

An efficient cyclization reaction allows for screening of large macrocycle libraries and yields a potent thrombin inhibitor.
Abstract
Macrocyclic compounds are an attractive modality for drug development, but the limited availability of large, structurally diverse macrocyclic libraries hampers the discovery of leads. Here, we describe the discovery of efficient macrocyclization reactions based on thiol-to-amine ligations using bis-electrophiles, their application to synthesize and screen large libraries of macrocyclic compounds, and the identification of potent small macrocyclic ligands. The thiol-to-amine cyclization reactions showed unexpectedly high yields for a wide substrate range, which obviated product purification and enabled the generation and screening of an 8988 macrocycle library with a comparatively small effort. X-ray structure analysis of an identified thrombin inhibitor (Ki = 42 ± 5 nM) revealed a snug fit with the target, validating the strategy of screening large libraries with a high skeletal diversity. The approach provides a route for screening large sub-kilodalton macrocyclic libraries and may be applied to many challenging drug targets.
INTRODUCTION
Macrocycles, consisting of 12 or more ring atoms, serve as the backbone for many important drugs, such as the immunosuppressant cyclosporine, the antibiotic vancomycin, or the oncology drug dactinomycin (1, 2). Macrocycles have received substantial attention for their ability to bind difficult targets containing flat, featureless surfaces considered “undruggable” by traditional small molecules, and because they can disrupt protein-protein interactions (3). Recent innovations in peptidic macrocycle development, based on mimicking protein epitopes (4) and screening large combinatorial cyclic peptide libraries using in vitro display (5, 6), have led to several clinical-stage macrocyclic peptides composed of typically 10 to 20 amino acids and directed against extracellular proteins.
An important lingering challenge is the generation of sub-kilodalton macrocyclic ligands that are thus small enough to be potentially orally available or cell permeable. The challenge of generating these ligands stems from a lack of sufficiently large libraries of small, structurally diverse macrocyclic compounds for high-throughput screening. Pharmaceutical company libraries, comprising typically between 1 and 2 million molecules, contain only a limited number of macrocycles and thus underrepresent this molecule class. A sub-kilodalton macrocycle compound library comprising as many as 14,400 macrocycles was developed by combinatorial chemistry, but it is based on only 16 different scaffolds and thus suffers from a small skeletal diversity (7). Approaches for generating larger libraries based on diversity-oriented synthesis (8) and DNA encoding (9, 10) have recently emerged and are promising.
In this work, we present a strategy for synthesizing libraries of thousands of small (<1 kDa), structurally highly diverse macrocycles and show that library screening yields macrocycles that fit perfectly to protein surfaces. The generation of large macrocycle libraries with a high skeletal diversity enabled efficient macrocyclization reactions based on thiol-to-amine ligations disclosed in this work.
RESULTS
We observed that short dipeptides, such as Gly-Cys, are efficiently cyclized by bridging the Cys thiol with the N-terminal amine using a range of bis-electrophiles (Fig. 1). Incubating Gly-Cys with excess (8 equiv) of, e.g., di(bromomethyl)pyridine (1) resulted in a 96% cyclized peptide yield and showed only one side product, a linker-dimerized peptide (4%; data S1). The first reaction step, the intermolecular alkylation of the thiol group with 1, proceeded with a high second-order rate constant of 51 ± 11 M−1 s−1 (Fig. 1A and fig. S1, A and B) that enabled a low peptide concentration (<1 mM) favoring cyclization over polymerization in the second step, the intramolecular reaction that proceeded at 4.1 × 10−4 ± 1.2 × 10−4 reactions/s (Fig. 1A and fig. S1, B and C). The rate of the second reaction was determined by incubating peptide with a high concentration of 1 to achieve formation of the intermediate product within seconds and by subsequent monitoring of the slower intramolecular reaction.
Fig. 1. Reaction rate, efficiency, and substrate scope of the thiol-to-amine macrocyclization reaction.
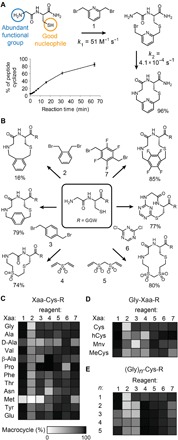
(A) Sample reaction of the dipeptide Gly-Cys and bis-electrophile linker 1. The higher thiol reactivity compared to amines enables stepwise reaction in the indicated order, forming a macrocycle as the main product with few side products. (B) Several bis-electrophiles efficiently cyclized the Gly-Cys substrate, adding between three and seven atoms to the macrocyclic backbone. (C to E) The substrate scope was assessed by quantifying the percent of linear substrate converted into the desired macrocycle. The N-terminal amino acid (C), the Cys side-chain length (D), and the number of amino acids were varied (E). R = Gly-Gly-Trp.
The high yield of the thiol-to-amine macrocyclization and the small number of side products removed the need for purification and thus enabled the synthesis of large combinatorial libraries by simply mixing linear peptides with a variety of bis-electrophile linkers of variable lengths, shapes, and chemical compositions. This created several macrocycles with different ring sizes from each peptide, thus accessing a large scaffold diversity with little effort. A particularly attractive feature of the thiol-to-amine cyclization strategy is that only one functional group, a thiol, needs to be built into the peptides as the second group, an amine, is present at the terminus of all peptides, meaning that small macrocycles can be produced. This is in contrast to other efficient macrocyclization reactions such as the thiol-to-thiol or the alkyne-to-azide cyclization that require incorporation of two functional groups via two extra amino acids, making the macrocyles larger.
We tested a panel of 15 additional bis-electrophiles (2 to 16; Fig. 1B and fig. S2, A and B) previously used for the cyclization of peptides via two thiol groups (11–14). One of the reagents, dichloroacetone (11), had been reported by Dawson and coworkers (13) to cyclize two 4–amino acid peptides via a thiol and an amino group. Of the 15 new linkers, seven showed yields greater than 70%, with only a limited number of side products that could be identified in most cases based on the mass (fig. S2C and data S1); namely, four other bis(bromomethyl)aryls that cyclize the substrate via an SN2 reaction (3, 7, 8, and 9), two acrylsulfones that react by Michael addition (4 and 5), and a di(chloro)triazine that follows an SNAr mechanism (6) (Fig. 1B and data S1).
