- (A2) injected rat. B1, B2, Immunofluorescence labeling showing the GFAP protein expression and DAPI in the same slices as shown in A1 and A2. C1, C2, Merged images of A1 and B1, A2 and B2, respectively. Scale bar, 20 μm. D, Representative Western blot analysis showing the Kir4.1 protein expression in the NS- and DHPG-injected eyes (top). Bar chart summarizing the Kir4.1 protein expression in the NS- and DHPG-injected eyes (below). n = 6, ***p < 0.001 versus NS. E, Representative Western blot analysis showing the GFAP protein expression in the NS- and DHPG-injected eyes (top). Bar chart summarizing the GFAP protein expression in the NS- and DHPG-injected eyes (below). n = 6, ***p < 0.001 versus NS. All data were normalized to those obtained in the NS eyes.
To address the question whether the DHPG-induced increase in GFAP expression in normal retina could be a consequence of reduced activity of Kir channels, we examined effects of DHPG on GFAP expression in Müller cells in normal retina when Kir channels were blocked by Ba2+. BaCl2 (200 μm) was intravitreally injected 1 d before DHPG (10 μm) injection. Five days after DHPG injection, expression of GFAP in retinal slices was examined by immunohistochemistry and retinal extracts by Western blot analysis. Figure 6A2 shows that GFAP expression was significantly increased in Müller cells in the BaCl2-injected retinal slice, compared with the control slice (Fig. 6A1). In the retina that received additional injection of DHPG (10 μm, 2 μl), however, no further increase in GFAP expression was seen (Fig. 6A3). Similarly, Western blotting revealed that total GFAP protein in BaCl2-injected retinas was profoundly increased to 171.7 ± 15.3% of NS-injected group (n = 6, p < 0.001; Fig. 6B), but it was not further increased by additional injection of DHPG (165.3 ± 17.0% of NS-injected retinas, n = 6, p = 0.875 vs BaCl2-injected retinas) (Fig. 6B). To exclude the possibility that Ba2+ may have nonspecific effects on the GFAP protein expression, we did control experiments by intravitreally injecting CoCl2, another toxic cation, which does not block Kir channels, using a similar procedure to that for Ba2+ injection. As shown in Figure 6C2, GFAP protein expression in Müller cells was not increased, but became even somewhat weaker, in the CoCl2 (50 μm, 2 μl)-injected retinal slice, as compared with that in the NS-injected retinal slice (Fig. 6C1). Moreover, additional injection of DHPG (10 μm, 2 μl) induced an increase in GFAP protein expression in the retina that had received injection of CoCl2 1 d before (Fig. 6C3). Consistently, Western blotting revealed that total GFAP protein in CoCl2-injected retinas was not different from that of NS-injected group (98.9 ± 9.3% of NS-injected retinas, n = 4, p > 0.05, Fig. 6D), but it was significantly increased by additional injection of DHPG (195.3 ± 18.9% of NS-injected retinas, n = 4, p < 0.001) (Fig. 6D). These results suggest that the elevated expression of GFAP in Müller cells by Ba2+ was not due to a nonspecific response of these cells to injection of toxic cation.
Figure 6.

Inhibition of Kir channels blocks DHPG-induced increase in GFAP expression in Müller cells. BaCl2 (200 μm), CoCl2 (50 μm), DHPG (10 μm), or vehicle (0.9% saline, NS) were injected into the vitreous space in a volume of 2 μl. BaCl2 or CoCl2 was injected 1 d before DHPG injection and the retinas were collected 5 d after the DHPG injection. A1–A3, Immunofluorescence labeling showing the GFAP protein expression and DAPI in retinal vertical slices taken from the NS- (A1), the BaCl2- (A2), and the BaCl2+DHPG- (A3) injected rats. B, Representative Western blot analysis showing the GFAP protein expression in NS-, BaCl2- and BaCl2+DHPG-injected eyes (top). Bar chart summarizing the GFAP protein expression in NS-, BaCl2-, and BaCl2+DHPG-injected eyes (below). n = 6. ***p < 0.001 versus NS. C1–C3, Immunofluorescence labeling showing the GFAP protein expression and DAPI in retinal vertical slices taken from NS- (C1), CoCl2- (C2), and CoCl2+DHPG- (C3) injected rats. D, Representative Western blot analysis showing the GFAP protein expression in NS-, CoCl2-, and CoCl2+DHPG-injected eyes (top). Bar chart summarizing the GFAP protein expression in NS-, CoCl2-, and CoCl2+DHPG-injected eyes (below). n = 6 for NS and 4 for CoCl2 and CoCl2+DHPG groups, respectively. ***p < 0.001 versus NS and ###p < 0.001 versus CoCl2 alone. All data were normalized to those obtained in the NS eyes. Scale bar, (for all the images) 20 μm.
Expression of Kir4.1 and GFAP in Müller cells in the COH model was altered by blocking mGluR5. NS or MPEP (10 μm) was injected intravitreally 3 d before the operation, and the expression of Kir4.1 and GFAP was examined in retinal sections 2 weeks after the operation. Injection of MPEP caused a significant increase in Kir4.1 expression and reduction in GFAP expression in Müller cells (Fig. 7A2,B2,C2), compared with those obtained in the NS-injected group (Fig. 7A1,B1,C1). Similar results were obtained in other five retinas. Western blotting further revealed that total Kir4.1 protein extracted from MPEP-injected retinas was increased to 152.1 ± 9.5% of NS-injected retinas (n = 6, p < 0.001; Fig. 7D), while total GFAP protein was decreased to 51.0 ± 4.8% of NS-injected retinas (n = 6, p < 0.001; Fig. 7E).
