

Abstract
Acute brain ischemia induces a local neuroinflammatory reaction and alters peripheral immune homeostasis at the same time. Recent evidence has suggested a key role of the gut microbiota in autoimmune diseases by modulating immune homeostasis. Therefore, we investigated the mechanistic link among acute brain ischemia, microbiota alterations, and the immune response after brain injury. Using two distinct models of acute middle cerebral artery occlusion, we show by next-generation sequencing that large stroke lesions cause gut microbiota dysbiosis, which in turn affects stroke outcome via immune-mediated mechanisms. Reduced species diversity and bacterial overgrowth of bacteroidetes were identified as hallmarks of poststroke dysbiosis, which was associated with intestinal barrier dysfunction and reduced intestinal motility as determined by in vivo intestinal bolus tracking. Recolonizing germ-free mice with dysbiotic poststroke microbiota exacerbates lesion volume and functional deficits after experimental stroke compared with the recolonization with a normal control microbiota. In addition, recolonization of mice with a dysbiotic microbiome induces a proinflammatory T-cell polarization in the intestinal immune compartment and in the ischemic brain. Using in vivo cell-tracking studies, we demonstrate the migration of intestinal lymphocytes to the ischemic brain. Therapeutic transplantation of fecal microbiota normalizes brain lesion-induced dysbiosis and improves stroke outcome. These results support a novel mechanism in which the gut microbiome is a target of stroke-induced systemic alterations and an effector with substantial impact on stroke outcome.
SIGNIFICANCE STATEMENT We have identified a bidirectional communication along the brain–gut microbiota–immune axis and show that the gut microbiota is a central regulator of immune homeostasis. Acute brain lesions induced dysbiosis of the microbiome and, in turn, changes in the gut microbiota affected neuroinflammatory and functional outcome after brain injury. The microbiota impact on immunity and stroke outcome was transmissible by microbiota transplantation. Our findings support an emerging concept in which the gut microbiota is a key regulator in priming the neuroinflammatory response to brain injury. These findings highlight the key role of microbiota as a potential therapeutic target to protect brain function after injury.
Keywords: inflammation, microbiota, stroke, T cells
Introduction
The inflammatory reaction to sterile tissue injury is a critical pathophysiological component in organ-specific injuries, including ischemic stroke (Iadecola and Anrather, 2011; Chamorro et al., 2012), myocardial infarction (Hofmann and Frantz, 2015), and other conditions (Huang et al., 2007). Previous reports have shown that T cells play a defining role in secondary neuroinflammation after brain ischemia (Shichita et al., 2009; Liesz et al., 2013c; Schwartz and Raposo, 2014). Experimental approaches using pharmacological or genetic lymphocyte depletion paradigms (Yilmaz et al., 2006; Kleinschnitz et al., 2010) or pharmacological approaches to block cerebral lymphocyte invasion (Liesz et al., 2011; Neumann et al., 2015) have established a primary role for lymphocytes, particularly T cells, as crucial mediators of an inflammatory collateral damage to the injured brain after stroke. Previous work from our group and others has proposed that Thelper cell subpopulations might have differential effects on stroke outcome: proinflammatory Th1, Th17, and γδ T cells have been associated with increased inflammatory damage and worse outcome, whereas regulatory T cells (Tregs) have been shown to suppress an overshooting neuroinflammatory reaction to brain injury (Liesz et al., 2009a; Shichita et al., 2009; Gelderblom et al., 2012; Liesz et al., 2013c). In contrast to primary autoimmune diseases of the CNS, acute brain ischemia induces a rapid local and peripheral immune activation (Iadecola and Anrather, 2011; Shichita et al., 2012; Liesz et al., 2015). T cells are recruited to the injured brain already within the first days after stroke (Gelderblom et al., 2009; Zhou et al., 2013) and peripheral T-cell activation can also be observed (Offner et al., 2006; Yilmaz et al., 2006; Liesz et al., 2009b; Vogelgesang et al., 2010). Nevertheless, the mechanisms of peripheral T-cell activation, specifically Thelper cell polarization, after acute brain injury are still elusive.
Several lines of evidence suggest that the gut microbiota is a key regulator of T-cell homeostasis and is intricately involved in the maturation of the immune system and maintaining the mutual coexistence of host and microbe (Ivanov et al., 2009; Hooper et al., 2012; Arpaia et al., 2013). Recently, the functional relation between the gut microbiota and brain function, termed the “gut–brain axis,” has become an emerging field in neuroscience and neuroimmunology (Collins et al., 2012; Cryan and Dinan, 2012). The gut microbiota has been attributed a decisive role in autoimmune diseases of the CNS (Berer et al., 2011; Lee et al., 2011). A recent report by l (Benakis et al. (2016) has demonstrated that antibiotic-treatment-induced dysbiosis of intestinal microbiota influences poststroke neuroinflammation and outcome in an experimental stroke model. However, the impact of stroke on the microbiota composition and the contribution of stroke-specific microbiota alterations on neuroinflammation were previously unknown. Therefore, the aim of this work was to investigate microbiota alterations after brain ischemia and their role in the poststroke neuroinflammatory reaction.
In this study, we observe dysbiosis of the gut microbiota via stress-mediated intestinal paralysis after stroke. In turn, microbiota dysbiosis is causally linked to changes in T-cell homeostasis, induction of a proinflammatory response, and deterioration of stroke outcome. Fecal microbiota transplantation to normalize poststroke dysbiosis is associated with an improved stroke outcome. Altogether, our data point to a novel and highly complex interplay between the brain and the gut microbiota after acute brain injury in which the microbiota is a target of stress-mediated pathways resulting dysbiosis, as well as an effector of immune homeostasis with profound impact on stroke outcome.
Materials and Methods
Animal experiments.
All animal experiments were performed consistent with the guidelines for the use of experimental animals and were approved by the governmental committee of Upper Bavaria (Regierungspraesidium Oberbayern #2532-65-2014). Wild-type C57BL/6J mice and Rag1−/− male mice were obtained from Charles River Laboratories. Germ-free (GF) C57BL/6J and GF Rag1−/− female mice were obtained from the Clean Mouse Facility, University of Bern, Switzerland. A priori sample size calculation was based on either variance and effect size from previous studies or on preliminary pilot experiments performed during this study. Data were excluded from all mice that died after surgery. Animals were randomized to treatment groups and all analyses were performed by investigators blinded to group allocation. Unblinding was performed after completion of statistical analyses. All animal experiments were performed and reported consistent with the ARRIVE guidelines (Kilkenny et al., 2010).
GF mouse handling and intestinal recolonization.
GF mice were housed in sterile HAN-gnotocages and received the same sterile water and pelleted food as conventional or recolonized mice. All surgical procedures, cage changes, and behavioral tests of GF mice were performed in a disinfected laminar flow microbiological safety cabinet. GF mice were recolonized with gut microbiota obtained from sham or post-filament middle cerebral artery (MCA) occlusion model (fMCAo) donor mice by gastric gavage of freshly prepared cecum microbiota. In brief, the donor mice were killed, cleaned with 70% ethanol, and the peritoneal cavity was opened to expose the cecum. The contents of the cecum were collected in sterile tubes, diluted in sterile water to 200 mg/ml, and centrifuged at 2000 rpm for 10 min to remove the large particles. The supernatants were collected into fresh sterile tubes and each recipient mouse received a single 200 μl bolus of the freshly prepared microbiota suspension.
Permanent distal MCA occlusion model (cMCAo).
Focal cerebral ischemia was induced as described previously by permanent occlusion of the MCA distal of the lenticulostriate arteries (Llovera et al., 2014). In brief, the mice were anesthetized with an intraperitoneal injection of fentanyl (0.05 mg/kg), midazolam (5 mg/kg), and medetomidine (0.5 mg/kg). The skull was exposed by skin incision, a burr hole was drilled in the temporal bone, and the MCA was permanently occluded using high-frequency electrocoagulation forceps. Immediately after surgery, anesthesia was antagonized by intraperitoneal injection of a combination of naloxon (1.2 mg/kg), flumazenil (0.5 mg/kg), and atipamezol (2.5 mg/kg). After recovery, mice were returned to their cages with ad libitum access to water and food. Sham surgery was performed by the same surgical procedures without coagulation of the exposed MCA. Throughout the surgical procedure, body temperature was maintained at 37°C using a feedback-controlled heating pad. The overall mortality rate in this experimental group was <5%. Exclusion criteria were subarachnoid hemorrhage or death during surgery.
Transient fMCAo.
