

Abstract
Recombinant adeno-associated virus (rAAV) vectors have recently been widely utilized for in in vivo gene therapy. The clinical dose definition of AAV vector requires the exact quantification as starting doses and for dose-escalation studies. Vector genome (vg) copies measured by quantitative PCR (qPCR) are commonly used for rAAV vector titration, and rAAV vector plasmids DNA is often used for qPCR standards, although the rAAV reference standard materials (RSMs) for serotypes 2 and 8 (rAAV2RSM and rAAV8RSM) are available from American Type Culture Collection. However, qPCR-based determination of the AAV vg is affected by the selection of the qPCR standard and the amplification target sites. In this study, we have developed a new PCR method, two-dimensional droplet digital PCR (2D ddPCR), for the absolute quantitation of target DNA and for evaluating the stability of the rAAV vector. The number of vg copies of rAAV2RSM determined by qPCR dramatically changed when standard plasmid DNAs with different conformations were treated with restriction enzymes, suggesting that qPCR amplification is significantly affected by the secondary structure of the standard. In contrast, the number of vg copies determined by ddPCR was unaffected by using primer probes for different positions of target sites or by the secondary structure conformation of the vg. Furthermore, the integrity of the AAV vg can be monitored using 2D ddPCR with fluorescein- and hexachloro-6-carboxy-fluorescine–labeled probes targeting different positions in the same rAAV genome. The titer of intact rAAV was highly correlated with rAAV activity in an accelerated (37°C) stability study. 2D ddPCR is a useful tool for rAAV vector quantitation and quality evaluation.
Keywords: droplet digital PCR, qPCR, two dimensional ddPCR, AAV, vector integrity
Introduction
Recombinant adeno-associated virus (rAAV) vectors have been used successfully in clinical trials to target various diseases, including Leber's congenital amaurosis (LCA)1 and hemophilia.2,3 The first gene therapy product approved by the European Medicines Agency was alipogene tiparvovec (Glybera) for treating adults with lipoprotein lipase deficiency,4,5 and the first gene therapy product approved in the United States was voretigene neparvovec (Luxturna) for treating LCA. The major advantages of the rAAV vector are that it is nonpathogenic for humans, as well as its long-term transgene expression in nondividing cells, its comparatively low immune profile, the finding that it elicits only limited inflammatory responses, and that, in some cases, it directs immune tolerance to transgene products.6
AAV is a nonenveloped single-stranded DNA virus. rAAV vectors contain transgene sequences flanked by AAV inverted terminal repeats (ITRs). Routine quality control conducted for preclinical and clinical rAAV vector preparations includes the determination of titer. Vector genome (vg) titer determined by quantitative PCR (qPCR) is the most common method used for the titration of rAAV vectors for clinical trial dosages because of the weak infectivity and expression of the transgene.7 The number of vg copies determined by qPCR assay is also used to evaluate biodistribution8 and viral shedding.9 The use of qPCR for determining AAV titer requires the preparation of a standard calibration curve using reference material, which is typically plasmid DNAs, though rAAV reference standard materials (RSMs) for serotypes 2 and 8 (rAAV2RSM and rAAV8RSM, respectively) have been established by the AAVRSM Working Group and are available for calibration.10,11 The secondary structure of reference plasmid DNA is known to affect the titration results obtained by qPCR. For example, the supercoiled conformation of circular plasmid suppressed annealing of the primer to the DNA template, resulting in regression line shift and overestimation of the titer.12 In addition, qPCR for different target sites in the same rAAV vg, for example, the ITR and the transgene, showed different titers even when the same plasmid DNA was used as a reference standard.13 Another disadvantage of qPCR is that the qPCR measurement does not reflect the activity (infectious activity) of the rAAV vector.10
Droplet digital PCR (ddPCR) is an improvement on traditional PCR methods that partitions a single PCR mixture into ∼20,000 nL-sized volume droplets. Fluorescent dye-based end-point PCRs occur separately and independently within these droplets, then the fluorescence of each droplet is scanned with a binary fluorescent of positive or negative. The combination of limiting dilution, PCR, and Poisson statistics results in ddPCR affords an absolute measurement of nucleic acid concentration without the need for a standard curve14 and thus could address the problem associated with the qPCR of rAAV already mentioned. ddPCR assay was superior to qPCR in rAAV titer in terms of both intra- and interassay precision and was more resistant to PCR inhibitors.15
In this article, we used rAAVRSM as a model to show that the problems associated with single-stranded rAAV vg titer determination using qPCR are resolved using ddPCR. Moreover, we demonstrate that the integrity of the rAAV vector can be monitored using two-dimensional qPCR (2D ddPCR) of fluorescein (FAM)- and hexachloro-6-carboxy-fluorescine (HEX)-labeled probes targeting different positions of the same rAAV genome. The titer of intact rAAV determined by 2D ddPCR was highly correlated with rAAV activity in stability test model. 2D ddPCR is also effective for evaluating the impurity of plasmid DNA used for vector production. Overall, we demonstrate that ddPCR is a useful tool for evaluating the titer and quality of rAAV vectors.