We then tested the substrate scope of seven of the cyclization reagents (1 to 7; Fig. 1, C and D, and data S1). Dipeptides with diverse N-terminal amino acids containing both l- and d-stereochemistry, β-branches (e.g., valine), N-methylations or cycles (e.g., proline), and β-amino acids (e.g., β-alanine) were efficiently cyclized by all bis-electrophiles (Fig. 1C). Methionine was the only intolerable amino acid due to the thioether side chain that reacted preferentially over the amine (fig. S2, D to F). Dipeptides containing cysteine homologs, such as homocysteine (hCys), 5-mercaptonorvaline (Mnv), and N-methylcysteine (MeCys), cyclized with good yields (Fig. 1D), as did longer peptides of the form (Gly)n-Cys (n = 2 to 4), although the yields were lower for some lengths (Fig. 1E). The substrate tolerance was much broader than in other macrocyclization reactions, e.g., in head-to-tail peptide cyclizations, and suggested that the thiol-to-amine cyclization strategy was suitable for the generation of large and structurally diverse libraries of macrocyclic compounds.
We initially synthesized a pilot-scale library comprising 1176 macrocycles and screened it against the proteases trypsin and thrombin (Fig. 2A). The macrocycles were generated by cyclizing tripeptides of the form X-Y-Z that each contained a cysteine or cysteine derivative hCys (25) or Mnv (26) in the last position (Z). We tailored the library for the trypsin and thrombin targets by incorporating arginine or homologous amino acids, known to bind to the S1 specificity pocket of trypsin-like serine proteases, into one of the amino acid positions (X or Y) (17 to 19). The remaining library position was occupied by structurally diverse building blocks, including β- and γ-amino acids that introduced skeletal diversity (20 to 24). The purified peptides (100 μM) were cyclized by simply mixing them with the seven linkers 1 to 7 (800 μM) in a combinatorial fashion resulting in a library containing 432 different macrocyclic backbones and thus a substantially larger skeletal diversity than those of reported macrocyclic libraries (backbone diversity illustrated in fig. S3 and on a poster provided in data S2). The reagents were pipetted to wells of 384 microplates using automated liquid-handling robots and disposable pipette tips. Screening the library against trypsin and thrombin identified several peptide/linker combinations that inhibited trypsin or thrombin (Fig. 2A). The activity of the macrocycles varied substantially based on the linker, indicating the importance of varying the backbone to tailor an inhibitor to a target of interest.
Fig. 2. Pilot-scale library of 1176 macrocycles screened against trypsin and thrombin.
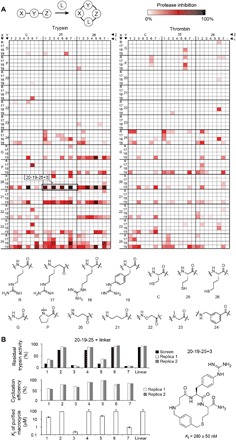
(A) As the library base, 168 tripeptides (X-Y-Z) were synthesized using the amino acid building blocks indicated in the matrix and cyclized at 100 μM with an eightfold molar excess of cyclization linker (L = 1 to 7). A control of noncyclized peptides was included (−). The extent of trypsin (left) or thrombin (right) inhibition was quantified in percentage of residual protease activity. (B) Macrocycles based on peptide 20-19-25 indicated by a frame in (A). The reproducibility of the synthesis and screening approach was assessed by repeating the cyclization of peptide 20-19-25 (replicas 1 and 2) twice. Cyclization efficiency was quantified by liquid chromatography–mass spectrometry and indicated in percentage of peptide added. Mean values and SD are indicated for the Ki values. The chemical structure and Ki of the best trypsin inhibitor are shown. *Ki > 100 μM.
Peptide 20-19-25 (numbers and letters indicate the building blocks in positions X-Y-Z, as shown in Fig. 2A) showed good inhibitory activity when cyclized with 1, 3, 5, and 7 and was analyzed in more detail (Fig. 2B and fig. S4A). Repeating the cyclization reactions for this peptide gave the same result, showing the reproducibility and robustness of the macrocyclization reaction in a microtiter format and the synthesis and screening approach in general (Fig. 2B, top). Liquid chromatography–mass spectrometry (LC-MS) analysis of these reactions showed that the macrocyclization yields for different linkers were in a relatively narrow range of 55 to 95%, suggesting that the different extents of protease inhibition in the screen stemmed from different macrocycle affinities and not from varying cyclization yields (Fig. 2B, middle). Analysis of the purified macrocycles showed potencies that correlated with the result of the nonpurified products in the screen (Fig. 2B, bottom, and fig. S4A). The best trypsin and thrombin inhibitors showed Kis of 280 ± 50 nM (Fig. 2B, right) and 7.3 ± 0.4 μM (fig. S4B), respectively.
We next generated and screened a library of 8988 slightly larger macrocycles containing five building blocks as opposed to four, with the goal of increasing the available peptide-target interactions to develop inhibitors of thrombin and tissue kallikrein 5 (KLK5), both important disease targets (15, 16). We did this by reacting 1284 different tetrapeptides with the same seven linkers as above, again in 384-well plates (Fig. 3A). The peptides were synthesized with a Gly-Gly-Trp appendix to purify them by simple precipitation in ether and to measure their concentration by tryptophan absorption. The library was designed to contain in the first three amino acid positions: (i) an arginine to target trypsin-like proteases, (ii) one of 107 amino acids with diverse side chains or backbone structures (table S1), and (iii) a glycine or β-alanine (20) to vary the macrocyclic backbone. The last position of the tetrapeptides was occupied by a cysteine in all peptides for cyclization. Screening against thrombin identified 178 hits with more than 50% protein inhibition and 5 hits with more than 95% inhibition (Fig. 3A). In the KLK5 screen, six hits showed more than 50% inhibition (fig. S5A).