Mice were anesthetized with isoflurane delivered in a mixture of 30% O2 and 70% N2O. An incision was made between the ear and the eye to expose the temporal bone. A laser Doppler probe was affixed to the skull above the MCA territory and the mice were placed in the supine position. An incision was made in the midline neck region and the common carotid artery and left external carotid artery were isolated and ligated; a 2 mm silicon-coated filament (catalog #701912PKRe; Doccol) was inserted into the internal carotid artery and MCA occlusion was confirmed by a corresponding decrease in blood flow (i.e., a decrease in the laser Doppler flow signal). After 60 min of occlusion, the animals were reanesthetized and the filament was removed. For the survival period, the mice were kept in their home cage with facilitated access to water and food. Sham-operated mice received the same surgical procedure except the filament was inserted and immediately removed. Body temperature was maintained throughout surgery using a feedback-controlled heating pad. The overall mortality rate in this group (excluding the sham-operated animals) was ∼10%. Exclusion criteria were as follows: insufficient MCA occlusion (a reduction in blood flow to >20% of the baseline value), mice that died during the surgery, and mice without induction of brain ischemia as quantified postmortem by histological analysis.
Cylinder test.
GF mice that were inoculated with a fecal suspension obtained from control and fMCAo-induced mice were tested for forepaw use and asymmetry by measuring behavioral asymmetry and general activity on the first and third days after cMCAo surgery. The test was performed in a sterile laminar flow safety cabinet. Mice were placed in a transparent acrylic glass cylinder (diameter: 8 cm; height: 25 cm) in front of two mirrors and videotaped (Llovera et al., 2014). To assess independent forelimb use, contact with one forelimb during full rear and landing with only one forelimb after full rear were scored by performing a frame-by-frame analysis of the recorded videos. All rearing movements during the trial were counted and used as a measure of the animal's overall activity.
Fecal microbiota transfer (FMT).
Freshly defecated feces were collected from restrained mice and placed in sterile tubes. Donor mice (Charles River) were housed under SPF conditions and the same ones were used for all recipient mice in a given group. After collection, the fecal pellets were immediately mixed in sterile water (to 200 mg/ml) and the suspension was centrifuged at 2000 rpm for 10 min. The supernatant was collected and 200 μl was administered to each mouse by gastric gavage. FMT treatment was administered daily for the survival period starting on the same day of stroke induction. For FMT experiments, the recipient mice were housed in cages with sterile bedding and ad libitum access to sterile water. For the fMCAo model, the mice did not have access to food to eliminate any confounding effects of differing food consumption between the fMCAo, sham-operated, and treatment groups. Therefore, fMCAo and FMT recipient mice received three daily oral doses of water-suspended ultra-fine ground chow containing the same composition and caloric intake as the pelleted food (equivalent to ∼2.5 mg of chow).
Infarct volumetry.
Perfused brains were removed at the indicated time points after stroke induction and frozen on powdered dry ice. Coronal cryosections (20 μm thick) were cut at 400 μm intervals. The sections were stained with cresyl violet consistent with standard protocols and scanned at 600 dpi. Infarct area was measured in each section using ImageJ software. For the fMCAo model, an edema correction for infarct volume was performed using the following formula: (ischemic area) = (direct lesion volume) − [(ipsilateral hemisphere) − (contralateral hemisphere)]. The total infarct volume was calculated by integrating the measured areas and intervals between the sections.
Preparation of cells from lymphoid organs.
Mice were deeply anesthetized with ketamine (120 mg/kg) and xylazine (16 mg/kg). Blood was obtained by intracardiac puncture and collected into EDTA test tubes; the plasma was isolated by centrifugation at 3000 rpm for 10 min and stored at −80°C until further use. The mice were then transcardially perfused with normal saline and the spleen and Peyer's patches (PPs) were removed into cold Hank's balanced salt solution. The organs were homogenized and filtered through 30–40 μm cell strainers; in the spleen samples, the erythrocytes were lysed using isotonic ammonium chloride buffer. The total cell counts per organ were measured using an automated cell counter (Bio-Rad). For the isolation of mucosal leukocytes, the distal ileum was dissected carefully to remove the muscle layer. The mucosal tissue was then minced into small pieces and incubated in RPMI medium containing 10 μg/ml DNase I and 0.5 mg/ml collagenase D (Roche) for 10 min at 37°C. The cell suspensions were then mechanically homogenized, filtered, and washed before flow cytometric analysis.
Isolation of brain-invading leukocytes.
Mice were deeply anesthetized with ketamine (120 mg/kg) and xylazine (16 mg/kg) and transcardially perfused with normal saline. Brains were carefully removed and ipsilateral hemispheres were used for preparation of cells as described previously (Liesz et al., 2011). Briefly, brains were cut into small pieces and digested in RPMI medium containing 10 μg of DNase I and 0.5 mg/ml collagenase D (Roche) for 10 min at 37°C. The cell suspension was mechanically homogenized and passed through a 40 μm cell strainer (Falcon). The cell suspension was resuspended with 40% Percoll solution (GE Healthcare) and overlayed on a 70% Percoll solution. After centrifugation at 2100 rpm for 25 min at room temperature, cells at the interphase were collected and washed in isotonic buffer.
Flow cytometry analysis.
The following mouse antigen-specific antibodies were purchased from eBioscience/BioLegend: CD3 FITC (17A2), CD4 PerCP Cy5 (clone RM4–5), CD45 eF450 (30-F11), CD11b PerCP Cy5.5 (M1/70), IFN-γ FITC (XMG1.2), and IL-17 APC (17B7). To quantify the various cell populations, cells were stained with specific antibodies in accordance with the manufacturer's protocols. For intracellular cytokine staining, cell suspensions from the spleen and PPs were resuspended in RPMI medium containing 10% heat-inactivated fetal calf serum, 1% penicillin/streptomycin, and 10 μm 2-mercaptoethanol. The cells were plated at a density of 3 × 105 cells/well on anti-CD3 antibody (clone 145-2C11; eBioscience)-coated 96-well plates, costimulated with soluble CD28 antibody (clone 37.51; eBioscience), and incubated for 40 h at 37°C and 5% CO2. After incubation, GolgiPlug (BD Biosciences) was added to the cells for 4 h, after which the cells were harvested for intracellular staining using the Foxp3/Transcription Factor Staining Kit (eBioscience). Stained cells were measured in a FACSverse flow cytometer (BD Biosciences) and analyzed using FlowJo version 10 (TreeStar).
FISH and histochemical staining.
The distal ileum was ligated on both sides with surgical sutures, removed, fixed in 4% paraformaldehyde, and 6-μm-thick transverse sections were prepared. The sections were mounted on glass slides and FISH was performed in an equilibrated humidity chamber at 46°C in hybridization buffer containing 0.9 m NaCl, 20 mm Tris-HCl, pH 7.2, 30% formamide, and 0.01% SDS for 3 h. Probes (EUB338: Cy5-GCT GCC TCC CGT AGG AGT-Cy5; EUB338-III: Cy5-GCT GCC ACC CGT AGG TGT-Cy5) were used at a final concentration of 5 ng/μl. Sections were hybridized in 20 μl of hybridization buffer containing the EUB338 or EUB338-III probe. The sections were then placed in prewarmed (48°C) wash buffer containing 100 mm NaCl, 20 mm Tris-HCl, pH 7.2, and 5 mm EDTA in autoclaved Milli-Q water (Millipore) for 10 min. SYTOX green (1:5000) was used to counterstain the nuclei. Images (1024 × 1024 pixels, 400 Hz scan rate) were acquired as z-stacks using a 63× glycerol objective on a confocal microscope (TCS SP5 X; Leica).
Immunohistology.
For analysis of labeled T cells from PPs, 12 μm coronal brain sections at bregma height were stained with anti-mouse CD3 polyclonal antibody (1:50; Abcam) and goat anti-rabbit Alexa Fluor 488 (1:200; Jackson Laboratories) antibodies. After nuclear staining with DAPI, sections were analyzed for the presence of CM-DiI+CD3+DAPI+ cells using confocal microscopy (TCS SP5 X; Leica).
Tracing migration of leukocytes from PPs to the brain.
Forty-eight hours after cMCAo or sham surgery, cells within PPs were labeled by microinjection of fluorescent-cell-staining dyes CFSE (25 μm in 2 μl of PBS per PP) or CM-DiI (5 μm in 2 μl of PBS per PP) (Life Technologies). Mice were killed 24 h after cell labeling and brains prepared either for flow cytometric analysis (CFSE labeling) or immunohistological analysis (CM-DiI labeling) as described above.
Postoperative ileus model.
Mice were anesthetized with isoflurane delivered in a mixture of 30% CO2 and 70% N2O. Before surgery, an injection of carprofen (5 mg/kg) was administered to the mice. After this, mice were fixed with surgical tape in the supine position on a feedback-regulated heating pad. The abdominal cavity was then opened 2 cm in length along the linea alba. With two moist sterile cotton applicators, the small intestine content was carefully touched from the pylorus to the cecum as described previously (Vilz et al., 2012). After surgery, the mice were sutured, monitored in a recovery chamber, and then returned to their home cages. Fecal samples were collected before and 3 d after the surgery for microbial DNA isolation and metagenomic analysis. Directly afterward, an oral bolus of FITC-dextran was administered for the gut motility test.
Gastrointestinal motility analysis.
