

Abstract
Spontaneous network activity (SNA) emerges in the spinal cord (SC) before the formation of peripheral sensory inputs and central descending inputs. SNA is characterized by recurrent giant depolarizing potentials (GDPs). Because GDPs in motoneurons (MNs) are mainly evoked by prolonged release of GABA, they likely necessitate sustained firing of interneurons. To address this issue we analyzed, as a model, embryonic Renshaw cell (V1R) activity at the onset of SNA (E12.5) in the embryonic mouse SC (both sexes). V1R are one of the interneurons known to contact MNs, which are generated early in the embryonic SC. Here, we show that V1R already produce GABA in E12.5 embryo, and that V1R make synaptic-like contacts with MNs and have putative extrasynaptic release sites, while paracrine release of GABA occurs at this developmental stage. In addition, we discovered that V1R are spontaneously active during SNA and can already generate several intrinsic activity patterns including repetitive-spiking and sodium-dependent plateau potential that rely on the presence of persistent sodium currents (INap). This is the first demonstration that INap is present in the embryonic SC and that this current can control intrinsic activation properties of newborn interneurons in the SC of mammalian embryos. Finally, we found that 5 μm riluzole, which is known to block INaP, altered SNA by reducing episode duration and increasing inter-episode interval. Because SNA is essential for neuronal maturation, axon pathfinding, and synaptogenesis, the presence of INaP in embryonic SC neurons may play a role in the early development of mammalian locomotor networks.
SIGNIFICANCE STATEMENT The developing spinal cord (SC) exhibits spontaneous network activity (SNA) involved in the building of nascent locomotor circuits in the embryo. Many studies suggest that SNA depends on the rhythmic release of GABA, yet intracellular recordings of GABAergic neurons have never been performed at the onset of SNA in the SC. We first discovered that embryonic Renshaw cells (V1R) are GABAergic at E12.5 and spontaneously active during SNA. We uncover a new role for persistent sodium currents (INaP) in driving plateau potential in V1R and in SNA patterning in the embryonic SC. Our study thus sheds light on a role for INaP in the excitability of V1R and the developing SC.
Keywords: development, excitability, mouse embryo, persistent sodium current, Renshaw cell, spontaneous network activity
Introduction
A remarkable feature of the developing CNS is its capacity to generate spontaneous network activity (SNA) at the end of neuronal migration. SNA occurs in the absence of any external inputs and is not experience-driven or use-dependent (Moody, 1998; Feller, 1999). SNA has been described as playing an essential role in several areas of the developing CNS, including the neocortex, the thalamus, the hippocampus, the locus ceruleus, the retina, and the spinal cord (Landmesser and O'Donovan, 1984; Ben-Ari et al., 1989; Garaschuk et al., 1998; Feller, 1999; Garaschuk et al., 2000; Gust et al., 2003; Corlew et al., 2004; Marder and Rehm, 2005; Myers et al., 2005; Gonzalez-Islas and Wenner, 2006; Hanson et al., 2008; Rockhill et al., 2009; Watt et al., 2009). SNA regulates the development of neural circuits by influencing synaptogenesis, neuronal maturation, axonal guidance and axonal pathfinding (Zhang and Poo, 2001; Hanson et al., 2008; Kirkby et al., 2013).
In the embryonic spinal cord (SC), SNA is characterized by long-lasting bursts of action potentials (APs) occurring every 2–4 min that can propagate along the cord (Hanson and Landmesser, 2003). SNA occurs at the onset of synaptogenesis at E12.5, before the emergence of functional neuromuscular junction and the formation of sensory and supraspinal inputs (Hanson and Landmesser, 2003). Bursts of APs recorded on ventral roots (Hanson and Landmesser, 2003) or in the whole SC (Yvert et al., 2004) are long-lasting episodes characterizing SNA in the embryonic SC. We previously showed that most individual motoneurons (MNs) only generate a single AP during each episode of SNA (Czarnecki et al., 2014) and we demonstrated that acetylcholine release could not directly synchronize MN firing during SNA as MNs do not express functional acetylcholine receptors (Czarnecki et al., 2014). Therefore, MNs must rely on other mechanisms to synchronize their firing during SNA. We have recently shown that MN activity occurring during SNA at the onset of synaptogenesis (E12.5) is generated by giant depolarizing potentials (GDPs) involving a massive and long-lasting release of GABA, as well as a moderate release of glutamate and glycine (Czarnecki et al., 2014). It is therefore likely that newborn GABAergic interneurons (INs) play an essential role in the generation of the sustained episodes of depolarization necessary to synchronize MN firing during SNA.
To gain insight into the excitability pattern of spinal GABAergic INs involved in the release of GABA during SNA, we used multiple approaches to examine the intrinsic activation properties of immature Renshaw cells (V1R) in the lumbar SC. Renshaw cells are known to regulate MN activity in the adult through recurrent synaptic inhibition (Eccles et al., 1956) and play an important role in the regulation of SC activity at late developmental stages in the chicken embryo (Wenner and O'Donovan, 2001). V1R are the first V1 INs to be generated during neurogenesis in the mouse embryo (Benito-Gonzalez and Alvarez, 2012). From E9.5 to E12.5, V1R migrate toward their final location between MN columns and the ventrolateral funiculus (Benito-Gonzalez and Alvarez, 2012; Alvarez et al., 2013). Because glycine, unlike GABA, is nearly absent in SC INs of E12.5 mouse embryo (Allain et al., 2004, 2006; Scain et al., 2010), V1R are likely to be GABAergic neurons at the early developmental stage.
Here, we show that, like MNs, V1R display GDPs during SNA at E12.5. However, we discovered that, unlike MNs, most V1R are able to produce repetitive spiking or sodium-dependent plateau potentials in response to GDPs (Czarnecki et al., 2014). Remarkably, these sustained discharges depend on the presence of a persistent sodium current (INap). In addition, we also demonstrate that INap already has important functions at the onset of SNA. Inhibition of INap alters SNA episode duration and inter-episode interval duration, which reveals that the ability of embryonic SC neurons to generate sustained discharge is required for a correct SNA pattern (Hanson and Landmesser, 2006).
Materials and Methods
Isolated spinal cord preparation.
These experiments were performed in accordance with European Community guiding principles on the care and use of animals (86/609/CEE, CE Off J no. L358, 18 December, 1986), French decree no. 97/748 of October 19, 1987 (J Off République Française, 20 October, 1987, pp. 12245–12248) and recommendations from the CNRS. We used GAD67eGFP knock-in mice to visualize putative GABAergic INs (Tamamaki et al., 2003). Briefly, a cDNA encoding enhanced GFP (eGFP) was targeted to the locus encoding the gene Gad1. GAD67 is a rate-limiting enzyme of GABA biosynthesis and is known to be a marker for GABAergic neurons (Le Corronc et al., 2011). To obtain E12.5 GAD67-eGFP embryos, 8- to 12-week-old wild-type Swiss female mice were crossed with heterozygous GAD67-eGFP Swiss male mice. HB9-eGFP mouse embryos were used to visualize MNs (Wichterle et al., 2002). To obtain E12.5 transgenic HB9-eGFP embryos, 8- to 12-week-old wild-type Swiss female mice were crossed with heterozygous HB9-eGFP C57BL/6Jrj male mice.
Embryos (269) obtained from 152 pregnant mice were used. Isolated embryonic mouse SCs were obtained as previously described (Delpy et al., 2008; Scain et al., 2010). Briefly, pregnant mice were anesthetized by intramuscular injection of a mix of ketamine and xylazine and killed using a lethal dose of CO2. Embryos of either sex were removed and the SC was isolated from eGFP-positive embryos. Whole SCs were maintained in an artificial CSF (ACSF) containing 135 mm NaCl, 25 mm NaHCO3, 1 mm NaH2PO4, 3 mm KCl, 11 mm glucose, 2 mm CaCl2, and 1 mm MgCl2 (307 mosmol/kg H2O), continuously bubbled with a 95% O2-5% CO2 gas mixture.
Whole-cell recordings and analysis.
The isolated SC was placed in a recording chamber and continuously perfused (2 ml/min) at room temperature (20–24°C) with the oxygenated ACSF described above. Whole-cell patch-clamp recordings of lumbar spinal V1R and MNs were performed under direct visualization using an infrared-sensitive CCD video camera.
In the SC of GAD67eGFP mouse embryos, eGFP neurons were detected using UV light. MNs were identified by their size, their location in the ventral area of the SC parenchyma (Czarnecki et al., 2014) and by the lack of eGFP expression. These neurons localized in this SC area express the MN transcription factors Islet1/2, as shown in a previous study (Scain et al., 2010). Recorded eGFP interneurons were localized in the ventrolateral area of the SC at the marginal zone between motor columns and the ventral funiculus, which is the known location of developing V1R (Stam et al., 2012). To confirm V1R identity, recorded cells were filled with neurobiotin (0.5–1 mg/ml) and stained with an antibody directed against Foxd3. Foxd3 is a specific transcription factor of V1 INs (Dottori et al., 2001; Stam et al., 2012) and the hallmark of V1R localized in the ventrolateral area of the embryonic SC (Carr et al., 1998; Benito-Gonzalez and Alvarez, 2012).
Whole-cell patch-clamp electrodes were pulled from thick-wall borosilicate glass using a Brown-Flaming puller (Sutter Instrument). The tip of the electrode was fire-polished using a microforge (Narishige). Patch-clamp electrodes had resistances of 4–7 MΩ. The electrode was filled with a solution containing the following (in mm): 96.4 K methanesulfonate, 33.6 KCl, 4 MgCl2, 4 Na2ATP, 0.3 Na3GTP, 10 EGTA, and 10 HEPES, pH 7.2 (290 mOsm/kg-H2O). Using these potassium methanesulfonate solutions, the equilibrium potential for chloride ions (ECl) ≈ −30 mV was close to the physiological values measured at E12.5 on spinal MNs (Delpy et al., 2008). The junction potential (6.6 mV) was systematically corrected offline. In some voltage-clamp experiments, the electrode contained the following: (in mm) 130 CsCl, 4 MgCl2, 4 Na2ATP, 10 EGTA, and 10 HEPES, pH 7.2 (290 mOsm/kg-H2O).
Signals were recorded using AxoPatch 200B or MultiClamp 700B amplifiers (Molecular Devices). Data were low-pass filtered (2 kHz), digitized (20 kHz) online using a Digidata 1440A interface, and acquired using PClamp 10.5 software (RRID:SCR_011323). Analyses were performed off-line using PClamp 10.5 software and AxoGraph X.1.6.4 (Molecular Devices; RRID:SCR_014284).
In voltage-clamp or current-clamp experiments, neurons were recorded at a holding potential (Vh) of −60 mV. Series resistance (10–20 MΩ) was monitored throughout the experiments and was 50–80% compensated. Data were discarded if series resistance varied by more than ≈30% from the initial value. In current-clamp mode, V1R intrinsic discharge patterns were elicited using depolarizing current steps (from 0 to ≈50 pA, 2–10 pA increments depending on the input resistance of the cell, 2 s) or depolarizing current triangular-ramp (from 0 to ≈50 pA, 5 pA increments, 20 s) with an 8 s interval to ensure that the membrane potential returned to baseline Vh. In voltage-clamp mode, whole-cell currents were elicited by a depolarizing voltage ramp. The 70 mV/s speed was chosen to elicit and measure persistent inward currents (INap; Huang and Trussell, 2008). Subtraction of the current evoked by the voltage ramp in the presence of 1 μm tetrodotoxin (TTX) to control voltage ramp-evoked current revealed INap.
