
Figure 1.
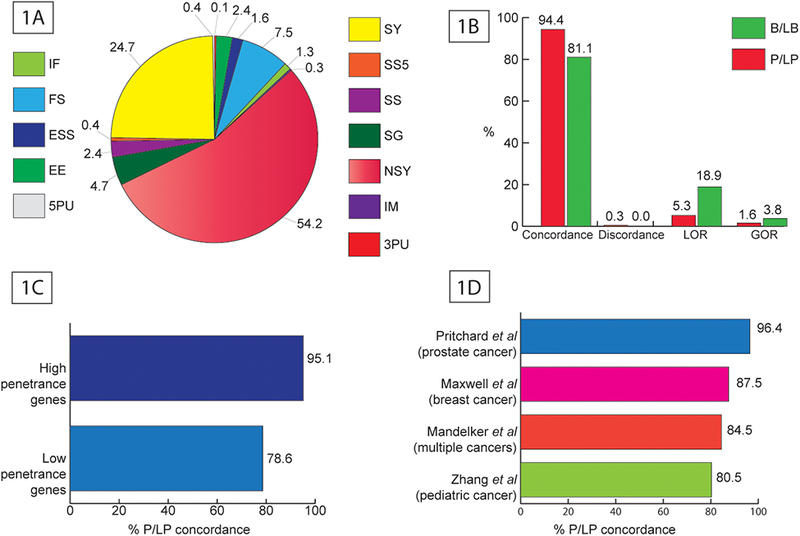
A: Distribution of 3513 variants in the test dataset by variant class (IF - Inframe insertion and/or deletion. It alters length but not frame of coding sequence, FS - Frameshifting insertion and/or deletion. It alters length and frame of coding sequence, ESS - Any variant that alters essential splice-site base (+1, +2, −1, −2), EE - Variant that alters the first or last three bases of an exon (i.e., the exon end), but not the frame of the coding sequence, 5PU - Any variant in 5′ untranslated region, SY - Synonymous variant. It does not alter amino acid or coding sequence length, SS5 - Any variant that alters +5 splice-site base but not an ESS base, SS - Any variant that alters splice-site base within the first eight intronic bases flanking exon (i.e., +8 to −8) but not an ESS or SS5 base, SG - Stop-gain (nonsense) variant caused by base substitution, NSY - Nonsynonymous variant. It alters amino acid(s) but not coding sequence length, IM - Variant that alters initiating methionine start codon, 3PU - Any variant in 3′ untranslated region). Figure 1B: Performance of PathoMAN’s variant classification against variant classification from three clinical laboratories – Ambry Genetics, Invitae and GeneDx. Figure 1C: Concordance of PathoMAN and clinical lab results for reported P/LP variants group by penetrance of the gene (1257 variants in 27 genes Supp table S5). Figure 1D: Concordance of PathoMAN and published reports for reported P/LP variants from four cancer studies (300 variants in 55 genes; Mandelker et al – 97 variants, Maxwell et al 40 variants, Pritchard et al – 56 variants and Zhang et al – 107 variants Supp table S7)