

Abstract
Ongoing tissue repair and formation and deposition of collagen-rich extracellular matrix in tissues and organs finally lead to fibrotic lesions and destruction of normal tissue/organ architecture and function. In the lung, scarring is observed in asthma, chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis to various degrees. At the cellular level immune cells, fibroblasts and epithelial cells are all involved in fibrotic processes. Mechanistically, fibroblast to myofibroblast transformation and epithelial to mesenchymal transition are major drivers of fibrosis. Amongst others, both processes are controlled by transforming growth factor beta-1 (TGFβ-1), a growth factor upregulated in idiopathic pulmonary fibrosis lungs. Phenotypic assays with primary human cells and complex disease-relevant readouts become increasingly important in modern drug discovery processes. We describe high-content screening based phenotypic assays with primary normal human lung fibroblasts and primary human airway epithelial cells. For both cell types, TGFβ-1 stimulation is used to induce fibrotic phenotypes in vitro, with alpha smooth muscle actin and collagen-I as readouts for FMT and E-cadherin as a readout for EMT. For each assay, a detailed image analysis protocols is described. Treatment of both cell types with TGFβ-1 and a transforming growth factor beta receptor inhibitor verifies the suitability of the assays for pharmacological interventions. In addition, the assays are compatible for siRNA and Cas9-ribonucleoprotein transfections, and thus are useful for genetic target identification/validation by modulating gene expression.
Keywords: FMT, EMT, Crispr-Cas9, siRNA, RNAi, Cas9-RNP, TGFβ-1
INTRODUCTION
Fibrosis is a process of continuous tissue repair characterized by the formation and deposition of fibrillary, collagen-rich extracellular matrix (ECM) leading to a progressive remodeling in most tissues and organs. Formation of fibrotic scar tissue is essential to restore and maintain tissue and organ integrity during wound healing after injury. However, when the scarring mechanisms exacerbate after repetitive injury, chronic fibrogenesis results in a shift from supportive fibrotic tissue to scar tissue. This coincides with an increased number and activity of extracellular matrix producing cells, which finally leads to destruction of normal tissue/organ architecture and function [1,2].
In the lung, tissue scarring/fibrosis occurs to a different extend in diseases like asthma, chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary disease (IPF). After tissue damage, activation of the immune system induces release of several cytokines and growth factors including transforming growth factor beta-1 (TGFβ-1) that signals the tissue to repair [3,4]. Mechanistically, epithelial to mesenchymal transition (EMT) is one of the major drivers of fibrosis, and approximately 30% of ECM-producing (myo-)fibroblasts derive from epithelial cells through EMT [5]. During EMT, tightly organized epithelial cells lose expression of the tight junction marker E-cadherin [6,7] and transform to mesenchymal cells with expression of the mesenchymal cell markers N-cadherin, vimentin and fibronectin. Furthermore, the cells gain migratory potential, enabling to migrate throughout the tissue [7-9]. Another important mechanism during fibrosis is fibroblast to myofibroblast transformation (FMT), when EMT-derived, resident and invading fibroblastoid cells are activated (e.g., by TGFβ-1) and start to differentiate towards myofibroblasts. Myofibroblasts are a heterogeneous population derived from different progenitors, characterized by formation of alpha smooth muscle actin (aSMA) containing stress fibers and the expression and secretion of fibrillar collagen, fibronectin and additional ECM components. Increased production and accumulation of ECM result in a permanently damaged fibrotic tissue with an aberrant architecture unable to function properly [1,7].
Phenotypic disease-relevant cellular assays are becoming more and more relevant in modern drug discovery. Phenotypic assays try to modulate disease-relevant phenotypes without an assumption about the specific target or mechanism of action [10]. For a successful phenotypic screen Vincent et al. [11] recently published the “rule of 3”: (1) The assay system needs to have a clear link to the disease either by the use of human primary or iPSC-derived cells; (2) The assay readout should be as close as possible to the disease pathobiology; and (3) as the use of an exogenous stimulus to induce a disease phenotype often results in identification of stimulus- instead of disease-modifying mechanisms, the authors recommend to use assays which are independent on exogenous stimuli [11]. For the phenotypic assays using either primary normal human lung fibroblasts (NHLF) to monitor FMT (collagen-I and aSMA readout) or primary human small airway epithelial cells (hSAEC) to model EMT (E-cadherin readout), the rules (1) and (2) are accomplished. The FMT and EMT assays described in the literature [12-17] rely on TGFβ-1 as a stimulus to induce FMT or EMT. However, as TGFβ-1 levels are higher in IPF lungs compared to lungs of healthy controls [18,19], and as TGFβ-1 is at least partially responsible for the exaggerated matrix deposition within the fibrotic lung [20-22], induction of both FMT and EMT with TGFβ-1 does not violate the third rule.
Phenotypic screening assays with primary human cells can be used for both pharmacological and genetic drug target identification and validation. Modulating gene expression in cells has been primarily achieved using RNAi technologies. RNAi mediated gene repression relies on either transfection of synthetic siRNAs, plasmid-encoded shRNAs or transduction of shRNA encoding viral particles. However, RNAi reagents are frequently prone to off-target effects. Therefore, hits from RNAi screens should be validated intensively, also by the use of orthogonal technologies, e.g., Crispr-Cas9 system in order to exclude potential off-target activities responsible for the observed phenotype [23]. So far, only a small number of studies successfully applied the Crispr-Cas9 technology to primary cells. Frequently, Cas9 and the sgRNA are virally delivered to the cells [13,24,25]. However, virally mediated Cas9-sgRNA delivery is time consuming with respect to virus generation and restricts its application to a lower throughput, unless a screen can be performed in a pooled format [26]. Recently, it has been shown that Cas9 and sgRNAs can be transfected in cells as Cas9-RNPs (ribonucleoproteins) [27,28]. Recombinant Cas9 and synthetic sgRNAs or cr:trRNAs are commercially available and thus enable the time effective screening of sg/cr:trRNA libraries by Cas9-RNP transfection.
We recently described a primary human lung fibroblast assay to study effects of lysyl oxidase (like) family member knock downs on TGFβ-1-dependent FMT with aSMA as a readout [29]. In the present study, we described a further optimized version of this assay, not only measuring aSMA as marker of myofibroblast formation, but also including ECM components, collagen-I and fibronectin as readouts. In addition, we delineated an EMT assay with E-cadherin as a readout. In combination with pharmacological and genetic methods, these assays are useful tools for compound characterization, target identification and validation in a higher throughput format. We showed that both assays exhibit a dose dependent responsiveness to TGFβ-1 and are pharmacologically modulated by TGFβ-1 receptor inhibitors (Alk5 inhibitor, Alk5i). In addition, they are compatible with siRNA and Cas9-RNP transfections to study gene function.