APs or plateau potentials were analyzed based on the following parameters during a 2 s current step: threshold potential, peak amplitude, half amplitude duration, rate of rise of events (by dividing the amplitude of the event by the duration from its onset to the peak; 2 s step width).
Boltzmann functions were used to describe INap activation on current evoked by voltage ramp. For the fit current (20 kHz sampling 2 kHz filter):
where G is conductance in nanosiemens, GMAX is the maximal conductance, V is the potential in millivolts, VHALF is the voltage for half-maximal activation in millivolts, and k is the slope factor in millivolts. Threshold for activation INap was determined by eye from Boltzmann curves.
Extracellular recordings.
In the same experimental conditions as for whole-cell recordings, spontaneous activity was recorded extracellularly from E12.5 SCs using glass electrodes. Extracellular electrodes were placed under visual control on the superficial part of the ventral horn at both the cervical and the lumbar levels. Targeted networks likely include MNs as well as surrounding INs (Czarnecki et al., 2014). Electrodes were connected to a high-gain AC amplifier (ISO-DAM8A-4 Bio-amplifier System, World Precision Instruments). Filtered (Filter cutoff frequency: 0.3–3 kHz) raw signals were integrated off-line and analyzed using Spike2 software (RRID:SCR_000903). Changes in burst duration and maximum instantaneous frequency were calculated from cervical and lumbar recordings.
Pharmacological agents.
During patch-clamp recordings, drugs were applied using 0.5 mm diameter quartz tubing positioned 50 μm away from the recording area under direct visual control. The quartz tubing was connected using a manifold to six solenoid valves linked with six reservoirs. Solutions were gravity-fed into the quartz tubing. Drug application was controlled using a VC-8 valve controller (Warner Instruments). The following pharmacological agents were used: TTX (1 μm, Alomone Labs) and riluzole (5–10 μm; Tocris Bioscience). Both were dissolved in the bath solution.
Immunohistochemistry.
All primary antibodies used and their respective dilutions are listed in Table 1. E12.5 embryos were collected from pregnant females. Once dissected out of their yolk sac, these embryos were immediately immersion-fixed in phosphate buffer (PB; 0.12 m) with 4% paraformaldehyde (PFA; freshly prepared in PB, pH 7.4) for 1 h at 4°C. Embryos were then rinsed with PB and cryoprotected in PB-15% sucrose at 4°C for 24 h and then in PB-30% sucrose at 4°C for 24 h. Embryos were embedded in OCT medium (VWR) and quickly frozen. Serial sections 20 μm thick were collected onto slides using a cryostat. Immunostaining was processed on SC transverse sections and on whole SC postrecording to confirm IN identity. To reveal neurobiotin-labeled cells, SCs were fixed for 1 h in 4% PFA and were then incubated in 0.12 m PB at 4°C until immunohistochemical studies. Tissues (embryo slices and whole SC) were thawed at room temperature, washed in PBS, incubated in NH4Cl (50 mm) diluted in PBS for 20 min and then permeabilized for 30 min in a blocking solution (10% goat serum in PBS) with 0.2% Triton X-100 and then for 48 h at 4°C with the primary antibodies, which were diluted in the 0.2% Triton X-100 blocking solution. Slices or whole SCs were then washed in PBS and incubated for 2 h at RT in the secondary antibodies (diluted at 1/1000 in the 0.2% Triton X-100 blocking solution): AlexaFluor 405 (ThermoFisher Scientific, catalog #A-31556; RRID:AB_221605) or 647 goat anti-rabbit (ThermoFisher Scientific, catalog #A-21244; RRID:AB_2535812); AlexaFluor 594 goat anti-guinea pig (ThermoFisher Scientific, catalog #A-11076; RRID:AB_2534120); AlexaFluor 649 donkey anti-guinea pig (Jackson ImmunoResearch, catalog #706-605-148; RRID:AB_2340476); AlexaFluor 594 goat anti-mouse (ThermoFisher Scientific, catalog #A-11005; RRID:AB_2534073); AlexaFluor 488 goat anti-chicken (ThermoFisher Scientific, catalog #A-11039; RRID:AB_2534096), and streptavidin-conjugated AlexaFluor 405 (1:1000; ThermoFisher Scientific, catalog #S32351). After washing in PBS, slides or whole SCs were dried and mounted in Mowiol medium (Millipore).
Table 1.
Primary antibodies
| Primary antibody | Company | Reference | Host/isotype | Dilution |
|---|---|---|---|---|
| Anti-calbindin-D28k | Swant, Switzerland | Swant, catalog #CB38; RRID:AB_2721225 | Rabbit polyclonal | 1:1500 |
| Anti-FoxD3 | Provided by C. Birchmeier, MDC Berlin, Germany | Storm, et al. (2009) | Guinea pig polyclonal | 1:5000 |
| Anti-MafB | Bethyl Laboratories | Bethyl, catalog #IHC-00351; RRID:AB_1279487 | Rabbit polyclonal | 1:1000 |
| Anti-GABA | Sigma-Aldrich | Sigma-Aldrich, catalog #A0310; RRID:AB_476667 | Mouse monoclonal | 1:800 |
| Anti-synaptophysin | Synaptic Systems | Synaptic Systems, catalog #101 011; RRID:AB_887824 | Mouse monoclonal | 1:1500 |
| Anti-GFP | Aves Labs | Aves Labs, catalog #GFP-1020; RRID:AB_10000240 | Chicken polyclonal | 1:1000 |
Confocal microscopy and image analysis.
Preparations were analyzed using a Leica SP5 confocal microscope. Immunostaining was observed using a 40× oil-immersion objective with a numerical aperture of 1.25, as well as with a 63× oil-immersion objective with a numerical aperture of 1.32 and a 4× digital zoom magnification. Serial optical sections were obtained with a Z-step of 1 μm (40×) and 0.2–0.3 μm (63×). Images (1024 × 1024; 12-bit color scale) were stored using Leica software LAS-AF and analyzed using ImageJ 1.5 software (Wright Cell Imaging Facility; RRID:SCR_008488). Colocalization of synaptophysin staining with calbindin staining, eGFP staining (MNs, HB9eGFP mouse embryos), or neurobiotin staining was assessed in three axes using a single confocal slice and x and y orthogonal views of the stack (ImageJ 1.5).
Sniffer recordings.
To make a “sniffer” electrode (Young and Poo, 1983) for GABA, an outside-out patch was pulled from a transfected HEK293 cell line expressing the GABAAR subunits α3β2γ2 (kindly provided by Michel Partiseti, Sanofi Advantis R&D LGCR/LIT, France). This GABAAR is highly specific for GABA and is characterized by slow desensitization, which makes it a good sensor for GABA paracrine release (Barberis et al., 2007). As a control, the electrode was first positioned outside the SC to verify any GABA contamination in the recording medium. The electrode was then pushed inside the SC and positioned within the motoneuronal area and the ventral funiculi. Outside-out patch-clamp electrodes (5–10 MΩ) were pulled from thick-wall borosilicate glass, fire-polished, and filled with (in mm): CsCl 130, MgCl2 4, Na2ATP 4, EGTA 10, HEPES 10, pH 7.2 (290 mOsm/kg-H2O). Single-channel currents were recorded using an AxoPatch 200B amplifier (Molecular Devices). Recordings were filtered at 10 kHz, sampled at 50 kHz, and stored on a PC computer. Membrane potential was held at −50 mV throughout the experiment. Analysis of the sniffer currents was performed using AxoGraph X.1.6.4 software.
Human embryonic kidney 293 cells (HEK293) were maintained in a 95% air-5% CO2 humidified incubator, at 35°C, in DMEM supplemented with 0.11 g/L sodium pyruvate, 6 g/L d-glucose, 10% (v/v) heat-inactivated fetal bovine serum (all from Invitrogen). Cells were passaged every 5–6 d (up to maximum 20 times). For electrophysiological recordings, cells were seeded onto glass coverslips coated with poly-l-ornithine (0.1 mg/ml).
Statistics.
All values were expressed as mean ± SD. Statistical significance was assessed by the nonparametric Kruskal–Wallis test with Dunn's posttests, Mann–Whitney test and Wilcoxon matched pairs test (GraphPad Prism 5.0 Software). Significance was determined as *p < 0.05, **p < 0.01, or ***p < 0.001.
Results
V1R are identified with calbindin, Foxd3, and MafB at the onset of SNA
In the present study, we used GAD67-(eGFP knock-in mice (Tamamaki et al., 2003) to visualize putative GABAergic INs (Figs. 1A1,B1,C1). V1R can be unambiguously identified by their characteristic position in the ventral horn both in the adult (Geiman et al., 2000) and in the embryo (Sapir et al., 2004; Stam et al., 2012). Most V1R were localized between the ventral border of MN columns and the ventral funiculi. To identify V1R in the lumbar SC of E12.5 GAD67eGFP embryos, we first performed immunostaining using anti-calbindin, anti-Foxd3 and anti-MafB antibodies (Fig. 1). Calbindin is a calcium-binding protein exclusively expressed by V1R when looking in the ventrolateral area of the SC between the MN columns and the funiculus (Stam et al., 2012). The forkhead transcription factor Foxd3 controls the early phase of V1R differentiation, whereas MafB is required to maintain V1R at later developmental stages, and both are therefore expressed at E12.5 in V1R (Stam et al., 2012). At E12.5 INs located in the ventrolateral marginal zone between the motor columns and the ventral funiculi express Foxd3 (Figs. 1A2,B2,C2) and calbindin (Figs. 1A3,B3), as previously described (Benito-Gonzalez et al., 2012; Stam et al., 2012). Foxd3+ INs were also observed in neurons positioned dorsally to the motor columns (Figs. 1A2,B2). However, these INs were not stained by calbindin antibody, hence this population of INs was not included in our study. In the V1R area, double immunostaining (Figs. 1A4,B4) indicated that 81.7 ± 15.0% of Foxd3+ INs localized in the ventral marginal zone were also immunoreactive to calbindin at E12.5 (n = 4 embryos, 2 sections/embryo). To ensure that all Foxd3+ INs localized in the area are V1R, we performed double immunostaining using anti-Foxd3 and anti-MafB antibodies (Figs. 1C). We found that 100% of Foxd3+ eGFP+ INs localized in the ventral marginal zone were also immunoreactive to anti-MafB antibody (n = 4 embryos, 2 sections/embryo), thus confirming their V1R fate (Fig. 1C4). It should also be noted that at this developmental stage we detected MafB+ eGFP− neurons localized within the motor columns (Figs. 1C1,C3) that are likely MNs (Stam et al., 2012). Therefore, we used Foxd3 immunostaining to confirm that the recorded IN belonged to the V1R population (Figs. 1D1–D4). Whereas Calbindin is a good marker for V1R, it was not used to identify recorded V1R because of cytoplasm-calbindin dilution complications during whole-cell recording (Müller et al., 2005) caused by the high nucleus/cytoplasm volume ratio in embryonic neurons.
Figure 1.

