

Abstract
Neuronal migration is a fundamental process during the development of the cerebral cortex and is regulated by cytoskeletal components. Microtubule dynamics can be modulated by posttranslational modifications to tubulin subunits. Acetylation of α-tubulin at lysine 40 is important in regulating microtubule properties, and this process is controlled by acetyltransferase and deacetylase. MEC-17 is a newly discovered α-tubulin acetyltransferase that has been found to play a major role in the acetylation of α-tubulin in different species in vivo. However, the physiological function of MEC-17 during neural development is largely unknown. Here, we report that MEC-17 is critical for the migration of cortical neurons in the rat. MEC-17 was strongly expressed in the cerebral cortex during development. MEC-17 deficiency caused migratory defects in the cortical projection neurons and interneurons, and perturbed the transition of projection neurons from the multipolar stage to the unipolar/bipolar stage in the intermediate zone of the cortex. Furthermore, knockdown of α-tubulin deacetylase HDAC6 or overexpression of tubulinK40Q to mimic acetylated α-tubulin could reduce the migratory and morphological defects caused by MEC-17 deficiency in cortical projection neurons. Thus, MEC-17, which regulates the acetylation of α-tubulin, appears to control the migration and morphological transition of cortical neurons. This finding reveals the importance of MEC-17 and α-tubulin acetylation in cortical development.
Introduction
During the development of the cerebral cortex, most neurons migrate from the site of their last mitotic division toward their final destination and generate the proper circuitry (Ayala et al., 2007; Marín et al., 2010). Two major cell migration strategies, radial and tangential migration, are adopted to form the complex structure in the developing cortex. Projection neurons are generated in the ventricular zone and migrate radially to the outer surface of the cortex. Interneurons arise from the subpallial telencephalon and migrate tangentially to their final destination (Marín and Rubenstein, 2001). Three steps are involved in the migration of cortical neurons: extension of the leading process, translocation of the nucleus to the extending process, and retraction of the trailing process (Ayala et al., 2007; Marín et al., 2010). Recent studies report that the transition from the multipolar to the unipolar/bipolar stage is critical for the migration of projection neurons (Tabata and Nakajima, 2003; LoTurco and Bai, 2006; Sarkisian et al., 2006). Importantly, the regulation of cytoskeletal components determines cell morphology and affects nuclear translocation (Tsai and Gleeson, 2005; Sarkisian et al., 2008; Jaglin and Chelly, 2009; Marín et al., 2010). Identification of new regulators of the cytoskeleton is helpful in understanding the molecular mechanisms behind neuronal migration.
In the CNS, proper regulation of microtubule dynamics is essential for neurite outgrowth and neuronal migration during development (Ayala et al., 2007; Métin et al., 2008; Witte et al., 2008). Microtubule-associated proteins and tubulin modifications combine together to regulate microtubule stability. LIS1 and doublecortin have been shown to stabilize microtubules (Gleeson et al., 1999) and regulate neuronal migration (Kappeler et al., 2006; Nasrallah et al., 2006; Ayala et al., 2007; Marín et al., 2010). However, it remains largely unclear how posttranslational modifications to tubulins regulate neuronal migration.
The α-tubulin acetylation is an important posttranslational modification for microtubules (Hammond et al., 2008; Perdiz et al., 2010; Janke and Bulinski, 2011). Acetylation of α-tubulin is controlled by balanced enzyme activity involving acetyltransferase and deacetylase. Two histone deacetylase-related enzymes, HDAC6 and SIRT2, have been found to deacetylate α-tubulin in vivo and in vitro (Hubbert et al., 2002; North et al., 2003). Several acetyltransferase complexes, including the elongator, NAT10, and ARD1-NAT1 complexes, have been found previously to regulate α-tubulin acetylation (Ohkawa et al., 2008; Creppe et al., 2009; Shen et al., 2009). ELP3, which is a catalytic subunit of the elongator complex, displayed only weak α-tubulin acetyltransferase activity.
MEC-17, which is a newly discovered acetyltransferase that is conserved from Tetrahymena to mammalian species, directly promotes α-tubulin acetylation in vitro and seems to be the major α-tubulin acetyltransferase in vivo (Akella et al., 2010; Shida et al., 2010). MEC-17 is required for touch sensation in Caenorhabditis elegans, normal embryonic development in zebrafish, and the rapid assembly of primary cilia in RPE-hTERT cells. However, the function of MEC-17 in the nervous system remains largely unknown. In the present study, we investigate the role and underlying mechanism of MEC-17 in neuronal migration within the cerebral cortex.
Materials and Methods
Animals.
All experiments were approved by the Committee of Use of Laboratory Animals and Common Facility of the Institute of Biochemistry and Cell Biology of the Chinese Academy of Sciences (Shanghai, China). Male adult (200–250 g), female time-pregnant, and postnatal day 0 (P0), P3, P7, and P14 Sprague Dawley rats of either sex were provided by Shanghai Laboratory Animal Center of the Chinese Academy of Sciences.
Plasmid construct.
The expression constructs of MEC-17 and α-tubulin were prepared using the PCR-amplified rat MEC-17 (NM_212498.1) and Tuba1a (NM_022298) coding sequences from the cDNA of the dorsal root ganglion of male adult rats. These were inserted into pCAG-IRES-eGFP vector. The constructs of untargeted, shRNA-resistant (R) MEC-17 (MEC-17R), acetylation-site mutant tubulinK40A and tubulinK40Q were prepared with KOD Plus (Toyobo). The following primers were used: MEC-17R, 5′-GCCGGCCCGTGTGGATCTCCAACAACAGATTATGACGATAGTAGATGAGCTGGGCAAGG-3′ and 5′-CCTTGCCCAGCTCATCTACTATCGTCATAATCTGTTGTTGGAGATCCACACGGGCCGGC-3′; tubulinK40A, 5′-GGCCAGATGCCAAGCGACGCGACCATTGGGGGAGGAGAT-3′ and 5′-ATCTCCTCCCCCAATGGTCGCGTCGCTTGGCATCTGGCC-3′; and tubulinK40Q, 5′-GGCCAGATGCCAAGCGACCAGACCATTGGGGGAGGAGAT-3′ and 5′-ATCTCCTCCCCCAATGGTCTGGTCGCTTGGCATCTGGCC-3′. The construct for in situ hybridization was generated by subcloning the PCR-amplified rat MEC-17 and inserting it into the pGEM-T easy vector (Promega). The following primers were used: 5′-CAGTATATGTTACAGAAAG-3′ and 5′-GGGTCAGTGGCCAGCAGAAG-3′. The short hairpin interfering RNA constructs were purchased from GenePharma. The MEC-17 shRNA-a and shRNA-b (targeting MEC-17 mRNA) sequences were 5′-GCAGCAAATCATGACTATTGT-3′ and 5′-GCAGGGAAAGGAGCCATTATT-3′, respectively. The HDAC6 shRNA (targeting HDAC6 mRNA) sequence was 5′-GGTACTTCCCATCGCCTATGA-3′.