Mice received a 100 μl oral bolus of 50 mg/ml FITC-dextran (70,000 kDa; Sigma-Aldrich) in 0.9% PBS. One hour after administration, the mice were killed and the entire intestinal tract from the stomach to the colon was removed and imaged using a chemiluminescence detection system (Fusion FX7). To quantify gastrointestinal transit (i.e., motility), the complete gastrointestinal tract was divided into segments, each segment was flushed with distilled water, and the fluorescence of the purified recovered flushing solution was measured using a fluorescence plate reader (Promega). The values obtained were normalized to blank controls and expressed as the percentage of fluorescence per intestinal segment.
ELISA for albumin quantification.
Mice were killed 3 d after fsham or fMCAo surgery and feces and plasma samples were collected for measurement of albumin concentrations. Sampled feces (50 mg/ml) and plasma (1:50000) were diluted in assay buffer (50 mm Tris, 0.1 m NaCl, 0.05% Tween 20, pH 7.4) and ELISA was performed with corresponding albumin standard dilutions. Albumin was quantified by ELISA and the ratio of feces to plasma albumin are presented (Bethyl Labratories).
Bacterial cultures.
Mice were deeply anesthetized and perfused transcardially with normal saline. The cecum's contents were then transferred to sterile tubes under strict aseptic conditions. A fixed amount of cecum content from each mouse was mixed in PBS to 50 mg/ml and dilutions were prepared. Two dilutions (1:40,000 and 1:400,000) from each sample were plated on blood agar plates (Oxoid) and incubated at 37°C for 48–72 h. To generate an anaerobic environment, the plates were placed in anaerobic jars containing AnaeroGen paper sachets (Oxoid). After the incubation period, the plates were examined for bacterial growth and the number of colonies was determined.
Microbial community analysis.
DNA from mouse feces was isolated using the QIAamp Fast DNA Stool Mini Kit (Qiagen). The total amount of DNA in each sample was measured using a Qubit fluorometer (Life Technologies). 16S rRNA amplicons were generated using primers corresponding to the hypervariable regions V1-V3 (primer 27F: AGAGTTTGATCCTGGCTCAG; primer 534R: ATTACCGCGGCTGCTGG), and the PCR products were purified. Libraries were prepared using the standard tagmentation procedure (Nextera XT; Illumina). All samples were sequenced on an Illumina MiSeq platform using a 300 bp paired-end approach. Amplicon sequencing datasets were analyzed using the Metagenomics (MG)-RAST pipeline (version 3.3.6) (Meyer et al., 2008). Low-quality reads (comprising <0.01% of all reads) were trimmed using the SolexaQA program; only high-quality reads were included in the subsequent analysis. 16S rRNA genes were identified by performing a BLAT (Kent, 2002) search, and amplicons were clustered at 97% identity with an e-value cutoff of 1e-5. A BLAT similarity search for the longest cluster representative was performed against the Ribosomal Database Project (RDP) database. Rarefaction curves, α diversity, and phylogenetic analysis were performed and phylogenetic trees were drawn using the MG-RAST platform. Read counts were normalized logarithmically for subsequent analysis. Principal component analysis plots were drawn using normalized values and Bray–Curtis distance. Differences between group means for genera were analyzed using ANOVA or Student's t test. p-values were adjusted for comparison of multiple comparisons using Bonferroni correction. The DNA sequences have been deposited in MG-RAST (http://metagenomics.anl.gov/) under accession number 9778. The linear discriminant analysis (LDA) effect size (LEfSe) method was used to identify microbial taxa showing a statistically different relative abundance between the microbiota after sham and fMCAo surgery (Segata et al., 2011). Experiments were performed with an α error set to 0.05 and the LDA threshold set to 2.0.
Quantitative RT-PCR.
Brain tissue from the ipsilateral and contralateral hemispheres was lysed in Qiazol Lysis Reagent (Qiagen) and total RNA was extracted using the MaXtract High Density kit (Qiagen). RNA was purified using the RNeasy Mini Kit in accordance with the manufacturer's instructions (Qiagen). Equal amounts of RNA from each sample were used to synthesize cDNA with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The quantitative expression of various cytokines was measured using predesigned RT2 qPCR Primer Assays and SYBR Green ROX qPCR Mastermix (Qiagen) in a LightCycler 480 II (Roche). A linear dilution-amplification curve was obtained from diluted pooled samples. Using this curve, the expression of each gene was measured relative to the expression of the housekeeping gene encoding peptidylprolyl isomerase A (i.e., cyclophilin). All assays were performed in duplicate.
Statistical analysis.
Data were analyzed using GraphPad Prism version 6.0. All summary data are expressed as the mean ± SD. All datasets were tested for normality using the Shapiro–Wilk normality test. The groups containing normally distributed data were tested using a two-way Student's t test (for two groups) or ANOVA (for more than two groups). The remaining data were analyzed using the Mann–Whitney U test. Similar variance was assured for all groups, which were compared statistically. Differences with a p-value <0.05 were considered to be statistically significant. p-values were adjusted for comparison of multiple comparisons using Bonferroni correction. More detailed methods for analyzing the amplicon sequencing results and the metabolomics data are provided in the respective Materials and Methods sections.
Results
Stroke induces intestinal microbiota dysbiosis
To investigate the impact of acute stroke on the gut microbiota, we used fMCAo, an experimental stroke model resulting in large hemispheric lesions (mean ± SD of all control experiments in this study: 72 ± 18 mm3; Fig. 1a). Next-generation sequencing of gut microbiota composition in mice after fMCAo and controls (naive and sham treatment) revealed substantial changes 3 d after severe stroke (Fig. 1b). We observed significant reduction in species diversity as a key feature of after stroke microbiota dysbiosis (Fig. 1c). Hallmarks of poststroke dysbiosis included changes in specific bacterial genera within the most abundant phyla: Firmicutes, Bacteroidetes, and Actinobacteria (Fig. 1d). We next performed an indicator taxa analysis using LEfSe algorithm analysis for the detection of specific features that were statistically different between sham- and fMCAo-operated mice. Using this analysis strategy, we identified several significantly changed species after stroke induction consistent with the above-shown phylogenetic tree analysis; however, we also identified corresponding changes in higher-order taxa of the respectively altered species (Fig. 1e). These results suggested a consistent and specific impact of brain injury on microbiota populations within the most abundant bacterial phyla.
Figure 1.
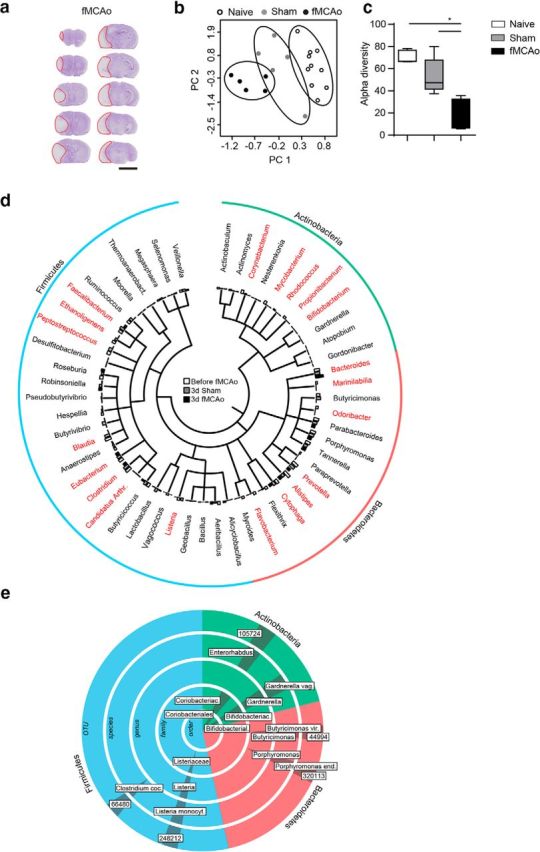
Severe stroke induces microbiota dysbiosis. a, Representative images of cresyl-violet-stained coronal brain sections 3 d after fMCAo. In each section, the infarct area is outlined in red 5 mm). b, Principal component (PC) analysis of the intestinal microbiota by taxonomic abundance patterns in naive mice (before sham or fMCAo surgery) and after sham and fMCAo surgery. c, Quantitative analysis of α diversity confirms significantly reduced species diversity of the gut microbiota after brain injury in the fMCAo model (Mann–Whitney U test). d, Phylogenetic tree illustrating the distribution of the identified bacterial genera within the most abundant phyla (Actinobacteria, Bacteroidetes, and Firmicutes) comparing post-fMCAo mice with sham-operated and before-fMCAo mice. Genera that were significantly altered in the fMCAo group compared with the sham and before-fMCAo groups are indicated in red (ANOVA, n = 5 mice per group). e, LEfSe algorithm analysis was performed for indicator taxa analysis identifying features that are statistically different between sham- and fMCAo-operated mice. Labels in boxes define significantly regulated operational taxonomic units (OTU, numbers) and higher order taxa, respectively.