V1R identification in the lumbar spinal cord of E12.5 embryos. A, Coronal slice of the lumbar spinal cord of E12.5 GA67-eGFP mouse embryo showing the distribution of eGFP neurons (A1), FoxD3 (A2), and calbindin (A3) -immunoreactive neurons. A4, Superimposed images showing the colocalization of eGFP, FoxD3 immunostaining, and calbindin immunostaining. B, Enlarged images from A showing that eGFP neurons localized in the ventrolateral area of the spinal cord (B1) are Foxd3-immunoreactive (B2), most of them being calbindin-immunoreactive (B3–B4). C, Coronal slice of the ventrolateral part of the lumbar spinal cord of E12.5 GA67-eGFP mouse embryo showing the distribution of eGFP neurons (C1), FoxD3 (C2), and MafB (C3)-immunoreactive neurons. C4, Superimposed images showing the colocalization of eGFP FoxD3 immunostaining and MafB immunostaining. Note that all FoxD3-immunoreactive neurons localized in the marginal zone of the ventrolateral area are also MafB-positive, indicating that they are V1R neurons. D1, Example of a neuron filled with neurobiotin during the recording at the lumbar level of an embryonic spinal cord open book preparation of GAD67-GFP mice at E12.5. This eGFP neuron (D2) was immunoreactive to Foxd3 antibody (D3) as shown in the merged images (D4). Each image corresponds to a single confocal section.
V1R already produce GABA at E12.5
As calbindin is localized within the cytoplasm of V1R, we used calbindin immunostaining to visualize V1R morphology (Stam et al., 2012). Calbindin+ neurons (96.6 ± 7.2%) in the ventrolateral marginal zone were also eGFP+ in E12.5 GAD67eGFP knock-in mouse embryos (n = 4 embryos, 2 sections/embryo), which could suggest that most V1R are likely to produce GABA at this developmental stage. To determine to what extent V1R already produce GABA at E12.5, we performed double immunostaining using anti-calbindin and anti-GABA antibodies (Figs. 2A,B). We found that 90.1 ± 10.5% (n = 8 embryos, 2 sections/embryo) of calbindin+ INs localized in the ventrolateral marginal zone (Figs. 2A2,B2) were also immunoreactive to GABA antibody (Figs. 2A3,A4,B3,B4), suggesting that V1R already produced enough GABA to be detected by immunohistochemistry at E12.5.
Figure 2.

V1R already produce GABA in the lumbar spinal cord of E12.5 embryos. A, Single confocal sections of coronal slice of the lumbar spinal cord of E12.5 GA67-eGFP mouse embryo showing the distribution of eGFP neurons (A1), calbindin (A2), and GABA (A3)-immunoreactive neurons. A4, Superimposed images showing the colocalization of eGFP, calbindin immunostaining, and GABA immunostaining. B1–B3, Enlarged images from A1, A2, and A3 showing eGFP neurons (B1), calbindin-immunoreactive neurons (B2), and GABA-immunoreactive neurons (B3) in the ventrolateral area of the spinal cord. B4, Superimposed images showing the colocalization of calbindin immunostaining and GABA immunostaining with z projections within a stack. Note the colocalization of calbindin immunostaining and GABA immunostaining in the three axes, indicating that V1R already produce GABA at E12.5.
It is important to note that many eGFP+ GABA− INs were detected within the ventral parenchyma at E12.5, as previously observed (Allain et al., 2004). This could result from the production of the GAD67 splice variant GAD25. At early developmental stages, GAD1 first encodes the truncated 25 kDa leader (GAD25) without GAD enzymatic activity and then the enzymatically active protein GAD44 (for review, see Le Corronc et al., 2011). These two GAD isoforms are downregulated during neuronal differentiation concomitantly with upregulation of GAD67 expression.
Synaptic-like contacts are detected between V1R and MNs at the onset of SNA
At E12.5, V1R axons are localized in ventral funiculi and extend for a few segments without forming collateral branches entering the motor columns (Alvarez et al., 2013). The first evidence for collateral V1R axons innervating the soma of lumbar MNs was found at E15.5 only while synaptic-like connections from MNs onto the V1R cell body were already observed at E12.5 (Alvarez et al., 2013). Accordingly, it was proposed that MNs can control V1R activity at E12.5 while V1R are unable to control MN activity at this time point (Alvarez et al., 2013). However, it should be noted that synaptophysin staining was found mainly in the ventral funiculus at E12.5 (Fig. 3A1; Alvarez et al., 2013; Czarnecki et al., 2014), where IN axonal projections and MN dendrite-like processes were observed (Czarnecki et al., 2014). Accordingly, we hypothesized that V1R already make some synaptic-like contacts with MN projections within the ventral funiculus.
Figure 3.
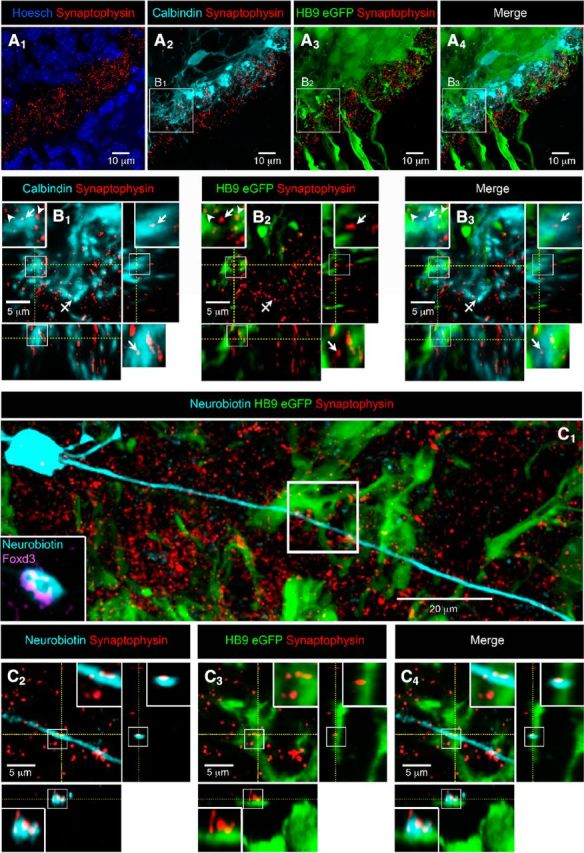
V1R make synaptic-like contacts with motoneurons (HB9-eGFP) at E12.5. A, Coronal slice of the lumbar spinal cord of E12.5 HB9-eGFP mouse embryo with cell nucleus staining (HOESCH) and synaptophysin immunostaining (A1). A1, Note that synaptophysin immunostaining is mainly restricted in the ventral funiculus. A2, Calbindin staining showing the distribution of V1R neurite extensions and synaptophysin immunostaining. A3, eGFP immunostaining showing the distribution of MN neurite extensions and synaptophysin immunostaining. Antibody against GFP was used to visualize MN morphology better (Czarnecki et al., 2014). A4, Superimposition of eGFP fluorescence, calbindin immunostaining and synaptophysin immunostaining (A1–A4 are confocal stacks). B, Single confocal sections with z projections of enlarged images from A2 to A4 showing the colocalization of synaptophysin punctates with calbindin immunostaining opposed to eGFP immunostaining (B1–B3; enlarged images in boxes). B2, Note that a synaptophysin punctate colocalized with calbindin immunostaining (B1, arrow) did not colocalize with eGFP immunostaining (B2, arrow). Note that synaptophysin punctate colocalized with eGFP immunostaining (B1, arrowheads) did not colocalize with calbindin immunostaining (B2, arrowhead). Barred arrow (B1) shows a colocalization of calbindin and synaptophysin immunostaining not opposed to eGFP immunostaining. B4, superimposed images (B1–B3) with z projections showing calbindin immunostaining and eGFP appositions. C1, Confocal stacks showing neurobiotin-injected Foxd3-immunoreactive V1R, HB9-eGFP immunostaining and synaptophysin immunostaining in an SC open book preparation. C2–C4, Single confocal sections with z projections of enlarged images from C1 (white box) showing the colocalization of synaptophysin punctates with neurobiotin staining (C2), the apposition of the same synaptophysin punctates to eGFP (C3) and the apposition of neurobiotin staining containing synaptophysin punctates to eGFP (C4; enlarged image in boxes in C2–C4), indicating the presence of a V1R synaptic-like contact on an MN neurite.
To address this issue we first performed multiple immunostaining using anti-synaptophysin antibody (putative release site), anti-calbindin antibody (V1R) and anti-eGFP antibody on coronal sections of the SC HB9eGFP mouse embryos to visualize MNs (Figs. 3A2–A4,B1–B3). As shown in Figure 3B3, many calbindin+ fibers were opposed to MN neurites (eGFP staining) within the ventral funiculus. We found 21.7 ± 8.4 synaptophysin+ punctates within calbindin+ fibers opposed to MN neurites per hemi section in coronal slices (n = 9; 5 embryos; Figs. 3B1–B3, insets, arrow). Conversely, synaptophysin punctates were also found in MN fibers (eGFP+) opposed to calbindin+ fibers (Figs. 3B1–B3, inset, arrowhead), which indicate the presence of MN putative release sites opposed to V1R within the ventral funiculus. We also found synaptophysin punctates within calbindin+ fibers that were not opposed to MN neurites (Figs. 3B1–B3, barred arrow). This may indicate the presence of nonsynaptic release sites and/or that V1R can make contact with fibers of neurons other than MNs. To further confirm the presence of V1R synaptic-like contacts on MNs, we performed multiple immunostaining using anti-synaptophysin antibody (putative release site) and anti-eGFP antibody on isolated SC after patch-clamp injection of neurobiotin in V1R (Figs. 3C). Recorded INs (eGFP-negative) in eGFP HB9 embryos were identified as V1R using Foxd3 immunostaining (Fig. 3C1, inset). We found 3–5 putative release sites per V1R (n = 4 cells) that were opposed to MN fibers within the ventral funiculus (Figs. 3C1–C4). We did not find any release site within the V1R axonal growth cone (data not shown).
Paracrine release of GABA occurred in the SC of E12.5 mouse embryos
Paracrine release of GABA may occur from nonsynaptic release sites and/or may reflect neurotransmitter spillover of GABA from immature synapses (Safiulina and Cherubini, 2009). Therefore, we hypothesized that GABA release can occur at this developmental stage. To detect the presence of GABA paracrine release, we used the sniffer technique (Scain et al., 2010). Outside-out patches from HEK cells expressing the GABAAR subunits α3β2γ2 were used as a sniffer to detect the presence of GABA in the extracellular space (Fig. 4A1). When inserting the sniffer electrode in the vicinity of MNs, we did not detect any GABAAR, thus indicating that basal GABA concentration is below the detection threshold of the sniffer patch (data not shown). The possibility of evoking GABA release, and hence GABAAR activation, by inducing a global cell membrane depolarization was tested using bath application of 30 mm KCl (Scain et al., 2010; Czarnecki et al., 2014). Increasing [K+]o evoked a significant GABAAR activation in the sniffer patch (n = 9), indicating that GABA can be released in the extracellular space in response to cell membrane depolarization (Fig. 4A1). We estimated the concentration of released GABA around MNs by comparing the peak amplitude of the sniffer response evoked by KCl application to sniffer responses evoked by exogenous application of 3, 10, and 30 μm GABA onto the same outside-out patch (n = 9). We found that the GABA concentration accounting for GABAAR activity evoked by KCl application was close to 3 μm (Fig. 4A2). Although the estimated concentration of nonsynaptic release of GABA was relatively low, the exogenous application of 3 μm GABA in the presence of 1 μm TTX was sufficient to evoke a 20.6 ± 6.1 mV MN depolarization (Vh = −60 mV; ECl = −30 mV; n = 7; Figs. 4B2).
Figure 4.