Figure 7.

Selective mGluR5 antagonist MPEP modulates Kir4.1 protein expression and inhibits GFAP expression in COH model. MPEP (1 μm) or vehicle (0.9% saline, NS) were injected into the vitreous space in a volume of 2 μl 3 d before the operation. A1, A2, Immunofluorescence labeling showing the Kir4.1 protein expression in retinal vertical slices taken from NS- (A1) or MPEP- (A2) injected rat at second week (G2w) after the operation. B1, B2, Immunofluorescence labeling showing the GFAP protein expression and DAPI in the same slices as shown in A1 and A2. C1, C2, Merged images of A1 and B1, A2 and B2, respectively. Scale bar, 20 μm. D, Representative Western blot analysis showing the Kir4.1 protein expression in NS- and MPEP-injected eyes (top). Bar chart summarizing the Kir4.1 protein expression in NS- and MPEP-injected eyes (below). n = 6, ***p < 0.001 versus NS. E, Representative Western blot analysis showing the GFAP protein expression in NS- and MPEP-injected eyes (top). Bar chart summarizing the GFAP protein expression in NS- and MPEP-injected eyes (below). n = 6, ***p < 0.001 versus NS. All data were normalized to those obtained in the NS eyes.
Ca2+-dependent PLC/IP3-ryanodine/PKC signaling pathway mediates the DHPG effect
We explored the intracellular signaling pathway that links the activation of mGluR I and the down-regulation of Kir currents in isolated Müller cells. Activation of mGluR I may stimulate adenylyl cyclase, which is followed by the activation of cAMP-dependent protein kinase PKA. While this signaling pathway is responsible for the inhibition of outward K+ currents in salamander Müller cells (Schwartz, 1993), it is unlikely involved in the DHPG-induced inhibition of Kir currents in rat Müller cells. Bath application of the adenylyl cyclase activator forskolin (5 μm) did not change the Kir current amplitudes (95.2 ± 6.2% of pretreatment levels, n = 6, p = 0.90; Fig. 8A). In addition, inhibition of intracellular cAMP by the membrane-permeable cAMP inhibitor Rp-cAMP (20 μm) hardly influenced the DHPG-induced suppression of Kir currents (Fig. 8B), and the peak current amplitudes obtained in the presence of Rp-cAMP were reduced to 52.8 ± 7.2% of control (n = 5, p < 0.001).
Figure 8.

PLC-PKC signaling pathway mediates the DHPG-induced inhibition of Kir currents. A, Plot of Kir inward currents as a function of time, showing that forskolin (5 μm) hardly changed the Kir currents. n = 6. The representative current traces (top) are taken from the time points as indicated. B, Plot of Kir inward currents evoked by a hyperpolarized voltage pulse from a holding potential of −80 mV to −160 mV as a function of time. Rp-cAMP (20 μm) failed to block the DHPG-induced reduction in Kir current amplitudes. The representative current traces (top) are used to generate the plot. n = 5. C, D, Plot of Kir inward currents as a function of time, showing that internal preinfusion of U73122 (5 μm) (C, n = 5), but not D609 (60 μm) (D, n = 6), blocked the inhibition effect of DHPG on Kir currents. The representative current traces (top) are taken from the time points as indicated. E, Kir current as a function of time, showing that intracellular pre-dialysis of Bis IV (5 μm) completely eliminated the DHPG effect on Kir currents (n = 6). The representative current traces are shown at the top. F, Kir current as a function of time, showing that extracellular application of chelerythrine chloride (5 μm) completely eliminated the DHPG effect on Kir currents (n = 9). All the current amplitudes are normalized to the amplitude of the first evoked event.
mGluR I is positively coupled to the PLC-PKC signaling pathway (Fagni et al., 2000; Hannan et al., 2001; Lüscher and Huber, 2010). To test possible involvement of PLC, isolated Müller cells were intracellularly dialyzed with U73122 (5 μm), a phosphatidylinositol (PI)-PLC inhibitor, through recording pipettes for at least 5 min, and then the effect of DHPG (10 μm) on Kir currents was examined. In the presence of U73122, DHPG no longer suppressed Kir currents with the peak current amplitudes at 3 min after DHPG application being 95.7 ± 10.7% of control (n = 5, p = 0.88; Fig. 8C). In contrast, intracellular dialysis with D609 (60 μm), a phosphatidylcholine (PC)-PLC inhibitor, did not block the DHPG-induced decrease in Kir currents, with the average peak current amplitudes, obtained at 3 min after DHPG application, being 45.2 ± 2.7% of control (n = 6, p < 0.001; Fig. 8D). Furthermore, consistent with previous studies conducted in a variety of cells (Henry et al., 1996; Karle et al., 2002; Park et al., 2005), intracellular dialysis with Bis IV (5 μm), a specific PKC inhibitor, fully eliminated the suppression of Kir currents by DHPG (101.2 ± 2.3% of control, n = 6, p = 0.82; Fig. 8E). Internal dialysis with chelerythrine chloride (5 μm), another PKC inhibitor, yielded similar results (104.2 ± 5.1% of control, n = 9, p = 0.92; Fig. 8F). These results suggest the involvement of the PLC-PKC signaling pathway in the DHPG effect on Kir currents.