Materials and Methods
Vectors
rAAV2RSM, rAAV8RSM, and vector plasmid pTR-UF11, each containing AAV2 ITR, GFP, and the SV40 polyA sequence (SV40), were purchased from ATCC.10
Primers and probes
The following primers and probes were used for qPCR and ddPCR. Universal ITR probe 5′-CACTCCCTCTCTGCGCGCTCG-3′, forward primer 5′-GGAACCCCTAGTGATGGAGTT-3′, reverse primer 5′-CGGCCTCAGTGAGCGA-3′16; SV40 probe 5′-AGCATTTTTTTCACTGCATTCTAGTTGTGGTTTGTC-3′, forward primer 5′-AGCAATAGCATCACAAATTTCACAA-3′, reverse primer 5′-CCAGACATGATAAGATACATTGATGAGTT-3′. For detection of replication origin of plasmid backbone, the following probe and primers were used: Ori probe 5′-CGCTCTGCTGAAGCCAGTTACCTTCGG-3′, forward primer 5′-GCGCGTAATCTGCTGCTTG-3′, and reverse primer 5′-CTACGGCTACACTAGAAGAACAGTA-3′. For qPCR, the 5′ ends of the ITR and SV40 probes were labeled with FAM. For ddPCR, the 5′ end of the ITR probe was labeled with HEX and the 5′ end of the SV40 probe was labeled with FAM. The 3′ ends of these probes were labeled with Black Hole Quencher-1.
rAAV genome extraction
For rAAV genome concentration detection, rAAV2RSM and rAAV8RSM were treated with DNase I (Takara Bio Inc., Tokyo, Japan) at a concentration of 250 U/mL at 37°C for 30 min, and subsequently, 20 mM EDTA was added to stop the reaction. For plasmid impurity detection, treatment of DNase I was ignored. Then, the same volume of protein lysis buffer from a SMITEST EX-R&D nucleic acid extraction kit (MBL Co, Nagoya, Japan) was added and the sample was treated at 56°C for 10 min. The obtained vg extractions were serially 10-fold diluted using TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). Vg titers were determined in duplicate by qPCR or ddPCR.