Fig. 3. Library of 8988 macrocycles screened against thrombin.
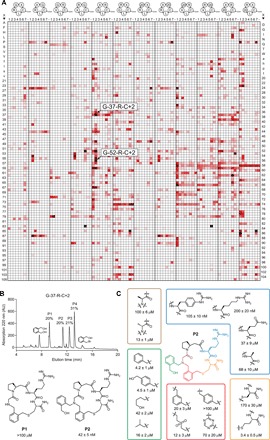
(A) Initially, 1284 tetrapeptides were synthesized with a C-terminal Gly-Gly-Trp tag (GGW) and cyclized with the seven bis-electrophiles 1 to 7 (L) to obtain macrocycles of the formats indicated above the matrix. X = any of 107 natural or unnatural amino acids (table S1), and 20 = β-alanine. The peptides G-37-R-C and G-52-R-C cyclized with 2, inhibiting thrombin >96%, are indicated. The extent of inhibition is indicated with color (black for 100% inhibition, red for 50% inhibition, and white for 0% inhibition; legend in Fig. 2A). (B) Reversed-phase high-performance liquid chromatography (RP-HPLC) chromatogram of 100 μM of G-37-R-C reacted with 800 μM of 2 showing four peptidic products (P1 to P4). P1 was identified as the expected macrocycle and P2 corresponds to macrocycle modified with a second linker 2 (at the Gly NH; second CH2Br is hydrolyzed). AU, arbitrary units. (C) Structure-activity relationship of P2. Mean values and SD are indicated for three measurements.
In the thrombin screen, the peptides G-37-R-C (37 = l-β-homoproline) and G-52-R-C (52 = d-β-homoproline) cyclized with 2 produced the lowest residual enzyme activity, namely, 3.1 and 3.4%, respectively, and were characterized in greater detail. Chromatographic analysis showed the formation of four major products in both reactions (fig. S5B). Resynthesis of these two peptides without the Gly-Gly-Trp tails and cyclization with 2 also yielded four products (Fig. 3B and fig. S5C). Inhibition assays showed that the second peak in each reaction (P2) was the active compound, inhibiting thrombin with Kis of 42 ± 5 nM (G-37-R-C) and 105 ± 6 nM (G-52-R-C). The other peaks had weak activities in the micromolar range or were inactive (Fig. 3B and fig. S6A). MS analysis revealed that the first peak (P1) with low activity was the expected macrocycle (structure P1; Fig. 3B) and that the highly active peak (P2) was the macrocycle with an additional linker 2 connected to the secondary amine in the backbone (structure P2, Fig. 3B). The second bromomethyl group of 2 was hydrolyzed to give an N-(2-(hydroxymethyl)benzyl) substituent. While not foreseen, linker 2 reacted twice with the peptide to generate a macrocycle with nanomolar potency. A structure-activity relationship analysis showed that all five building blocks of the macrocycle and the unforeseen hydroxymethyl-benzyl group, the precise backbone structure, and the amino acid stereocenters were all essential for binding (Fig. 3C and fig. S6B), indicating that the macrocycle needs a well-defined structure to fit the target and underscoring the importance of including skeletal diversity in macrocyclic libraries. Analysis of hits from the KLK5 screen identified macrocycles with Kis in the high nanomolar to low micromolar range (fig. S6C).
A specificity profiling of the thrombin inhibitor P2 showed that the most similar proteases, structurally and functionally, were not inhibited, demonstrating a high target selectivity (Fig. 4A). As expected, P2 inhibited coagulation in blood plasma, as seen by a prolonged activated partial thromboplastin time [aPTT; effective concentration that doubles clotting time (EC2×) = 4.5 μM] and prothrombin time (PT; EC2× = 4.2 μM) (Fig. 4B). X-ray crystallographic analysis of P2 bound to thrombin revealed that the macrocycle occupies a 201 Å2 region of the non-prime substrate-binding space [resolution, 2.3 Å; Protein Data Bank (PDB) 6GWE; Fig. 4, C and D, fig. S7, and tables S2 and S3]. P2 binding displaced Glu217 from its preferred position by around 2.6 Å, creating a new hydrophobic cavity that was occupied by the benzyl group of 2 (Fig. 4, E and F). The snug fit of the specific backbone to the target validated the importance of screening a large skeletal diversity because a precise shape that would have been difficult to plan by rational design was required to fit this pocket.
Fig. 4. Specificity profiling, activity in human plasma, and x-ray structure.
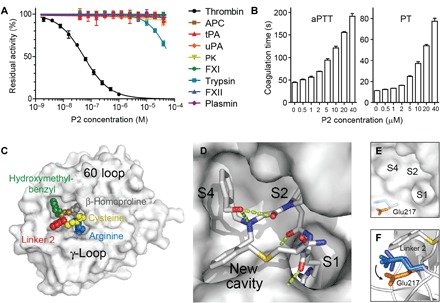
(A) Inhibition of homologous proteases by P2. APC, activated protein C; tPA, tissue plasminogen activator; uPA, urokinase plasminogen activator; PK, plasma kallikrein. (B) Inhibition of coagulation in human blood plasma. (C) X-ray structure of P2 bound to thrombin at a resolution of 2.3 Å (PDB entry 6GWE). The 60 and γ-loops are shown for orientation. Colors correspond to those in Fig. 3C. (D) Binding to non-prime subsites S1, S2, S4, and a new cavity. H-bond interactions are shown as green dashed lines. (E) Non-prime site of thrombin without P2 bound (PDB 1SG8). (F) Side chain of Glu217 before (blue; from thrombin structures ASG8, 4HFP, and 1HGT) and after P2 binding (orange). Linker 2 of P2 is shown in the figure.