Real-time PCR.
The mRNA was isolated from different tissues of male adult rats and the P0, P3, P7, and P14 rats of either sex using TRIzol reagent (Invitrogen), and total cDNA was synthesized using a cDNA synthesis kit (Invitrogen). The primers used to amplify the MEC-17 cDNA were 5′-GTTACAGAAAGAGCGAGTGGA-3′ and 5′-GGTGTTGATGGGCAAAGAA-3′. For the control, glyceraldehyde 3-phosphate dehydrogenase cDNA was amplified with 5′-GGCAAGTTCAACGGCACAG-3′ and 5′-CGCCAGTAGACTCCACGAC-3′ as primers.
In situ hybridization. In situ hybridization was performed as described previously (Wang et al., 2010). In brief, antisense probes were amplified with PCR primers for rat MEC-17. Probes were labeled with digoxigenin (Roche). Rat brains from male adult rats and the P0, P3, P7, and P14 rats of either sex were fixed with 4% paraformaldehyde and then dehydrated consecutively in 20 and 30% sucrose at 4°C overnight. Cryostat sections of 50 μm were cut with a freezing microtome (Leica). The sections were hybridized with probes for 18 h at 67°C. The hybridization signal was detected with alkaline phosphatase-coupled antibodies against digoxigenin (1:2000; Roche) and nitrobluetetrazolium/5-bromo-4-chloro-3-indolyl phosphate (Roche) as color reaction substrates. The images were acquired with a Neurolucida microscope (MBF bioscience).
Cell culture and transfection.
The ND7-23 cells (European Collection of Animal Cell Cultures) were maintained in DMEM (without glutamine; Invitrogen) supplemented with 2 mm l-glutamine (Invitrogen) and 10% fetal bovine serum (Biochrom). The ND7-23 cells were transfected with plasmid using LipofectAMINE 2000 (Invitrogen). The cells were then used in different assays 2 d after transfection.
Primary cortical neurons were prepared from rat cortex at embryonic day 18.5 (E18.5), incubated with 0.125% trypsin-EDTA (Invitrogen) for 20 min at 37°C and then triturated. The indicated plasmids were transiently transfected into the dissociated cortical neurons by electroporation with the Nucleofector II (Amaxa) using the Rat Neuron Nucleofector Solution Kit and program O-003. The dissociated neurons were seeded onto poly-d-lysine-coated (Sigma) dishes and cultured in Neurobasal medium supplemented with 2 mm l-glutamine and 2% B-27 (Invitrogen). The cells were cultured for 12 h and fixed with 4% paraformaldehyde for further immunostaining.
In utero electroporation. In utero electroporation was performed as described previously (Saito, 2006; Chen et al., 2008). In brief, uteruses of rats at gestation day 16 were exposed, and then 3 μg of indicated plasmid mixed with 1 μg of the GFP plasmid and 0.1 μl of fast green (2 mg/ml; Sigma) was injected via transuterine pressure into the lateral ventricle of the embryo. Next, electric pulses were generated by an ElectroSquireportator T830 (BTX) and applied to the cerebral wall over five repetitions of 50 V for 50 ms with an interval of 1 s. The uterine horns were then replaced in the abdominal cavity, and the abdominal wall and skin were sutured using a surgical needle and thread. After the indicated length of time, the rats were processed for tissue analysis.
Immunostaining.
Transcardial perfusion with 4% paraformaldehyde was performed on the rats. The brains of the E18.5, P0, P7, and P30 rats of either sex were removed and postfixed in the same fixative and then dehydrated in 20 and 30% sucrose at 4°C overnight. The cryostat sections of 50 μm were permeabilized and blocked in PBS supplemented with 1% Triton X-100 and 5% bovine serum albumin for 10 min. The primary antibodies used for single immunofluorescence staining were rabbit antibodies against GFP (1:1000; Invitrogen), cleaved-caspase 3 (1:500; Cell Signaling Technology), Ki-67 (1:500; Abcam), or mouse antibodies against nestin (1:500; Millipore Bioscience Research Reagents). The sections were then labeled with donkey secondary antibodies against rabbit or mouse conjugated with FITC or Cy3 (1:100; Jackson ImmunoResearch), counterstained with DAPI (1:2000; Invitrogen), and analyzed with a TCS SP5 II confocal microscope (Leica).
For analysis of neuron position in the cortical wall, three to five brains were used for each experimental condition with ∼500 cells counted in each brain. The different subregions of the cortical wall were identified based on cell density after staining with DAPI. For analysis of neuronal morphology in the intermediate zone (IZ) at P0, 50–150 cells were selected from each brain, and at least three brains were used in each experimental condition. Furthermore, the percentages of neurons in different subregions or neurons in different stages (multipolar, unipolar/bipolar, or unipolar/bipolar stage with highly branched leading processes) were calculated for each brain.
Explant culture.
The medial ganglionic eminence (MGE) was dissected from the brains of E14.5 rats. The plasmid expressing MEC-17 shRNA (3 μg) was mixed with the plasmid expressing GFP (1 μg), and this plasmid mixture was injected into the dissected MGE tissue. Next, electric pulses (generated by an ElectroSquireportator T830) of 100 V were applied to the tissue twice for 100 ms, with an interval of 1 s. The MGE tissue was then cut into small pieces under the stereomicroscope, and the pieces were embedded in Matrigel in glass-bottom dishes. The explants were cultured for 2 d in Neurobasal medium supplemented with 2 mm l-glutamine and 2% B-27 at 37°C. Images were captured with an IX71 stereomicroscope (Olympus). For cell counting, three tissue samples were analyzed from each MGE, and four MGEs were used in each experimental condition. The percentage of GFP-expressing cells out of the explant for each MGE and the distance of migrating GFP-expressing cells from the explant were calculated.
Immunoblotting.