Activation of mGluR I may increase intracellular Ca2+ concentrations ([Ca2+]i) through IP3- and/or ryanodine-sensitive Ca2+ stores in neurons (Fagni et al., 2000; Morikawa et al., 2003; Sohn et al., 2011). When recording pipettes were filled with internal Ca2+-free solution containing 10 mm BAPTA, a calcium chelator, bath application of 10 μm DHPG did not change the Kir currents (Fig. 9A), suggesting the involvement of intracellular calcium stores. Furthermore, by blocking IP3 receptors with intracellular dialysis of heparin (5 mg/ml), a competitive IP3 receptor antagonist, DHPG of 10 μm caused a moderate suppression of the Kir currents, which was much less than that obtained without dialysis of heparin (84.3 ± 6.2% vs 39.1 ± 5.9% of control, n = 5, p = 0.024) (Fig. 9B). Experiments with caffeine, a ryanodine receptor agonist, yielded similar results. Bath application of 1 mm caffeine reduced the Kir currents by 18.2 ± 6.8% (n = 6) (Fig. 9C). It seems that activation of mGluR I-induced increase in [Ca2+]i in rat Müller cells could be due to Ca2+ release from both IP3- and ryanodine-sensitive Ca2+ stores.
Figure 9.

Ca2+ release from intracellular stores is involved in the DHPG effect on Kir currents. A, Plot of Kir inward currents as a function of time, showing that internal preperfusion of BAPTA (10 mm) to chelate intracellular Ca2+ eliminated fully the DHPG effect on Kir currents (n = 6). The representative current traces recorded at different time points are shown at the top as indicated. B, Plot of Kir inward currents as a function of time, showing that intracellular preinfusion of heparin (5 mg/ml) partially rescued the DHPG-induced inhibition in Kir currents (n = 5). The representative current traces (top) are taken from the time points as indicated. C, Plot of Kir inward currents as a function of time, showing that extracellular perfusion of caffeine (1 mm) induced a minor reduction in Kir currents with a long time delay (n = 6). The representative current traces are shown at the top. All the current amplitudes are normalized to the amplitude of the first evoked event.
Changes in calcium/calmodulin-dependent protein kinase II (CaMKII) activity, which have been reported in various types of central neurons (Choe and Wang, 2001; Mockett et al., 2011), were unlikely involved in DHPG-induced suppression of Kir currents in Müller cells. Bath application of CaMKII inhibitors KN-62 (5 μm)/KN-93 (10 μm) did not change the Kir currents. In the presence of KN-62/KN-93, addition of 10 μm DHPG suppressed the Kir currents, which was not much different from that observed in NS. Following application DHPG for 300 s, the average current amplitudes were 49.7 ± 16.3% (for KN-62, n = 5, p < 0.001) (Fig. 10A) and 44.8 ± 5.5% of the level obtained before DHPG application (for KN-93, n = 9, p < 0.001) (Fig. 10B).
Figure 10.

No involvement of CaMKII in DHPG-induced inhibition of Kir currents. A, B, Plots of Kir currents as a function of time, showing that extracellular application of CaMKII inhibitors KN-62 (5 μm) (A, n = 5) or KN-93 (10 μm) (B, n = 9) did not block the DHPG effect on Kir currents. The representative current traces (top) are taken from the time points as indicated. All the current amplitudes are normalized to the amplitude of the first evoked event.
Discussion
Kir4.1 channels in rat Müller cells
Rat Müller cells express various subtypes of Kir channels, including Kir2.1, Kir4.1, and Kir 5.1 (Kofuji et al., 2002; Ishii et al., 2003; Iandiev et al., 2006; Wurm et al., 2006). These channels with high K+ permeability are essential for Müller cells to maintain homeostasis of [K+]o (Kofuji et al., 2002; Bringmann et al., 2006). In the present study, the currents evoked by voltage steps/ramps recorded from isolated rat Müller cells were highly dependent on [K+]o (Fig. 1B), exhibiting a weakly rectifying I–V relationship (Fig. 1D,E). Moreover, the currents were largely suppressed by Ba2+. All these results suggest that Kir4.1 may be a primary protein component mediating the currents (Kofuji et al., 2002; Bringmann et al., 2006). It should be noted that the currents recorded under our experimental conditions may involve a smaller component contributed by members of the tandem-pore domain (2P-domain) K+ channel, such as TASK-1, TASK-2, and TASK-3, which are expressed in rat Müller cells (Kofuji et al., 2002; Skatchkov et al., 2006).
Kir channels and Müller cell gliosis in experimental glaucoma
Although down-regulation or loss of Müller cell Kir currents commonly occurs in many retinal injuries and diseases, there is only limited evidence, indicating that this is the case in glaucoma and experimental glaucoma (Francke et al., 1997; Bolz et al., 2008). In our rat COH model we clearly revealed that Kir currents were significantly reduced, which is consistent with the observation in patients with secondary glaucoma, showing large decrease or loss of Kir currents of Müller cells (Francke et al., 1997). The decreased Kir currents may be due to the reduced expression of Kir4.1 proteins, as shown by Western blotting (Fig. 3G). Meanwhile, a progressive increase in GFAP expression in Müller cells was observed (Fig. 3H), suggesting the occurrence of Müller cell gliosis. We tend to consider a possibility that the enhanced GFAP expression may be a consequence of the reduced activity of Kir4.1 channels. A strong argument in favor of this possibility is that intravitreal injection of BaCl2, which is supposed to block Kir channels, increased GFAP expression in normal retina and no further increase in GFAP expression was seen even when mGluR I was activated by DHPG (Fig. 6). It should be noted that during 4–6 weeks after the operation the inhibition of Kir currents was attenuated (Fig. 2), but GFAP expression did not match such changes. A possible explanation is that Müller cell gliosis lasts for a long period of time even when the stimulating factor (changes in Kir currents) is attenuated. In retinal diseases other than glaucoma it has been shown that alteration of Kir currents may be associated with the extent of gliosis. That is, no significant change in Kir currents is found in some slow retinal degenerative diseases, such as observed in DBA/2J mice (Bolz et al., 2008), where Müller cells undergo a nonproliferative gliosis, moderate reduction of Kir currents is seen in conservative gliosis, and dramatic down-regulation is related to proliferative gliosis (Bringmann et al., 2000).