DISCUSSION
In conclusion, we report thiol-to-amine macrocyclization reactions that allow for the generation of potentially ten- to hundreds of thousands of macrocycles with a high backbone diversity. In a proof-of-concept study, we identified a selective nanomolar inhibitor that perfectly fits the active site of the target. Sampling a sufficiently large skeletal diversity provided a macrocycle with the optimal size, shape, and charge complementarity for the target. The ease of purification-free macrocycle production will allow for larger library screens to identify macrocyclic ligands for many targets. Macrocycles with a molecular mass below 1 kDa have a high chance to be cell permeable or offer a good starting point for making cell-permeable compounds by reducing the polar surface.
MATERIALS AND METHODS
Chemical reagents
All chemical linker reagents are commercially available. Reagents 1 to 4, 8, 10, and 13 were purchased from Sigma-Aldrich. Reagents 5, 6, and 16 were purchased from Fluorochem. Reagents 7 and 9 were bought from abcr. Reagents 11 and 12 were purchased from Acros and Apollo Scientific, respectively. Reagents 14 and 15 were purchased from MolMall.
Peptide synthesis and purification
Peptides were synthesized on an Intavis MultiPep RSi parallel peptide synthesizer by standard Fmoc (fluorenylmethyloxycarbonyl)solid-phase chemistry on Rink amide 4-methylbenzhydrylamine (MBHA) resin (resin, 0.3 mmol/g; scale, 0.05 mmol). The coupling was carried out twice for each amino acid [4 equivalents (equiv)] using hexafluorophosphate azabenzotriazole tetramethyl uronium (HATU;4 equiv) and N-methylmorpholine (NMM; 10 equiv) in 0.9 ml of N,N´-dimethylformamide (DMF) with shaking at 400 rpm for 45 min. The resin was washed once with 2 ml of DMF, and unreacted amino groups were capped by incubation with 5% acetic anhydride and 6% 2,6-lutidine in 0.8 ml of DMF with shaking for 5 min. The resin was washed seven times with 2 ml of DMF, and Fmoc groups were removed by incubation twice with 0.8 ml of 20% (v/v) piperidine in DMF with shaking at 400 rpm for 5 min. The resin was washed seven times with 2 ml of DMF. At the end of the synthesis, the resin was washed twice with 0.6 ml of dichloromethane (DCM). In all washing steps, the resin was shaken at 400 rpm for 1 min.
Peptides were cleaved from the resin, and the protecting groups were removed under reducing conditions by incubation in 5 ml of cleavage solution [90% trifluoroacetic acid (TFA), 2.5% H2O, 2.5% thioanisole, 2.5% phenol, and 2.5% 1,2-ethanedithiol (EDT)] with shaking for 2 hours at room temperature (RT). The resin was removed by vacuum filtration, and peptides were precipitated with cold diethyl ether (45 ml), incubated for 30 min at −20°C, and pelleted by centrifugation at 4000g for 30 min.
Peptides were purified with a high-performance liquid chromatography (HPLC) system (Prep LC 2535 HPLC, Waters) using a preparative C18 reversed-phase column (10 μm, 100 Å, 19 mm by 250 mm; SunFire prep C18 OBD, Waters), applying a flow rate of 20 ml/min and a linear gradient of 0 to 30% (v/v) solvent B for 30 min [solvent A: 99.9% (v/v) H2O and 0.1% (v/v) TFA; solvent B: 99.9% (v/v) acetonitrile (ACN) and 0.1% (v/v) TFA]. Fractions containing the desired peptide were lyophilized.
The purity was assessed by analyzing around 10 μg of peptide by reversed-phase HPLC (RP-HPLC; 1260 HPLC system, Agilent) using a C18 column (5 μm, 300 Å, 4.6 mm by 250 mm; ZORBAX 300SB-C18, Agilent). Peptides were run at a flow rate of 1 ml/min with a linear gradient of 0 to 50% (v/v) solvent B for 15 min [solvent A: 94.9% (v/v) H2O, 5% (v/v) ACN, and 0.1% (v/v) TFA; solvent B: 94.9% (v/v) ACN, 5% (v/v) H2O, and 0.1% (v/v) TFA]. The mass of purified peptides was determined by electrospray ionization MS (ESI-MS) in positive ion mode on a single quadrupole LC-MS (LCMS-2020, Shimadzu).
Determination of reaction kinetics
The inter- and intramolecular reactions of the thiol-to-amine macrocyclization were followed by quantifying substrate, intermediates, and product concentrations by analytical RP-HPLC (1260 HPLC system, Agilent). The samples were analyzed using a C18 column (5 μm, 300 Å, 4.6 mm by 250 mm; ZORBAX 300SB-C18, Agilent) and a linear gradient of 0 to 50% solvent B for 15 min [solvent A: 94.9% (v/v) H2O, 5% (v/v) ACN, and 0.1% (v/v) TFA; solvent B: 94.9% (v/v) ACN, 5% (v/v) H2O, and 0.1% (v/v) TFA] at a flow rate of 1 ml/min. The identity of isolated products was determined by MS (ESI-MS) in positive ion mode on a single quadrupole LC-MS (LCMS-2020, Shimadzu).
For determining the second-order rate constant k1 of the intermolecular reaction, the peptide Ac-GCGW (10 μM) was incubated with linker 1 (10, 20, 40, and 80 μM) in aqueous buffer [60 mM NH4HCO3 (pH 8)] containing 20% ACN for 5 min at 30°C, quenched by addition of benzyl mercaptan (800 μM), and analyzed by RP-HPLC. The rate of constant k1 of the reaction was calculated on the basis of the quantity of peptide linker thiobenzyl adduct formed using the equation Δ[peptide]/Δtime = k1[peptide][linker], assuming second-order reaction kinetics.