The samples were processed for SDS-PAGE, transferred, and probed with mouse antibodies against acetylated-α-tubulin (Ac-tubulin; 1:50,000; Sigma) or α-tubulin (1:50,000; Sigma), or with rabbit antibodies against HDAC6 (1:1000; Millipore). The experiment was repeated at least three times.
Statistical analysis.
All data are represented as the means ± SEM. Statistical significance was calculated using an unpaired Student's t test, and differences were considered significant at a level of p < 0.05.
Results
MEC-17 is highly expressed in the cerebral cortex during development
To investigate the expression pattern of MEC-17, we analyzed MEC-17 expression in different tissues in adult rats. Real-time PCR showed that MEC-17 was expressed broadly in all tissues and at a relatively high level in the CNS (Fig. 1A). We further analyzed the expression of MEC-17 in the cerebral cortex at different developmental stages in rats, and we found that the expression level of MEC-17 increased from E14.5 to E18.5 and decreased rapidly after P0 (Fig. 1B). Furthermore, in situ hybridization showed that MEC-17 was primarily expressed through the cortical wall at E14.5, but was highly expressed in the cortical plate (CP) at E16.5, E18.5, and P0 (Fig. 1C,D). The expression of MEC-17 was downregulated in the IZ at both E18.5 and P0 (Fig. 1C,D). After P7, MEC-17 expression in cerebral cortex decreased rapidly and then remained at a low level in the brains of adult rats (Fig. 1C,D). These data suggest that MEC-17 is highly expressed in the cerebral cortex during development.
Figure 1.

MEC-17 is expressed in adult rats and the developing cerebral cortex. A, B, Relative expression levels of MEC-17 in different tissues in adult rats (A) and the cerebral cortex at different developmental stages (B) as measured by real-time PCR. The data were normalized to the adult cerebral cortex. C, D, MEC-17 transcript distribution in the frontal section of the telencephalon (C) and cerebral cortex (D) at different developmental stages. Scale bars: C, 1 mm; D, left, 50 μm; right, 200 μm. All data represent the means ± SEM.
Loss of MEC-17 results in migratory and morphological defects in cortical projection neurons
Because most cortical projection neurons in the CP region migrate during the period between E18.5 and P0, we explored whether MEC-17 was involved in the radial migration of these neurons. Two shRNA constructs were used to knock down endogenously expressed MEC-17. The MEC-17 mRNA level decreased significantly after transfection with the plasmid expressing MEC-17 shRNA in ND7–23 cells, which are derived from rat dorsal root ganglion/mouse neuroblastoma hybrid cells (Fig. 2A). Therefore, it appears that these two shRNA constructs efficiently attenuated the endogenous expression of MEC-17. E16.5 rats were then electroporated with the plasmid expressing control or MEC-17 shRNA constructs. Four days later, we analyzed cell position in the cortical wall of the developing somatosensory cortex and found that at P0, 51% of neurons expressing the control construct migrated to the CP region, whereas only 49% of neurons remained in the IZ, ventricular zone (VZ), and subventricular zone (SVZ) (Fig. 2B,E). In contrast, 98% of MEC-17-deficient neurons remained in IZ and VZ/SVZ (Fig. 2B,E). At P7, the cortical neurons completed radial migration, whereas 36% of MEC-17-deficient neurons remained in the white matter (WM), and only 39% of MEC-17-deficient neurons reached their terminal destination in layer II/III (Fig. 2C,F). Moreover, coexpression of MEC-17R, which was refractory to silencing by MEC-17 shRNA, corrected the migratory defect in both the P0 and P7 cortexes (Fig. 2B,C,E,F), indicating that the migratory defect caused by silencing MEC-17 is not due to the off-target effect. Even as late as P30, 33% of the MEC-17-deficient neurons could not reach their terminal destination (Fig. 2D,G), suggesting that loss of MEC-17 blocks radial migration of the cortical projection neurons. Together, these results indicate that MEC-17 is critical for radial migration of the cortical projection neurons.
Figure 2.

MEC-17 deficiency alters the radial migration of the cortical projection neurons. A, Relative expression levels of MEC-17 in ND7–23 cells transfected with control (Ctrl) shRNA or two shRNAs targeting MEC-17 mRNA (MEC-17 shRNA-a and shRNA-b) as measured by real-time PCR. The data were normalized to the control group. B–D, Coronal sections of rat cortex at P0 (B), P7 (C), and P30 (D) electroporated at E16.5 with a GFP-expressing plasmid along with control shRNA, MEC-17 shRNAs, or MEC-17 shRNA-a together with untargeted MEC-17 (MEC-17R). Sections were counterstained with DAPI (blue). E–G, Histograms show the distribution of transfected cortical neurons in electroporated brains at P0 (E), P7 (F), and P30 (G) across different cortical regions. Scale bars: 200 μm. **p < 0.01, ***p < 0.001 versus control shRNA in the corresponding region; ##p < 0.01, ###p < 0.001 versus MEC-17 shRNA-a in the corresponding region. All data represent the means ± SEM.
Loss of MEC-17 did not appear to disrupt the nestin-labeled radial glia scaffold along which cortical projection neurons migrate (Fig. 3A). MEC-17 deficiency did not induce apoptosis of newly generated neurons or proliferation of neuronal progenitor cells, which were labeled with the cell apoptosis marker cleaved Caspase-3 and the cell division marker Ki-67, respectively (Fig. 3B,C). Thus, MEC-17 is not required for radial glia organization, survival, or cell division in vivo.
Figure 3.

MEC-17 deficiency affects the morphology of cortical projection neurons in the IZ region but does not affect radial glia organization, survival, or cell division. A–C, Coronal sections of rat cortex at E18.5, which were electroporated at E16.5 with control shRNA or MEC-17 shRNA-a along with a GFP-expressing plasmid, were immunostained for the indicated markers (red) including the radial glia organization marker nestin (A), the cell apoptosis marker cleaved Caspase-3 (B), and the cell division marker Ki-67 (C). D, Coronal sections of rat cortex in the IZ region and histogram showing the distribution of neurons at the multipolar stage, unipolar/bipolar stage (unbranched), and unipolar/bipolar stage with highly branched leading processes (branched) at P0 after MEC-17 depletion. Scale bars: A, 200 μm; B–D, 50 μm. **p < 0.01, ***p < 0.001 versus control shRNA; #p < 0.05, ##p < 0.01, ###p < 0.001 versus MEC-17 shRNA-a. All data represent the means ± SEM.