Extensive brain injury impairs gastrointestinal function
Next, we investigated which mechanisms link acute brain injury and microbiota dysbiosis. It was reported previously that patients with severe brain injuries experience reduced gastrointestinal motility (Olsen et al., 2013; Engler et al., 2014). Consistent with these clinical observations, we detected severe gastrointestinal paralysis after large infarcts induced by the fMCAo stroke model using a gastric fluorescent bolus tracking technique (Fig. 2a,b). Reduced gastrointestinal motility was associated with overgrowth of intestinal bacteria after severe experimental stroke compared with sham surgery (Fig. 2c). Due to this functional gastrointestinal impairment, we further investigated the impact of stroke on intestinal barrier function. We did not observe bacterial translocation or transmucosal bacterial invasion after stroke by in situ hybridization with a general bacteria (EUB 388) probe (Fig. 2d). However, we measured a significantly increased protein leakage by feces/plasma albumin ratio after fMCAo compared with sham-treated controls (Fig. 2e). These results suggested a functionally, but not morphologically, impaired intestinal barrier after fMCAo in association with poststroke intestinal paralysis. Previous reports from our group and others have identified dysregulation of the autonomous nervous system and a pronounced stress response after stroke as one of the key pathways involved in mediating systemic effects on remote organ function after acute brain injury (Meisel et al., 2005; Chamorro et al., 2007; Chamorro et al., 2012; Liesz et al., 2013b; Mracsko et al., 2014). We detected increased blood catecholamine levels after extensive stroke in the fMCAo model to be associated with poststroke intestinal paralysis (Fig. 2f). Consistent with our results, a recent report by Houlden et al. (2016) demonstrated poststroke intestinal changes as a consequence of the catecholaminergic stress response. It was shown previously that the severity of the brain injury is correlated with the extent of secondary systemic immunomodulation (Meisel et al., 2005; Liesz et al., 2009b). Consistent with these previous immunological findings, induction of only small cortical lesions in a second stroke model (cMCAo, mean infarct volume in this model: 8 ± 2 mm3) caused no significant change in the microbiota composition and species diversity (Fig. 2g,h). Small brain lesions in the cMCAo model also did not induce gastrointestinal paralysis (Fig. 2i). These results indicate that infarct severity might be the key factor determining intestinal dysfunction, although we cannot ultimately exclude the possibility that other factors differing between the investigated stroke model also contributed to this effect.
Figure 2.

Lesion severity determines gastrointestinal dysfunction. a, Intestinal motility was measured 24 h after fMCAo or sham surgery. Representative fluorescence images of the complete gastrointestinal tract 60 min after gastric instillation of FITC-dextran showing retention of the fluorescent bolus in the upper gastrointestinal tract as a marker of severely impaired motility after fMCAo compared with sham mice. Arrowheads indicate cecum. b, Quantification of fluorescence intensity in intestinal segments. Note the retention of fluorescence signal in the upper gastrointestinal tract after fMCAo (n = 8 per group, 3 individual experiments). c, Number of colony forming units (CFUs) per milligram of murine cecal content cultured under anaerobic conditions after sham or fMCAo surgery (n = 8–9 mice per group, 2 independent experiments). d, No bacterial invasion into lamina propria was found in transverse sections of mouse ileum, which were hybridized using a general bacteria-specific EUB338 probe with Sytox green counterstaining. Left panels: Magnification, 20×; scale bars, 100 μm; right panels: magnification, 63×, scale bars, 10 μm. e, Albumin concentrations were determined by ELISA and are represented as the ratio of the concentrations in feces and plasma from sham- and fMCAo-operated mice (n = 5 mice per group, 2 individual experiments). f, Plasma catecholamine levels, represented as catecholamine metabolite concentrations of metanephrine and normetanephrine, were significantly increased 24 h after fMCAo compared with sham surgery (n = 10 per group, 3 individual experiments). g, Representative cresyl-violet-stained coronal brain sections 3 d after stroke induction in the cMCAo model. The small cortical lesions are outlined in red. h, Principal component (PC) analysis (left) for fecal taxonomic abundance after csham and cMCAo compared with fMCAo, and α diversity for the indicated groups (right) illustrate that small cortical lesions do not affect microbiota compositon. i, Representative fluorescence images (left) and quantitative analysis (right, n = 6) of gastrointestinal motility after csham and cMCAo surgery corresponding to data shown in a and b. Bar graph: *p < 0.05, mean ± SD.
To investigate the causality of gut motility changes after stroke and observed microbiota dysbiosis, we further determined effects of inducing gastrointestinal paralysis by physical intestinal manipulation, a model for postsurgical ileus (Vilz et al., 2012). Postsurgical ileus mimicked the disturbed gastrointestinal motility pattern of stroke animals (Fig. 3a) and recapitulated general features of microbiota dysbiosis such as reduced α diversity (Fig. 3b,c). Despite this, in-depth phylogenetic analysis also revealed substantial differences in specifically regulated taxa between stroke and mechanically induced dysbiosis (Fig. 3d). Collectively, these results suggest stress-mediated paralytic ileus as a potential cause of poststroke microbiota dysbiosis, although alternative CNS-specific pathways could be involved in poststroke dysbiosis.
Figure 3.
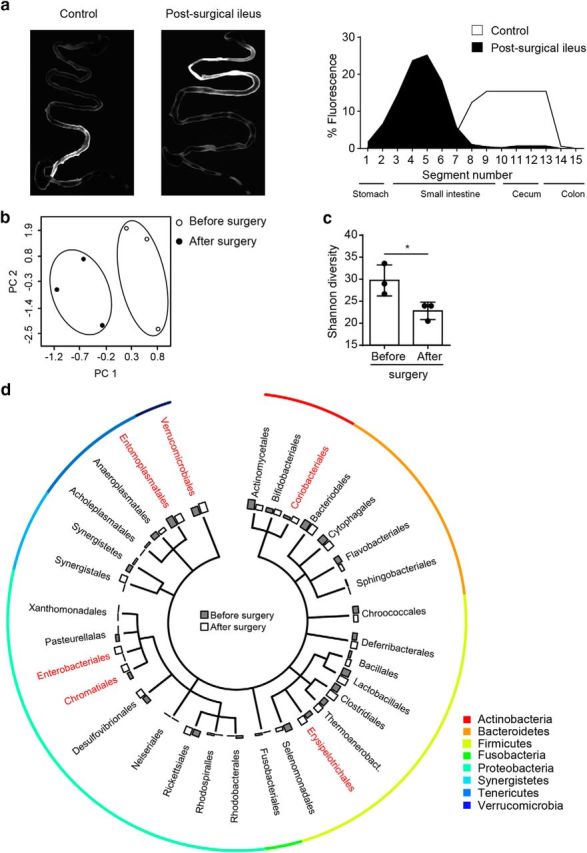
Postsurgical ileus induces intestinal motility dysfunction and dysbiosis of the gut microbiota. a, Intestinal motility was measured 3 d after surgical ileus induction or sham surgery. Shown are representative images of the gastrointestinal tract 60 min after an oral dose of FITC-dextran showing retention of fluorescence signal in the upper gastrointestinal tract (left). Corresponding quantification of fluorescence intensity per intestinal segment (n = 3 per group, right panel). b, Principal component (PC) analysis of the microbiota composition in mice before and 3 d after mechanical manipulation of the ileus reveals microbiota alteration. c, Analysis of Shannon diversity index show a significantly reduced species diversity induced by the postsurgical ileus model. d, Shown is a phylogenetic tree illustrating the distribution of the identified bacterial orders comparing microbiota before and 3 d after surgery. Orders that significantly differed are highlighted in red (n = 3 mice per group, t test [unpaired]). All bar graphs: *p < 0.05, mean ± SD.