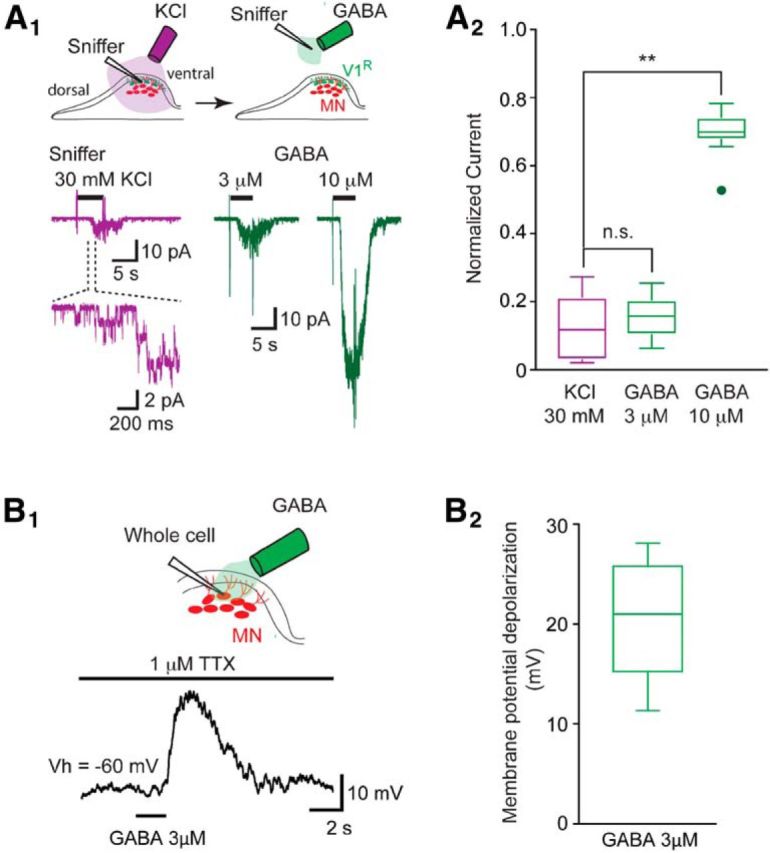
Paracrine release of GABA detected by a sniffer outside-out patch. A1, Top drawing shows the location of the outside-out sniffer to detect paracrine release of GABA (left) and to obtain outside-out currents in response to GABA application (left). Bottom traces (purple): example of outside-out sniffer current evoked by the application of 30 mm KCl when the sniffer electrode was positioned in the dorsal area of the SC close to motor columns. Enlarged trace shows single-channel currents at the onset of the sniffer current. Right traces (green) show outside-out current evoked by the application of 3 and 10 μm GABA to a sniffer patch positioned outside the spinal cord. Purple and green traces are from the same outside-out patch. A2, Box plots of normalized maximum outside-out current evoked by KCl application (purple left) and by the application of 3 or 10 μm GABA (green right) on the same outside-out sniffer patch (n = 9). Amplitudes of the outside-out currents evoked by the application of 30 mm KCl, 3 or 10 μm GABA, were normalized to the amplitude of the outside-out currents evoked by the application of 30 μm GABA (data not shown). Note that the normalized amplitude of the outside-out current evoked by 30 mm KCl application (0.118 ± 0.097) was not significantly different (p > 0.9) from the normalized amplitude of the outside-out current evoked by the application of 3 μm GABA (0.152 ± 0.062). Normalized amplitudes of the outside-out currents evoked by the application of 30 mm KCl or of 3 μm GABA were significantly different (KCl: p = 0.0029; 3 μm GABA: p = 0.00665) from the normalized amplitude of the outside-out currents evoked by the application of 10 μm GABA (0.697 ± 0.073). **p < 0.01. B1, Example of motoneuron membrane potential depolarization evoked by the application of 3 μm GABA in the presence of 1 μm TTX (current-clamp recording: Vh = −60 mV; ECl = −30 mV). B2, The application of 3 μm GABA evoked a depolarizing response of 20.6 ± 6.1 mV (n = 7).
V1R exhibit heterogeneous excitability patterns at the onset of SNA
Spontaneous or evoked V1R activity was assessed using whole-cell current-clamp recordings and the open-book SC preparation (Scain et al., 2010). To determine the identity of recorded INs, GAD67eGFP neurons were filled with neurobiotin (≈1 mg/ml) in combination with post hoc Foxd3 staining (Figs. 1D). At E12.5, V1R had a whole-cell capacitance of 13.7 ± 3.8 pF (n = 192) and an input resistance of 1242 ± 620 MΩ (n = 192). Spontaneous activity recorded in 37 V1R was characterized by recurrent GDPs (Fig. 5), occurring every 3.7 ± 2.4 min (n = 30), similar to what has been observed in MNs (∼3 min; Czarnecki et al., 2014). Interestingly, in 21.6% (n = 8/37) of the recorded V1R, GDPs evoked long-lasting depolarizing plateau potentials (Fig. 5A), whereas 37.8% (n = 14/37) of the recorded V1R had the ability to spike repetitively during GDPs (Fig. 5B). Plateau potentials had an absolute amplitude of 53.4 ± 11.5 mV (n = 8) and a half-amplitude duration ranging from 0.12 to 0.86 s (0.44 ± 0.22 s; n = 8). In 16.2% (n = 6/37) of the recorded V1R, GDPs evoked a single spike or a doublet (Fig. 5C), while the remaining 24.3% (n = 9/37) of the recorded V1R did not spike during GDPs (data not shown). These data show that V1R can generate various spontaneous activity patterns and raise the question of their intrinsic activation properties.
Figure 5.

V1R display plateau potential, repetitive firing or generate a single AP during episodes of SNA in E12.5 spinal cord. Examples of spontaneous activities recorded in V1R being characterized by GDPs displaying plateau potential (A), repetitive firing (B), or a single AP (C) activity (Vh = −60 mV). Recordings shown in A–C are from different cells.
To analyze the intrinsic activation properties of embryonic V1R, we depolarized the cells from a holding potential of −60 mV using 2 s step current pulses or 20 s depolarizing current ramps. At E12.5, all V1R were excitable (n = 165), but depolarizing current injections triggered various and complex excitability patterns (Fig. 6). 28.7% (n = 47/165) of the analyzed V1R were able to generate 1–3 APs in response to suprathreshold depolarizing steps (Fig. 6A). When a depolarizing ramp was applied, only fast membrane potential oscillations of small amplitude were observed. We called these V1R, SS-V1R (“single” spiking V1R). In contrast, 21.2% (n = 36/165) of the recorded V1R were able to generate long-lasting events defined as plateau potentials in response to depolarizing current pulses and in response to depolarizing current ramps. These plateau potentials were all-or-none events (Fig. 6B). They can be evoked by long depolarizing pulses (2 s; Fig. 6B) and short depolarizing current pulses (Fig. 6F2). They had a half amplitude duration of ≈ 700 ms and an absolute peak amplitude of ≈3 mV (Table 2). Remarkably, these plateau potentials were sodium-dependent events as they were fully blocked by the application of 1 μm TTX (Fig. 6F1). Unlike SS-V1R, these V1R can generate repetitive plateau potentials when depolarized by a suprathreshold depolarizing current ramp (Fig. 6B). We called these V1R, PP-V1R (plateau potential V1R). A third group of V1R (41.5%; n = 68/165) was identified based on their ability to generate repetitive AP firing in response to suprathreshold depolarizing current steps or to depolarizing current ramps. In these cells, APs had a half amplitude duration ≈ 13 ms (Table 2). We called these V1R, RS-V1R (repetitive spiking V1R; Fig. 6C). Finally, a fourth group of cells (8.5%; n = 14/165) was determined according to their ability to generate both APs and plateau potential-like events (Fig. 6D). We called these V1R, ME-V1R (mixed event V1R). SS-V1R are unlikely to be dying neurons. Developmental cell death of V1 INs occurs after E14.5 in mouse embryos (Prasad et al., 2008) and SS-V1R passive electrical properties did not differ from those of more active V1R (Table 2).
Figure 6.

V1R display different excitability patterns in E12.5 embryonic spinal cords. Excitability patterns were analyzed using depolarizing current step (2 s) and depolarizing current ramp (20 s). A–D, Representative traces of voltage responses showing single-spiking activity (A), plateau potential activity (B), repetitive AP firing (C), and mixed repetitive-spiking/plateau potential activity (D). E, Proportions of V1R subtypes according to the observed discharge patterns. 28.6% of V1R could not sustain repetitive spiking, 41.7% were repetitive-spiking V1R, 8.5% were mixed V1R, 21.2% were plateau potential V1R (n = 164). F, Plateau potentials are blocked by TTX (1 μm) application (n = 5/5; F1, F2). F2, Plateau potentials are evoked by short (100 ms) pulses of depolarizing current.
Table 2.
Intrinsic functional properties of V1R
| SS-V1R | RS-V1R | PP-V1R | |
|---|---|---|---|
| Whole cell capacitance, pF | 13.3 ± 2.8 | 15.3 ± 4.5 | 12.4 ± 3.6 |
| N = 82 | N = 60 | N = 50 | |
| Input resistance, MΩ | 1298 ± 682 | 1156 ± 616 | 1254 ± 508 |
| N = 82 | N = 60 | N = 50 | |
| Threshold, mV | −34.1 ± 3.3 | −33.6 ± 4.9 | −35.9 ± 6.0 |
| N = 46 | N = 53 | N = 36 | |
| Peak amplitude, mV | −6.8 ± 6.1 | 3.2 ± 6.7 | 3.4 ± 10.3 |
| N = 46 | N = 53 | N = 36 | |
| Half-width, ms | 11.2 ± 8.2 | 12.8 ± 6.9 | 705.7 ± 631.7 |
| N = 46 | N = 53 | N = 36 | |
| Rate of rise, mV/ms | 8.4 ± 3.8 | 13.4 ± 9.4 | 6.2 ± 3.7 |
| N = 27 | N = 27 | N = 19 |
SS-V1R, Single-spiking Renshaw cells; RS-V1R, repetitive-spiking Renshaw cells; PP-V1R, plateau potential Renshaw cells. Threshold values represent the averaged AP threshold for single-spiking V1R and repetitive spiking V1R and the averaged plateau potential threshold for plateau potential V1R. Peak amplitude values represent the averaged peak amplitude of the AP for single-spiking V1R, the averaged peak amplitude of the first AP in a train for repetitive spiking V1R and the averaged peak amplitude of plateau potentials for plateau potential V1R. Half-width values represent the averaged half width of the AP for single-spiking V1R, the averaged half-width of the first AP in a train for repetitive spiking V1R and the averaged half width of plateau potentials for plateau potential V1R. Rate of rise values represent the averaged rate of rise of the AP for single-spiking V1R, the average rate of rise of the first AP in a train for repetitive spiking V1R and the average rate of rise of plateau potentials for plateau potential V1R. Values are expressed as mean ± SD. Input resistance of SS-V1R, input resistance of RS-V1R, and input resistance of PP-V1R were not significantly different (p = 0.17).
Considering that the different types of intrinsic activation pattern may reflect a continuum between a single spike and a plateau potential, we chose to focus our study on the three clear patterns of intrinsic activation, SS-V1R, RS-V1R and PP-V1R, and to not study the mixed regime in depth. Table 2 summarizes passive and active intrinsic electrical properties of recorded SS-V1R, RS-V1R and PP-V1R. Note that the difference in intrinsic excitability in these different classes of V1R is not due to difference in their membrane resistance (p > 0.1).
These results reveal that V1R can have different regimes of activity in the embryonic SC at the onset of synaptogenesis and SNA (E12.5; Scain et al., 2010; Czarnecki et al., 2014).
Sustained discharge in embryonic V1R depends on persistent sodium current
A key element in neurons, which exhibit sustained discharges, is the expression of subthreshold slow-inactivating inward currents. Among these inward currents, the TTX-sensitive persistent inward current (INap) plays a fundamental role in controlling sustained discharge in INs and MNs in the SC at postnatal developmental stages (Lee and Heckman, 1998; Kuo et al., 2006; Theiss et al., 2007; Ziskind-Conhaim et al., 2008).