It was previously reported that the expression of Kir4.1 protein in Müller cells was strongly downregulated by retinal ischemia-reperfusion, while the Kir2.1 protein expression was not altered (Iandiev et al., 2006). In the present study, the weakly rectifying I–V relationships of the Kir channel currents and the suppression of the currents by Ba2+ all suggest that Kir4.1 may be a primary component mediating these currents in our conditions (Fig. 1). Moreover, in glaucomatous eyes the decrease in Kir4.1 protein expression was observed in all parts of the Müller cells (Fig. 3), while the changes caused by ischemia occurred in specific regions of Müller cells (Iandiev et al., 2006).
A question that remains to be addressed is how activation of mGluR I leads to down-regulation of Kir4.1 protein expression in Müller cells. There may be two possible pathways. The first possible pathway could be that persistent glutamate exposure activates mGluR I-mediated intracellular signaling, which may decrease the mRNA level of Kir4.1 by down-regulating transcription. It has been shown that retinal ischemia-induced decrease in Kir4.1 protein expression is accompanied by a down-regulation of the mRNA level for Kir4.1 (Iandiev et al., 2006). An alternative possibility is that activation of mGluR I increases the activity of PKC, thus leading to Kir4.1 protein internalization. Although there are no data about Kir4.1 channel internalization, PKC-dependent ATP-sensitive potassium channel (KATP) internalization and lysosomal degradation have been reported (Hu et al., 2003; Manna et al., 2010; Aziz et al., 2012).
Activation of mGluR I may contribute to Müller cell gliosis by suppressing Kir channels
As shown in Figure 4, activation of mGluR5 by DHPG suppressed Kir channels. Unlike mGluR I receptor-mediated regulation of outward K+ currents in Müller cells due to activation of the cAMP-PKA pathway (Schwartz, 1993), mGluR I-mediated suppression of Kir currents in rat Müller cells depends on the intracellular Ca2+-dependent PI-PLC/PKC signaling pathway, but without the involvement of PKA and CaMKII signaling pathways. Meanwhile, activation of mGluR I enhanced the expression of GFAP in Müller cells. It was evident that the changes in Kir and GFAP expression induced by mGluR I activation mimicked the changes induced by COH. Moreover, as shown in Figure 6, suppression of Kir channels by Ba2+ blocked DHPG-induced Müller cells gliosis. The enhanced Kir4.1 expression and the reduced GFAP expression in Müller cells by MPEP in the COH rats are suggestive of the involvement of mGluR5 in Müller cell gliosis (Fig. 7). It is recently shown that functional expression of Kir4.1 is sufficient to inhibit proliferation in glioma cells by shifting the cells from G2/M phase into the quiescent G0/G1 stage of the cell cycle (Higashimori and Sontheimer, 2007). It should be indicated that whether glutamate levels are increased in glaucomatous eyes is a controversial issue (Levkovitch-Verbin et al., 2002; Dreyer et al., 1996). However, growing evidence suggests that the excess of extracellular glutamate may be a potential risk factor for retinal injury in glaucoma, as evidenced by prevention of ganglion cell death due to the administration of NMDA receptor antagonists MK-801/memantine in rat experimental glaucoma models (Guo et al., 2006; Salt and Cordeiro, 2006; Seki and Lipton, 2008). Indeed, glutamate transporters were found to be significantly reduced in rat glaucoma models, indicating frustration of the effective buffering of extracellular glutamate (Vorwerk et al., 2000; Martin et al., 2002; Harada et al., 2007). In case extracellular glutamate levels are elevated in glaucoma, mGluR5 may be overactivated, thus causing a suppression of Kir channels of Müller cells, which results in enhanced GFAP expression (gliosis).
Elevated retinal glutamate levels may also lead to an overactivation of NMDA receptors in general. However, many studies show no significant glutamate-induced Ca2+ transient in mammalian (rat, guinea pig) retinal Müller cells (Newman and Zahs, 1997; Bringmann et al., 2002b; Newman, 2005; Rillich et al., 2009). Because Müller cells have a relatively hyperpolarized resting membrane potential, the NMDA channels in these cells may be blocked by Mg2+ under physiological conditions.