Quantitative PCR
qPCR was performed using the optimized qPCR method previously described, with some modification.10 One microliter rAAV genome extraction sample was added to 25 μL reaction mixture along with QuantiTect Probe PCR master mixture (Qiagen, Hilden, Germany) with 0.4 μM primers and 0.2 μM probe (all final concentrations). The vector plasmid of rAAV2RSM, pTR-UF11 (MBA-331, ATCC), was chosen as a reference standard plasmid according to the standard operating procedure (SOP) of ATCC. Circular plasmid was purified from Escherichia coli competent cells by miniprep (Qiagen). Linearized plasmid was obtained by digesting pTR-UF11 with ScaI (within the ampicillin-resistant gene in the plasmid backbone), and plasmid without the ITR structure was prepared by digestion with SmaI (within the ITR region of rAAV) (Takara Bio Inc.). Spectrophotometric system bias was prevented by directly using plasmid DNA samples diluted appropriately using TE buffer and digested by restriction enzyme, rather than by purifying the DNA samples using a QIAquick PCR purification kit (Qiagen) as described in a previous method.10 Calibration curves were obtained with 10-fold diluted plasmid from 106 to 100 copies/μL. PCR amplification was performed using an Applied Biosystems 7500 Real-Time PCR System, and two-step cycling (40 cycles: 95°C for 15 s, 60°C for 1 min) preceded by a 10-min incubation at 95°C. Data were analyzed using 7500 software v2.0.6 (Applied Biosystems). Standard curves were plotted as threshold cycle (Ct) on the y-axis versus the log (number of copies) on the x-axis. Coefficient of correlation R2 obtained for the standard curve should be >0.99. When ITR quantitation of rAAV were determined using pTR-UF11 as a reference standard, a correction factor of 2 must been introduced, because pTR-UF11 is a double-strand DNA and contains two ITR amplification sites (one in positive strand and the other in negative strand).16
ddPCR
The rAAV genome extraction sample was diluted from 20,000 to 100 copies/μL to determine the dynamic linear range of the ddPCR assay. The same dilution buffers were used as in the qPCR assays except for adding 0.05% Pluronic F-68 (Gibco, Invitrogen, Grand Island, NY). The reaction mixtures were assembled using a ddPCR Supermix for Residual DNA detection kit (BIO-RAD, Hercules, CA) with 0.9 μM primers and 0.1 μM probe, in a final volume of 20 μL as stated in the manufacturer's protocol. Because the secondary structure of the plasmid is more complex than that of rAAV genomes, plasmid samples were treated with SmaI to remove the ITR and plasmid tail before ddPCR as described in ddPCR amplification guide (Bio-Rad 6407). Furthermore, stable detection results were obtained by addition of 0.5 M betaine (Sigma-Aldrich, St Louis, MO) into the ddPCR reaction mixtures. Then, test samples were emulsified with droplet generator oil using a QX-100 droplet generator (BIO-RAD) according to the manufacturer's instructions. PCR amplification of the droplets was performed using a conventional thermal cycler with the following parameters: 95°C for 10 min, followed by 36 cycles of 95°C for 30 s, 60°C for 1 min, and 72°C for 15 s followed by a final 98°C heat treatment for 10 min. The PCR plate was subsequently scanned using a QX200 droplet reader (BIO-RAD) and the data were analyzed with QuantaSoft software (BIO-RAD). For 2D ddPCR, two sets of primers and probes labeled with FAM and HEX, respectively, were added to each reaction mixture, and ddPCR was performed in the same tube. The average volume of the droplets was 0.85 nL.17 According to the Poisson distribution, the concentration of the template is calculated using the following formula:
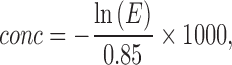 |
where conc is the number of copies of the target per microliter of reaction solution and E is the frequency of droplets that contain no nucleic acid template. Percentage for each group was calculated from the ratio of the concentration of target group to the concentration sum of total droplets with fluorescence signals.
AAV accelerated stability study
rAAV2RSM stock was incubated at 37°C for 0, 1, 3, or 7 days, and 10 μL of the treated rAAV2RSM was inoculated to Vero cells. Remaining infectious activity of the treated rAAV2RSM was determined by FACS analysis of GFP-expressed Vero cells 48 h postinfection and shown as transduction unit per milliliter.18 Remaining virus genome of the treated rAAV2RSM was detected by qPCR and 2D ddPCR as already described.
Result
The variability in qPCR titration of rAAV vg is caused by differences in the secondary structures of rAAV and the reference plasmid
We demonstrated the problems associated with the qPCR titration of rAAV by using two sets of primers and probes against the ITR and SV40-polyA region (SV40) chosen as target sequences for comparison (Fig. 1A). AAV2 contains two 145 base-long ITRs at the termini that contain two hairpin loops to form a T-shaped self-priming structure.19 Quantification of rAAV by ITR qPCR requires extended denaturation conditions to mitigate extensive secondary hairpin structure formation by AAV2 ITR.20 We, therefore, compare the amplification efficiency of rAAV2RSM and three conformations of vector plasmid (circular, linearized and SmaI-digested plasmid DNA, pTR-UF11) as reference standards. There are two SmaI sites within one hairpin loop in the ITR structure and thus both the SmaI-digested plasmid DNA and the rAAV genome lost their ITR secondary structures during the denaturation step in the PCR. As shown in Fig. 1B, when rAAV2RSM was used as a reference standard, the vg titer of rAAV2RSM as determined by either ITR qPCR or SV40 qPCR fell within the 95% confidence intervals of the published values.10 In contrast, the qPCR titer of the rAAV2 genome differed dramatically when circular, linearized, or SmaI-digested plasmid DNAs were used as the qPCR reference standards. The standard curves are shown in the following order from top to bottom: circular, linearized plasmid, rAAV2RSM, and SmaI-digested plasmid DNA (Fig. 1C). The standard curves shifted more dramatically when ITR was used as an amplification target compared with SV40.