For determining the first-order rate constant k2 of the intramolecular reaction, the peptide GCGGW (100 μM) was incubated with linker 1 (800 μM) in aqueous buffer [60 mM NH4HCO3 (pH 8)] containing 20% ACN at 30°C. The samples were collected at different time points, quenched by benzyl mercaptan (8 mM), and analyzed by analytical RP-HPLC. The rate constant of the reaction was calculated on the basis of the formation of cyclized peptide using the equation Δ(peptide)/Δ(time) = k2(peptide), assuming (i) that the thiol-alkylation reaction is much faster than the cyclization reaction at the applied peptide and linker concentrations and (ii) that the intramolecular cyclization reaction follows first-order kinetics.
Determination of substrate scope
Short peptides containing an N-terminal primary or secondary amino group, an amino acid with a thiol group in the side chain and either a Gly-Gly-Trp or a Gly-Trp appendix at the C terminus, were used. The tryptophan residues facilitated HPLC analysis of the peptides and reaction products due to the strong absorption at 280 nm. Using a 0.5-ml reaction tube, the following reagents were added sequentially and incubated for 1 hour at 37°C: 78 μl of 60 mM NH4HCO3 (pH 8), 2 μl of peptide in H2O (5 mM), and 20 μl of linker reagent in ACN (4 mM). The final concentrations of peptide and linker reagent were 100 and 800 μM, respectively. The reactions were analyzed by LC-MS (LCMS-2020, Shimadzu) [injection volume of 4 μl; Kinetix C18 column, 2.6 μm, 100 Å, 50 mm by 2.1 mm (Phenomenex); gradient: solvent A (99.95% H2O and 0.05% formic acid) and 0 to 40% solvent B (99.95% ACN and 0.05% formic acid) for 5 min; flow, 1 ml/min; MS analysis, positive mode].
Pilot library synthesis and screening
Peptides were synthesized as described above at a 0.05 mmol scale. The peptide pellets were dissolved in 10 ml of water and directly purified by RP-HPLC on Waters HPLC system (Prep LC 2535 HPLC) using a semipreparative C18 column (Waters XBridge BEH300 prep C18, 5 μm, 10 mm by 250 mm), applying a linear gradient of 0 to 30% solvent B for more than 30 min [solvent A: 99.9% (v/v) H2O and 0.1% (v/v) TFA; solvent B: 99.9% (v/v) ACN and 0.1% (v/v) TFA] at a flow rate of 6 ml/min. Fractions containing the peptides were lyophilized and dissolved in H2O to prepare 5 mM stock solutions.
The peptides were cyclized with linkers in a combinatorial fashion in a 384-deep-well plate (3347, Corning) as follows. Seventy-eight microliters of 60 mM NH4HCO3 buffer (pH 8) was added to the wells by an automated dispenser (Multidrop Dispenser, Thermo Fisher Scientific) using eight lines and a small cassette. Two microliters of 5 mM purified linear peptides was transferred by a Biomek FXP laboratory automation workstation from a v-bottom 96-well plate (6018321, Ratiolab) containing different peptides in 88 wells and water in eight wells to eight quadrants of two 384-deep-well plates using a 96 pipetting head and 100-μl disposable plastic tips (AP96 P20, 717255, Beckman). Twenty microliters of 4 mM linker 1 to 7 (4 mM in ACN) was transferred from 96-well plates [v-bottom polypropylene (PP), 6018321, Ratiolab] to the two 384-deep-well plates using a 96 pipetting head and 100-μl disposable plastic tips (AP96 P20, 717255, Beckman) using the same workstation. In each transfer, one of the seven linkers placed in 88 wells of the 96-well plate was transferred along with ACN in eight wells. The plates were incubated at 37°C for 2 hours.
The reaction products were screened for inhibition of trypsin and thrombin in low volume flat bottom non-binding surface (NBS) 384-well plates (3820, Corning) as follows. The reaction mixture (1.5 μl) was transferred from the reaction plate to the assay plate using the Biomek FXP laboratory automation workstation and a 384 pipetting head using 50-μl disposable plastic tips (AP384 P30XL, A22288, Beckman). Human cationic trypsin (8.5 μl, 0.18 nM) or human α thrombin (3.53 nM) in assay buffer [10 mM tris-Cl (pH 7.4), 150 mM NaCl, 10 mM MgCl2, 1 mM CaCl2, 0.1% (w/v) bovine serum albumin (BSA), and 0.01% (v/v) Triton X-100] was dispensed with the BioTek MultiFlo dispenser to each well to reach final concentrations of 0.1 nM for trypsin and 2 nM for thrombin and incubated at RT for 15 min. Five microliters of fluorogenic protease substrates in assay buffer containing 3% dimethyl sulfoxide (DMSO) was dispensed to each well using the same dispenser but different cassettes for enzyme and substrate. For both enzymes, the substrate Z-Gly-Gly-Arg-AMC (150 μM) was added to reach a final concentration of 50 μM. The residual protease activity was measured by monitoring the change in fluorescence intensity over time. The fluorescence intensity was measured with a fluorescence plate reader (excitation, 360 nm; emission, 465 nm; Tecan Infinite F500) at 25°C for 30 min with a reading every 3 min. The extent of inhibition (%) was calculated by comparing the residual activity to a control without macrocycle.
Preparative synthesis of macrocycles
Cyclic peptides were produced in milligram scale by cyclizing purified linear peptide as follows. Around 4 mg of peptide was dissolved in 80 ml of 60 mM NH4HCO3 (pH 8) to reach a concentration of 125 μM. Twenty milliliters of 2 mM linker reagent in ACN was added to reach a final concentration of 100 μM peptide and 400 μM linker reagent. The reaction was incubated for 2 hours at 37°C. The reaction was lyophilized, and the products were dissolved sequentially in 1 ml of DMSO and 9 ml of H2O and purified by preparative RP-HPLC.