Pathophysiological implication of Müller cell gliosis in glaucoma
Effects caused by Müller cell gliosis on retinal neurons may be cytoprotective or cytotoxic (for review, see Bringmann et al., 2006). Among them, Müller cell gliosis in experimental glaucoma may result in dysregulation of the glio-neuronal interaction in the retina (Bringmann et al., 2000; Bringmann and Reichenbach, 2001). It is also noteworthy that the reduced activity of Kir channels due to the overactivation of mGluR I may lead to a depolarization of Müller cells, thus reducing glutamate uptake of Müller cells, as glutamate transport into the cells is strongly voltage-dependent (Sarantis and Attwell, 1990). Such a change in glutamate transport could cause an increased accumulation of extracellular glutamate, resulting in a further activation of mGluR I. Moreover, down-regulation of Kir channels may also change the water transport into the cells by impairing spatial potassium buffering, which is suggested to contribute to the development of retinal edema (Nagelhus et al., 1999; Pannicke et al., 2004). The present study provides evidence, suggesting that appropriate reduction of retinal mGluR I activity in glaucoma, which could alleviate Müller cell gliosis, may be an effective way for preventing the loss of retinal ganglion cells. It should be noted that our glaucoma model, exhibiting a rapid increase in IOP, is different from the one in DBA/2J mice that have a much longer time course of IOP elevation before elevation can be seen. Whether mechanisms underlying Müller cell gliosis in a chronic glaucoma model, like in DBA/2J mice, which is more pertinent to human glaucoma, are similar or different needs to be further explored.
Footnotes
This work was supported by grants from the National Program of Basic Research sponsored by the Ministry of Science and Technology of China (2011CB504602), the National Natural Science Foundation of China (31070966; 30900427; 30973264; 30930034), the Key Research Program of Science and Technology Commissions of Shanghai Municipality (11JC1401200), Research Fund for the Doctoral Program of Higher Education of China (20110071110031), and the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry.
References
- Aziz Q, Thomas AM, Khambra T, Tinker A. Regulation of the ATP-sensitive potassium channel subunit, Kir6.2, by a Ca2+-dependent protein kinase C. J Biol Chem. 2012;287:6196–6207. doi: 10.1074/jbc.M111.243923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolz S, Schuettauf F, Fries JE, Thaler S, Reichenbach A, Pannicke T. K+ currents fail to change in reactive retinal glial cells in a mouse model of glaucoma. Graefes Arch Clin Exp Ophthalmol. 2008;246:1249–1254. doi: 10.1007/s00417-008-0872-x. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Reichenbach A. Role of Muller cells in retinal degenerations. Front Biosci. 2001;6:E72–E92. doi: 10.2741/bringman. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Francke M, Pannicke T, Biedermann B, Kodal H, Faude F, Reichelt W, Reichenbach A. Role of glial K+ channels in outogeny and gliosis: a hypothesis based upon studies on Müller cells. Glia. 2000;29:35–44. doi: 10.1002/(sici)1098-1136(20000101)29:1<35::aid-glia4>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Pannicke T, Uhlmann S, Kohen L, Wiedemann P, Reichenbach A. Membrane conductance of Müller glial cells in proliferative diabetic retinopathy. Can J Ophthalmol. 2002a;37:221–227. doi: 10.1016/s0008-4182(02)80113-2. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Pannicke T, Weick M, Biedermann B, Uhlmann S, Kohen L, Wiedemann P, Reichenbach A. Activation of P2Y receptors stimulates potassium and cation currents in acutely isolated human Muller (glial) cells. Glia. 2002b;37:139–152. doi: 10.1002/glia.10025. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, Skatchkov SN, Osborne NN, Reichenbach A. Müller cells in the health and diseased retina. Prog Retin Eye Res. 2006;25:397–424. doi: 10.1016/j.preteyeres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Iandiev I, Pannicke T, Wurm A, Hollborn M, Wiedemann P, Osborne NN, Reichenbach A. Cellular signaling and factors involved in Müller cell gliosis: neuroprotective and detrimental effects. Prog Retin Eye Res. 2009;28:423–451. doi: 10.1016/j.preteyeres.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Butt AM, Kalsi A. Inwardly rectifying potassium channels (Kir) in central nervous system glia: a special role for Kir4.1 in glial functions. J Cell Mol Med. 2006;10:33–44. doi: 10.1111/j.1582-4934.2006.tb00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caraci F, Battaglia G, Sortino MA, Spampinato S, Molinaro G, Copani A, Nicoletti F, Bruno V. Metabotropic glutamate receptors in neurodegeneration/neuroprotection: Still a hot topic? Neurochem Int. 2012 doi: 10.1016/j.neuint.2012.01.017. [DOI] [PubMed] [Google Scholar]