In the lower IZ and SVZ regions of developing cortex, neurons generally display multipolar morphology with multiple and complex neurites (Tabata and Nakajima, 2003; LoTurco and Bai, 2006). In the IZ and CP regions, migrating projection neurons exhibit a bipolar morphology, with a leading process directed toward the pia and a trailing process extending below (LoTurco and Bai, 2006). Thus, during cortical development, it is important for the projection neurons to become unipolar/bipolar as they migrate from the progenitor region toward the IZ region (Tabata and Nakajima, 2003; Noctor et al., 2004). Recent studies indicated that the depletion of several genes, such as Filamin A, LIS1, or doublecortin, can cause accumulation of multipolar neurons and migratory defects in cortical projection neurons (Bai et al., 2003; Nagano et al., 2004; Tsai et al., 2005). We further examined the morphology of MEC-17-deficient neurons, and we found that these neurons remained in the multipolar stage or developed to the unipolar/bipolar stage with highly branched leading processes in the IZ region (Fig. 3D). Such defects could be corrected by expressing MEC-17R (Fig. 3D). Previous studies have shown that unipolar/bipolar neurons with highly branched leading processes can also result from perturbing the transition of projection neurons from the multipolar stage to the unipolar/bipolar stage (Elias et al., 2007; Heng et al., 2008). Thus, the migratory defect induced by MEC-17 deficiency in cortical neurons could be due to the attenuation of neuronal polarization.
MEC-17 deficiency leads to a migratory defect in cortical interneurons
Unlike cortical projection neurons, the majority of cortical GABAergic interneurons originate from regions in the subpallial telencephalon that includes the lateral ganglionic eminence, MGE, and anterior entopeduncular area (Marín and Rubenstein, 2001; Ayala et al., 2007) and migrate tangentially to reach their destination (Anderson et al., 1997). In situ hybridization showed that MEC-17 was expressed in the MGE at E14.5 and E16.5 (Fig. 1C). To test whether MEC-17 is involved in the migration of cortical interneurons, we examined migration of MGE interneurons in vitro. Quantitative analysis in isolated explants of electroporated E14.5 MGE cells showed that only 9.1% of MEC-17-deficient cells migrated out of the explants, whereas 44.5% of cells expressing control shRNA were found to migrate (Fig. 4A, B). Moreover, the migration distance of MEC-17-deficient cells was much shorter than that of the control cells (Fig. 4A,C), indicating that the migratory ability of MEC-17-deficient interneuron precursors was impaired. Thus, MEC-17 is also critical for regulating the migration of cortical interneurons.
Figure 4.
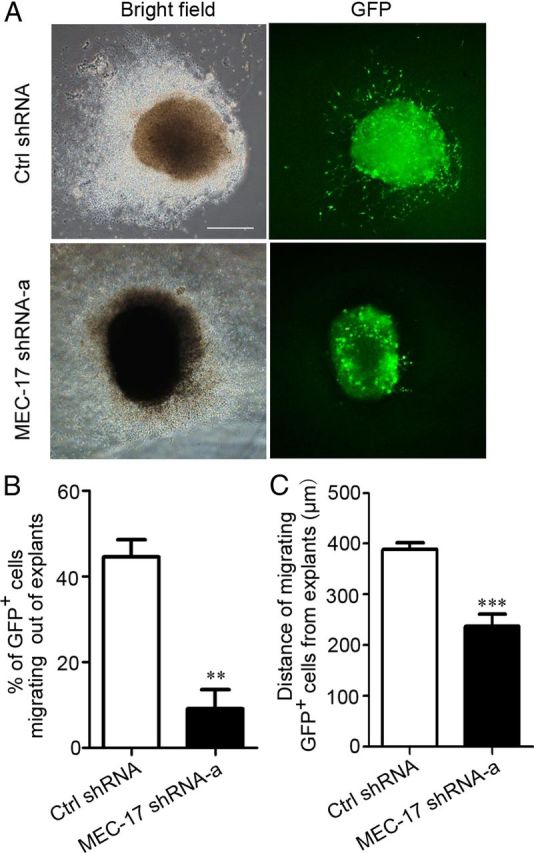
MEC-17 deficiency impairs interneuron migration from the MGE in vitro. A, The MGE explants were isolated from E14.5 rats, electroporated with a GFP-expressing plasmid along with control shRNA or MEC-17 shRNA-a, and cultured for 48 h. B, C, Histograms illustrating the percentage of GFP-expressing cells out of the explants (B) and the distance of migrating GFP-expressing cells from the explants (C). Scale bar, 500 μm. **p < 0.01, *** p < 0.001 versus control shRNA. All data represent the means ± SEM.
MEC-17 deficiency reduces α-tubulin acetylation and causes more marked defects in neuronal migration than tubulinK40A overexpression
MEC-17 has been found to be a major α-tubulin acetyltransferase (Akella et al., 2010; Shida et al., 2010). We examined the α-tubulin acetylation in both the brain slices of E18.5 rats in utero electroporated at E16.5 (Fig. 5A) and cultured cortical neurons (Fig. 5C). The immunostaining intensity of acetylated α-tubulin was decreased in the MEC-17-deficient neurons (Fig. 5A, C), suggesting that MEC-17 controls α-tubulin acetylation in cortical neurons. Immunoblotting also showed that MEC-17 deficiency markedly reduced the level of acetylated α-tubulin in ND7–23 cells (Fig. 5B). A previous study showed that overexpression of the acetylation-site mutant tubulinK40A impaired radial migration of cortical projection neurons in E17.5 mouse brains (Creppe et al., 2009). We then examined whether overexpression of tubulinK40A affected α-tubulin acetylation in cortical neurons. Immunoblotting showed that overexpressing tubulinK40A did not reduce the level of acetylated α-tubulin in ND7–23 cells compared to overexpressing wild-type tubulin (Fig. 5B). In cultured cortical neurons, overexpressing tubulinK40A did not change the immunostaining intensity of acetylated α-tubulin compared to overexpressing wild-type tubulin (Fig. 5C). Thus, MEC-17 deficiency, but not tubulinK40A overexpression, reduces α-tubulin acetylation.
Figure 5.