Figure 1.

Measured values of qPCR titer depend on the secondary structure of the reference standards. (A) Genome structure of rAAV2RSM. Thin arrows and thick arrows indicate the positions of the amplification target sites on ITR and SV40, respectively. (B) Vector genome titer of rAAV2RSM determined by qPCR showed variation when using reference standards with different secondary structures in pTR-UF11 plasmid DNA. Circular plasmid DNA; linearized plasmid DNA linearized by ScaI digestion; SmaI digested plasmid DNA. A 95% CI of rAAV2RSM determined by the AAVRSM Working Group is 2.7–4.75 × 1010 vg/mL.10 (C) Calibration curves for ITR qPCR (upper panel) and SV40 qPCR (lower panel) obtained using reference standards with different structures. R2 of each calibration curve was >0.99. qPCR of RSM was performed after DNase I and proteinase K treatment as described in Material and Methods section. Because SDS in proteinase K buffer inhibits the qPCR and thus the signal could not be detected when the sample was diluted <100-fold, thus only five points were measured in the RSM sample. CI, confidence interval; Ct, threshold cycle; ITR, inverted terminal repeat; qPCR, quantitative PCR; rAAV, recombinant adeno-associated virus; rAAV2RSM, rAAV reference standard material for serotype 2; RSM, reference standard material; SDS, sodium dodecyl sulfate; vg, vector genome.
We, therefore, hypothesized that the hairpin structure of ITR and the “plasmid tail” attached to ITR inhibited annealing of the primer and probes to the DNA template, thus shifting the standard curve and affecting the qPCR value. In addition to Hou's theory,12 we speculated that the annealing of ITR primers to the rAAV genome is easier than annealing to plasmid DNA because the “plasmid tail” sticking to the ITR 5′-end forms a more complex structure than the rAAV genome, inhibiting the annealing of ITR primers to the target sequence near the ITR structure in plasmid DNAs (Fig. 2A). Therefore, removal of the plasmid tail by SmaI digestion would allow rapid annealing of the ITR primer to the target sequence, shifting down the standard curve of the plasmid DNA, resulting in the underestimation of rAAV as shown in Fig. 1B. In contrast, when using linearized or circular plasmid DNA as a reference standard, the standard curve shifts up, resulting in an overestimation of the rAAV genome (Fig. 2B). In contrast, when qPCR titer is determined using an amplification target far from the ITR structure (e.g., SV40), it is less affected by the secondary structures of ITR and the plasmid tail.
Figure 2.

The secondary structure of the plasmid DNA tail disturbed the amplification of ITR by qPCR. (A) The illustrations show the left part of the rAAV2RSM positive strand and the different secondary structures of pTR-UF11 plasmid DNAs. Thin arrows and thick arrows indicate the position of ITR and SV40 target sites, respectively. The arrowhead indicates the cutting site of SmaI. The treatment of pTR-UF11 with SmaI disrupted the ITR secondary structure. qPCR amplification was more efficient in the absence of hindrance by the plasmid tail, in SmaI-digested plasmid, and in the rAAV genome compared with linearized plasmid or circular plasmid. This resulted in up shift or down shift of the standard curves and thus overestimation or underestimation of the values measured by qPCR as shown in (B).