- Chen J, Miao Y, Wang XH, Wang Z. Elevation of p-NR2AS1232 by Cdk5/p35 contributes to retinal ganglion cell apoptosis in a rat experimental glaucoma model. Neurobiol Dis. 2011;43:455–464. doi: 10.1016/j.nbd.2011.04.019. [DOI] [PubMed] [Google Scholar]
- Choe ES, Wang JQ. Group I metabotropic glutamate receptors control phosphorylation of CREB, Elk-1 and ERK via a CaMKII-dependent pathway in rat striatum. Neurosci Lett. 2001;313:129–132. doi: 10.1016/s0304-3940(01)02258-3. [DOI] [PubMed] [Google Scholar]
- Dreyer EB, Zurakowski D, Schumer RA, Podos SM, Lipton SA. Elevated glutamate levels in the vitreous body of humans and monkeys with glaucoma. Arch Ophthalmol. 1996;114:299–305. doi: 10.1001/archopht.1996.01100130295012. [DOI] [PubMed] [Google Scholar]
- Fagni L, Chavis P, Ango F, Bockaert J. Complex interactions between mGluRs, Intracellular Ca2+ stores and ion channels in neurons. Trends Neurosci. 2000;23:80–88. doi: 10.1016/s0166-2236(99)01492-7. [DOI] [PubMed] [Google Scholar]
- Francke M, Pannicke T, Biedermann B, Faude F, Wiedemann P, Reichenbach A, Reichelt W. Loss of inwardly rectifying potassium currents by human retinal glial cells in diseases of the eye. Glia. 1997;20:210–218. doi: 10.1002/(sici)1098-1136(199707)20:3<210::aid-glia5>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Francke M, Faude F, Pannicke T, Bringmann A, Eckstein P, Reichelt W, Wiedemann P, Reichenbach A. Electrophysiology of rabbit Müller (glial) cells in experimental retinal detachment and PVR. Invest Ophthalmol Vis Sci. 2001;42:1072–1079. [PubMed] [Google Scholar]
- Goureau O, Régnier-Ricard F, Courtois Y. Requirement for nitric oxide in retinal neuronal cell death induced by activated Müller glial cells. J Neurochem. 1999;72:2506–2515. doi: 10.1046/j.1471-4159.1999.0722506.x. [DOI] [PubMed] [Google Scholar]
- Guo L, Salt TE, Maass A, Luong V, Moss SE, Fitzke FW, Cordeiro MF. Assessment of neuroprotective effects of glutamate modulation on glaucoma-related retinal ganglion cell apoptosis in vivo. Invest Ophthalmol Vis Sci. 2006;47:626–633. doi: 10.1167/iovs.05-0754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan AJ, Blakemore C, Katsnelson A, Vitalis T, Huber KM, Bear M, Roder J, Kim D, Shin HS, Kind PC. PLC-beta1, activated via mGluRs, mediates activity-dependent differentiation in cerebral cortex. Nat Neurosci. 2001;4:282–288. doi: 10.1038/85132. [DOI] [PubMed] [Google Scholar]
- Harada T, Harada C, Nakamura K, Quah HM, Okumura A, Namekata K, Saeki T, Aihara M, Yoshida H, Mitani A, Tanaka K. The potential role of glutamate transporters in the pathogenesis of normal tension glaucoma. J Clin Invest. 2007;117:1763–1770. doi: 10.1172/JCI30178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry P, Pearson WL, Nichols CG. Protein kinase C inhibition of cloned inward rectifier (HRK1/KIR2.3) K+ channels expressed in Xenopus oocytes. J Physiol. 1996;495:681–688. doi: 10.1113/jphysiol.1996.sp021625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashimori H, Sontheimer H. Role of Kir4.1 channels in growth control of glia. Glia. 2007;55:1668–1679. doi: 10.1002/glia.20574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu K, Huang CS, Jan YN, Jan LY. ATP-sensitive potassium channel traffic regulation by adenosine and protein kinase C. Neuron. 2003;38:417–432. doi: 10.1016/s0896-6273(03)00256-3. [DOI] [PubMed] [Google Scholar]
- Iandiev I, Tenckhoff S, Pannicke T, Biedermann B, Hollborn M, Wiedemann P, Reichenbach A, Bringmann A. Differential regulation of Kir4.1 and Kir2.1 expression in the ischemic rat retina. Neurosci Lett. 2006;396:97–101. doi: 10.1016/j.neulet.2005.11.016. [DOI] [PubMed] [Google Scholar]
- Inman DM, Horner PJ. Reactive nonproliferative gliosis predominates in a chronic mouse model of glaucoma. Glia. 2007;55:942–953. doi: 10.1002/glia.20516. [DOI] [PubMed] [Google Scholar]
- Ishii M, Horio Y, Tada Y, Hibino H, Inanobe A, Ito M, Yamada M, Gotow T, Uchiyama Y, Kurachi Y. Expression and clustered distribution of an inwardly rectifying potassium channel, KAB-2/Kir4.1, on mammalian retinal Müller cell membrane: their regulation by insulin and laminin signals. J Neurosci. 1997;17:7725–7735. doi: 10.1523/JNEUROSCI.17-20-07725.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii M, Fujita A, Iwai K, Kusaka S, Higashi K, Inanobe A, Hibino H, Kurachi Y. Differential expression and distribution of Kir5.1 and Kir4.1 inwardly rectifying K+ channels in retina. Am J Physiol Cell Physiol. 2003;285:C260–C267. doi: 10.1152/ajpcell.00560.2002. [DOI] [PubMed] [Google Scholar]
- Karle CA, Zitron E, Zhang W, Wendt-Nordahl G, Kathöfer S, Thomas D, Gut B, Scholz E, Vahl CF, Katus HA, Kiehn J. Human cardiac inwardly-rectifying K+ channel Kir(2.1b) is inhibited by direct protein kinase C-dependent regulation in human isolated cardiomyocytes and in an expression system. Circulation. 2002;106:1493–1499. doi: 10.1161/01.cir.0000029747.53262.5c. [DOI] [PubMed] [Google Scholar]