Measurement of inhibition constants
Inhibition constants (Ki) were determined by measuring the residual activities of protease in the presence of different dilutions of inhibitor (twofold dilutions) using fluorogenic substrates. Activities were measured at 25°C in buffer containing 10 mM tris-Cl (pH 7.4), 150 mM NaCl, 10 mM MgCl2, 1 mM CaCl2, 0.1% (w/v) BSA, and 0.01% (v/v) Triton X-100 by monitoring the change of fluorescence intensity (excitation, 368 nm; emission, 467 nm) or absorption (405 nm) for 30 min using a Tecan Infinite M200 Pro plate reader. Proteases were of human origin and were purchased from Molecular Innovations or Innovative Research. KLK5 was expressed recombinantly as described elsewhere. The following protease concentrations were used: 0.1 nM trypsin, 2 nM thrombin, 5 nM KLK5, 0.6 nM protein C, 7.5 nM tPA (tissue plasminogen activator), 1.5 nM uPA (urokinase plasminogen activator), 0.25 nM plasma kallikrein, 1 nM factor XIa, 2 nM factor XIIa, and 2.5 nM plasmin. The following fluorogenic substrates were used at a final concentration of 50 μM: Z-Gly-Gly-Arg-AMC for trypsin, thrombin, tPA and uPA, Z-Phe-Arg-AMC for plasma kallikrein, Boc-Phe-Ser-Arg-AMC for factor XIa, Boc-Gln-Gly-Arg-AMC for factor XIIa, and H-D-Val-Leu-Lys-AMC for plasmin. The fluorogenic substrate Val-Pro-Arg-AMC for KLK5 was used at a final concentration of 100 μM. The chromogenic substrate Pyr-Pro-Arg-pNA was used at a final concentration of 0.48 mM to measure protein C activity. The median inhibitory concentration (IC50) values were determined by fitting sigmoidal curves to the data using the following four-parameter logistic equation used for dose response curves
x is the logarithm of the concentration of the peptide, y is the percentage activity relative to the reaction without peptide, and IC50 values were derived from the fitted curve. p is the Hill coefficient. The Ki values were calculated on the basis of the IC50s using the Cheng-Prusoff equation. The Km for trypsin and thrombin for the substrate Z-Gly-Gly-Arg-AMC were determined to be 90 and 168 μM, respectively, and the Km of KLK5 for Val-Pro-Arg-AMC was 216.3 μM.
Large-scale library synthesis and screening
Peptides were synthesized by Fmoc chemistry on Rink amide MBHA resin (resin, 0.3 mmol/g) at a scale of 5 μmol in 96-well reaction plates (32.410, Intavis) using an automated peptide synthesizer (Intavis MultiPep RSi). The coupling was carried out twice for each amino acid (4 equiv) using HATU (4 equiv) and NMM (10 equiv) in 130 μl of DMF without shaking for 45 min. The resin was washed twice with 225 μl of DMF, and unreacted amino groups were capped by incubation with 5% acetic anhydride and 6% 2,6-lutidine in 100 μl of DMF without shaking for 5 min. The resin was washed six times with 225 μl of DMF, and Fmoc groups were removed by incubation twice with 120 μl of 20% (v/v) piperidine in DMF without shaking for 5 min. The resin was washed eight times with 200 μl of DMF. At the end of the synthesis, the resin was washed twice with 200 μl of DCM. In all washing steps, the resin was incubated for 1 min.
Peptides were cleaved from the resin, and the protecting groups were removed under reducing conditions as follows. Volumes of 300, 100, and 100 μl of cleavage solution (90% TFA, 2.5% H2O, 2.5% thioanisole, 2.5% phenol, and 2.5% EDT) were added sequentially to wells of the reaction plate and incubated each time for 30 min to pass the resin by gravity flow. The flow-through was collected in a 96-deep-well plate (32.296, Intavis). Another 100 μl of cleavage solution was added, vacuum was applied, and the volume of the reaction solution was reduced by evaporation to 0.2 ml. Peptides were precipitated by addition of 1.5 ml of cold diethyl ether to each well and incubation for 1 hour at −20°C and pelleted by centrifugation at 4000g for 30 min. The peptide pellets were dissolved in ACN/H2O (1:1) and lyophilized, and the peptide was redissolved in H2O. The concentration was determined by measuring absorption at 280 nM using a nanodrop 8000 device (Thermo Scientific). The concentration was adjusted by addition of H2O to obtain stock solutions of 5 mM in v-shaped PP 96-well plates. The peptides were cyclized with linkers in a combinatorial fashion in 384-deep-well plates, as described above for the pilot-scale macrocycle library.
The reaction products were screened for inhibition of thrombin and KLK5 in low volume flat bottom NBS 384-well plates (Corning 3820) as follows. Reaction mixture (2 μl) was transferred from the reaction plate to the assay plate using the Biomek FXP laboratory automation workstation and a 384 pipetting head using 50-μl disposable plastic tips (AP384 P30XL, A22288). Eight microliters of human α thrombin (3.75 nM) or KLK5 (3.38 nM) in assay buffer [10 mM tris-Cl (pH 7.4), 150 mM NaCl, 10 mM MgCl2, 1 mM CaCl2, 0.1% (w/v) BSA, and 0.01% (v/v) Triton X-100] were dispensed with the BioTek MultiFlo dispenser to each well to reach final concentrations of 2 nM for thrombin and 5 nM for KLK5 and incubated at RT for 15 min. Five microliters of fluorogenic protease substrates in assay buffer containing 3% DMSO were dispensed to each well using the same dispenser. For thrombin, the substrate Z-Gly-Gly-Arg-AMC (150 μM) was added to reach a final concentration of 50 μM, and for KLK5, Val-Pro-Arg-AMC (300 μM) was added to reach 100 μM.
The residual protease activity was measured by monitoring the change in fluorescence intensity over time. The fluorescence intensity was measured with a fluorescence plate reader (excitation, 360 nm; emission, 465 nm; Tecan Infinite F500) at 25°C for 30 min with a reading every 3 min. The extent of inhibition (%) was calculated by comparing the residual activity to a control without macrocycle.