Dysbiosis is causally linked to deteriorated stroke outcome
We next investigated whether poststroke dysbiosis has a functional impact on stroke outcome. To investigate this mechanism, we used a microbiota-transfer model into GF mice. We transplanted microbiota obtained from sham- and fMCAo-operated mice into GF recipient mice; 3 d later, we induced cortical lesions by the cMCAo model, which does not alter microbiota composition per se, as shown above (Fig. 4a). Metagenomic analysis of recipients' feces samples at the time point of cMCAo induction (i.e., 3 d after transplantation) confirmed establishment of distinct microbiota in the recipient mice, which resembled the specific pattern of the microbiota donors (Fig. 4b). Remarkably, mice that had received microbiota from brain-injured animals developed significantly larger infarct volumes after cortical lesions in the cMCAo stroke model compared with littermates that were recolonized with sham-surgery microbiota (Fig. 4c). In addition, functional impairment in recipients of the dysbiotic microbiota was exacerbated, as determined by forelimb use asymmetry and overall activity (rearing efforts) in the established cylinder test for this stroke model (Fig. 4d). We next analyzed the expression of the cytokines IL-17 and IFN-γ and of the transcription factor Foxp3 as markers of Thelper cell polarization in brains 5 d after cMCAo. Recipients of post-fMCAo microbiota exhibited massively increased expression of the proinflammatory IFN-γ and IL-17 cytokines, which are markers of Th1 and Th17 T cell polarization, respectively, and have previously been associated with worse outcome in experimental stroke models (Yilmaz et al., 2006; Liesz et al., 2009a; Shichita et al., 2009; Gelderblom et al., 2012). In contrast, Foxp3 expression, as a marker for neuroprotective Treg cells, did not differ significantly between sham and fMCAo microbiota recipients (Fig. 4e). In addition, we performed flow cytometry analysis for intracellular cytokine expression of T cells in PPs to investigate the effect of the distinct microbiota on intestinal T-cell polarization. Here, we detected a >4-fold increased expression of proinflammatory Th17 (IL-17+) and Th1 (IFN-γ+) cells in PPs of fMCAo microbiota compared with sham microbiota recipients, consistent with results obtained from brain tissue (Fig. 4f). These results indicate that the dysbiotic microbiota after large (fMCAo) brain infarcts induces a predominantly proinflammatory Th1 and Th17 response, which is associated with increased infarct volume. This finding is consistent with previous studies showing that T-cell polarization can be induced by selected members of Bacteroidetes and Firmicutes (Round and Mazmanian, 2010; Magrone and Jirillo, 2013). In addition to lymphocyte polarization/activation, microbial products can regulate innate immunity and contribute to intestinal monocyte/macrophage maturation and activation (Bain and Mowat, 2014). Here, we found increased number of mucosal CD11b+ monocytes in mice that were recipients of post-fMCAo microbiota (Fig. 4g). Together, these results indicate an activation of intestinal innate and adaptive immune cells in response to poststroke dysbiotic microbiota.
Figure 4.

Brain ischemia-induced dysbiosis alters the poststroke immune reaction and exacerbates stroke outcome. a, Experimental design for recolonizing GF mice with gut microbiota from sham- or fMCAo- operated SPF donor mice. Three days after recolonization, mice underwent cMCAo induction and were killed another 5 d later for subsequent analysis. b, Principal component analysis of the microbiota composition in donor mice after sham and fMCAo surgery and microbiota composition in GF recipient mice 3 d after transplantation with donor microbiota. Analysis revealed a distinct pattern of the two donor populations before and after transplantation to recipient GF mice. c, Comparison of brain infarct volumes (cMCAo) in the recolonized recipient mice 5 d after cMCAo induction. d, Recolonizing mice with the gut microbiota obtained from post-fMCAo donors significantly increased brain lesion volumes and significantly reduced behavioral performance assessed by measuring forelimb use asymmetry (left) and total rearing activity (right) in the cylinder test (n = 5–8 per group, 2 individual experiments). e, Relative gene expression levels of IL-17, IFN-γ, and Foxp3 in the ipsilateral (ipsi) and contralateral (contra) hemispheres of GF recipient mice (n = 5–8 per group, 2 individual experiments). Recolonization with microbiota from fMCAo donor mice massively increased Th17 (IL-17) and Th1 (IFN-γ) expression compared with recipients of sham-microbiota. f, Representative dot plots with gating strategy for the flow cytometry analysis of Th17 (IL-17) and Th1 (IFN-γ) T cells in PPs (left). Percentages of IL-17+ and IFN-γ + T cells were significantly increased in PPs of mice recolonized with the fMCAo-microbiota (right panel). g, Percentage of CD11b+ monocytes/macrophages in the mucosal layer of the small intestine (terminal ileum) was significantly increased in mice receiving the microbiota from fMCAo donors compared with sham microbiota. All bar graphs: *p < 0.05, mean ± SD.
T cells migrate from the intestine to the poststroke brain
Interestingly, the peripheral immune responses coincided precisely with the massively increased cerebral gene expression for proinflammatory cytokines, suggesting that peripherally activated and/or polarized T cells may migrate to the injured brain after cMCAo in GF recipient mice. To investigate potential T-cell migration from PPs to the brain, we used a fluorescent labeling technique by microinjection of either CFSE or CM-DiI in all detectable PPs of the mice intestines after cMCAo or sham surgery (Fig. 5a). We confirmed that this microinjection technique labeled cells only locally in PPs, but no systemic cell labeling by potentially diffusing dye occurred in blood or spleen (Fig. 5b). Consistent with a previous report (Benakis et al., 2016), T cells and monocytes fluorescently labeled after microinjection in PPs were detected in the ischemic hemisphere 3 d after cMCAo consistent with the previously demonstrated kinetics of poststroke leukocyte invasion (Gelderblom et al., 2009; Liesz et al., 2011) (Fig. 5c,d). CFSE-positive cells derived from labeled PPs accounted for ∼25% of total T cells and the Thelper cell subpopulation 3 d after cMCAo (Fig. 5e). We confirmed these findings in an independent experiment using Cm-DiI as a lipophilic labeling dye and subsequent histological analysis (Fig. 5f). Here, superimposing localization of T cells from five mice on one coronal section map, we detected brain-invading T cells surrounding the ischemic core consistent with previous reports using the same stroke model (Liesz et al., 2011; Zhou et al., 2013); moreover, the percentage of PP-derived T cells was consistent with results obtained from the above-shown flow cytometric analysis (Fig. 5f,g). These results unequivocally demonstrate the invasion of considerable numbers of T cells from the intestine to the periinfarct tissue at risk in the postischemic brain with a remarkably fast kinetics.
Figure 5.

Lymphocytes migrate from PPs to the brain after stroke. a, Experimental design for tracing the migration of PP-derived lymphocytes in mice after cMCAo or sham surgery. CSFE or CM-DiI was injected in PP 2 d after the respective surgery and, 24 h later, brain and lymphoid organs were dissected and analyzed for dye-positive T cells. b, Validation of site-specific T-cell labeling with CFSE in PP 3 h after microinjection; T-cell labeling was not detectable in blood and spleen. c, Representative histogram for CFSE+ T cells (gated for CD45+CD3+ expression) from flow cytometry analysis of brain homogenates of the ipsilateral hemisphere 24 h after CFSE microinjection in all detectable PPs. d, Quantification of flow cytometry analysis shows increased numbers of CFSE+ total T cells (CD3+) and Thelper cells (CD4+) and monocytes (CD11b+) in ischemic hemispheres 3 d after cMCAo compared with sham control. e, Percentage of CFSE-labeled CD3+ and CD4+ T cells identified in ischemic hemispheres 3 d after cMCAo and 24 h after PP labeling. f, Brain-invading CM-DiI+ T cells derived from PPs were identified in the peri-infarct region and are illustrated as a cumulative map from five mice on one topographical coronal brain section at the bregma level g, Quantification of CM-DiI- and CM-DiI+ T cells (CD3+) per one histological brain section used to generate the cumulative map shown in f.
Fecal microbiota transplantation is neuroprotective after stroke
Based on our results, we hypothesized that poststroke dysbiosis is a key novel target for modulating the systemic immune response after stroke. Therefore, we tested the use of FMT as a therapeutic approach to restore a healthy microbiome in animals after stroke. Indeed, FMT treatment starting at the day of stroke induction and performed once daily during the survival period significantly reduced the lesion after large stroke using the fMCAo stroke model for severe brain infarctions (Fig. 6a). This improvement in stroke outcome was associated with increased numbers of Foxp3+ Treg cells in peripheral immune organs and in the ischemic brain after stroke (Fig. 6b). To further elucidate the contribution of lymphocytes to this effect, we examined FMT in a model of lymphocyte-deficient Rag1−/− mice. FMT had no effect on lesion size in Rag1−/− mice, supporting the notion that microbiome-mediated effects on brain injury are mediated by lymphocytes (Fig. 6c). Metagenomic analysis of microbial communities showed a normalization of stroke-induced dysbiosis and partially restored microbiota diversity by the FMT treatment in mice with extensive infarcts (Fig. 6d,e). More in-depth analysis revealed that FMT treatment increased abundance of several of the very same taxa, which were reduced by stroke-induction compared with sham treatment (Fig. 6f). These findings suggest that FMT therapy normalizes stroke-induced changes to microbial communities efficiently and might thereby exert a neuroprotective function after stroke.
Figure 6.