- Kashiwagi K, Iizuka Y, Araie M, Suzuki Y, Tsukahara S. Effects of retinal glial cells on isolated rat retinal ganglion cells. Invest Ophthalmol Vis Sci. 2001;42:2686–2694. [PubMed] [Google Scholar]
- Kofuji P, Biedermann B, Siddharthan V, Raap M, Iandiev I, Milenkovic I, Thomzig A, Veh RW, Bringmann A, Reichenbach A. Kir potassium channel subunit expression in retinal glial cells: implications for spatial potassium buffering. Glia. 2002;39:292–303. doi: 10.1002/glia.10112. [DOI] [PubMed] [Google Scholar]
- Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010;460:525–542. doi: 10.1007/s00424-010-0809-1. [DOI] [PubMed] [Google Scholar]
- Levkovitch-Verbin H, Martin KR, Quigley HA, Baumrind LA, Pease ME, Valenta D. Measurement of amino acid levels in the vitreous humor of rats after chronic intraocular pressure elevation or optic nerve transection. J Glaucoma. 2002;11:396–405. doi: 10.1097/00061198-200210000-00005. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Huber KM. Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron. 2010;65:445–459. doi: 10.1016/j.neuron.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna PT, Smith AJ, Taneja TK, Howell GJ, Lippiat JD, Sivaprasadarao A. Constitutive endocytic recycling and protein kinase C-mediated lysosomal degradation control KATP channel surface density. J Biol Chem. 2010;285:5963–5973. doi: 10.1074/jbc.M109.066902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KR, Levkovitch-Verbin H, Valenta D, Baumrind L, Pease ME, Quigley HA. Retinal glutamate transporter changes in experimental glaucoma and after optic nerve transection in the rat. Invest Ophthalmol Vis Sci. 2002;43:2236–2243. [PubMed] [Google Scholar]
- Mockett BG, Guévremont D, Wutte M, Hulme SR, Williams JM, Abraham WC. Calcium/calmodulin-dependent protein kinase II mediates group I metabotropic glutamate receptor-dependent protein synthesis and long-term depression in rat hippocampus. J Neurosci. 2011;31:7380–7391. doi: 10.1523/JNEUROSCI.6656-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa H, Khodakhah K, Williams JT. Two intracellular pathways mediate metabotropic glutamate receptor-induced Ca2+ mobilization in dopamine neurons. J Neurosci. 2003;23:149–157. doi: 10.1523/JNEUROSCI.23-01-00149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagelhus EA, Horio Y, Inanobe A, Fujita A, Haug FM, Nielsen S, Kurachi Y, Ottersen OP. Immunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Müller cells is mediated by a coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia. 1999;26:47–54. doi: 10.1002/(sici)1098-1136(199903)26:1<47::aid-glia5>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Naka M, Kanamori A, Negi A, Nakamura M. Reduced expression of aquaporin-9 in rat optic nerve head and retina following elevated intraocular pressure. Invest Ophthalmol Vis Sci. 2010;51:4618–4626. doi: 10.1167/iovs.09-4712. [DOI] [PubMed] [Google Scholar]
- Newman EA. Inward-rectifying potassium channels in retinal glial (Müller) cells. J Neurosci. 1993;13:3333–3345. doi: 10.1523/JNEUROSCI.13-08-03333.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Calcium increases in retinal glial cells evoked by light-induced neuronal activity. J Neurosci. 2005;25:5502–5510. doi: 10.1523/JNEUROSCI.1354-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA, Zahs KR. Calcium waves in retinal glial cells. Science. 1997;275:844–847. doi: 10.1126/science.275.5301.844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoletti F, Bruno V, Catania MV, Battaglia G, Copani A, Barbagallo G, Ceña V, Sanchez-Prieto J, Spano PF, Pizzi M. Group-I metabotropic glutamate receptors: hypotheses to explain their dual role in neurotoxicity and neuroprotection. Neuropharmacology. 1999;38:1477–1484. doi: 10.1016/s0028-3908(99)00102-1. [DOI] [PubMed] [Google Scholar]
- Pannicke T, Faude F, Reichenbach A, Reichelt W. A function of delayed rectifier potassium channels in glial cells: maintenance of an auxiliary membrane potential under pathological conditions. Brain Res. 2000;862:187–193. doi: 10.1016/s0006-8993(00)02144-2. [DOI] [PubMed] [Google Scholar]
- Pannicke T, Iandiev I, Uckermann O, Biedermann B, Kutzera F, Wiedemann P, Wolburg H, Reichenbach A, Bringmann A. A potassium channel-linked mechanism of glial cell swelling in the postischemic retina. Mol Cell Neurosci. 2004;26:493–502. doi: 10.1016/j.mcn.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Pannicke T, Iandiev I, Wurm A, Uckermann O, vom Hagen F, Reichenbach A, Wiedemann P, Hammes HP, Bringmann A. Diabetes alters osmotic swelling and membrane characteristics of glial cells in rat retina. Diabetes. 2006;55:633–639. doi: 10.2337/diabetes.55.03.06.db05-1349. [DOI] [PubMed] [Google Scholar]
- Park WS, Han J, Kim N, Youm JB, Joo H, Kim HK, Ko JH, Earm YE. Endothelin-1 inhibits inward rectifier K+ channels in rabbit coronary arterial smooth muscle cells through protein kinase C. J Cardiovasc Pharmacol. 2005;46:681–689. doi: 10.1097/01.fjc.0000182846.08357.ed. [DOI] [PubMed] [Google Scholar]