MATERIALS AND METHODS
Materials
Normal human lung fibroblasts (NHLF), human small airway epithelial cells (hSAEC) and cell culture media (FBM and SABM media plus corresponding SingleQuots) were from Lonza. The supplier guarantees informed consent from the donors to use the cells for research purposes. Phosphate buffered saline (PBS), Triton-X-100, formaldehyde, carbonate solution, vitamin C, bovine serum albumin (BSA), methanol, mouse monoclonal anti-collagen-I and anti-alpha smooth muscle actin antibodies and tumor growth factor beta-1 were from Sigma. Sigma also provided the Alk5i SB-525334. HEPES, HBSS, OptiMEM, mouse anti-E-cadherin antibody, secondary goat anti-mouse-IgG1-AF568 and anti-mouse-IgG2a-AF647 antibodies, RNAiMAX siRNA transfection reagent, TaqMan gene expression kits, High-Capacity cDNA Reverse Transcription Kit, AmpliTaq Gold 360 Master Mix, HCS Cell Mask Green and Hoechst 33342 nuclear dye were purchased from ThermoFisher. The anti-fibronectin antibody ab6328 was delivered by Abcam. Integrated DNA Technologies (IDT) provided recombinant Cas9 protein and the Cas9 electroporation enhancer, GE Healthcare Ficoll 70 and 100. From Dharmacon, synthetic Edit-R-crispr-, -tracrRNAs (Edit-R Human TGFBR1 crRNA: CM-003929-03-0002.; Edit-R Human ACTA2 crRNA: CR-003450-02; Edit-R DNMT3B Control Kit: U-007010-05; Edit-R CRISPR-Cas9 Synthetic tracrRNA: U-002005-05) and synthetic ON-Traget Plus SMART Pool siRNAs (ON-TARGETplus Non-targeting Pool: D-001810-10-05; ON-TARGETplus TGFBR1: L-003929-00-0005; ON-TARGETplus ACTA2: L-003450-00-0005) were purchased. Edit-R lentiviral Basticidin-Cas9-Nuclease expressing particles were obtained from Dharmacon. Poly-D-lysine coated 384 well microtiter plates suitable for image acquisition and analysis were delivered by PerkinElmer. For Cas9-RNP electroporation, the Nucleofector System and 96-well shuttle device together with the Nucleofector Primary Solution P3 from Lonza was used. RNeasy Plus 96 Kit was from Qiagen. Direct PCR Lysis Reagent was from Peqlab, proteinase K from Macherey Nagel. Poly-D-lysine coated 384 well CellCarrier microtiter plates were purchased from Perkin Elmer.
Fibroblast to myofibroblast transformation assay
At day 0, for compound testing (e.g., Alk5i) 1000 NHLF cells were plated in full medium (FMB plus Single Quots) in poly-D-lysine coated 384 well CellCarrier microtiter plates. 24 h later (day 1), cells were washed 3 times with HBSS/10 mM Hepes/0.0375% carbonate buffer and starved in medium without SingleQuots for 24 h. At day 2 post seeding, medium was replaced with FBM medium containing TGFβ-1, 200 μM vitamin C, 37.5 mg/ml Ficoll 70 and 25 mg/ml Ficoll 400. Where indicated the Alk5i diluted in medium with 0.1% DMSO was included. In siRNA and Cas9-RNP transfection experiments, 0.1% FCS was included in the starvation buffer throughout the assay. At the end of the assay (day 5), cells were fixed for 30 min on ice with methanol, 3 times washed with PBS and permeabilized with PBS/1% Triton-X-100 for 20 min at ambient temperature. Followed by 3 wash steps with PBS, non-specific antibody binding was reduced by a blocking step with 3%BSA in PBS for 30 min at ambient temperature. Antibodies targeting collagen-I and aSMA, each diluted 1:1000 were added to the cells for 1 h at 37°C after an additional wash step with PBS. The anti-fibronectin antibody was used in a final concentration of 0.05 μg/ml. Primary antibodies were removed by 3 wash steps with PBS and the secondary antibodies labeled with AF568 for the anti-collagen-I- and anti-fibronectin-IgG1 and AF647 for the anti-aSMA-IgG2a diluted 1:1000 were incubated for 30 min at 37°C together with the nuclear dye Hoechst. Antibodies were removed by 3 washes with PBS and cell cytoplasm was stained by addition of 1:50000 diluted HCS CellMaskGreen dye for 30 min at ambient temperature. After an additional PBS washing step, plates were sealed and images were acquired with an IN Cell Analyzer 2200. Nuclear, cytoplasm, aSMA and collagen-I or aSMA and fibronectin images were acquired in the DAPI, FITC, Cy7 and Cy5 channels, respectively.
Images were transferred to Perkin Elmer’s Columbus Image Storage and Analysis system. Image analysis was performed using a custom made image analysis protocol (Table S1), with total number of cells, number of fiber-like αSMA structures/cell and total collagen/fibronectin area/total number of cells as parameters used to quantify effects of compounds (e.g., Alk5i), siRNAs and Cas9-RNPs.
Epithelial to mesenchymal transition assay
For the EMT assay, 4000 hSAECs/well were seeded in a poly-D-lysine coated 384 CellCarrier microtiter plate at day 0. Day 1 post seeding medium was exchanged after 3 washes with HBSS/10 mM Hepes/0.0375% carbonate buffer. 24 h later (day 2), medium was replaced with medium containing TGFβ-1. Where the Alk5i was indicated was included in medium with 0.1% DMSO. After 72 h, cells were fixed with 2% formaldehyde containing 1 μM Hoechst 33342 to stain nuclei for 30 min at ambient temperature. Following 3 washes with PBS, non-specific antibody binding was blocked by incubation with 2%BSA in PBS for 30 min at ambient temperature. The blocking solution was removed by 3 wash steps with PBS, and the cells were incubated with the primary mouse-anti-E-cadherin antibody (final concentration 0.4 μg/ml) for 1 h at 37°C. Three washes with PBS were followed by an incubation with the secondary AF568-labed goat anti-mouse-IgG1 (1:1000 diluted) for 1 h at 37°C. Following 3 PBS wash steps, cells were permeabilized with 0.1% Triton-X-100 for 20 min at ambient temperature, washed again and incubated for 30 min at ambient temperature with HCS CellMask Green (1:50000 diluted). Images were acquired with a PerkinElmer Opera Phenix High Content Screening System, after a final wash step and plate sealing. Nuclear, cytoplasm and Ecadherin images were acquired in the Hoechst, AF488 and AF568 channels, respectively.
Images were transferred to Perkin Elmer’s Columbus Image Storage and Analysis system. Image analysis was performed using a custom made image analysis protocol (Table S2), with total number of cells, (total E-cadherin intensity × total E-cadherin area) / (total cell number) as parameters used to quantify effects of compounds (e.g., Alk5i), siRNAs and Cas9-RNPs.
siRNA transfections
Transfection of siRNAs in NHLF cells for the FMT assay was performed 6 h after cell seeding. A transfection mix for one well in a 384 well mictrotiter plate with 2000 cells in 70 μl FBM medium with SingleQuots consisted of 0.2 μl RNAiMAX, 56 nM siRNA in 20 μl FBM medium, resulting in a final siRNA concentration of 16 nM/well. The 4000 hSAECs per well for the EMT assay were transfected with a similar transfection cocktail, in which FBM medium was replaced by OptiMEM. All transfection mixtures were incubated for 20 min at ambient temperature to form transfection complexes before addition to the cells. 24 h after transfection, the transfection complexes were removed and the assays were continued as described above.
Gene expression analysis
For determination of siRNA mediated gene knock down, cells were lysed and RNA was prepared using the RNeasy Plus 96 Kit according to the manufacture’s protocol. 2 μg of total RNA was reverse transcribed with the High-Capacity cDNA Reverse Transcription Kit as described in the supplier’s protocol. qPCR with 2μl of cDNA and gene-specific TaqMan Assays (TGFBR1: Hs00610320_m1; ACTA2: Hs00909449_m1, RNA-polymerase II amplification primers: GCAAGCGGATTCCATTTGG and TCTCAGGCCCGTAGTCATCCT, probe: AAGCACCGGACTCTTGCCTCACTTCATC) was performed as suggested in the manual. The relative gene-specific knock down was calculated as percentage knock down compared to control siRNA treated cells after normalization to RNA polymerase II expression.