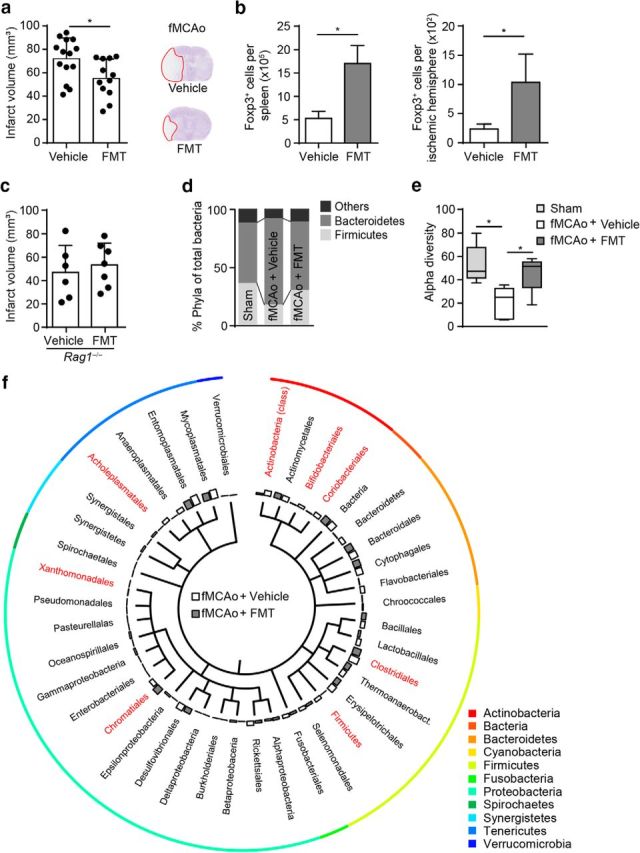
Fecal microbiota transplantation improves stroke outcome. a, Brain infarct volume 3 d after fMCAo was compared between mice receiving vehicle or FMT; FMT treatment significantly reduced brain lesion volume (three independent experiments). b, Flow cytometric analysis shows increased Foxp3+ Treg cell counts in spleens and ischemic hemispheres after FMT treatment compared with controls 3 d after fMCAo (n = 5 per group). c, Brain infarct volume 3 d after fMCAo in lymphocyte-deficient Rag1−/− mice did not differ between vehicle and FMT treatment, suggesting a lymphocyte-mediated effect (two independent experiments). d, FMT treatment normalized microbiota composition after fMCAo-induced dysbiosis as demonstrated by the taxonomic abundance of eubacterial phyla. Note the normalization in the abundance ratio between Firmicutes, Bacteroidetes, and the less abundant phyla (others) in the FMT-treated group. e, Analysis of the corresponding α diversity by the Shannon diversity index showing that FMT treatment partially reversed the reduced species diversity induced by fMCAo (n = 5 mice per group). f, Phylogenetic tree illustrating the distribution of the identified bacterial orders comparing vehicle- and FMT-treated mice 3 d after fMCAo. Orders that significantly differed between groups are highlighted in red (n = 5 mice per group, t test [unpaired]). All bar graphs: *p < 0.05, mean ± SD.
Discussion
Our results support the concept of a bidirectional communication along the brain–gut–microbiome–immune axis. Recent reports have suggested that the microbiota plays an evident role in developmental and autoimmune brain disorders (Berer et al., 2011; Cryan and Dinan, 2012) and that antibiotic treatment-induced dysbiosis affects stroke outcome (Benakis et al., 2016). Here, we report that stroke itself markedly affects the intestinal microbial composition and that these changes in turn can determine stroke outcome.
In our study, we have demonstrated the substantial effects of brain injury on microbiota composition; these effects included reduction in microbiota species diversity and intestinal bacterial overgrowth with a preferential expansion of the Bacteroidetes phylum. In addition, we identified more specific stroke-induced changes on the bacterial genus and even the species level. These findings on poststroke dysbiosis are consistent with recent reports describing changes in intestinal microbiota composition in stroke patients (Karlsson et al., 2012; Swidsinski et al., 2012; Yin et al., 2015). Several of these features of microbiota alterations are of direct pathophysiological relevance; specifically, high microbiota diversity has been suggested as a key feature of a healthy microbiome (Claesson et al., 2012; Human Microbiome Project, 2012). Our analyses also revealed that the surgical procedure itself (i.e., sham surgery) also induced apparent changes in microbiota composition, thus highlighting the sensitivity of microbiota composition to stress responses (Carabotti et al., 2015). However, brain injury compared with sham surgery imposed an additional and substantial impact on microbiota composition and we were able to detect specific species and their corresponding higher-order taxa most significantly associated with acute brain injury compared with sham surgery using indicator taxa analysis.
Our results suggested that microbiota dysbiosis after stroke is associated with reduced gastrointestinal motility and intestinal paralysis in a postsurgical ileus model recapitulated several key features of poststroke dysbiosis. These findings have broad clinical implications; specifically, the intestinal dysfunction revealed by our animal model was recently reported in patients after acute brain injury (Bansal et al., 2009; Olsen et al., 2013). However, a decisive causality among poststroke stress response, impaired motility, and dysbiosis could not be clarified in this study. In addition, we cannot exclude other direct mediators released from the necrotic brain tissue affecting microbiota composition via currently unknown mechanisms. One potential alternative mechanism not investigated in this study is the release of proinflammatory alarmins such as ATP, HMGB1, and S100 proteins from the injured brain with potential direct effects on intestinal immunity and microbiota composition (Liesz et al., 2015; Singh et al., 2016).
Our results demonstrate that microbiota dysbiosis is an important factor in determining poststroke inflammation and thereby stroke outcome in an experimental stroke model. Numerous reports over the last decade have highlighted the importance of the secondary inflammatory response to brain injury as a key pathophysiological mechanism (Iadecola and Anrather, 2011; Chamorro et al., 2012). Moreover, immunotherapeutic strategies emerge to be tested progressively in human stroke patients and first studies report a beneficial effect of inhibiting cerebral lymphocyte invasion in stroke patients (Fu et al., 2014; Zhu et al., 2015). However, the role of the gut microbiota in stroke patients was elusive until the very recent publication of an elegant report by Benakis et al. (2016), who observed alteration of lymphocyte populations in the intestinal immune compartment after induction of dysbiosis by antibiotic treatment. Dysbiosis-induced changes in the peripheral immune system had a striking impact on stroke outcome with changes in infarct volume by 60% between treatment groups. In contrast to our study, antibiosis-induced intestinal dysbiosis had a neuroprotective effect with reduced infarct volumes and improved functional outcome after amoxicillin treatment. Nevertheless, the underlying mechanisms of microbiota-brain communication identified in the study by Benakis et al. (2016) and our report are largely identical, such as alteration in T-cell homeostasis, changes in the Treg/Th17 ratio, and migration of intestinal lymphocytes to the ischemic brain. The at-first-sight contradictory impact of dysbiosis on final stroke outcome might depend on the differential immune polarization by specific bacterial species. In fact, whereas antibiotic treatment induced Treg expansion and reduction of Th17 cells (Benakis et al., 2016), poststroke dysbiosis investigated in our study itself favors an opposing pattern with predominant expansion of proinflammatory T-cell subpopulations. Future studies will be required to analyze the contribution of specific microbial species in this highly complex interplay to identify neuroprotective or harmful bacteria in stroke.
Results from our microbiota transplantation experiments to GF animals using microbiota from donors undergoing stroke induction or sham surgery have clearly demonstrated a causal link between poststroke microbiota dysbiosis and changes in peripheral immunity and finally a worse outcome after stroke. A potential limitation of this experiment was the only short recolonization time of 3 d from transplantation to stroke induction or 8 d from transplantation to analysis, because previous reports have suggested that recolonization of GF mice with a complete fecal microbial induces pronounced immune activation with a peak at ∼4 d after the start of colonization (El Aidy et al., 2012). This short time period for recolonization was nevertheless chosen deliberately to avoid shifts in microbiota composition during longer recolonization periods. Therefore, whereas the results from this specific recolonization experiment might overestimate immunological differences, they nevertheless provide a first proof-of-concept for the causality between dysbiosis and poststroke immune alterations.
In our study, we have observed a substantial induction of proinflammatory Th1 and Th17 Thelper cell polarization by transfer of a dysbiotic microbiome. A consistent cytokine expression pattern with markedly increased IFN-γ and IL-17 levels were observed in brains of mice receiving the dysbiotic microbiota. Moreover, we could determine in a cell-tracking experiment from intestinal PPs to the ischemic brain that at least one-fourth of all brain-invading T cells within the acute phase after stroke are derived from the intestinal immune compartment, which is consistent with a previous report (Benakis et al., 2016). This observation of T-cell invasion and activation is consistent with numerous reports on the surprisingly fast kinetics of poststroke adaptive immunity (Schroeter et al., 1994; Jander et al., 1995; Gelderblom et al., 2009; Liesz et al., 2011; Chamorro et al., 2012). The central neuroinflammatory reaction starts as early as hours after stroke, with T cells invading the brain in substantial numbers between 3 and 5 d after brain injury (Gelderblom et al., 2009; Chamorro et al., 2012). Furthermore, we have detected previously substantial clonal expansion of T cells using spectratype analysis within the first week after acute brain injury (Liesz et al., 2013a). Therefore, initiation of an adaptive immune response and particularly activation of T cells occurs within few days after stroke, consistent with results presented in this study. Specifically, it was shown previously that Thelper cell polarization occurs very early after brain injury and the Treg/Th17 balance contributes to outcome within the first 3 d after stroke (Liesz et al., 2009a; Shichita et al., 2009; Kleinschnitz et al., 2010).