Loss of MEC-17, but not tubulinK40A overexpression, results in a reduction of α-tubulin acetylation in cortical neurons. A, Coronal sections of rat cortex at E18.5, which were electroporated at E16.5 with a GFP-expressing plasmid along with control shRNA or MEC-17 shRNA-a, were stained with antibody against Ac-tubulin (red). A white-dashed profile indicates a neuron electroporated with control shRNA or MEC-17 shRNA-a. B, Immunoblots and histogram showing the level of α-tubulin acetylation in ND7–23 cells expressing control shRNA, MEC-17 shRNA-a, wild-type (WT) tubulin, and tubulinK40A, respectively. **p < 0.01 versus control shRNA. All data represent the means ± SEM. C, Cultured cortical neurons from E18.5 rats were electroporated with different constructs as indicated and immunostained with antibody against Ac-tubulin (red). A white-dashed profile indicates a neuron electroporated with control shRNA, MEC-17 shRNA-a, tubulin, and tubulinK40A, respectively. Scale bars: A, 5 μm; C, 10 μm.
To compare the migratory defect caused by MEC-17 deficiency with that induced by tubulinK40A overexpression in cortical projection neurons, we electroporated the E16.5 rat brain with the plasmid-expressing GFP together with the plasmid-expressing wild-type tubulin or tubulinK40A. At P0, tubulinK40A overexpression caused a slight migratory defect in cortical projection neurons compared to wild-type tubulin overexpression (Fig. 6A). At P7, all tubulinK40A-expressing neurons reached layer II/III (Fig. 6B). This finding suggests that overexpression of tubulinK40A results in only a mild migratory defect and does not block the migration of neurons to their final destination. Together, the migratory defect in MEC-17-deficient cortical neurons is more pronounced than that induced by tubulinK40A overexpression.
Figure 6.

TubulinK40A overexpression causes a slight migratory defect in the cortical projection neurons. A, B, Coronal sections of the cerebral cortex at P0 (A) and P7 (C) in rats electroporated at E16.5 with a GFP-expressing plasmid along with tubulin or tubulinK40A. The sections were counterstained with DAPI (blue). The histograms show the distribution of transfected cortical neurons in electroporated brain at P0 (B) and P7 (D) across different cortical regions. Scale bars: 200 μm. *p < 0.05 versus tubulin in the corresponding region. All data represent the means ± SEM.
The reduction of α-tubulin acetylation is involved in the migratory defects induced by MEC-17 deficiency
To test whether the migratory defect in cortical neurons caused by MEC-17 deficiency is due to the reduction of α-tubulin acetylation, the shRNA construct of α-tubulin deacetylase HDAC6 was used to increase α-tubulin acetylation (Hubbert et al., 2002). Immunoblotting showed that the reduction in α-tubulin acetylation caused by MEC-17 deficiency was partially reversed by transfection of the plasmid expressing HDAC6 shRNA in ND7-23 cells (Fig. 7A). However, coexpression of MEC-17R completely recovered the reduced acetylation level of α-tubulin caused by MEC-17 shRNA (Fig. 7A). Importantly, an HDAC6 deficiency could reduce the migratory defect caused by loss of MEC-17 in both the P0 and P7 cortexes (Fig. 7B,C), suggesting that the migratory defect could be due to the reduced acetylation of α-tubulin. Furthermore, the morphological defects in the MEC-17-deficient neurons could be largely corrected by knocking down HDAC6 in the P0 cortex (Fig. 7D). These data suggest that the migratory and morphological defects caused by loss of MEC-17 in cortical neurons could be due to a reduction in α-tubulin acetylation.
Figure 7.
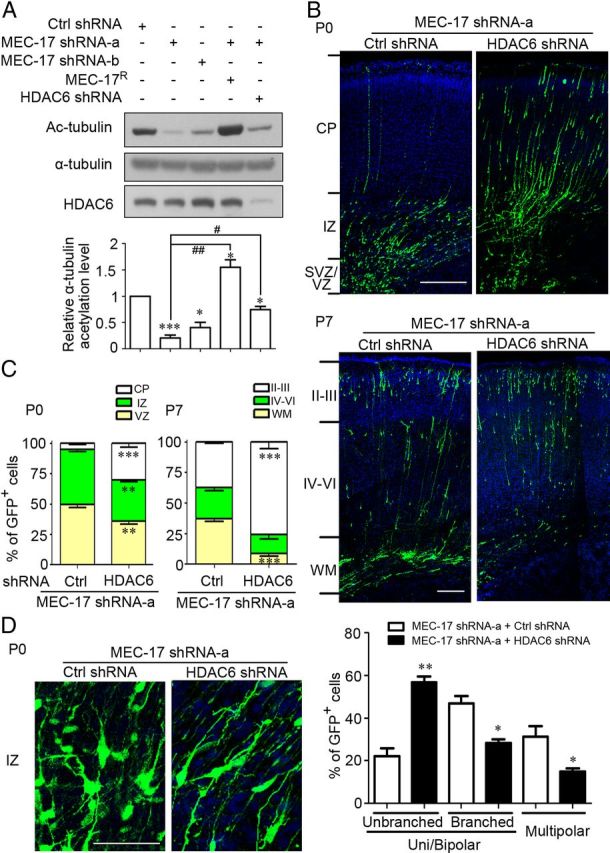
Loss of HDAC6 reduces the defects in MEC-17-deficient cortical projection neurons. A, Immunoblots and histogram showing the acetylation level of α-tubulin in ND7–23 cells transfected with different constructs as indicated. *p < 0.05, ***p < 0.001 versus control shRNA; #p < 0.05, ##p < 0.01 versus MEC-17 shRNA-a. B, C, Coronal sections of rat cortex at P0 (B, top) and P7 (bottom) electroporated at E16.5 with a GFP-expressing plasmid along with MEC-17 shRNA-a or MEC-17 shRNA-a together with HDAC6 shRNA. Sections were counterstained with DAPI (blue). Histograms show the distribution of transfected cortical projection neurons in electroporated brain across different cortical regions at P0 (C, left) and P7 (right). **p < 0.01, ***p < 0.001 versus those coexpressing MEC-17 shRNA-a with control shRNA in the corresponding region. D, Coronal sections of rat cortex in the IZ region and histogram showing the distribution of neurons expressing MEC-17 shRNA-a or MEC-17 shRNA-a and HDAC6 shRNA at the multipolar stage, unipolar/bipolar stage (unbranched), or unipolar/bipolar stage with highly branched leading processes (branched) at P0. *p < 0.05, **p < 0.01 versus those coexpressing MEC-17 shRNA-a with control shRNA. Scale bars: B, 200 μm; D, 50 μm. All data represent the means ± SEM.