Cas9-RNP transfections
For Cas9-RNP transfections, each 200 μM Edit-R-crRNA and Edit-R-trRNA were hybridized by incubation for 5 min at 95°C and cooling to ambient temperature. The RNPs were formed by incubating 20.74 μM recombinant Cas9 protein, with 24 μM cr:trRNA duplexes in a total volume of 5 μl. Complex formation was achieved by incubation for 20 min at ambient temperature. For controls without Cas9 protein, cr:trRNA duplexes were incubated with 20.74 μM BSA instead of Cas9. Next, the RNP complexes were mixed with either 100000 NHLF cells or hSAECs in 20 μl Nucleofector Solution P3 plus 1 μl electroporation enhancer, and electroporated with Nucleofector program 96-EH-100 or 96-DC-100 for NHLF and hSAEC cells, respectively. Electroporated NHLF cells and hSAECs were resuspended in 375 μl of their respective medium with SingleQuots and 5000 (NHLF) or 10000 (hSAEC) cells were seeded in one well of a 384 microtiter plate. Again, 24 h after transfection the transfection complexes were removed and the assays were continued as described above.
Generation and transfection of Cas9-expressing NHLF cells
NHLF cells constitutively expressing Cas9 were generated by transduction of 100000 NHLF cells with Edit-R lentiviral EF1a-Basticidin-Cas9-Nuclease expressing particles at a MOI of 1. 24 h after transduction, medium was exchanged and selection for genomic integration of the viral genome was started by addition of 8 μg/ml Blasticidin S. After 4 passages, cell stocks were prepared and used for FMT assays essentially as described above, with the duplexed cr:trRNAs directly transfected without prior RNP formation.
Gene editing analysis
Gene editing efficiency was determined by PCR amplification of the edited genomic loci, followed by Sanger sequencing and the use of the Inference of CRISPR editing (ICE) tool [30]. After removal of cell culture medium, 15 μl of PCR lysis reagent containing 0.2 mg/ml Proteinase K was added to each well of a 384 microtiter plate and incubated for 2 h at 55°C under constant agitation. The lysis reaction was stopped by heating the reaction to 85°C for 45 min. 5 μl crude cell lysate was amplified with 10 μM of each forward and reverse PCR primers (TGFBR1_F: AAATTCAGTGACTTTTGGTG, TGFBR1_R: GAGTTTCAGCAATGATATGT; ACTA2_F: TTATTTGGAGGCAATTCAGG, ACTA2_R: TGTGGTGTTCTGTATCAGAGA; DMNT3B_F: TGAGAAGGAGCCACTTGCTT, DMNT3B_R: GACCAAGAACGGGAAAGTCA) using AmpliTaq Gold 360 MasterMix for 35 cycles with an annealing temperature of 50°C. The amplicons were 425, 406 and 544 bp in length. Purified PCR products were Sanger sequenced using the PCR forward primers.
Data analysis
All data obtained were further processed and analyzed using GraphPad Prism7®, version 7.03. Statistical significance was calculated using a Student’s t-test.
RESULTS
TGFβ-1-mediated fibroblast-to-myofibroblast transition assay
We used normal primary human lung fibroblasts to establish a TGFβ-1-dependent FMT assay with aSMA, collagen-I and fibronectin as high content screening based readouts (Fig. 1A).
Figure 1.
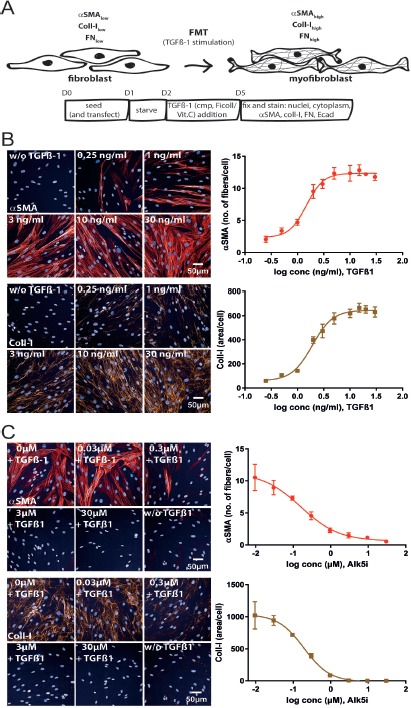
Phenotypic FMT assay. A. Schematic representation of the FMT assay with time points of treatment and readouts. At the end of the assay cells were prepared for high content imaging for alpha smooth muscle actin (αSMA) and collagen I (Coll-I) expression. B. Images of NHLF cells treated with increasing doses of TGFβ-1, stained for nuclei, αSMA and Coll-I. Images were acquired with an IN Cell 2200 high content imager (left). Number of αSMA expressing fibers/cell and Coll-I area/cell was quantified with the described custom protocols using the Columbus software and plotted against cumulating doses of TGFβ-1 (right). The TGFβ-1 EC50 was 1.5 and 2 ng/ml for αSMA and Coll-I, respectively. C. Immunofluorescence images of NHLF cells stimulated with 5 ng/ml TGFβ-1 and increasing concentrations of the Alk5i SB-525334 (left). The Alk5i dose-dependently inhibited αSMA and Coll-I expression (right) with an IC50 of 0.16 μM and 0.19 μM for αSMA and Coll-I, respectively.
A specific image analysis script was developed as described in the Materials and Methods section (see also Table S1). The aSMA signal was not evenly dispersed throughout the whole cytoplasm, but seen in fiber like structures, reflecting the incorporation of newly translated aSMA into the F-actin cytoskeleton. Rising concentrations of TGFβ-1 induced a dose-dependent increase in aSMA and collagen-I with an EC50 of 1.5 and 2 ng/ml (Fig. 1B). An effect of TGFβ-1 on cell numbers was not observed (Fig. S1A). TGFβ-1-mediated collagen-I expression is only detectable, if the cells were treated with the molecular crowding agent Ficoll in the presence of vitamin C (Fig. S1B). A TGFβ-1 concentration of 5 ng/ml reproducibly induced aSMA and collagen-I expression submaximally (approximately EC90), and was chosen for all further experiments. Calculation of z’-values as a statistical parameter for assay quality resulted in z’-values of 0.7 ± 0.047, 0.69 ± 0.035 and 0.2 ± 0.134 for aSMA, collagen-I and fibronectin, respectively (for n = 10 different plates with n = 38 non-TGFβ-1- and n = 24 5 ng/ml TGFβ-1-treated wells). To validate the TGFβ-1-mediated effects on FMT, cells were preincubated with a TGF receptor kinase inhibitor (Alk5i) before stimulation with TGFβ-1. As expected, the Alk5i dose-dependently inhibited aSMA, collagen-I and fibronectin expression (Fig. 1C and Fig. S1C) with an IC50 value of 0.16 μM, 0.19 μM and 0.6 μM for aSMA, collagen-I and fibronectin, respectively. Alk5i induced inhibition of TGFβ-1-mediated aSMA expression was tested in 3 additional donors and the potency was comparable between the different donors. However, donor 1 showed a less prominent induction of aSMA expression after stimulation with TGFβ-1 (Fig. S1D). Due to the lower z’-value for fibronectin and the co-staining of either aSMA and fibronectin or aSMA and collagen-I, collagen-I was selected as the extracellular matrix component to be analyzed in all further experiments.