To determine whether INap is already expressed by V1R in E12.5 mouse embryo, we made whole-cell voltage-clamp recordings. In this set of experiments, the electrodes were filled with a Cs-based intracellular solution to minimize contamination of the recorded current by potassium currents. A test pulse from −100 to −20 mV evoked TTX-sensitive sodium currents: a large amplitude transient current (INaT) followed by a smaller amplitude current persisting over 0.5 s was also fully blocked by TTX (Fig. 7A1, inset), which indicates the presence of INap. This also indicates that sodium-independent voltage-gated inward currents were not detected at this developmental stage. To characterize INap better, we used a slow voltage ramp (70 mV/s) from −100 to +20 mV designed to inactivate transient currents (Huang and Trussell, 2008). Subtraction of the trace after TTX application revealed the INap on the I–V curve (Fig. 7A1). INap was observed in all recorded V1R. We further investigated the activation properties of the INap. The Boltzmann fit of the voltage dependence of the INap generated by voltage ramps revealed a Vhalf of −22.9 ± 5.7 mV and a slope factor (k) of 7.0 ± 1.8 mV in V1R (n = 8; Fig. 7A2). In V1R, the INap activated at Vh = −67.4 ± 6.2 mV (n = 8) and the mean persistent sodium conductance density (gNaP/pF) was 41.8 ± 22.7 pS/pF (n = 12; Fig. 7B).
Figure 7.

INap is already expressed in V1R. A1, Representative trace of INap evoked by a slow depolarizing voltage ramp in a V1R (CsCl intracellular solution). INap (black trace) was isolated by subtracting the current elicited by a voltage ramp (70 mV/s) in the presence of TTX (inset, green trace) from the control current (inset, black trace). TTX-sensitive current was blocked by 5 μm riluzole (red trace). Left, Inset shows the protocol to generate voltage-dependent slow inward currents in control conditions (black), after 5 μm riluzole application (red) or 1 μm TTX application (green). Right, Inset shows the current evoked by a depolarizing voltage step from −100 to 20 mV in the absence and in the presence of 5 μm riluzole. A2, Voltage dependence of INap conductance calculated from the trace shown in A1. The activation curve was obtained by transforming the current evoked by a depolarizing voltage ramp from −100 to 20 mV (70 mV/s) using the following equation: GNaP = − INap/[(−Vh)+ENa+] where Vh is the holding potential at time t during a depolarizing voltage ramp and ENa+ is the equilibrium potential for sodium (ENa+ = 60 mV). The GNaP/Vh curve was fitted with a Boltzmann function (see Materials and Methods), where Vhalf is the Vh value for INap half activation, k the slope factor of the curve and Gmax the maximum conductance. B, Box plot showing Gmax density in V1R (n = 12). C, Box plots showing the variation of the percentage INap block by 5 μm riluzole in V1R.
To further confirm that this persistent inward current truly reflects the presence of INap, we tested the effect of a low concentration of riluzole (5 μm), which is known to block INap (Urbani and Belluzzi, 2000). Indeed, 5 μm riluzole suppressed 81.5 ± 14.6% (n = 15; p = 0.0005) of the INap in V1R (Fig. 7C), whereas at this low concentration riluzole only slightly reduced the peak amplitude of INaT (see red arrows in Fig. 7A1, inset). We confirmed on MNs that this low concentration had a minimal effect on the sodium-dependent AP, which was not the case when riluzole concentration was increased to 10 μm (Figs. 8A,B1–B3). AP amplitude was significantly reduced to 83.2 ± 12.6% of control in the presence of 5 μm riluzole (n = 7; p = 0.008), but was reduced to 67.5 ± 17.0% of control when riluzole concentration was increased to 10 μm (n = 7, p = 0.0078). AP threshold potential was significantly reduced in the presence of 10 μM riluzole (p = 0.0156) and AP half-width was significantly reduced in the presence of 5 μm (p = 0.0313) and 10 μm riluzole (p = 0.0313). We then reasoned that the lack of repetitive spiking or plateau potential activity observed in some V1R might depend on a reduced level of INap. To test this hypothesis, we compared the presence of INap in V1R that cannot generate repetitive spiking (SS-V1R) with V1R that can generate repetitive spiking (RS-V1R) and with V1R generating plateau potentials (PP-V1R). We developed a voltage- and current-clamp recording protocol in a given neuron allowing us to associate the amplitude of the INap with the excitability pattern in each type of spinal neuron (K-methanesulfonate intracellular solution; Fig. 9A1–A4). The same voltage ramp (70 mV/s) was used as described above. The INap current was revealed by subtracting ramp-evoked current in the presence of 1 μm TTX from controls. In these conditions, SS-V1R had a significantly lower gNaP density (22.1 ± 12.6 pS/pF, n = 13) compared with RS-V1R (57.8 ± 17.4 pS/pF, n = 7) and to PP-V1R (70.6 ± 34.0 pS/pF, n = 11; p < 0.0001; Fig. 9A4). These results highlight a direct relationship between the strength of INap and the ability of V1R to sustain repetitive spiking and plateau potential activity.
Figure 8.
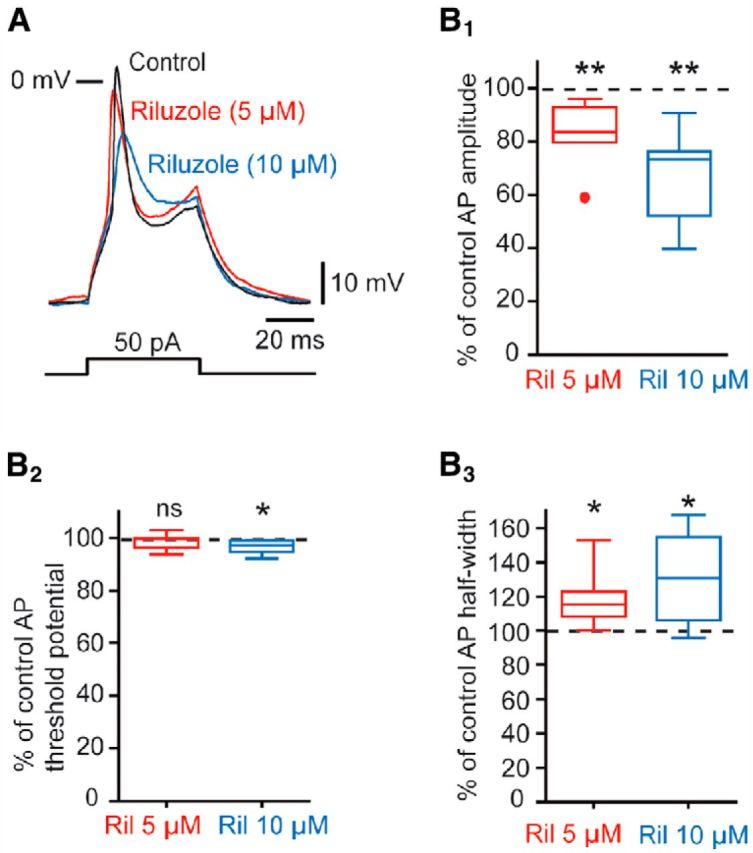
Effect of 5 and 10 μm riluzole on APs. A, Effect of 5 μm (red trace) and 10 μm (blue trace) riluzole on the AP evoked by a depolarizing current step in an MN. B1, Box plot showing the percentage changes in AP amplitude (% control) in the presence of 5 μm riluzole and 10 μm riluzole. B2, Box plot showing the percentage changes in AP threshold (% control) in the presence of 5 and 10 μm riluzole. B3, Box plot showing the percentage changes in AP half-width (% control) in the presence of 5 and 10 μm riluzole. *p < 0.05, **p < 0.01.
Figure 9.

Sustained discharge in embryonic V1R depends on INap. A, Representative traces of INap recorded in V1R that cannot sustain repetitive spiking (SS-V1R; A1), in a repetitive-spiking V1R (RS-V1R; A2) and in a plateau potential V1R (PP-V1R; A3). INap was isolated by subtracting the current elicited by a slow voltage ramp (−100 to + 20 mV; 70 mV/s) in the presence of 1 μm TTX from the current evoked in the absence of TTX. A4, Box plots showing Gmax density in SS-V1R (n = 13), RS-V1R (n = 8) and PP-V1R (n = 11). Note that Gmax density is significantly lower in SS-V1R (p < 0.01). B, Representative traces showing the effect of riluzole application (5 μm) on the intrinsic activity pattern evoked by suprathreshold current steps (left traces) or a suprathreshold current ramp (right traces) in an RS-V1R. Note that riluzole blocks repetitive spiking (n = 10/10). C, Representative traces showing the effect of riluzole application on plateau potential evoked by suprathreshold current steps (left traces) or by a suprathreshold current ramp (right traces) in a V1R. Note that riluzole blocks plateau potential activity (n = 7/7). ***p < 0.001.
To determine whether INap is required for repetitive spiking in embryonic V1R, we assessed the effect of riluzole on V1R that can sustain AP firing or plateau potential activity. The application of 5 μm riluzole turned all RS-V1R into SS-V1R (n = 10; Fig. 9B). In the presence of 5 μm riluzole, the depolarizing current step failed to trigger more than a doublet in these cells. 5 μm riluzole did not significantly change the AP threshold (−37.2 ± 3.2 mV vs −36.8 ± 6.0 mV with 5 μm riluzole, n = 10, p = 0.70). In all PP-V1R, riluzole prevented the plateau potential activity (n = 7; Fig. 9C). Increasing the amplitude of the injected current failed to evoke any AP, indicating that PP-V1R were turned into nonexcitable neurons when INap was blocked.
Altogether, these results indicate that INap already plays an important role in V1R intrinsic activation properties at this early embryonic developmental stage of SC neuronal networks.
Blocking persistent sodium current alters SNA pattern and SNA propagation along the cord
Synchronous activation of MNs and INs was proposed to drive episodes of activity during SNA (Hanson and Landmesser, 2003; Czarnecki et al., 2014). If intrinsic activation properties of the few INs that produce GABA including V1R, rely on the INap, we expected that blocking INap would alter episodes of activity during SNA in a manner similar to that observed in the presence of GABAAR antagonists (Hanson and Landmesser, 2003; Czarnecki et al., 2014). Blocking GABA neurotransmission during SNA evoked an increase in inter-episode interval, but had little effect on the duration of episode of activity and on their propagation along the cord (Hanson and Landmesser, 2003). In addition, we previously showed that blocking GABA neurotransmission decreased the amplitude of spontaneous giant inward currents (sGICs) recorded in MNs during SNA (Czarnecki et al., 2014).
To determine the involvement of INap in SNA, we first tested the effect of 5 μm riluzole on the spontaneous activity of MNs using whole-cell voltage-clamp recordings. In control conditions, SGICs occurred every 4 min (4.0 ± 0.8 min, n = 9), had mean amplitude of 291 ± 200 pA (n = 9) and a half-width of 0.6 ± 0.3 s (Vh = −60 mV; ECl = 3 mV; Fig. 10A1–A2). The time course of the effect of riluzole on sGIC occurrence was variable among MNs. In one of nine recorded MNs, riluzole abolished GIC activity in <10 min. In other MNs, one to two GICs could still be observed during the 20–30 min application of riluzole. (Fig. 10A2). During this transitory period, riluzole significantly reduced the amplitude of GICs (30.8 ± 32.7% reduction; n = 8; p = 0.039; Fig. 10B1) and the half-duration of GICs (44.0 ± 27.2% reduction; n = 8; p = 0.0078; Fig. 10B2). To assess the effect of riluzole on SNA, we made long-lasting extracellular recordings of SC activity using two electrodes positioned on the cervical and lumbar superficial part of the ventral horn of the SC at E12.5 (Czarnecki et al., 2014; Fig. 10C1).