HDAC6 is mainly distributed in the cytoplasm, and α-tubulin is not the only substrate of this deacetylase (Kovacs et al., 2005; Zhang et al., 2007). Previous studies reported that substitution of the acetylation amino acid lysine with glutamine in α-tubulin mimics constitutive acetylated protein because glutamine is hydrophilic and uncharged, similar to the acetylated lysine (Scroggins et al., 2007; Chu et al., 2011). We generated an acetylation-mimicking mutant tubulinK40Q to reproduce the function of acetylated α-tubulin. Overexpression of tubulinK40Q did not affect the radial migration of the cortical projection neurons at P0 (Fig. 8A,C) and P7 (B,C), suggesting that the level of α-tubulin acetylation in a physiological condition is sufficient for microtubule function. Moreover, overexpression of tubulinK40Q in the cortical neurons reduced the migratory and morphological defects caused by the loss of MEC-17 at P0 (Fig. 8D,F,G) and P7 (E–G). However, the rescue effect of MEC-17R on the neuronal migration in the MEC-17-deficient neurons was more pronounced than that induced by tubulinK40Q overexpression or HDAC6 depletion (Fig. 2E,F, 7C, 8F). Together, these results indicate that the reduced acetylation of α-tubulin is involved in the migratory defects caused by MEC-17 deficiency.
Figure 8.

TubulinK40Q overexpression reduces the defects in MEC-17-deficient cortical projection neurons. A, B, Coronal sections of rat cortex at P0 (A) and P7 (B) electroporated at E16.5 with a GFP-expressing plasmid along with tubulin or tubulinK40Q. D, E, Coronal sections of rat cortex at P0 (D) and P7 (E) electroporated at E16.5 with a GFP-expressing plasmid and MEC-17 shRNA-a along with tubulin or tubulinK40Q. C, F, Histograms show the distribution of transfected cortical projection neurons in electroporated brain across different cortical regions at both P0 and P7. G, Coronal sections of rat cortex in the IZ region and histogram showing the distribution of neurons coexpressing MEC-17 shRNA-a with tubulin or tubulinK40Q at the multipolar stage, unipolar/bipolar stage (unbranched), and unipolar/bipolar stage with highly branched leading processes (branched) at P0. *p < 0.05, **p < 0.01, ***p < 0.001 versus those coexpressing MEC-17 shRNA-a with tubulin in the corresponding region. Scale bars: A, B, D, E, 200 μm; G, 50 μm. All data represent the means ± SEM.
Discussion
Posttranslational modifications to tubulin subunits regulate microtubule dynamics. MEC-17 is a newly discovered α-tubulin acetyltransferase and plays a major role in α-tubulin acetylation at lysine 40 in vivo. We found that MEC-17 was critical for regulating neuronal migration in the cerebral cortex of the rat. MEC-17-deficient projection neurons remained in a multipolar stage or developed to the unipolar/bipolar stage with highly branched leading processes in the IZ region. Therefore, the migratory defect seen in these cortical neurons may result from an abnormal neuronal polarization. Importantly, a MEC-17 deficiency leads to a loss of α-tubulin acetylation in cortical neurons. The migratory and morphological defects in MEC-17-deficient neurons could be reduced by knockdown of HDAC6 or overexpression of tubulinK40Q (which mimics tubulin acetylation). Thus, MEC-17-induced α-tubulin acetylation is required for the morphological transition and migration of cortical neurons during development.
The presence of MEC-17 is required for the migration of cortical neurons
MEC-17 is highly expressed in the cerebral cortex during development. We observed that MEC-17 deficiency led to 98% of the cortical projection neurons remaining in the IZ and VZ/SVZ regions at P0. At P7, an age by which neurons have completed radial migration, only 39% of MEC-17-deficient neurons reached their terminal destination in layer II/III, suggesting that MEC-17 is critical for the radial migration of cortical projection neurons. Furthermore, loss of MEC-17 may also attenuate the migration of cortical interneurons because the migration of MEC-17-deficient cells was blocked in MGE explants. Previous research showed that low doses of nocodazole impaired leading process morphogenesis and cell directionality but did not affect migration speed, indicating that microtubules control cell morphology rather than cell motility in cortical interneurons (Baudoin et al., 2008; Métin et al., 2008). However, high doses of nocodazole impaired the migration speed of cortical interneurons (Baudoin et al., 2008). Blockage of interneuron migration via MEC-17 deficiency offers support for the important role of microtubules in regulating the migration speed of the cortical interneurons. The cellular mechanisms for this MEC-17 regulation should be further investigated.
A previous study reported that the elongator complex controls the radial migration of the cortical projection neurons through the acetylation of α-tubulin (Creppe et al., 2009). However, the regulatory effect of the elongator complex on α-tubulin acetylation is controversial. Recent research showed that a deficiency in ELP3, which is the catalytic subunit of elongator complex, did not decrease the level of α-tubulin acetylation in cell lines and cortical neurons (Miœkiewicz et al., 2011). Whether α-tubulin acetylation underlies the migratory defect in ELP3-deficient neurons needs to be further clarified. Moreover, loss of ELP1, which is the scaffold subunit, caused the neurons to reach their final destination at P2 (Creppe et al., 2009), suggesting that the elongator complex only delayed the migration of the cortical projection neurons. In the present study, we showed that 33% of MEC-17-deficient projection neurons did not reach layer II/III at P30, indicating that MEC-17 is more critical for neuronal migration in the cerebral cortex as an acetyltransferase.
α-Tubulin acetylation is involved in MEC-17 functions in developing cortical neurons
MEC-17 has been found to directly and specifically promote α-tubulin acetylation at lysine 40 in vitro and in vivo (Akella et al., 2010; Shida et al., 2010). MEC-17 deficiency in Tetrahymena, C. elegans, zebrafish, or HeLa cells leads to a loss of α-tubulin acetylation (Akella et al., 2010; Shida et al., 2010). We also found a marked reduction of α-tubulin acetylation in MEC-17-deficient cortical neurons and ND7–23 cells. Both loss of tubulin deacetylase HDAC6 and overexpression of tubulinK40Q largely restored the morphological transition and migration of MEC-17-deficient cortical neurons. These results indicate that the tubulin is indeed the relevant target of MEC-17 in cortical projection neurons. In fact, the MEC-17R-induced rescue effect on the migratory defects was found to be more pronounced than that induced by HDAC6 deficiency or tubulinK40Q overexpression. This finding may be due to a relatively low expression level of tubulinK40Q compared to endogenous tubulin. Moreover, the level of tubulin acetylation following HDAC6 deficiency was lower than that induced by MEC-17R overexpression. In addition, other proteins may be substrates of MEC-17, and acetylation of these proteins could also be involved in the regulation of neuronal migration.