TGFβ-1-mediated epithelial-to-mesenchymal transition assay
Primary human small airway epithelial cells were used for the TGFβ-1-dependent EMT assay with E-cadherin (Ecad) as a high content screening based readout (Fig. 2A). The corresponding image analysis script is described in Material and Methods section and Table S2. In the EMT assay, TGFβ-1 stimulation led to a signal decrease. Increasing doses of TGFβ-1 gradually reduced Ecad expression, reaching a minimum of 10 ng/ml. Therefore, 10 ng/ml TGFβ-1 was used as the standard concentration for all further experiments. Preincubation with 10 μM Alk5i before addition of TGFβ-1 restored Ecad expression to levels even higher than in non-TGFβ1-treated cells (Fig. 2B). Analysis of additional donors revealed similar effects of TGFβ-1 and the Alk5i plus TGFβ-1 (Fig. S2). The calculated Alk5i-IC50 for inhibition of Ecad expression determined from a dose-response curve was 0.12 μM (Fig. 2C) which was comparable to its effects on TGFβ-1-stimualted FMT. The assay quality parameter z’-value was 0.68 for this specific experiment, and generally was in a similar range (0.6 ± 0.08, for n = 9 different plates with n = 32 non-TGFβ-1- and n = 52 10 ng/ml TGFβ-1-treated wells).
Figure 2.
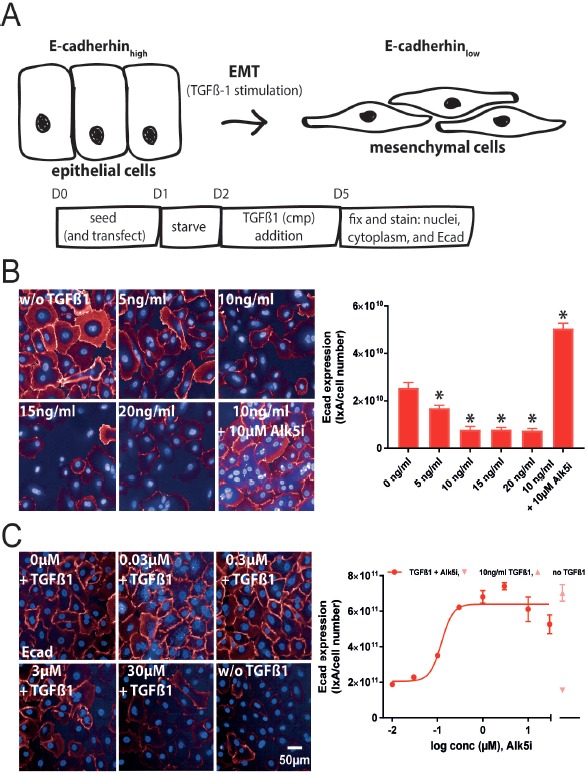
Phenotypic EMT assay. A. Schematic representation of the EMT assay with time points of treatment and readouts. At the end of the assay cells were prepared for high content imaging on E-cadherin expression. B. Images of hSAEC treated with increasing doses of TGFβ-1 and stained for nuclei and Ecad. Images were acquired with an Opera Phenix High Content Screening System (right). Ecad/ total cell numbers was quantified with a custom protocol using Columbus software and plotted against cumulating doses of TGFβ-1. From this data 10 ng/ml TGFβ-1 for all further experiments was chosen. C. Immunofluorescence images of hSAECs stimulated with 10 ng/ml TGFβ-1 and progressive concentrations of the Alk5i SB-525334 (right). The Alk5i dose-dependently inhibited the TGFβ-1-mediated reduction of Ecad expression an IC50 of 0.12 μM (left). *P < 0.05 versus non-TGFβ-1 treated cells.
Genetic modulation of gene expression in phenotypic FMT and EMT assays with siRNAs
To test whether the described human primary cell-based FMT and EMT phenotypic assays could be used to modify gene expression levels by siRNA transfection, we performed proof-of-concept assays, in which we either reduced TGFBR1 (FMT and EMT) or ACTA2 (FMT) expression. siRNA-mediated knock down of TGFBR1, one of the signaling receptor kinases of TGFβ-1, should result in a similar reduction of the aSMA/collagen-I expression for FMT and Ecad expression for EMT as observed by an Alk5i treatment of the cells. In the FMT assay, knock down of TGFBR1 led to a complete absence of aSMA and collagen-I expression. In addition to the TGFβ-1 receptor knock down, we also reduced ACTA2 expression with corresponding siRNAs. Again, the knock down completely abolished aSMA expression with only marginal effects on collagen-I expression. For both genes the knock down efficiency was more than 90% (Fig. 3A). In the EMT assay in the absence of TGFβ-1, the knock down of TGFBR1 already induced Ecad expression, comparable to an Alk5i. TGFβ-1 treatment reduced Ecad expression only in control siRNA (siCT) transfected and non-transfected cells, but not in TGFBR1 siRNA transfected cells, again showing the specificity of the siRNA and the TGFβ-1-depencency of the assay (Fig. 3B). The knock down efficiency of TGFBR1 in the EMT was determined to be higher than 90%.
Figure 3.
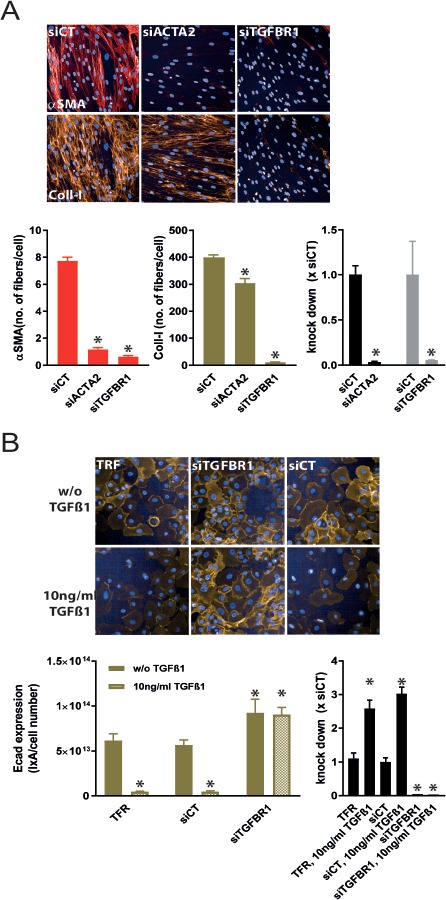
siRNA mediated knock down of ACTA2 and/or TGFBR1 in primary human cell phenotypic assays. A. NHLF cells were transfected with siRNAs targeting ACTA2 (siACTA2) and TGFBR1 (siTGFBR1). ACTA2 and TGFBR1 KD reduced αSMA expression in the presence of TGFβ-1 stimulation. TGFBR1 KD, but not ACTA2 decreased Coll-I expression. Both siRNAs efficiently reduced their mRNA expression by more than 90% compared to control siRNA (siCT) transfected cells. B. Non-treated and TGFβ-1-treated hSAECs were incubated with transfection reagent only (TFR) or transfection reagent plus siTGFBR1 or siCT and stained for Ecad expression. TGFBR1-KD completely abolished TGFβ-1-mediated reduction of Ecad expression. TGFBR1 mRNA expression was reduced by > 90% by siTGFBR1 transfection. Note, TGFβ-1 increases TGFBR1 expression about 3-fold. *P < 0.05 versus siCT.