Preparative synthesis of P2 and derivatives
Peptides were synthesized on the Intavis MultiPep RSi parallel peptide synthesizer by standard Fmoc solid-phase chemistry on Rink amide MBHA resin (resin, 0.3 mmol/g; scale, 0.025 mmol) as described above. N-terminal acylation of peptide was carried out manually by treating the free N-terminal resin-bound peptide with bromoacetic acid (0.4 M, 170 μmol, 6.8 equiv) in DMF (425 μl), followed by addition of N,N′-diisopropylcarbodiimide (2 M, 200 μmol, 8 equiv) in DMF (100 μl). The reaction mixture was shaken at 400 rpm for 30 min. The resin was washed seven times with 0.6 ml of DMF. SN2 displacement reactions were performed by treatment of primary amine (2.5 M, 1.0 mmol, 40 equiv) in DMSO (0.5 ml), followed by shaking for 1 hour. Peptides were washed, cleaved, cyclized, and purified as described above.
Determination of PT and aPTT
Coagulation times were determined in human plasma using a STAGO STart 4 coagulation analyzer (Diagnostica). Human single donor plasma was used (Innovative Research). For the extrinsic coagulation, 50 μl of plasma with or without inhibitor was placed in the incubating chamber of the instrument for 2 min at 37°C. After incubation, 100 μl of innovin (recombinant human tissue factor, synthetic phospholipids, and calcium in stabilized Hepes buffer system; Dade Behring/Siemens) was added using the pipet connected to the instrument. Upon addition of this reagent, the electromagnetically induced movement of a steel ball in the plasma was monitored. The time until the ball stops moving was recorded as coagulation time. For the intrinsic coagulation, 100 μl of plasma with or without inhibitor was incubated with 100 μl of Pathromtin SL (silicon dioxide particles and plant phospholipids in Hepes buffer system, Siemens) for 2 min at 37°C. Subsequently, the coagulation was triggered by addition of 100 μl of CaCl2 solution (25 mM, Siemens).
Crystallization, data collection, and structure determination
Human α-thrombin was purchased from Haematologic Technologies (catalog no., HCT-0020). The protein-stabilizing agent was removed by using PD-10 desalting column (GE Healthcare) equilibrated with 20 mM tris-HCl and 200 mM NaCl (pH 8.0). Buffer-exchanged human α-thrombin was incubated with the macrocycle P2 at a molar ratio of 1:3 and subsequently concentrated to 7.5 mg/ml by using a 3000 MWCO Vivaspin ultrafiltration device (Sartorius Stedim Biotech GmbH). Further P2 macrocycle was added during the concentration procedure to ensure that the inhibitor was present at a molar excess of at least threefold.
Crystallization trials of the complex were carried out at 293 K in a 96-well two-drop MRC (Medical Research Council) crystallization plate (Hampton Research, CA, USA) using the sitting-drop vapor diffusion method and the JCSG-plus, PACT premiere, Morpheus, and LMB crystallization screens (Molecular Dimensions Ltd., Suffolk, UK). Droplets of 600-nl volume (with a 1:1 protein:precipitant ratio) were set up using an Oryx 8 crystallization robot (Douglas Instruments Ltd., Berkshire, UK) and equilibrated against 80 μl of reservoir solution. Crystals appeared within 2 to 3 days in two different conditions: (i) 100 mM sodium citrate and 20% (w/v) polyethylene glycol, molecular weight 3000 (PEG 3000) (pH 5.5) and (ii) 100 mM MES and 15% (w/v) PEG 3350 (pH 6.2). Further attempts to optimize the conditions were performed by varying the protein complex concentration, the drop volume, and the protein:precipitant ratio. Best crystals were obtained using 100 mM MES and 15% (w/v) PEG 3350 (pH 6.2) as precipitant agent. For x-ray data collection, crystals were mounted on CryoLoops (Hampton Research, CA, USA), soaked in cryoprotectant solution [25% ethylene glycol, 100 mM MES, and 15% (w/v) PEG 3350 (pH 6.2)], and flash cooled in liquid nitrogen.
X-ray diffraction data of human α-thrombin in complex with P2 were collected at the ID29 beamline of the European Synchrotron Radiation Facility (Grenoble, France). The best crystals diffracted to a maximum solution of 2.30 Å. Crystals belong to the P212121 space group with unit cell dimensions a = 55.94 Å, b = 80.91 Å, and c = 159.43 Å. The asymmetric unit contains two molecules, corresponding to a Matthews coefficient of 2.67 Å3/Da and a solvent content of about 54% of the crystal volume. Frames were indexed and integrated with software XIA2 via the automatic processing protocol available at the beamline, merged, and scaled with AIMLESS (CCP4i2 crystallographic package).
The structure was solved by molecular replacement with the software PHASER, and the model 3U69 was used as a template. Refinement was carried out using REFMAC and Phenix. Rebuilding and fitting of the macrocycle were performed manually with graphic software COOT. Since the first cycles of refinement, the electron density corresponding to a portion of the bound peptide was visible in the electron density map. The refined model contains 4975 protein atoms, 141 P2 ligand atoms, 150 water molecules, and 48 atoms of other molecules. The final crystallographic R factor is 0.20 (Rfree, 0.236). Geometrical parameters of the model are as expected or better for this resolution. The solvent-excluded volume and the corresponding buried surface were calculated by the 3V web server using a spherical probe of 1.5 Å radius. Intramolecular and intermolecular hydrogen bond interactions were analyzed by ProFunc, LigPlot+, and PyMOL software.
Supplementary Material
Acknowledgments
Funding: This work was supported by the Swiss National Science Foundation (grant 157842, NCCR Chemical Biology, and grant 166929). Author contributions: C.H., S.S.K., M.B.-B., M.G.K., M.V.P., J.F.M., and K.D. developed and studied the macrocyclization reactions. S.S.K., M.G.K, Y.W., J.B., J.V., and G.T. performed the screens. Y.W. identified P2. P.G. expressed KLK5. A.A., L.C., and X.-D.K. solved and analyzed the x-ray structure. All authors analyzed the data, discussed the results, and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors upon request. Crystallographic data are available from the PDB under accession no. 6GWE.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaaw2851/DC1
Supplementary Results
Fig. S1. Reaction kinetics of the thiol-to-amine cyclization reaction.