Our study provides evidence that the level of α-tubulin acetylation is critical for the migration of cortical neurons. MEC-17 deficiency resulted in a loss of α-tubulin acetylation and defects in neuronal migration in the cerebral cortex. Both MEC-17R overexpression and HDAC6 deficiency increased the level of α-tubulin acetylation, resulting in phenotypic recovery of MEC-17-deficient neurons. However, MEC-17 overexpression (data not shown) and tubulinK40Q overexpression did not significantly influence the radial migration of cortical projection neurons, indicating that α-tubulin acetylation in a normal physiological condition is sufficient for regulating microtubule dynamics. However, considering the necessity for very delicate control of axonal and dendritic development, it is also important to temporally and spatially regulate α-tubulin acetylation in the subcellular domains of neurons. Future studies might focus on investigating the distribution and fine regulation of MEC-17 and HDAC6 in the subcellular domains of neurons.
Manipulation of MEC-17 provides a useful tool for studying the functions of α-tubulin acetylation
The function of α-tubulin acetylation in Tetrahymena was easily evaluated by knocking in α-tubulinK40R, which could abolish the acetylation of all α-tubulins because the Tetrahymena genome contains only one α-tubulin gene (Gaertig et al., 1995). However, knocking in α-tubulinK40R could not fully reveal the role of α-tubulin acetylation in Mammalia because mammalian genomes have multiple genes encoding α-tubulin. Overexpressing an acetylation-site mutant α-tubulinK40A is another way to study the function of α-tubulin acetylation, and a previous study showed that α-tubulinK40A overexpression delayed the migration of cortical projection neurons (Creppe et al., 2009). We observed that α-tubulinK40A overexpression caused a slight migratory defect in cortical neurons in the P0 brain but not in the P7 brain. It is possible that the expression level of exogenous tubulin could be lower than that of endogenous tubulin, and overexpressing α-tubulinK40A may not be sufficient to markedly reduce α-tubulin acetylation in neurons. Accordingly, the expressed α-tubulinK40A may integrate into endogenous microtubules and only produce a slight effect on neuronal migration. Therefore, the migratory defect induced by MEC-17 deficiency was stronger than that created by α-tubulinK40A overexpression. Manipulating MEC-17 expression could be a useful tool for studying the physiological roles of α-tubulin acetylation in the nervous system.
Footnotes
This work was supported by National Natural Science Foundation of China Grant 30930044 and National Basic Research Program of China Grant 2010CB912001. We thank Dr. Qing-Feng Wu, Liu Yang, and Dr. Shuai Li for discussions on this project.
The authors declare no competing financial interests.
References
- Akella JS, Wloga D, Kim J, Starostina NG, Lyons-Abbott S, Morrissette NS, Dougan ST, Kipreos ET, Gaertig J. MEC-17 is an α-tubulin acetyltransferase. Nature. 2010;467:218–222. doi: 10.1038/nature09324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SA, Eisenstat DD, Shi L, Rubenstein JL. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1997;278:474–476. doi: 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- Ayala R, Shu T, Tsai LH. Trekking across the brain: the journey of neuronal migration. Cell. 2007;128:29–43. doi: 10.1016/j.cell.2006.12.021. [DOI] [PubMed] [Google Scholar]
- Bai J, Ramos RL, Ackman JB, Thomas AM, Lee RV, LoTurco JJ. RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat Neurosci. 2003;6:1277–1283. doi: 10.1038/nn1153. [DOI] [PubMed] [Google Scholar]
- Baudoin JP, Alvarez C, Gaspar P, Métin C. Nocodazole-induced changes in microtubule dynamics impair the morphology and directionality of migrating medial ganglionic eminence cells. Dev Neurosci. 2008;30:132–143. doi: 10.1159/000109858. [DOI] [PubMed] [Google Scholar]
- Chen G, Sima J, Jin M, Wang KY, Xue XJ, Zheng W, Ding YQ, Yuan XB. Semaphorin-3A guides radial migration of cortical neurons during development. Nat Neurosci. 2008;11:36–44. doi: 10.1038/nn2018. [DOI] [PubMed] [Google Scholar]
- Chu CW, Hou F, Zhang J, Phu L, Loktev AV, Kirkpatrick DS, Jackson PK, Zhao Y, Zou H. A novel acetylation of β-tubulin by San modulates microtubule polymerization via down-regulating tubulin incorporation. Mol Biol Cell. 2011;22:448–456. doi: 10.1091/mbc.E10-03-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creppe C, Malinouskaya L, Volvert ML, Gillard M, Close P, Malaise O, Laguesse S, Cornez I, Rahmouni S, Ormenese S, Belachew S, Malgrange B, Chapelle JP, Siebenlist U, Moonen G, Chariot A, Nguyen L. Elongator controls the migration and differentiation of cortical neurons through acetylation of α-tubulin. Cell. 2009;136:551–564. doi: 10.1016/j.cell.2008.11.043. [DOI] [PubMed] [Google Scholar]
- Elias LA, Wang DD, Kriegstein AR. Gap junction adhesion is necessary for radial migration in the neocortex. Nature. 2007;448:901–907. doi: 10.1038/nature06063. [DOI] [PubMed] [Google Scholar]
- Gaertig J, Cruz MA, Bowen J, Gu L, Pennock DG, Gorovsky MA. Acetylation of lysine 40 in α-tubulin is not essential in Tetrahymena thermophila. J Cell Biol. 1995;129:1301–1310. doi: 10.1083/jcb.129.5.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson JG, Lin PT, Flanagan LA, Walsh CA. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999;23:257–271. doi: 10.1016/s0896-6273(00)80778-3. [DOI] [PubMed] [Google Scholar]
- Hammond JW, Cai D, Verhey KJ. Tubulin modifications and their cellular functions. Curr Opin Cell Biol. 2008;20:71–76. doi: 10.1016/j.ceb.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng JI, Nguyen L, Castro DS, Zimmer C, Wildner H, Armant O, Skowronska-Krawczyk D, Bedogni F, Matter JM, Hevner R, Guillemot F. Neurogenin 2 controls cortical neuron migration through regulation of Rnd2. Nature. 2008;455:114–118. doi: 10.1038/nature07198. [DOI] [PubMed] [Google Scholar]
- Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- Jaglin XH, Chelly J. Tubulin-related cortical dysgeneses: microtubule dysfunction underlying neuronal migration defects. Trends Genet. 2009;25:555–566. doi: 10.1016/j.tig.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Janke C, Bulinski JC. Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat Rev Mol Cell Biol. 2011;12:773–786. doi: 10.1038/nrm3227. [DOI] [PubMed] [Google Scholar]
- Kappeler C, Saillour Y, Baudoin JP, Tuy FP, Alvarez C, Houbron C, Gaspar P, Hamard G, Chelly J, Métin C, Francis F. Branching and nucleokinesis defects in migrating interneurons derived from doublecortin knockout mice. Hum Mol Genet. 2006;15:1387–1400. doi: 10.1093/hmg/ddl062. [DOI] [PubMed] [Google Scholar]
- Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- LoTurco JJ, Bai J. The multipolar stage and disruptions in neuronal migration. Trends Neurosci. 2006;29:407–413. doi: 10.1016/j.tins.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Marín O, Rubenstein JL. A long, remarkable journey: tangential migration in the telencephalon. Nat Rev Neurosci. 2001;2:780–790. doi: 10.1038/35097509. [DOI] [PubMed] [Google Scholar]
- Marín O, Valiente M, Ge X, Tsai LH. Guiding neuronal cell migrations. Cold Spring Harb Perspect Biol. 2010;2:a001834. doi: 10.1101/cshperspect.a001834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Métin C, Vallee RB, Rakic P, Bhide PG. Modes and mishaps of neuronal migration in the mammalian brain. J Neurosci. 2008;28:11746–11752. doi: 10.1523/JNEUROSCI.3860-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miœkiewicz K, Jose LE, Bento-Abreu A, Fislage M, Taes I, Kasprowicz J, Swerts J, Sigrist S, Versees W, Robberecht W, Verstreken P. ELP3 controls active zone morphology by acetylating the ELKS family member bruchpilot. Neuron. 2011;72:776–788. doi: 10.1016/j.neuron.2011.10.010. [DOI] [PubMed] [Google Scholar]
- Nagano T, Morikubo S, Sato M. Filamin A and FILIP (Filamin A-Interacting Protein) regulate cell polarity and motility in neocortical subventricular and intermediate zones during radial migration. J Neurosci. 2004;24:9648–9657. doi: 10.1523/JNEUROSCI.2363-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasrallah IM, McManus MF, Pancoast MM, Wynshaw-Boris A, Golden JA. Analysis of non-radial interneuron migration dynamics and its disruption in Lis1+/− mice. J Comp Neurol. 2006;496:847–858. doi: 10.1002/cne.20966. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martínez-Cerdeño V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- Ohkawa N, Sugisaki S, Tokunaga E, Fujitani K, Hayasaka T, Setou M, Inokuchi K. N-acetyltransferase ARD1-NAT1 regulates neuronal dendritic development. Genes Cells. 2008;13:1171–1183. doi: 10.1111/j.1365-2443.2008.01235.x. [DOI] [PubMed] [Google Scholar]
- Perdiz D, Mackeh R, Poüs C, Baillet A. The ins and outs of tubulin acetylation: more than just a post-translational modification? Cell Signal. 2010;23:763–771. doi: 10.1016/j.cellsig.2010.10.014. [DOI] [PubMed] [Google Scholar]
- Saito T. In vivo electroporation in the embryonic mouse central nervous system. Nat Protoc. 2006;1:1552–1558. doi: 10.1038/nprot.2006.276. [DOI] [PubMed] [Google Scholar]
- Sarkisian MR, Bartley CM, Chi H, Nakamura F, Hashimoto-Torii K, Torii M, Flavell RA, Rakic P. MEKK4 signaling regulates filamin expression and neuronal migration. Neuron. 2006;52:789–801. doi: 10.1016/j.neuron.2006.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkisian MR, Bartley CM, Rakic P. Trouble making the first move: interpreting arrested neuronal migration in the cerebral cortex. Trends Neurosci. 2008;31:54–61. doi: 10.1016/j.tins.2007.11.009. [DOI] [PubMed] [Google Scholar]
- Scroggins BT, Robzyk K, Wang D, Marcu MG, Tsutsumi S, Beebe K, Cotter RJ, Felts S, Toft D, Karnitz L, Rosen N, Neckers L. An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol Cell. 2007;25:151–159. doi: 10.1016/j.molcel.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Zheng X, McNutt MA, Guang L, Sun Y, Wang J, Gong Y, Hou L, Zhang B. NAT10, a nucleolar protein, localizes to the midbody and regulates cytokinesis and acetylation of microtubules. Exp Cell Res. 2009;315:1653–1667. doi: 10.1016/j.yexcr.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Shida T, Cueva JG, Xu Z, Goodman MB, Nachury MV. The major α-tubulin K40 acetyltransferase αTAT1 promotes rapid ciliogenesis and efficient mechanosensation. Proc Natl Acad Sci U S A. 2010;107:21517–21522. doi: 10.1073/pnas.1013728107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Multipolar migration: the third mode of radial neuronal migration in the developing cerebral cortex. J Neurosci. 2003;23:9996–10001. doi: 10.1523/JNEUROSCI.23-31-09996.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai JW, Chen Y, Kriegstein AR, Vallee RB. LIS1 RNA interference blocks neural stem cell division, morphogenesis, and motility at multiple stages. J Cell Biol. 2005;170:935–945. doi: 10.1083/jcb.200505166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai LH, Gleeson JG. Nucleokinesis in neuronal migration. Neuron. 2005;46:383–388. doi: 10.1016/j.neuron.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Wang HB, Zhao B, Zhong YQ, Li KC, Li ZY, Wang Q, Lu YJ, Zhang ZN, He SQ, Zheng HC, Wu SX, Hökfelt TG, Bao L, Zhang X. Coexpression of δ- and μ-opioid receptors in nociceptive sensory neurons. Proc Natl Acad Sci U S A. 2010;107:13117–13122. doi: 10.1073/pnas.1008382107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte H, Neukirchen D, Bradke F. Microtubule stabilization specifies initial neuronal polarization. J Cell Biol. 2008;180:619–632. doi: 10.1083/jcb.200707042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yuan Z, Zhang Y, Yong S, Salas-Burgos A, Koomen J, Olashaw N, Parsons JT, Yang XJ, Dent SR, Yao TP, Lane WS, Seto E. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell. 2007;27:197–213. doi: 10.1016/j.molcel.2007.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]