Genetic modulation of gene expression in phenotypic FMT and EMT assays using Crispr-Cas9
Next, we established an orthogonal technology to siRNA based modulation of gene expression. Therefore, we investigated if Cas9-RNP-transfections are compatible with our FMT and EMT assays. We used synthetic crRNAs hybridized to a synthetic trRNA and incorporated in a ribonucleoprotein complex together with recombinant Cas9 protein. To assess specificity cells were transfected with hybridized cr:trRNAs incubated with BSA instead of Cas9 protein. In the FMT assay, TGFBR1-Cas9-RNPs strongly reduce the aSMA and collagen-I signal. Similar to the siACTA2 transfection, the transfection of a Cas9-RNP targeting ACTA2 resulted in a strong reduction of aSMA expression with only minor effects on collagen-I expression. Control transfections with BSA instead of Cas9 were without any effects on both aSMA and coll-I expression (Fig. 4A and 4B). Determination of Cas9-RNP-mediated gene editing by an ICE analysis revealed an editing efficiency of around 70% for both ACTA2- and TGFBR1-Cas9-RNPs (Fig. 4B). Although the editing efficiency is less compared to the siRNA mediated knock down efficiency of both ACTA2 and TGFBR1, the effects on aSMA and collagen-I expression are similar. In addition to Cas9-RNP transfections, we generated a pool of recombinant NHLF cells by lentiviral transduction, in which Cas9 was stably expressed. After transfection of cr:trRNA duplexes targeting ACTA2 and TGFBR1, the effects on TGFβ-1-mediated FMT was analyzed. Again, ACTA2 and TGFBR1 cr:trRNAs reduced aSMA expression. Collagen-I expression was only affected in TGFBR1-targeted cell. However, the level of aSMA and collagen reduction was not as pronounced as in siRNA or Cas9-RNP transfected cells. Compared to Cas9-RNP transfections, the editing efficiencies after cr:trRNA transfection in Cas9-expressing cells was lower (Fig. 4C).
Figure 4.
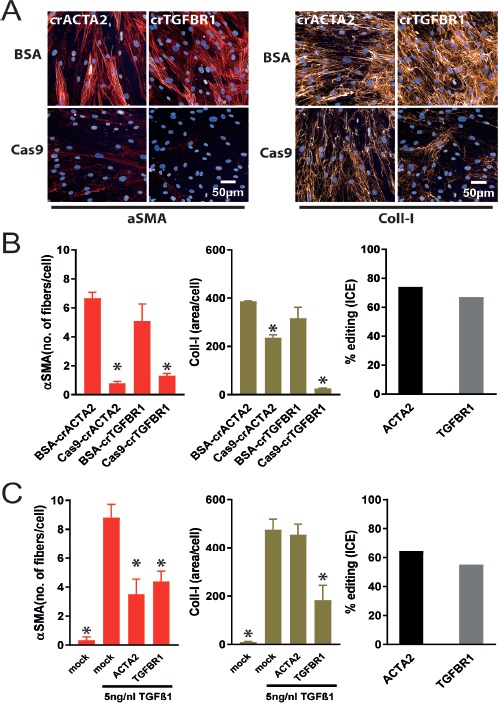
Crispr-Cas9 mediated knock out of ACTA2 and/or TGFBR1 in the FMT assay. A. NHLF cells were transfected with Cas9-cr:trRNA-RNPs targeting ACTA2 (crACTA2) and TGFBR1 (crTGFBR1). ACTA2 and TGFBR1 knock out reducedαSMA expression, whereas Coll-I expression was diminished only by Cas9-RNPs targeting TGFBR1, but not ACTA2. B. Quantitative analysis of images shown in (A) (right and middle) and editing efficiency calculated in ICE (left). Editing efficiencies for ACTA2 and TGFBR1-RNP are both > 80%. *P < 0.05 versus BSA-crRNA transfected cells. C. ACTA2 and TGFBR1 cr:trRNA transfection in NHLF cells constitutively expressing Cas9. In contrast to TGFβ-1-treated NHLF cells, mock transfected NHLF cells without TGFβ-1 are devoid of αSMA and Coll-I expression. ACTA2- and TGFBR1-cr:trRNA transfection reduced αSMA expression less than the corresponding Cas9-RNP transfections. Again Coll-I expression was not affected by an ACTA2 knock out. TGFBR1-cr:trRNA transfection diminished Coll-I expression (right and middle). Editing efficiencies are at least 50% in Cas9 expressing cells, which is lower as in Cas9-RNP transfected cells. *P < 0.05 versus mock transfected cells treated with TGFβ-1.
Transfections in hSAECs were done with Cas9-RNPs targeting either TGFBR1 or DMNT3B, a positive control for gene editing sold by Dharmacon. In mock transfected cells, 10 ng/ml TGFβ-1 lead to a complete loss of Ecad expression. Transfection with Cas9-RNPs targeting DMNT3B or the respective BSA control did not interfere with TGFβ-1-driven EMT (as seen by complete absence of Ecad staining). However, Cas9-RNPs targeting TGFBR1 abolished EMT with an even higher expression of Ecad compared to mock and BSA controls. This is similar to the effects of an Alk5i and siRNAs targeting TGFBR1. For the TGFBR1 and DMNT3B Cas9-RNPs, the editing efficiencies were greater than 90% and 60%, respectively (Fig. 5).
Figure 5.
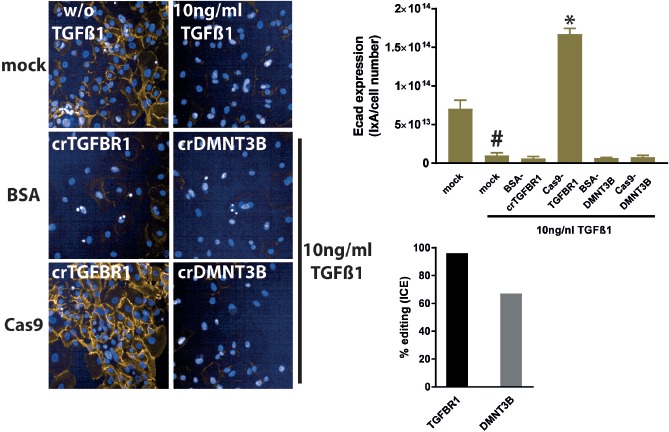
Crispr-Cas9 mediated knock out of TGFBR1 and DMNT3B in the EMT assay. hSAECs were either mock transfected (Cas9 replaced by BSA) or with Cas9-cr:trRNA-RNPs targeting TGFBR1 (crTGFBR1) and DMNT3B (crDMNT3B). In mock transfected cells TGFβ-1 repressed Ecad expression. TGFBR1 knock out abolished Ecad repression completely, whereas a DMNT3B knock out did not alter Ecad expression. Editing efficiency for TGFBR1- and DMNT3B-RNPs were at least 60%. *P < 0.05 versus BSA-crRNA transfected cells, #: mock transfected, TGFβ-1 treated versus mock-transfected non-TGFβ-1-treated cells.