The assumption of intestinal priming of Thelper cells by the microbiota and rapid translocation to the ischemic brain after stroke was further corroborated by the results obtained in experiments in which we treated mice after extensive brain ischemia (fMCAo model) by FMT from healthy donors. FMT treatment reduced lesion size and was associated with expansion of regulatory (neuroprotective) T cells in the peripheral immune system and in the ischemic brain. These findings are consistent with numerous previous reports demonstrating a key role for differential Thelper cell priming in stroke pathophysiology, in which proinflammatory Th1, Th17, and γδ T cells deteriorate poststroke inflammation, whereas Treg cells limit the inflammatory collateral damage (Shichita et al., 2009; Iadecola and Anrather, 2011; Chamorro et al., 2012).
Interestingly, the protective effect of FMT treatment was absent in lymphocyte-deficient Rag1−/− mice, supporting a key role of lympyhocytes in mediating the microbiota's effect on brain function. However, the specific mechanisms underlying this complex phenomenon are still unclear and will need further investigations. Specifically, the interaction of microbiota-dependently activated T cells with other immunocompetent cells of the poststroke brain (microglia, astrocytes, endothelium, invading leukocyte subpopulations) requires further analyses. A previous report by Erny et al. demonstrated a key role of the gut microbiota in microglial function and proposed microbiota-derived short chain fatty acids as the underlying mediator of microbiota–microglia communication (Erny et al., 2015). Therefore, it remains to be elucidated whether the important role of microbiota-dependent T-cell polarization after stroke shown by us and others (Benakis et al., 2016) affects neuronal protection/toxicity directly or if it affects stroke outcome indirectly via microglial function, as was shown previously to occur by brain-invading lymphocytes (Appel, 2009; Lucin and Wyss-Coray, 2009; Liesz et al., 2011). In addition, we have also observed activation of intestinal monocytes/macrophages by a dysbiotic poststroke microbiome. Moreover, intestinal monocytes were detected to invade the brain in the acute phase after stroke. Therefore, in addition to T-cell polarization, brain-invading monocytes could also potentially play a role in microbiota-mediated effects on stroke outcome.
Our results suggest a novel concept in which dysbiosis of the gut microbiota is a consequence of acute brain injury and a key effector in poststroke immune alterations with considerable impact on stroke outcome. Our findings suggest that restoring the health and balance of the intestinal microbiome could add to the treatment of stroke patients.
Footnotes
This work was supported by the Excellence Cluster of the German Research Foundation “Munich Cluster for Systems Neurology (SyNergy)” to A.L. and by the SPP1656 “Intestinal Microbiota” (STE 1971/4-1) to B.S. All DNA sequences used for microbial community analyses have been deposited in MG-RAST (http://metagenomics.anl.gov/) under accession number 9778. We thank Kerstin Hofmann for providing technical assistance, Rainer Malik (Institute for Stroke and Dementia Research, Munich, Germany) for advice regarding the sequencing analysis, Sandrine Brugiroux (Max-von-Pettenkofer Institute, Munich, Germany) for technical advice regarding fluorescence in situ hybridization, and Kathleen McCoy (University of Bern, Switzerland) for providing GF mice.
The authors declare no competing financial interests.
References
- Appel SH. CD4+ T cells mediate cytotoxicity in neurodegenerative diseases. J Clin Invest. 2009;119:13–15. doi: 10.1172/JCI38096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, Rudensky AY. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451–455. doi: 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain CC, Mowat AM. Macrophages in intestinal homeostasis and inflammation. Immunol Rev. 2014;260:102–117. doi: 10.1111/imr.12192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal V, Costantini T, Kroll L, Peterson C, Loomis W, Eliceiri B, Baird A, Wolf P, Coimbra R. Traumatic brain injury and intestinal dysfunction: uncovering the neuro-enteric axis. J Neurotrauma. 2009;26:1353–1359. doi: 10.1089/neu.2008.0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M, Sita G, Racchumi G, Ling L, Pamer EG, Iadecola C, Anrather J. Commensal microbiota affects ischemic stroke outcome by regulating intestinal gammadelta T cells. Nat Med. 2016 doi: 10.1038/nm.4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, Wekerle H, Krishnamoorthy G. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. 2011;479:538–541. doi: 10.1038/nature10554. [DOI] [PubMed] [Google Scholar]
- Carabotti M, Scirocco A, Maselli MA, Severi C. The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol. 2015;28:203–209. [PMC free article] [PubMed] [Google Scholar]
- Chamorro A, Amaro S, Vargas M, Obach V, Cervera A, Gómez-Choco M, Torres F, Planas AM. Catecholamines, infection, and death in acute ischemic stroke. J Neurol Sci. 2007;252:29–35. doi: 10.1016/j.jns.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Chamorro Á, Meisel A, Planas AM, Urra X, van de Beek D, Veltkamp R. The immunology of acute stroke. Nat Rev Neurol. 2012;8:401–410. doi: 10.1038/nrneurol.2012.98. [DOI] [PubMed] [Google Scholar]
- Claesson MJ, Jeffery IB, Conde S, Power SE, O'Connor EM, Cusack S, Harris HM, Coakley M, Lakshminarayanan B, O'Sullivan O, Fitzgerald GF, Deane J, O'Connor M, Harnedy N, O'Connor K, O'Mahony D, van Sinderen D, Wallace M, Brennan L, Stanton C, et al. Gut microbiota composition correlates with diet and health in the elderly. Nature. 2012;488:178–184. doi: 10.1038/nature11319. [DOI] [PubMed] [Google Scholar]
- Collins SM, Surette M, Bercik P. The interplay between the intestinal microbiota and the brain. Nat Rev Microbiol. 2012;10:735–742. doi: 10.1038/nrmicro2876. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. 2012;13:701–712. doi: 10.1038/nrn3346. [DOI] [PubMed] [Google Scholar]
- El Aidy S, van Baarlen P, Derrien M, Lindenbergh-Kortleve DJ, Hooiveld G, Levenez F, Doré J, Dekker J, Samsom JN, Nieuwenhuis EE, Kleerebezem M. Temporal and spatial interplay of microbiota and intestinal mucosa drive establishment of immune homeostasis in conventionalized mice. Mucosal Immunol. 2012;5:567–579. doi: 10.1038/mi.2012.32. [DOI] [PubMed] [Google Scholar]
- Engler TM, Dourado CC, Amâncio TG, Farage L, de Mello PA, Padula MP. Stroke: bowel dysfunction in patients admitted for rehabilitation. Open Nurs J. 2014;8:43–47. doi: 10.2174/1874434601408010043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erny D, Hrabe de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, Keren-Shaul H, Mahlakoiv T, Jakobshagen K, Buch T, Schwierzeck V, Utermohlen O, Chun E, Garrett WS, McCoy KD, Diefenbach A, Staeheli P, Stecher B, Amit I, Prinz M. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. 2015;18:965–977. doi: 10.1038/nn.4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Zhang N, Ren L, Yan Y, Sun N, Li YJ, Han W, Xue R, Liu Q, Hao J, Yu C, Shi FD. Impact of an immune modulator fingolimod on acute ischemic stroke. Proc Natl Acad Sci U S A. 2014;111:18315–18320. doi: 10.1073/pnas.1416166111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, Arumugam TV, Orthey E, Gerloff C, Tolosa E, Magnus T. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40:1849–1857. doi: 10.1161/STROKEAHA.108.534503. [DOI] [PubMed] [Google Scholar]
- Gelderblom M, Weymar A, Bernreuther C, Velden J, Arunachalam P, Steinbach K, Orthey E, Arumugam TV, Leypoldt F, Simova O, Thom V, Friese MA, Prinz I, Hölscher C, Glatzel M, Korn T, Gerloff C, Tolosa E, Magnus T. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood. 2012;120:3793–3802. doi: 10.1182/blood-2012-02-412726. [DOI] [PubMed] [Google Scholar]
- Hofmann U, Frantz S. Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction. Circ Res. 2015;116:354–367. doi: 10.1161/CIRCRESAHA.116.304072. [DOI] [PubMed] [Google Scholar]
- Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houlden A, Goldrick M, Brough D, Vizi ES, Lenart N, Martinecz B, Roberts IS, Denes A. Brain injury induces specific changes in the caecal microbiota of mice via altered autonomic activity and mucoprotein production. Brain Behav Immun. 2016 doi: 10.1016/j.bbi.2016.04.003. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Rabb H, Womer KL. Ischemia-reperfusion and immediate T cell responses. Cell Immunol. 2007;248:4–11. doi: 10.1016/j.cellimm.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umesaki Y, Honda K, Littman DR. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jander S, Kraemer M, Schroeter M, Witte OW, Stoll G. Lymphocytic infiltration and expression of intercellular adhesion molecule-1 in photochemically induced ischemia of the rat cortex. J Cereb Blood Flow Metab. 1995;15:42–51. doi: 10.1038/jcbfm.1995.5. [DOI] [PubMed] [Google Scholar]
- Karlsson FH, Fåk F, Nookaew I, Tremaroli V, Fagerberg B, Petranovic D, Bäckhed F, Nielsen J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun. 2012;3:1245. doi: 10.1038/ncomms2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8:e1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinschnitz C, Schwab N, Kraft P, Hagedorn I, Dreykluft A, Schwarz T, Austinat M, Nieswandt B, Wiendl H, Stoll G. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood. 2010;115:3835–3842. doi: 10.1182/blood-2009-10-249078. [DOI] [PubMed] [Google Scholar]
- Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2011;108:4615–4622. doi: 10.1073/pnas.1000082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, Giese T, Veltkamp R. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009a;15:192–199. doi: 10.1038/nm.1927. [DOI] [PubMed] [Google Scholar]
- Liesz A, Hagmann S, Zschoche C, Adamek J, Zhou W, Sun L, Hug A, Zorn M, Dalpke A, Nawroth P, Veltkamp R. The spectrum of systemic immune alterations after murine focal ischemia: immunodepression versus immunomodulation. Stroke. 2009b;40:2849–2858. doi: 10.1161/STROKEAHA.109.549618. [DOI] [PubMed] [Google Scholar]
- Liesz A, Zhou W, Mracskó É, Karcher S, Bauer H, Schwarting S, Sun L, Bruder D, Stegemann S, Cerwenka A, Sommer C, Dalpke AH, Veltkamp R. Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain. 2011;134:704–720. doi: 10.1093/brain/awr008. [DOI] [PubMed] [Google Scholar]
- Liesz A, Karcher S, Veltkamp R. Spectratype analysis of clonal T cell expansion in murine experimental stroke. J Neuroimmunol. 2013a;257:46–52. doi: 10.1016/j.jneuroim.2013.01.013. [DOI] [PubMed] [Google Scholar]
- Liesz A, Rüger H, Purrucker J, Zorn M, Dalpke A, Möhlenbruch M, Englert S, Nawroth PP, Veltkamp R. Stress mediators and immune dysfunction in patients with acute cerebrovascular diseases. PLoS One. 2013b;8:e74839. doi: 10.1371/journal.pone.0074839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesz A, Zhou W, Na SY, Hämmerling GJ, Garbi N, Karcher S, Mracsko E, Backs J, Rivest S, Veltkamp R. Boosting regulatory T cells limits neuroinflammation in permanent cortical stroke. J Neurosci. 2013c;33:17350–17362. doi: 10.1523/JNEUROSCI.4901-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesz A, Dalpke A, Mracsko E, Antoine DJ, Roth S, Zhou W, Yang H, Na SY, Akhisaroglu M, Fleming T, Eigenbrod T, Nawroth PP, Tracey KJ, Veltkamp R. DAMP signaling is a key pathway inducing immune modulation after brain injury. J Neurosci. 2015;35:583–598. doi: 10.1523/JNEUROSCI.2439-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llovera G, Roth S, Plesnila N, Veltkamp R, Liesz A. Modeling stroke in mice: permanent coagulation of the distal middle cerebral artery. J Vis Exp. 2014:e51729. doi: 10.3791/51729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron. 2009;64:110–122. doi: 10.1016/j.neuron.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrone T, Jirillo E. The interaction between gut microbiota and age-related changes in immune function and inflammation. Immun Ageing. 2013;10:31. doi: 10.1186/1742-4933-10-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci. 2005;6:775–786. doi: 10.1038/nrn1765. [DOI] [PubMed] [Google Scholar]
- Meyer F, Paarmann D, D'Souza M, Olson R, Glass EM, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A, Wilkening J, Edwards RA. The metagenomics RAST server: a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mracsko E, Liesz A, Karcher S, Zorn M, Bari F, Veltkamp R. Differential effects of sympathetic nervous system and hypothalamic-pituitary-adrenal axis on systemic immune cells after severe experimental stroke. Brain Behav Immun. 2014;41:200–209. doi: 10.1016/j.bbi.2014.05.015. [DOI] [PubMed] [Google Scholar]
- Neumann J, Riek-Burchardt M, Herz J, Doeppner TR, König R, Hütten H, Etemire E, Männ L, Klingberg A, Fischer T, Görtler MW, Heinze HJ, Reichardt P, Schraven B, Hermann DM, Reymann KG, Gunzer M. Very-late-antigen-4 (VLA-4)-mediated brain invasion by neutrophils leads to interactions with microglia, increased ischemic injury and impaired behavior in experimental stroke. Acta Neuropath. 2015;129:259–277. doi: 10.1007/s00401-014-1355-2. [DOI] [PubMed] [Google Scholar]
- Offner H, Subramanian S, Parker SM, Wang C, Afentoulis ME, Lewis A, Vandenbark AA, Hurn PD. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J Immunol. 2006;176:6523–6531. doi: 10.4049/jimmunol.176.11.6523. [DOI] [PubMed] [Google Scholar]
- Olsen AB, Hetz RA, Xue H, Aroom KR, Bhattarai D, Johnson E, Bedi S, Cox CS, Jr, Uray K. Effects of traumatic brain injury on intestinal contractility. Neurogastroenterol Motil. 2013;25:593–e463. doi: 10.1111/nmo.12121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A. 2010;107:12204–12209. doi: 10.1073/pnas.0909122107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeter M, Jander S, Witte OW, Stoll G. Local immune responses in the rat cerebral cortex after middle cerebral artery occlusion. J Neuroimmunol. 1994;55:195–203. doi: 10.1016/0165-5728(94)90010-8. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Raposo C. Protective Autoimmunity: A Unifying Model for the Immune Network Involved in CNS Repair. Neuroscientist. 2014;20:343–358. doi: 10.1177/1073858413516799. [DOI] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, Iwaki T, Okada Y, Iida M, Cua DJ, Iwakura Y, Yoshimura A. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat Med. 2009;15:946–950. doi: 10.1038/nm.1999. [DOI] [PubMed] [Google Scholar]
- Shichita T, Hasegawa E, Kimura A, Morita R, Sakaguchi R, Takada I, Sekiya T, Ooboshi H, Kitazono T, Yanagawa T, Ishii T, Takahashi H, Mori S, Nishibori M, Kuroda K, Akira S, Miyake K, Yoshimura A. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat Med. 2012;18:911–917. doi: 10.1038/nm.2749. [DOI] [PubMed] [Google Scholar]
- Singh V, Roth S, Veltkamp R, Liesz A. HMGB1 as a key mediator of immune mechanisms in ischemic stroke. Antioxid Redox Signal. 2016;24:635–651. doi: 10.1089/ars.2015.6397. [DOI] [PubMed] [Google Scholar]
- Swidsinski A, Loening-Baucke V, Krüger M, Kirsch S. Central nervous system and the colonic bioreactor: analysis of colonic microbiota in patients with stroke unravels unknown mechanisms of the host defense after brain injury. Intestinal Research. 2012;10:332–342. doi: 10.5217/ir.2012.10.4.332. [DOI] [Google Scholar]
- Vilz TO, Overhaus M, Stoffels B, Websky M, Kalff JC, Wehner S. Functional assessment of intestinal motility and gut wall inflammation in rodents: analyses in a standardized model of intestinal manipulation. J Vis Exp. 2012;67:4086. doi: 10.3791/4086. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelgesang A, May VE, Grunwald U, Bakkeboe M, Langner S, Wallaschofski H, Kessler C, Bröker BM, Dressel A. Functional status of peripheral blood T-cells in ischemic stroke patients. PLoS One. 2010;5:e8718–e8718. doi: 10.1371/journal.pone.0008718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation. 2006;113:2105–2112. doi: 10.1161/CIRCULATIONAHA.105.593046. [DOI] [PubMed] [Google Scholar]
- Yin J, Liao SX, He Y, Wang S, Xia GH, Liu FT, Zhu JJ, You C, Chen Q, Zhou L, Pan SY, Zhou HW. Dysbiosis of gut microbiota with reduced trimethylamine-N-oxide level in patients with large-artery atherosclerotic stroke or transient ischemic attack. J Am Heart Assoc. 2015;4:e002699. doi: 10.1161/JAHA.115.002699. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Liesz A, Bauer H, Sommer C, Lahrmann B, Valous N, Grabe N, Veltkamp R. Postischemic brain infiltration of leukocyte subpopulations differs among murine permanent and transient focal cerebral ischemia models. Brain Pathol. 2013;23:34–44. doi: 10.1111/j.1750-3639.2012.00614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Fu Y, Tian D, Sun N, Han W, Chang G, Dong Y, Xu X, Liu Q, Huang D, Shi FD. Combination of the immune modulator fingolimod with alteplase in acute ischemic stroke: a pilot trial. Circulation. 2015;132:1104–1112. doi: 10.1161/CIRCULATIONAHA.115.016371. [DOI] [PMC free article] [PubMed] [Google Scholar]