- Rillich K, Gentsch J, Reichenbach A, Bringmann A, Weick M. Light stimulation evokes two different calcium responses in Müller glial cells of the guinea pig retina. Eur J Neurosci. 2009;29:1165–1176. doi: 10.1111/j.1460-9568.2009.06682.x. [DOI] [PubMed] [Google Scholar]
- Salt TE, Cordeiro MF. Glutamate excitotoxicity in glaucoma: throwing the baby out with the bathwater? Eye. 2006;20:730–731. doi: 10.1038/sj.eye.6701967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarantis M, Attwell D. Glutamate uptake in mammalian retinal glia is voltage- and potassium-dependent. Brain Res. 1990;516:322–325. doi: 10.1016/0006-8993(90)90935-5. [DOI] [PubMed] [Google Scholar]
- Schwartz EA. L-glutamate conditionally modulates the K+ current of Muller glial cells. Neuron. 1993;10:1141–1149. doi: 10.1016/0896-6273(93)90062-v. [DOI] [PubMed] [Google Scholar]
- Seki M, Lipton SA. Targeting excitotoxic/free radical signaling pathways for therapeutic intervention in glaucoma. Prog Brain Res. 2008;173:495–510. doi: 10.1016/S0079-6123(08)01134-5. [DOI] [PubMed] [Google Scholar]
- Skatchkov SN, Eaton MJ, Shuba YM, Kucheryavykh YV, Derst C, Veh RW, Wurm A, Iandiev I, Pannicke T, Bringmann A, Reichenbach A. Tandem-pore domain potassium channels are functionally expressed in retinal (Müller) glial cells. Glia. 2006;53:266–276. doi: 10.1002/glia.20280. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Reactive astrocytes in neural repair and protection. Neuroscientist. 2005;11:400–407. doi: 10.1177/1073858405278321. [DOI] [PubMed] [Google Scholar]
- Sohn JW, Yu WJ, Lee D, Shin HS, Lee SH, Ho WK. Cyclic ADP ribose-dependent Ca2+ release by group I metabotropic glutamate receptors in acutely dissociated rat hippocampal neurons. PLoS One. 2011;6:e26625. doi: 10.1371/journal.pone.0026625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada Y, Horio Y, Kurachi Y. Inwardly rectifying K+ channel in retinal Müller cells: comparison with the KAB-2/Kir4.1 channel expressed in HEK293T cells. Jpn J Physiol. 1998;48:71–80. doi: 10.2170/jjphysiol.48.71. [DOI] [PubMed] [Google Scholar]
- Tezel G, Wax MB. Glial modulation of retinal ganglion cell death in glaucoma. J Glaucoma. 2003;12:63–68. doi: 10.1097/00061198-200302000-00014. [DOI] [PubMed] [Google Scholar]
- Tezel G, Li LY, Patil RV, Wax MB. Tumor neurosis factor-alpha and its receptor-1 in the retina of normal and glaucomatous eyes. Invest Ophthalmol Vis Sci. 2001;42:1787–1794. [PubMed] [Google Scholar]
- Vorwerk CK, Naskar R, Schuettauf F, Quinto K, Zurakowski D, Gochenauer G, Robinson MB, Mackler SA, Dreyer EB. Depression of retinal glutamate transporter function leads to elevated intravitreal glutamate levels and ganglion cell death. Invest Ophthalmol Vis Sci. 2000;41:3615–3621. [PubMed] [Google Scholar]
- Wang X, Tay SS, Ng YK. An immunohistochemical study of neuronal and glial cell reactions in retinae of rats with experimental glaucoma. Exp Brain Res. 2000;132:476–484. doi: 10.1007/s002210000360. [DOI] [PubMed] [Google Scholar]
- Woldemussie E, Wijono M, Ruiz G. Müller cell response to laser-induced increase in intraocular pressure in rats. Glia. 2004;47:109–119. doi: 10.1002/glia.20000. [DOI] [PubMed] [Google Scholar]
- Wu J, Zhang S, Sun X. Neuroprotective effect of upregulated sonic Hedgehog in retinal ganglion cells following chronic ocular hypertension. Invest Ophthalmol Vis Sci. 2010;51:2986–2992. doi: 10.1167/iovs.09-4151. [DOI] [PubMed] [Google Scholar]
- Wurm A, Pannicke T, Iandiev I, Wiedemann P, Reichenbach A, Bringmann A. The developmental expression of K+ channels in retinal glial cells is associated with a decrease of osmotic cell swelling. Glia. 2006;54:411–423. doi: 10.1002/glia.20391. [DOI] [PubMed] [Google Scholar]
- Xue LP, Lu J, Cao Q, Hu S, Ding P, Ling EA. Müller glial cells express nestin coupled with glial fibrillary acidic protein in experimentally induced glaucoma in the rat retina. Neuroscience. 2006;139:723–732. doi: 10.1016/j.neuroscience.2005.12.032. [DOI] [PubMed] [Google Scholar]
- Yang XF, Miao Y, Ping Y, Wu HJ, Yang XL, Wang Z. Melatonin inhibits tetraethylammonium-sensitive potassium channels of rod ON type bipolar cells via MT2 receptors in rat retina. Neuroscience. 2011;173:19–29. doi: 10.1016/j.neuroscience.2010.11.028. [DOI] [PubMed] [Google Scholar]
- Zhang S, Wang H, Lu Q, Qing G, Wang N, Wang Y, Li S, Yang D, Yan F. Detection of early neuron degeneration and accompanying glial responses in the visual pathway in a rat model of acute intraocular hypertension. Brain Res. 2009;1303:131–143. doi: 10.1016/j.brainres.2009.09.029. [DOI] [PubMed] [Google Scholar]
- Zhao WJ, Zhang M, Miao Y, Yang XL, Wang Z. Melatonin potentiates glycine currents through a PLC/PKC signaling pathway in rat retinal ganglion cells. J Physiol. 2010;588:2605–2619. doi: 10.1113/jphysiol.2010.187641. [DOI] [PMC free article] [PubMed] [Google Scholar]