DISCUSSION
Amongst a plethora of different cell types and mechanisms contributing to fibrogenesis, fibroblasts and FMT and epithelial cells transforming to mesenchymal cells, a mechanism named EMT are of major importance in fibrotic diseases [2,31,32]. Phenotypic screening assays with disease relevant readouts become increasingly popular in the last years as it is thought that they are more predictive for the clinical therapeutic response [10,33,34]. Indeed, in a recent phenotypic screen aiming to identify antifibrotic compounds, a compound identified in the cellular screen showed activity in a preclinical animal model of fibrosis [35]. In the present study, we described two assays using human primary cells (NHLF and hSAEC) in combination with phenotypic readouts (aSMA, collagen-I, fibronectin and E-cadherin) to study the fibrotic mechanisms FMT and EMT in vitro. We stimulated both cell types with the profibrotic growth factor TGFβ-1 and investigated the performance of both assays as screening tools for identification of antifibrotic compounds and as screening assays for genetic target identification and validation assays. For the latter, we used either siRNA mediated knock down or Crispr-Cas9 mediated knock out approaches.
Our first goal was to establish and pharmacologically validate a FMT and an EMT assay with primary human pulmonary cells compatible with high content screening readouts and sophisticated image analysis tools to obtain robust screening assays. Several recently described FMT assays [13,35,36] simply measure aSMA fluorescence intensity. As for myofibroblast contraction the incorporation of aSMA in F-actin fibers is required [37], we developed a protocol enabling the quantification of aSMA incorporated in the F-actin stress fiber cytoskeleton as fiber-like structures per cells, which resulted in a very robust assay window with excellent assay statistics (i.e., z’-value). A similar image analysis script was published recently [38]. These authors described a FMT assay which measured the expression of procollagen, but not mature collagen. We included in our FMT assay, apart from aSMA and fibronectin mature collagen. Usually, fibrogenic cells in monolayer cell culture do not produce significant amounts of mature collagen albeit full procollagen expression [39]. However, addition of macromolecules like dextran sulfate of Ficoll enhanced mature collagen production via an artificial crowding effect [39-41]. In our FMT assay, we used a mixture of Ficoll 70 and 400 as a macromolecular crowding agent. For both fibronectin and collagen-I quantification, we developed an image analysis protocol detecting the total fibronectin/collagen-I area normalized to the total cell number. At least for collagen-I expression, we obtained very good assay statistics. The final image analysis protocol for the EMT assay quantified the total Ecad fluorescence × total area of Ecad staining per total cell number. This value delivered a highly robust assay statistic. With the optimized assays and analysis protocols, we clearly showed that both assays can be modulated by different TGFβ-1 concentrations and that a TGFβ-1 receptor kinase inhibitor (the Alk5i SB-525334) dose-dependently reduced aSMA and collagen-I (and fibronectin) expression in the FMT assay. In the EMT assay, the Alk5i dose-dependently inhibited the TGFβ-1-mediated reduction in Ecad expression. In addition, we observed that the Alk5i is able to restore the amount of Ecad to levels higher than in non-TGFβ-1 treated cells. The reason for this could be that TGFβ-1 in the fetal calf serum which is included in the hSAEC medium already dampened Ecad expression in non-stimulated cells. When this medium-derived TGFβ-1 activity is inhibited together with the exogeneously added TGFβ-1 activity, Ecad levels might rise even higher compared to non-TGFβ-1 treated cells. This is in line with experiments in which the TGFBR1 was ablated by either siRNA or Cas9-RNPs resulting in Ecad levels higher than in those observed in non-TGFβ-1 treated cells. The submicromolar potency of the Alk5i SB-525334 in this study is comparable to published potencies in other fibrotic assays [35,36,42]. With the robustness, also with respect to the relative donor-independent reproducibility, it should be feasible to run at least medium throughput phenotypic fibrosis screens to identify either new chemical matter for further optimization or new targets in case annotated libraries.
For genetic target identification or validation, we could show that the assays can be used to transfect both siRNAs and Cas9-RNPs. We recently used siRNA transfections in the FMT assay with aSMA as a readout to study the role lysyl oxidase (like) family members in pulmonary fibrosis [29]. In this study, we extended the previously described FMT assay by the collagen-I and fibronectin readouts and described an advanced image analysis protocol for the 3 different readouts using the Columbus software instead of the IN Cell Developer Software which we used before [29]. Implementation of automated siRNA transfection would enable siRNA screens up to a genome-wide format in primary human fibroblasts of both healthy and diseased donors, comparable to the screen described in Oh et al., using IMR-90 fibroblast cells. The EMT assay is again a high throughput amenable phenotypic assay in primary human airway epithelial cells, and when combined with siRNA transfections should enable target identification/validation screens comparable to successfully described siRNA screens in non-airway epithelial cell lines [43,44].
Having shown that the primary cell based FMT and EMT assays can be used to genetically interfere with gene expression by RNAi technologies, we were also interested in if we could adapt the assays for use of the Crispr-Cas9 gene editing technology. Recently, adenoviral mediated introduction of the both Cas9 and sgRNAs in primary human fibroblasts and epithelial cells in the context of FMT and EMT was published [13]. However, we wanted to transfect the primary cells with commercially available reagents. Therefore, electroporation conditions were established for both NHLF cells and hSAEC to transfect recombinant Cas9 protein complexed in a RNP with a cr:trRNA duplex. We obtained editing efficiencies by ICE analysis of ≥ 60% for 3 different genes in the 2 different cell types. This is several fold higher compared to published indel frequencies, which were determined by T7 mismatch assays after Cas9-RNP transfection or adenoviral Cas9-sgRNA delivery in primary cells [13,45]. Surprisingly, both the phenotypic effects and the editing efficiencies of NHLF cells stably expressing Cas9 after cr:trRNA transfection were smaller compared to cells transfected with Cas9-RNPs. Similar, Seki and Rutz [46] showed superior effects of Cas9-RNPs compared to lentivirally delivered sgRNA-Cas9 all-in-one vector constructs. Different gene editing efficiencies observed between RNP transfected and lentivirally delivered sgRNAs might be related to the higher Cas9 protein amount on a per cell basis. However, Cas9 protein levels were not measured in our experiments. Apart from the higher editing efficiencies, Cas9-RNPs are associated with reduced off-target effects due to the shorter half-life of the Cas9-RNP in comparison to e.g., sgRNA-Cas9 lentiviral transductions [47]. To complement the genetic toolbox with phenotypic readouts, the CrisprA and CrisprI technology for transcriptional activation and inhibition [26,48] could be established for primary human cell assays.
In summary, in this study we showed that primary human pulmonary cells from different donors can be used to successfully establish FMT and EMT phenotypic assays for identification and characterization of pro- and anti-fibrotic compounds. Both assays were amenable to genetic modulation with RNAi and Crispr-Cas9 technologies. Combined with a FACS-able readout, both assays might also be modified to run pooled genetic screens [26]. A FMT assay with FACS-able aSMA readout in primary human fibroblasts was already published [49].
Supplementary Material
Figure S1. A. TGFβ-1 effects on cell numbers in FMT assay. B. Alk5i effect on fibronectin expression in FMT assay. C. Effect of Ficoll on collagen-I expression in FMT assay D. FMT assay with different donors.
Figure S2. Different hSAEC donors in EMT assay.