Figure 10.
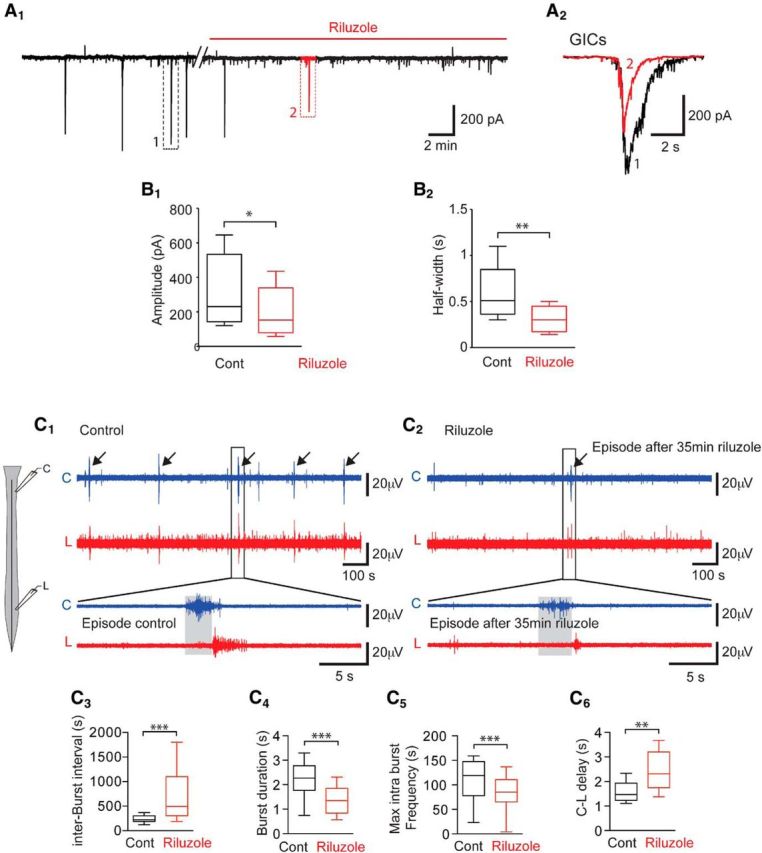
Riluzole dramatically decreases the frequency and duration of episodes of SNA in E12.5 embryonic spinal cord. A1, Application of 5 μm riluzole inhibits spontaneous GIC activity in MNs (voltage-clamp recordings; Vh = −60 mV; ECl = −30 mV). A2, Enlarged trace from (A1) showing GIC before (1, black) and at the onset of riluzole application (2, red). Note that the amplitude and the duration of GICs were decreased on riluzole application. B1, Box plots showing the amplitude of the GIC in a control and on riluzole application (n = 8). B2, Box plots showing half amplitude durations of the GIC in control and on riluzole application (n = 8). Note that the amplitude (p < 0.05) and the duration (p < 0.01) of GICs were significantly reduced in the presence of riluzole. C, SNA recorded at the cervical (C) and lumbar (L) levels (see schematic drawing on the left) in extracellular configuration before (C1) and after 5 μm riluzole (C2). Note that one episode still occurred 35 min after riluzole application. C3–C6, Box plots illustrating interburst interval, burst duration, intraburst spike frequency, and cervical-lumbar delay of episode propagation. *p < 0.05, **p < 0.01, ***p < 0.001.
These recordings allowed us to monitor SC neuron activity related to GIC activity recorded in MNs (Czarnecki et al., 2014). We found that a 30–40 min application of 5 μm riluzole significantly increased the inter-episode interval from 4.18 ± 1.93 min in control conditions to 12.34 ± 9.08 min in the presence of riluzole (p < 0.001, n = 12 paired SCs; 196.3 ± 203% increase; Fig. 10C2,C3), revealing a strong involvement of the INap current in the rhythmicity of early SC network activity. We therefore analyzed individual bursts of AP exhibited by the cervical (C) and lumbar (L) SC networks in the presence of riluzole. We found that the duration of bursts (both C and L), compared before (2.31 ± 0.89 s) and after the riluzole application (1.44 ± 0.7 s), was significantly reduced (36.5 ± 18.3% reduction; n = 22; p < 0.001; Fig. 10C4). In addition, the instantaneous AP frequency within a burst at both the cervical level (C level) and the lumbar level (L level) was significantly reduced by 25.3 ± 11.9% (p < 0.001) in the presence of riluzole (112.4 ± 39.6 Hz in control conditions and 83.3 ± 34.4 Hz with riluzole, n = 22; Fig. 10C5). We also observed a reduction in the speed of propagation of SNA episodes between the C level and L level of the SC in the presence of riluzole. Propagation of SNA along the cord was investigated by analysis of the time needed for a burst of AP recorded at the C level to reach L level (distance between the 2 recording levels ≈4 mm; Yvert et al., 2004). We found that the C-L time was significantly increased (p < 0.001) from 1.56 ± 0.42 s in control conditions to 2.42 ± 0.77 s in the presence of riluzole (56.3 ± 36.1% increase; n = 10; Fig. 10C6).
Riluzole had a stronger effect on SNA than previously observed in the presence of GABAAR antagonists (Hanson and Landmesser, 2003; Czarnecki et al., 2014). Targeted networks during extracellular recordings likely include MNs as well as surrounding INs (Hanson and Landmesser, 2003; Czarnecki et al., 2014) including V1R GABAergic INs. Because most GABAergic V1R are able to generate repetitive firing in response to GDP, which is not the case for MNs (Czarnecki et al., 2014), this apparent discrepancy could be explained if blocking GABA neurotransmission does not strongly alter GABAergic INs firing, just as riluzole does. We cannot, however, exclude that INap is also present in neurons other than V1R in the SC at E12.5. Anyhow, these results indicate that INap plays an important role in SNA at the onset of synaptogenesis (E12.5) by regulating neuron excitability in the mouse embryonic SC.
Discussion
How excitability of neurons evolves during SC development was extensively studied at developmental stages at which central pattern generators are already functional. At these developmental stages (P0–P5), INap is present in MNs and in SC INs (Tazerart et al., 2007; Zhong et al., 2007) and is required to generate fictive locomotion (Zhong et al., 2007). Here, we show that V1R already express INap at the onset of synaptogenesis in the embryonic (E12.5) SC (Scain et al., 2010). Our results reveal that INap already closely controls the V1R excitability pattern. In addition, we found that low concentrations of riluzole, an INap blocker, dramatically altered the SC SNA pattern, suggesting that INap is already expressed by neurons in the mammalian embryonic SC well before the control of muscle contraction by MNs (Sanes and Lichtman, 1999).
V1R display different excitability patterns at the onset of SNA
Repetitive firing of presynaptic neurons is required for long-lasting neurotransmitter-dependent episodes, as GDPs observed in MNs at E12.5. Previous studies have examined the development of passive and active membrane properties of MNs and INs in late embryonic and newborn rats or mice (Ziskind-Conhaim, 1988; Gao and Ziskind-Conhaim, 1998; Vinay et al., 2000; Theiss et al., 2007; Perry et al., 2015; Bikoff et al., 2016), but there is no information about the excitability of INs at early mammalian embryonic developmental stages when SNA first arises. We found that a majority of V1R (≈60%) can already generate repetitive AP firing or long-lasting sodium-dependent plateau potentials at the onset of SNA, indicating that these cells are already active at this early embryonic developmental stage. Our analysis revealed a strong heterogeneity in V1R excitability pattern, which can be separated into different independent classes, but such functional diversity is unlikely to persist in the adult. In the adult, V1R were found to generate two different excitability patterns only, a prominent low-threshold depolarization and burst firing followed by continuous firing dependent on Vh (Perry et al., 2015; Bikoff et al., 2016). It is therefore likely that the functional heterogeneity we observed at E12.5 reflects an immature form of V1R excitability (Perry et al., 2015; Bikoff et al., 2016). Sodium-dependent plateau potentials were also observed in ipsilateral caudal SC INs of zebrafish embryos at the appearance of functional neuromuscular junctions and then disappeared at more mature stages (Tong and McDearmid, 2012).
INap already regulate excitability patterns of V1R at early developmental stages
In this study, we reveal the presence of INap in neurons of mammalian embryos at the onset of SNA. Persistent inward currents (PICs) are present in many types of neurons. PICs can be calcium- and/or sodium-dependent depending on the neuron subtypes. We did not find any evidence for a significant calcium component in recorded PICs in V1R at E12.5. Indeed, the PIC was fully blocked by TTX in V1R, which contrasts with what is known about more mature spinal neurons like MNs (Hounsgaard and Kiehn, 1989; Bui et al., 2006; Carlin et al., 2009). Accordingly, we concluded that immature V1R at the onset of SNA generate a pure sodium-dependent PIC. INap recorded in V1R of the mouse embryo has an activation onset (≈−65 mV) similar to that of INap at postnatal developmental stages in the rat and mouse SC (Kuo et al., 2006; Tazerart et al., 2007; Theiss et al., 2007; Tazerart et al., 2008; Dai and Jordan, 2010). The Boltzmann constant of activation calculated for INap in V1R (≈7 mV) is in the range of the values known for INap in INs and MNs (≈6 mV) of neonatal rats (Bouhadfane et al., 2013) or in the calyx of Held (≈ 8 mV; Huang and Trussell, 2008). Small differences in the voltage dependency of INap activation may reflect differences in sodium channel α-subunit and/or β-subunit combination expression (Isom et al., 1994; Qu et al., 2001).
INap likely has diverse functions and plays an important function in locomotor pattern generation in neonatal rats (Tazerart, S. et al., 2007). In neonate SC, INap is present in both INs and MNs (Kuo et al., 2006; Tazerart et al., 2007) and is known to generate pacemaker activities in central pattern generator INs (Tazerart et al., 2008). Remarkably, INap also contributes to lumbar MN activity related to plateau potential in rat neonates (Bouhadfane et al., 2013). However, while in neonate MNs, INap is essential for self-sustained firing only during calcium-dependent plateau potential depolarization (Carlin et al., 2009; Bouhadfane et al., 2013), plateau potential activity in V1R at E12.5.is fully INap-dependent.
Does INap already participate in SC activity at the onset of neuronal network formation?
INap plays a crucial role in the regulation of locomotor pattern generation in rat neonates (Tazerart et al., 2007; Zhong et al., 2007; Bouhadfane et al., 2013) and in the SC of the zebrafish embryo during the cooling stage (Tong and McDearmid, 2012). Here, we demonstrated that a low concentration of riluzole dramatically reduced the amplitude and the duration of GICs recorded on MNs and strongly altered SC activity (extracellular recordings) at the onset of SNA.
In our experiments, we used a concentration of riluzole that has a minimal effect on the sodium AP waveform (Fig. 6). However, beside its effect on INap, riluzole is known to inhibit the release of glutamate (Chéramy et al., 1992), to inhibit AMPA receptor activation (Albo et al., 2004), GABAA and glycine receptor activation (Mohammadi et al., 2001) and to block several voltage-gated channels including calcium channels (Huang et al., 1997; Ahn et al., 2006). With the exception of AMPA receptor inhibition (Albo et al., 2004), these side effects of riluzole occur at concentrations ≥ 10 μm (Chéramy et al., 1992; Huang et al., 1997; Mohammadi et al., 2001; Ahn et al., 2006). Because the inhibition of glutamate neurotransmission did not alter SNA in the mouse embryonic SC at E12.5 (Czarnecki et al., 2014), it is unlikely that the alteration of SNA we observed in the presence of 5 μm riluzole is caused by the inhibition of glutamatergic synaptic activity (Albo et al., 2004). Accordingly, we propose that INap already regulates the SNA pattern at the onset of synaptogenesis in mammalian embryos before the formation of the locomotor SC network and of functional neuromuscular junctions. However, we cannot exclude that spinal neurons other than V1R exhibit INap-dependent sustain discharge at this developmental stage.