Fig. S2. Thiol-to-nitrogen cyclization reagents and side products.
Fig. S3. Scaffold diversity in pilot-scale macrocycle library.
Fig. S4. Characterization of trypsin and thrombin hits.
Fig. S5. KLK5 screen and HPLC analysis of thrombin hits.
Fig. S6. Structures, HPLC analysis, and activities of macrocycles.
Fig. S7. Chemical structure of P2.
Table S1. Chemical structures and codes of amino acids.
Table S2. Statistics on x-ray structure data collection and refinement.
Table S3. Interactions between P2 and thrombin.
Data S1. Raw data of peptide macrocyclization reactions.
Data S2. Poster showing all 432 different macrocycle scaffolds in the pilot-scale library.
REFERENCES AND NOTES
- 1.Driggers E. M., Hale S. P., Lee J., Terrett N. K., The exploration of macrocycles for drug discovery — An underexploited structural class. Nat. Rev. Drug Discov. 7, 608–624 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Giordanetto F., Kihlberg J., Macrocyclic drugs and clinical candidates: What can medicinal chemists learn from their properties? J. Med. Chem. 57, 278–295 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Villar E. A., Beglov D., Chennamadhavuni S., Porco J. A., Kozakov D., Vajda S., Whitty A., How proteins bind macrocycles. Nat. Chem. Biol. 10, 723–731 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luther A., Moehle K., Chevalier E., Dale G., Obrecht D., Protein epitope mimetic macrocycles as biopharmaceuticals. Curr. Opin. Chem. Biol. 38, 45–51 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Deyle K., Kong X.-D., Heinis C., Phage selection of cyclic peptides for application in research and drug development. Acc. Chem. Res. 50, 1866–1874 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Taylor R. D., Rey-Carrizo M., Passioura T., Suga H., Identification of nonstandard macrocyclic peptide ligands through display screening. Drug Discov. Today Technol. 26, 17–23 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Marcaurelle L. A., Comer E., Dandapani S., Duvall J. R., Gerard B., Kesavan S., Lee M. D. IV, Liu H., Lowe J. T., Marie J. C., Mulrooney C. A., Pandya B. A., Rowley A., Ryba T. D., Suh B. C., Wei J., Young D. W., Akella L. B., Ross N. T., Zhang Y. L., Fass D. M., Reis S. A., Zhao W. N., Haggarty S. J., Palmer M., Foley M. A., An aldol-based build/couple/pair strategy for the synthesis of medium- and large-sized rings: Discovery of macrocyclic histone deacetylase inhibitors. J. Am. Chem. Soc. 132, 16962–16976 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Isidro-Llobet A., Murillo T., Bello P., Cilibrizzi A., Hodgkinson J. T., Galloway W. R. J. D., Bender A., Welch M., Spring D. R., Diversity-oriented synthesis of macrocyclic peptidomimetics. Proc. Natl. Acad. Sci. U.S.A. 108, 6793–6798 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Usanov D. L., Chan A. I., Maianti J. P., Liu D. R., Second-generation DNA-templated macrocycle libraries for the discovery of bioactive small molecules. Nat. Chem. 10, 704–714 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y., De Luca R., Cazzamalli S., Pretto F., Bajic D., Scheuermann J., Neri D., Versatile protein recognition by the encoded display of multiple chemical elements on a constant macrocyclic scaffold. Nat. Chem. 10, 441–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jo H., Meinhardt N., Wu Y., Kulkarni S., Hu X., Low K. E., Davies P. L., DeGrado W. F., Greenbaum D. C., Development of α-helical calpain probes by mimicking a natural protein–protein interaction. J. Am. Chem. Soc. 134, 17704–17713 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Timmerman P., Beld J., Puijk W. C., Meloen R. H., Rapid and quantitative cyclization of multiple peptide loops onto synthetic scaffolds for structural mimicry of protein surfaces. Chembiochem 6, 821–824 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Assem N., Ferreira D. J., Wolan D. W., Dawson P. E., Acetone-linked peptides: A convergent approach for peptide macrocyclization and labeling. Angew. Chem. Int. Ed. Engl. 54, 8665–8668 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kale S. S., Villequey C., Kong X.-D., Zorzi A., Deyle K., Heinis C., Cyclization of peptides with two chemical bridges affords large scaffold diversities. Nat. Chem. 10, 715–723 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Prassas I., Eissa A., Poda G., Diamandis E. P., Unleashing the therapeutic potential of human kallikrein-related serine proteases. Nat. Rev. Drug Discov. 14, 183–202 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Arsenault K. A., Hirsh J., Whitlock R. P., Eikelboom J. W., Direct thrombin inhibitors in cardiovascular disease. Nat. Rev. Cardiol. 9, 402–414 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaaw2851/DC1
Supplementary Results
Fig. S1. Reaction kinetics of the thiol-to-amine cyclization reaction.
Fig. S2. Thiol-to-nitrogen cyclization reagents and side products.
Fig. S3. Scaffold diversity in pilot-scale macrocycle library.
Fig. S4. Characterization of trypsin and thrombin hits.
Fig. S5. KLK5 screen and HPLC analysis of thrombin hits.
Fig. S6. Structures, HPLC analysis, and activities of macrocycles.
Fig. S7. Chemical structure of P2.
Table S1. Chemical structures and codes of amino acids.
Table S2. Statistics on x-ray structure data collection and refinement.
Table S3. Interactions between P2 and thrombin.
Data S1. Raw data of peptide macrocyclization reactions.
Data S2. Poster showing all 432 different macrocycle scaffolds in the pilot-scale library.