Table S1. FMT assay image analysis protocol.
Table S2. EMT assay image analysis protocol.
Supplementary information of this article can be found online at http://www.jbmethods.org/jbm/rt/suppFiles/285.
Acknowledgments
We acknowledge Martin Lenter for discussions. Achim Kirsch from PerkinElmer is acknowledged for his support during image analysis protocol development.
References
- 1.Pakshir P, Hinz B. (2018) The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol 68-69: 81-93. doi: 10.1016/j.matbio.2018.01.019. PMID: [DOI] [PubMed] [Google Scholar]
- 2.Weiskirchen R, Weiskirchen S, Tacke F. (2018) Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol Aspects Med 65: 2-15. doi: 10.1016/j.mam.2018.06.003. PMID: [DOI] [PubMed] [Google Scholar]
- 3.Yasukawa A, Hosoki K, Toda M, Miyake Y, Matsushima Y, et al. (2013) Eosinophils promote epithelial to mesenchymal transition of bronchial epithelial cells. PLoS One 8: e64281. doi: 10.1371/journal.pone.0064281. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang X, Liang L, Zong C, Lai F, Zhu P, et al. (2015) Kupffer cells-dependent inflammation in the injured liver increases recruitment of mesenchymal stem cells in aging mice. Oncotarget 7:1084-1095. doi: 10.18632/oncotarget.6744. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalluri R, Neilson EG. (2003) Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest 112: 1776-1784. doi: 10.1172/JCI20530. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karicheva O, Rodriguez-Vargas JM, Wadier N, Martin-Hernandez K, Vauchelles R, et al. (2016) PARP3 controls TGFβ and ROS driven epithelial-to-mesenchymal transition and stemness by stimulating a TG2-Snail-E-cadherin axis. Oncotarget 7: 64109-64123. doi: 10.18632/oncotarget.11627. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rout-Pitt N, Farrow N, Parsons D, Donnelley M. (2018) Epithelial mesenchymal transition (EMT): a universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respir Res 19: 136. doi: 10.1186/s12931-018-0834-8. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson JR, Nishioka M, Chakir J, Risse PA, Almaghlouth I, et al. (2013) IL-22 contributes to TGF-beta1-mediated epithelial-mesenchymal transition in asthmatic bronchial epithelial cells. Respir Res 14: 118. doi: 10.1186/1465-9921-14-118. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jonsdottir HR, Arason AJ, Palsson R, Franzdottir SR, Gudbjartsson T, et al. (2015) Basal cells of the human airways acquire mesenchymal traits in idiopathic pulmonary fibrosis and in culture. Lab Invest 95: 1418-1428. doi: 10.1038/labinvest.2015.114. PMID: [DOI] [PubMed] [Google Scholar]
- 10.Moffat JG, Vincent F, Lee JA, Eder J, Prunotto M. (2017) Opportunities and challenges in phenotypic drug discovery: an industry perspective. Nat Rev Drug Discov 16: 531-543. doi: 10.1038/nrd.2017.111. PMID: [DOI] [PubMed] [Google Scholar]
- 11.Vincent F, Loria P, Pregel M, Stanton R, Kitching L, et al. (2015) Developing predictive assays: the phenotypic screening “rule of 3". Sci Transl Med 7: 293. doi: 10.1126/scitranslmed.aab1201. PMID: [DOI] [PubMed] [Google Scholar]
- 12.Câmara J, Jarai G. (2010) Epithelial-mesenchymal transition in primary human bronchial epithelial cells is Smad-dependent and enhanced by fibronectin and TNF-alpha. Fibrogenesis Tissue Repair 3: 2. doi: 10.1186/1755-1536-3-2. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Voets O, Tielen F, Elstak E, Benschop J, Grimbergen M, et al. (2017) Highly efficient gene inactivation by adenoviral CRISPR/Cas9 in human primary cells. PLoS One 12: e0182974. Epub PubMed Central PMCID: doi: 10.1371/journal.pone.0182974. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao L, Xu Y, Tao L, Yang Y, Shen X, et al. (2018) Oxymatrine inhibits transforming growth factor beta1 (TGF-beta1)-induced cardiac fibroblast-to-myofibroblast transformation (FMT) by mediating the Notch signaling pathway in vitro. Med Sci Monit 24: 6280-6288. doi: 10.12659/MSM.910142. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jung H, Lee DS, Park SK, Choi JS, Jung WK, et al. (2018) Fucoxanthin Inhibits Myofibroblast Differentiation and Extracellular Matrix Production in Nasal Polyp-Derived Fibroblasts via Modulation of Smad-Dependent and Smad-Independent Signaling Pathways. Mar Drugs 16. doi: 10.3390/md16090323. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qu MH, Han C, Srivastava AK, Cui T, Zou N, et al. (2016) miR-93 promotes TGF-beta-induced epithelial-to-mesenchymal transition through downregulation of NEDD4L in lung cancer cells. Tumour Biol 37: 5645-5651. doi: 10.1007/s13277-015-4328-8. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toba-Ichihashi Y, Yamaoka T, Ohmori T, Ohba M. (2015) Up-regulation of Syndecan-4 contributes to TGF-β1-induced epithelial to mesenchymal transition in lung adenocarcinoma A549 cells. Biochem Biophys Rep 5: 1-7. doi: 10.1016/j.bbrep.2015.11.021. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khalil N, O'Connor RN, Flanders KC, Unruh H. (1996) TGF-beta 1, but not TGF-beta 2 or TGF-beta 3, is differentially present in epithelial cells of advanced pulmonary fibrosis: an immunohistochemical study. Am J Respir Cell Mol Biol 14: 131-138. doi: 10.1165/ajrcmb.14.2.8630262. PMID: [DOI] [PubMed] [Google Scholar]
- 19.Coker RK, Laurent GJ, Shahzeidi S, Lympany PA, du Bois RM, et al. (1997) Transforming growth factors-beta 1, -beta 2, and -beta 3 stimulate fibroblast procollagen production in vitro but are differentially expressed during bleomycin-induced lung fibrosis. Am J Pathol 150:981-991. PMID: [PMC free article] [PubMed] [Google Scholar]
- 20.King TE, Jr, Pardo A, Selman M. (2011) Idiopathic pulmonary fibrosis. Lancet 378: 1949-1961. doi: 10.1016/S0140-6736(11)60052-4. PMID: [DOI] [PubMed] [Google Scholar]
- 21.Selman M, Pardo A. (2014) Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. an integral model. Am J Respir Crit Care Med 189: 1161-1172. doi: 10.1164/rccm.201312-2221PP. PMID: [DOI] [PubMed] [Google Scholar]
- 22.Wuyts WA, Agostini C, Antoniou KM, Bouros D, Chambers RC, et al. (2013) The pathogenesis of pulmonary fibrosis: a moving target. Eur Respir J 41: 1207-1218. doi: 10.1183/09031936.00073012. PMID: [DOI] [PubMed] [Google Scholar]
- 23.Mohr SE, Smith JA, Shamu CE, Neumüller RA, Perrimon N. (2014) RNAi screening comes of age: improved techniques and complementary approaches. Nat Rev Mol Cell Biol 15: 591-600. doi: 10.1038/nrm3860. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kabadi AM, Ousterout DG, Hilton IB, Gersbach CA. (2014) Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res 42: e147. doi: 10.1093/nar/gku749. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bellec J, Bacchetta M, Losa D, Anegon I, Chanson M, et al. (2015) CFTR inactivation by lentiviral vector-mediated RNA interference and CRISPR-Cas9 genome editing in human airway epithelial cells. Curr Gene Ther 15: 447-459. doi: 10.2174/1566523215666150812115939. PMID: [DOI] [PubMed] [Google Scholar]