Because V1R already produce GABA at E12.5 and make synaptic-like contacts with MNs while MNs make synaptic-like contacts with V1R (see also (Alvarez et al., 2013), it is likely that V1R participate in early SC SNA. Although correlative, our data reinforce the hypothesis that a primitive V1R-MN recurrent-like circuit may exist at the onset of synaptogenesis in the mouse embryo. This primitive V1R-MN recurrent circuit differs in several ways from the adult RC-MN recurrent circuits. Evoked MN spiking did not trigger a recurrent synaptic response at E12.5 (Le Bras et al., 2014), which suggests that although V1R projects on MNs and vice versa, it is unlikely that V1R projecting on MNs receive inputs from these MNs and vice versa. It is also unlikely that MN release sites apposed on V1R are mixed glutamatergic and cholinergic inputs as observed at postnatal stages (Nishimaru et al., 2005; Lamotte d'Incamps et al., 2017). Glutamatergic vesicular transporters were not observed within MNs at E12.5 (Czarnecki et al., 2014). But we cannot completely exclude that some immature motor axon synapses on V1R already release aspartate, as suggested in the adult (Richards et al., 2014). However, contrary to what is observed for GABAergic and cholinergic networks, the spontaneous activation of glutamate receptors had a minor role only in the generation of SNA episodes at E12.5 (Czarnecki et al., 2014).
We clearly show that paracrine release of GABA can occur in the embryonic SC, which may explain the smooth shape of GABAergic-dependent GDPs observed in MNs during SNA (Czarnecki et al., 2014). Since V1R are GABAergic at this embryonic age and possess putative release sites, V1R may be one of the sources for the paracrine release of GABA we observed. Accordingly, we propose that V1R-MN interactions occur through synaptic and paracrine release and participate in the synchronization of neuronal assembly required for the generation of the propagating waves of activity characterizing SC SNA at E12.5 (Momose-Sato and Sato, 2013).
Conclusions
Together, our findings demonstrate that INap is already present in developing SC neurons at an early developmental stage and governs V1R excitability. INap plays an important role in the regulation of locomotor pattern generation at postnatal developmental stages in rodents (Tazerart et al., 2008). Because the application of a low concentration of riluzole altered SNA, we propose that INap contributes to the patterning of embryonic SC activity at the onset of synaptogenesis. Accordingly, the capacity of SC neurons to generate sustained firing must be crucial for correct embryonic SC patterned activity at the onset of synaptogenesis, which is required for the correct development of MN projections toward their peripheral targets (Hanson and Landmesser, 2006).
Footnotes
This work was supported by FRM Grant DEQ20160334891, AFM Grant 18564, the Institut National de la Santé et de la Recherche Médicale, and the Centre National de la Recherche Scientifique. We thank Elim Hong for helpful discussion. We thank Laure Lecoin and Julien Bouvier for assistance in MafB antibodies and Thomas Müller et Carmen Birchmeier for assistance in Foxd3 antibodies.
The authors declare no competing financial interests.
References
- Ahn HS, Kim SE, Jang HJ, Kim MJ, Rhie DJ, Yoon SH, Jo YH, Kim MS, Sung KW, Hahn SJ (2006) Interaction of riluzole with the closed inactivated state of Kv4.3 channels. J Pharmacol Exp Ther 319:323–331. 10.1124/jpet.106.106724 [DOI] [PubMed] [Google Scholar]
- Albo F, Pieri M, Zona C (2004) Modulation of AMPA receptors in spinal motor neurons by the neuroprotective agent riluzole. J Neurosci Res 78:200–207. 10.1002/jnr.20244 [DOI] [PubMed] [Google Scholar]
- Allain AE, Baïri A, Meyrand P, Branchereau P (2004) Ontogenic changes of the GABAergic system in the embryonic mouse spinal cord. Brain Res 1000:134–147. 10.1016/j.brainres.2003.11.071 [DOI] [PubMed] [Google Scholar]
- Allain AE, Baïri A, Meyrand P, Branchereau P (2006) Expression of the glycinergic system during the course of embryonic development in the mouse spinal cord and its co-localization with GABA immunoreactivity. J Comp Neurol 496:832–846. 10.1002/cne.20967 [DOI] [PubMed] [Google Scholar]
- Alvarez FJ, Benito-Gonzalez A, Siembab VC (2013) Principles of interneuron development learned from Renshaw cells and the motoneuron recurrent inhibitory circuit. Ann N Y Acad Sci 1279:22–31. 10.1111/nyas.12084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberis A, Mozrzymas JW, Ortinski PI, Vicini S (2007) Desensitization and binding properties determine distinct α1β2γ2 and α3β2γ2 GABA(A) receptor-channel kinetic behavior. Eur J Neurosci 25:2726–2740. 10.1111/j.1460-9568.2007.05530.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Cherubini E, Corradetti R, Gaiarsa JL (1989) Giant synaptic potentials in immature rat CA3 hippocampal neurones. J Physiol 416:303–325. 10.1113/jphysiol.1989.sp017762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito-Gonzalez A, Alvarez FJ (2012) Renshaw cells and ia inhibitory interneurons are generated at different times from p1 progenitors and differentiate shortly after exiting the cell cycle. J Neurosci 32:1156–1170. 10.1523/JNEUROSCI.3630-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikoff JB, Gabitto MI, Rivard AF, Drobac E, Machado TA, Miri A, Brenner-Morton S, Famojure E, Diaz C, Alvarez FJ, Mentis GZ, Jessell TM (2016) Spinal inhibitory interneuron diversity delineates variant motor microcircuits. Cell 165:207–219. 10.1016/j.cell.2016.01.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhadfane M, Tazerart S, Moqrich A, Vinay L, Brocard F (2013) Sodium-mediated plateau potentials in lumbar motoneurons of neonatal rats. J Neurosci 33:15626–15641. 10.1523/JNEUROSCI.1483-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui TV, Ter-Mikaelian M, Bedrossian D, Rose PK (2006) Computational estimation of the distribution of L-type Ca(2+) channels in motoneurons based on variable threshold of activation of persistent inward currents. J Neurophysiol 95:225–241. 10.1152/jn.00646.2005 [DOI] [PubMed] [Google Scholar]
- Carlin KP, Bui TV, Dai Y, Brownstone RM (2009) Staircase currents in motoneurons: insight into the spatial arrangement of calcium channels in the dendritic tree. J Neurosci 29:5343–5353. 10.1523/JNEUROSCI.5458-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr PA, Alvarez FJ, Leman EA, Fyffe RE (1998) Calbindin D28k expression in immunohistochemically identified Renshaw cells. Neuroreport 9:2657–2661. 10.1097/00001756-199808030-00043 [DOI] [PubMed] [Google Scholar]
- Chéramy A, Barbeito L, Godeheu G, Glowinski J (1992) Riluzole inhibits the release of glutamate in the caudate nucleus of the cat in vivo. Neurosci Lett 147:209–212. 10.1016/0304-3940(92)90597-Z [DOI] [PubMed] [Google Scholar]
- Corlew R, Bosma MM, Moody WJ (2004) Spontaneous, synchronous electrical activity in neonatal mouse cortical neurones. J Physiol 560:377–390. 10.1113/jphysiol.2004.071621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czarnecki A, Le Corronc H, Rigato C, Le Bras B, Couraud F, Scain AL, Allain AE, Mouffle C, Bullier E, Mangin JM, Branchereau P, Legendre P (2014) Acetylcholine controls GABA-, glutamate-, and glycine-dependent giant depolarizing potentials that govern spontaneous motoneuron activity at the onset of synaptogenesis in the mouse embryonic spinal cord. J Neurosci 34:6389–6404. 10.1523/JNEUROSCI.2664-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Jordan LM (2010) Multiple patterns and components of persistent inward current with serotonergic modulation in locomotor activity-related neurons in cfos-EGFP mice. J Neurophysiol 103:1712–1727. 10.1152/jn.01111.2009 [DOI] [PubMed] [Google Scholar]
- Delpy A, Allain AE, Meyrand P, Branchereau P (2008) NKCC1 cotransporter inactivation underlies embryonic development of chloride-mediated inhibition in mouse spinal motoneuron. J Physiol 586:1059–1075. 10.1113/jphysiol.2007.146993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dottori M, Gross MK, Labosky P, Goulding M (2001) The winged-helix transcription factor Foxd3 suppresses interneuron differentiation and promotes neural crest cell fate. Development 128:4127–4138. [DOI] [PubMed] [Google Scholar]
- Eccles JC, Fatt P, Landgren S (1956) The inhibitory pathway to motoneurones. Progr Neurobiol 2:72–82. [PubMed] [Google Scholar]
- Feller MB. (1999) Spontaneous correlated activity in developing neural circuits. Neuron 22:653–656. 10.1016/S0896-6273(00)80724-2 [DOI] [PubMed] [Google Scholar]
- Gao BX, Ziskind-Conhaim L (1998) Development of ionic currents underlying changes in action potential waveforms in rat spinal motoneurons. J Neurophysiol 80:3047–3061. 10.1152/jn.1998.80.6.3047 [DOI] [PubMed] [Google Scholar]
- Garaschuk O, Hanse E, Konnerth A (1998) Developmental profile and synaptic origin of early network oscillations in the CA1 region of rat neonatal hippocampus. J Physiol 507:219–236. 10.1111/j.1469-7793.1998.219bu.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaschuk O, Linn J, Eilers J, Konnerth A (2000) Large-scale oscillatory calcium waves in the immature cortex. Nat Neurosci 3:452–459. 10.1038/74823 [DOI] [PubMed] [Google Scholar]
- Geiman EJ, Knox MC, Alvarez FJ (2000) Postnatal maturation of gephyrin/glycine receptor clusters on developing Renshaw cells. J Comp Neurol 426:130–142. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Islas C, Wenner P (2006) Spontaneous network activity in the embryonic spinal cord regulates AMPAergic and GABAergic synaptic strength. Neuron 49:563–575. 10.1016/j.neuron.2006.01.017 [DOI] [PubMed] [Google Scholar]
- Gust J, Wright JJ, Pratt EB, Bosma MM (2003) Development of synchronized activity of cranial motor neurons in the segmented embryonic mouse hindbrain. J Physiol 550:123–133. 10.1113/jphysiol.2002.038737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MG, Landmesser LT (2003) Characterization of the circuits that generate spontaneous episodes of activity in the early embryonic mouse spinal cord. J Neurosci 23:587–600. 10.1523/JNEUROSCI.23-02-00587.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MG, Landmesser LT (2006) Increasing the frequency of spontaneous rhythmic activity disrupts pool-specific axon fasciculation and pathfinding of embryonic spinal motoneurons. J Neurosci 26:12769–12780. 10.1523/JNEUROSCI.4170-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MG, Milner LD, Landmesser LT (2008) Spontaneous rhythmic activity in early chick spinal cord influences distinct motor axon pathfinding decisions. Brain Res Rev 57:77–85. 10.1016/j.brainresrev.2007.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hounsgaard J, Kiehn O (1989) Serotonin-induced bistability of turtle motoneurones caused by a nifedipine-sensitive calcium plateau potential. J Physiol 414:265–282. 10.1113/jphysiol.1989.sp017687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CS, Song JH, Nagata K, Yeh JZ, Narahashi T (1997) Effects of the neuroprotective agent riluzole on the high voltage-activated calcium channels of rat dorsal root ganglion neurons. J Pharmacol Exp Ther 282:1280–1290. [PubMed] [Google Scholar]
- Huang H, Trussell LO (2008) Control of presynaptic function by a persistent Na(+) current. Neuron 60:975–979. 10.1016/j.neuron.2008.10.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom LL, De Jongh KS, Catterall WA (1994) Auxiliary subunits of voltage-gated ion channels. Neuron 12:1183–1194. 10.1016/0896-6273(94)90436-7 [DOI] [PubMed] [Google Scholar]
- Kirkby LA, Sack GS, Firl A, Feller MB (2013) A role for correlated spontaneous activity in the assembly of neural circuits. Neuron 80:1129–1144. 10.1016/j.neuron.2013.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo JJ, Lee RH, Zhang L, Heckman CJ (2006) Essential role of the persistent sodium current in spike initiation during slowly rising inputs in mouse spinal neurones. J Physiol 574:819–834. 10.1113/jphysiol.2006.107094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamotte d'Incamps B, Bhumbra GS, Foster JD, Beato M, Ascher P (2017) Segregation of glutamatergic and cholinergic transmission at the mixed motoneuron Renshaw cell synapse. Scientific reports 7:4037. 10.1038/s41598-017-04266-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landmesser LT, O'Donovan MJ (1984) Activation patterns of embryonic chick hind limb muscles recorded in ovo and in an isolated spinal cord preparation. J Physiol 347:189–204. 10.1113/jphysiol.1984.sp015061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bras B, Fréal A, Czarnecki A, Legendre P, Bullier E, Komada M, Brophy PJ, Davenne M, Couraud F (2014) In vivo assembly of the axon initial segment in motor neurons. Brain Struct FuncT 219:1433–1450. 10.1007/s00429-013-0578-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Corronc H, Rigo JM, Branchereau P, Legendre P (2011) GABA(A) receptor and glycine receptor activation by paracrine/autocrine release of endogenous agonists: more than a simple communication pathway. Mol Neurobiol 44:28–52. 10.1007/s12035-011-8185-1 [DOI] [PubMed] [Google Scholar]
- Lee RH, Heckman CJ (1998) Bistability in spinal motoneurons in vivo: systematic variations in persistent inward currents. J Neurophysiol 80:583–593. 10.1152/jn.1998.80.2.583 [DOI] [PubMed] [Google Scholar]
- Marder E, Rehm KJ (2005) Development of central pattern generating circuits. Curr Opin Neurobiol 15:86–93. 10.1016/j.conb.2005.01.011 [DOI] [PubMed] [Google Scholar]
- Mohammadi B, Krampfl K, Moschref H, Dengler R, Bufler J (2001) Interaction of the neuroprotective drug riluzole with GABA(A) and glycine receptor channels. Eur J Pharmacol 415:135–140. 10.1016/S0014-2999(01)00847-0 [DOI] [PubMed] [Google Scholar]
- Momose-Sato Y, Sato K (2013) Optical imaging of the spontaneous depolarization wave in the mouse embryo: origins and pharmacological nature. Ann N Y Acad Sci 1279:60–70. 10.1111/j.1749-6632.2012.06806.x [DOI] [PubMed] [Google Scholar]
- Moody WJ. (1998) Control of spontaneous activity during development. J Neurobiol 37:97–109. [DOI] [PubMed] [Google Scholar]
- Müller A, Kukley M, Stausberg P, Beck H, Müller W, Dietrich D (2005) Endogenous Ca2+ buffer concentration and Ca2+ microdomains in hippocampal neurons. J Neurosci 25:558–565. 10.1523/JNEUROSCI.3799-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CP, Lewcock JW, Hanson MG, Gosgnach S, Aimone JB, Gage FH, Lee KF, Landmesser LT, Pfaff SL (2005) Cholinergic input is required during embryonic development to mediate proper assembly of spinal locomotor circuits. Neuron 46:37–49. 10.1016/j.neuron.2005.02.022 [DOI] [PubMed] [Google Scholar]
- Nishimaru H, Restrepo CE, Ryge J, Yanagawa Y, Kiehn O (2005) Mammalian motor neurons corelease glutamate and acetylcholine at central synapses. Proc Natl Acad Sci U S A 102:5245–5249. 10.1073/pnas.0501331102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry S, Gezelius H, Larhammar M, Hilscher MM, Lamotte d'Incamps B, Leao KE, Kullander K (2015) Firing properties of Renshaw cells defined by Chrna2 are modulated by hyperpolarizing and small conductance ion currents ih and ISK. Eur J Neurosci 41:889–900. 10.1111/ejn.12852 [DOI] [PubMed] [Google Scholar]
- Prasad T, Wang X, Gray PA, Weiner JA (2008) A differential developmental pattern of spinal interneuron apoptosis during synaptogenesis: insights from genetic analyses of the protocadherin-gamma gene cluster. Development 135:4153–4164. 10.1242/dev.026807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Curtis R, Lawson D, Gilbride K, Ge P, DiStefano PS, Silos-Santiago I, Catterall WA, Scheuer T (2001) Differential modulation of sodium channel gating and persistent sodium currents by the β1, β2, and β3 subunits. Mol Cell Neurosci 18:570–580. 10.1006/mcne.2001.1039 [DOI] [PubMed] [Google Scholar]
- Richards DS, Griffith RW, Romer SH, Alvarez FJ (2014) Motor axon synapses on Renshaw cells contain higher levels of aspartate than glutamate. PloS One 9:e97240. 10.1371/journal.pone.0097240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockhill W, Kirkman JL, Bosma MM (2009) Spontaneous activity in the developing mouse midbrain driven by an external pacemaker. Dev Neurobiol 69:689–704. 10.1002/dneu.20725 [DOI] [PubMed] [Google Scholar]
- Safiulina VF, Cherubini E (2009) At immature mossy fibers-CA3 connections, activation of presynaptic GABA(B) receptors by endogenously released GABA contributes to synapses silencing. Front Cell Neurosci 3:1. 10.3389/neuro.03.001.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW (1999) Development of the vertebrate neuromuscular junction. Annu Rev Neurosci 22:389–442. 10.1146/annurev.neuro.22.1.389 [DOI] [PubMed] [Google Scholar]
- Sapir T, Geiman EJ, Wang Z, Velasquez T, Mitsui S, Yoshihara Y, Frank E, Alvarez FJ, Goulding M (2004) Pax6 and engrailed 1 regulate two distinct aspects of Renshaw cell development. J Neurosci 24:1255–1264. 10.1523/JNEUROSCI.3187-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scain AL, Le Corronc H, Allain AE, Muller E, Rigo JM, Meyrand P, Branchereau P, Legendre P (2010) Glycine release from radial cells modulates the spontaneous activity and its propagation during early spinal cord development. J Neurosci 30:390–403. 10.1523/JNEUROSCI.2115-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stam FJ, Hendricks TJ, Zhang J, Geiman EJ, Francius C, Labosky PA, Clotman F, Goulding M (2012) Renshaw cell interneuron specialization is controlled by a temporally restricted transcription factor program. Development 139:179–190. 10.1242/dev.071134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm R, Cholewa-Waclaw J, Reuter K, Bröhl D, Sieber M, Treier M, Müller T, Birchmeier C (2009) The bHLH transcription factor Olig3 marks the dorsal neuroepithelium of the hindbrain and is essential for the development of brainstem nuclei. Development 136:295–305. 10.1242/dev.027193 [DOI] [PubMed] [Google Scholar]
- Tamamaki N, Yanagawa Y, Tomioka R, Miyazaki J, Obata K, Kaneko T (2003) Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J Comp Neurol 467:60–79. 10.1002/cne.10905 [DOI] [PubMed] [Google Scholar]
- Tazerart S, Viemari JC, Darbon P, Vinay L, Brocard F (2007) Contribution of persistent sodium current to locomotor pattern generation in neonatal rats. J Neurophysiol 98:613–628. 10.1152/jn.00316.2007 [DOI] [PubMed] [Google Scholar]
- Tazerart S, Vinay L, Brocard F (2008) The persistent sodium current generates pacemaker activities in the central pattern generator for locomotion and regulates the locomotor rhythm. J Neurosci 28:8577–8589. 10.1523/JNEUROSCI.1437-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theiss RD, Kuo JJ, Heckman CJ (2007) Persistent inward currents in rat ventral horn neurones. J Physiol 580:507–522. 10.1113/jphysiol.2006.124123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong H, McDearmid JR (2012) Pacemaker and plateau potentials shape output of a developing locomotor network. Curr Biol 22:2285–2293. 10.1016/j.cub.2012.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbani A, Belluzzi O (2000) Riluzole inhibits the persistent sodium current in mammalian CNS neurons. Eur J Neurosci 12:3567–3574. 10.1046/j.1460-9568.2000.00242.x [DOI] [PubMed] [Google Scholar]
- Vinay L, Brocard F, Pflieger JF, Simeoni-Alias J, Clarac F (2000) Perinatal development of lumbar motoneurons and their inputs in the rat. Brain Res Bull 53:635–647. 10.1016/S0361-9230(00)00397-X [DOI] [PubMed] [Google Scholar]
- Watt AJ, Cuntz H, Mori M, Nusser Z, Sjöström PJ, Häusser M (2009) Traveling waves in developing cerebellar cortex mediated by asymmetrical Purkinje cell connectivity. Nat Neurosci 12:463–473. 10.1038/nn.2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenner P, O'Donovan MJ (2001) Mechanisms that initiate spontaneous network activity in the developing chick spinal cord. J Neurophysiol 86:1481–1498. 10.1152/jn.2001.86.3.1481 [DOI] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, Jessell TM (2002) Directed differentiation of embryonic stem cells into motor neurons. Cell 110:385–397. 10.1016/S0092-8674(02)00835-8 [DOI] [PubMed] [Google Scholar]
- Young SH, Poo MM (1983) Spontaneous release of transmitter from growth cones of embryonic neurones. Nature 305:634–637. 10.1038/305634a0 [DOI] [PubMed] [Google Scholar]
- Yvert B, Branchereau P, Meyrand P (2004) Multiple spontaneous rhythmic activity patterns generated by the embryonic mouse spinal cord occur within a specific developmental time window. J Neurophysiol 91:2101–2109. 10.1152/jn.01095.2003 [DOI] [PubMed] [Google Scholar]
- Zhang LI, Poo MM (2001) Electrical activity and development of neural circuits. Nat Neurosci 4:1207–1214. 10.1038/nn753 [DOI] [PubMed] [Google Scholar]
- Zhong G, Masino MA, Harris-Warrick RM (2007) Persistent sodium currents participate in fictive locomotion generation in neonatal mouse spinal cord. J Neurosci 27:4507–4518. 10.1523/JNEUROSCI.0124-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziskind-Conhaim L. (1988) Electrical properties of motoneurons in the spinal cord of rat embryos. Dev Biol 128:21–29. 10.1016/0012-1606(88)90262-X [DOI] [PubMed] [Google Scholar]
- Ziskind-Conhaim L, Wu L, Wiesner EP (2008) Persistent sodium current contributes to induced voltage oscillations in locomotor-related Hb9 interneurons in the mouse spinal cord. J Neurophysiol 100:2254–2264. 10.1152/jn.90437.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]