- 26.Schuster A, Erasimus H, Fritah S, Nazarov PV, van Dyck E, et al. (2018) RNAi/CRISPR Screens: from a Pool to a Valid Hit. Trends Biotechnol 37: 38-55. doi: 10.1016/j.tibtech.2018.08.002. PMID: [DOI] [PubMed] [Google Scholar]
- 27.Kim S, Kim D, Cho SW, Kim J, Kim J. (2014) Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24: 1012-1019. doi: 10.1101/gr.171322.113. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu J, Gaj T, Yang Y, Wang N, Shui S, et al. (2015) Efficient delivery of nuclease proteins for genome editing in human stem cells and primary cells. Nat Protoc 10: 1842-1859. doi: 10.1038/nprot.2015.117. PMID: [DOI] [PubMed] [Google Scholar]
- 29.Aumiller V, Strobel B, Romeike M, Schuler M, Stierstorfer BE, et al. (2017) Comparative analysis of lysyl oxidase (like) family members in pulmonary fibrosis. Sci Rep 7: 149. doi: 10.1038/s41598-017-00270-0. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsiau T, Conant D, Maures T, Waite K, Yang J, et al. (2018) Inference of CRISPR Edits from Sanger Trace Data. bioRxiv. doi: 10.1101/251082. [DOI] [PubMed] [Google Scholar]
- 31.Ijaz T, Pazdrak K, Kalita M, Konig R, Choudhary S, et al. (2014) Systems biology approaches to understanding Epithelial Mesenchymal Transition (EMT) in mucosal remodeling and signaling in asthma. World Allergy Organ J 7: 13. doi: 10.1186/1939-4551-7-13. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zent J, Guo L. (2018) Signaling mechanisms of myofibroblastic activation: Outside-in and inside-out. Cell Physiol Biochem 49: 848-868. doi: 10.1159/000493217. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Swinney DC, Anthony J. (2011) How were new medicines discovered? Nat Rev Drug Discov 10: 507-519. doi: 10.1038/nrd3480. PMID: [DOI] [PubMed] [Google Scholar]
- 34.Scannell JW, Bosley J. (2016) When quality beats quantity: Decision theory, drug discovery, and the reproducibility crisis. PLoS One 11: e0147215. doi: 10.1371/journal.pone.0147215. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bollong MJ, Yang B, Vergani N, Beyer BA, Chin EN, et al. (2017) Small molecule-mediated inhibition of myofibroblast transdifferentiation for the treatment of fibrosis. Proc Natl Acad Sci USA 114: 4679-4684. doi: 10.1073/pnas.1702750114. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang XT, Sun XJ, Li C, Liu Y, Zhang L, et al. (2018) Establishing a cell-based high-content screening assay for TCM compounds with anti-renal fibrosis effects. Evid Based Complement Alternat Med 2018: 7942614. doi: 10.1155/2018/7942614. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Talele NP, Fradette J, Davies JE, Kapus A, Hinz B. (2015) Expression of alpha-smooth muscle actin determines the fate of mesenchymal stromal cells. Stem Cell Reports 4: 1016-1030. doi: 10.1016/j.stemcr.2015.05.004. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oh RS, Haak AJ, Smith KMJ, Ligresti G, Choi KM, et al. (2018) RNAi screening identifies a mechanosensitive ROCK-JAK2-STAT3 network central to myofibroblast activation. J Cell Sci 131. doi: 10.1242/jcs.209932. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lareu RR, Arsianti I, Subramhanya HK, Yanxian P, Raghunath M. (2007) In vitro enhancement of collagen matrix formation and crosslinking for applications in tissue engineering: a preliminary study. Tissue Eng 13: 385-391. doi: 10.1089/ten.2006.0224. PMID: [DOI] [PubMed] [Google Scholar]
- 40.Lareu RR, Subramhanya KH, Peng Y, Benny P, Chen C, et al. (2007) Collagen matrix deposition is dramatically enhanced in vitro when crowded with charged macromolecules: the biological relevance of the excluded volume effect. FEBS Lett 581: 2709-2014. doi: 10.1016/j.febslet.2007.05.020. PMID: [DOI] [PubMed] [Google Scholar]
- 41.Chen CZC, Peng YX, Wang ZB, Fish PV, Kaar JL, et al. (2009) The Scar-in-a-Jar: studying potential antifibrotic compounds from the epigenetic to extracellular level in a single well. Br J Pharmacol 158: 1196-1209. doi: 10.1111/j.1476-5381.2009.00387.x. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carthy JM, Stöter M, Bellomo C, Vanlandewijck M, Heldin A, et al. (2016) Chemical regulators of epithelial plasticity reveal a nuclear receptor pathway controlling myofibroblast differentiation. Sci Rep 6: 29868. doi: 10.1038/srep29868. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lotz-Jenne C, Lüthi U, Ackerknecht S, Lehembre F, Fink T, et al. (2016) A high-content EMT screen identifies multiple receptor tyrosine kinase inhibitors with activity on TGFβ receptor. Oncotarget 7: 25983-26002. doi: 10.18632/oncotarget.8418. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pavan S, Meyer-Schaller N, Diepenbruck M, Kalathur RKR, Saxena M, et al. (2018) A kinome-wide high-content siRNA screen identifies MEK5-ERK5 signaling as critical for breast cancer cell EMT and metastasis. Oncogene 37: 4197-4213. doi: 10.1038/s41388-018-0270-8. PMID: [DOI] [PubMed] [Google Scholar]
- 45.Liang X, Potter J, Kumar S, Zou Y, Quintanilla R, et al. (2015) Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J Biotechnol 208: 44-53. doi: 10.1016/j.jbiotec.2015.04.024. PMID: [DOI] [PubMed] [Google Scholar]
- 46.Seki A, Rutz S. (2018) Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J Exp Med 215: 985-997. doi: 10.1084/jem.20171626. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Farboud B, Jarvis E, Roth TL, Shin J, Corn JE, et al. (2018) Enhanced Genome Editing with Cas9 Ribonucleoprotein in Diverse Cells and Organisms. J Vis Exp. doi: 10.3791/57350. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dominguez AA, Lim WA, Qi LS. (2015) Beyond editing: repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat Rev Mol Cell Biol 17: 5-15. doi: 10.1038/nrm.2015.2. PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Michalik M, Pierzchalska M, Włodarczyk A, Wójcik KA, Czyż J, et al. (2011) Transition of asthmatic bronchial fibroblasts to myofibroblasts is inhibited by cell-cell contacts. Respir Med 105: 1467-1475. doi: 10.1016/j.rmed.2011.04.009. PMID: [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. A. TGFβ-1 effects on cell numbers in FMT assay. B. Alk5i effect on fibronectin expression in FMT assay. C. Effect of Ficoll on collagen-I expression in FMT assay D. FMT assay with different donors.
Figure S2. Different hSAEC donors in EMT assay.
Table S1. FMT assay image analysis protocol.
Table S2. EMT assay image analysis protocol.
Supplementary information of this article can be found online at http://www.jbmethods.org/jbm/rt/suppFiles/285.