The use of ddPCR offers reproducible titer measurements and more detailed information regarding the AAV genome
ddPCR can be used to measure the absolute values of vg copies without a reference standard and thus we used this approach for rAAV2RSM titration. The high linearity of the ddPCR titer, regardless of whether ITR or SV40 was targeted, allowed detection in the range between 20,000 and 100 copies/reaction with coefficient of determination R2 values of 0.9986 and 0.9968, respectively (Fig. 3A). Furthermore, we confirmed that the ddPCR titer is not affected by conformational changes of the rAAV genome by treating with or without SmaI and then subjecting the product to qPCR and ddPCR. As shown in Fig. 3B, the qPCR titer of rAAV determined using the ITR target was increased by SmaI digestion, whereas the titer determined by SV40 qPCR was less affected by SmaI digestion. In contrast, ddPCR analysis provided reproducible titers regardless of the target site selection (ITR or SV40) and conformational changes resulting from SmaI digestion (Fig. 3B).
Figure 3.

ddPCR detection of rAAV2RSM was not affected by the secondary structure of the vector genome or by the selection of the amplification target site. (A) ddPCR detection range of rAAV2RSM using ITR and SV40 as target sites. Eight ddPCR data points, corresponding to number of droplets ranging from 14,000 and 16,000, were plotted. The CV% values were <2.9% and 3.1% for ITR and SV40, respectively. (B) Comparison of the titer of rAAV2RSM vector genome treated with SmaI or not, as determined by qPCR and ddPCR. Linearized plasmid DNA was used as a reference standard for qPCR. Values are shown as mean ± SD from two independent experiments. CV%, percent coefficient of variation; ddPCR, droplet digital PCR; SD, standard deviation.
We have developed a multiplex detection methodology for ddPCR (2D ddPCR), which provides information about the integrity of the rAAV vg from a 2D plot of droplet fluorescence. Using this technique, we can analyze the quality of the rAAV2 vg by simultaneous detection with SV40 and ITR primers and probe, labeled with FAM (detected by channel 1) and HEX (detected by channel 2), respectively. As shown in Fig. 4, the distribution of droplets with FAM and HEX signals is separated into four groups: one double positive group (SV40+/ITR+ group, containing intact vg), two single positive groups (SV40+/ITR− group and SV40−/ITR+ group, containing incomplete vg), and one double negative group (SV40−/ITR− group, droplets containing no PCR product). After converting the number of droplets in each group to concentration according to the Poisson distribution, we determined that 59.32%, 11.88%, and 28.81% of the droplets were SV40+/ITR+, SV40+/ITR−, and SV40−/ITR+, respectively (Fig. 4A), showing that 40% of the rAAV2RSM templates contained incomplete vg. In contrast, SV40 and ITR 2D ddPCR detection of SmaI-digested vector plasmid were also divided into four groups, and the droplet ratio of SV40+/ITR+, SV40+/ITR−, and SV40−/ITR+ was 96.83%, 1.47% and 1.70%, respectively (Fig. 4C).
Figure 4.

2D ddPCR detection to evaluate the integrity of rAAV2RSM. Dot plot profiles of FAM-labeled SV40 primer and probe (channel 1, amplitude) and HEX-labeled ITR primer and probe (channel 2, amplitude). Droplets emitting 2D signals were separated into four groups as indicated. The number of droplets in each single or double positive group was calculated based on the Poisson distribution. The ratios of each group are indicated as percentages. (A) rAAV2RSM. (C), SmaI-digested pTR-UF11. (B) The dynamic linear range of each group was confirmed by detecting emission from a series of diluted samples. 2D, two-dimensional; FAM, fluorescein; HEX, hexachloro-6-carboxy-fluorescine.
The singleplex ddPCR system was reported that the dynamic range for absolute quantitation spans from a single copy up to ∼100,000 copies.14 However, for our 2D ddPCR, amplification targets, SV40 and ITR, exist on the same rAAV genome. That is, a single droplet containing more than one template DNA would overestimate the percentage of intact rAAV by our 2D ddPCR. Serial dilutions of rAAV2RSM from 20,000 to 100 copies/reaction were analyzed by ITR and SV40 2D ddPCR plots to determine the precise dynamic range. As shown in Fig. 4B, the detection values of the major amplicons (SV40+/ITR+ group) retained linearity throughout the entire range. However, the values of the minor amplicons (SV40+/ITR− or SV40−/ITR+ groups) showed leveling off near the upper limit and tailing at low concentrations, resulting in underestimation near the upper limit and overestimation at the lower limit. The ratio between each group was constant from 10,000 to 400 copies/reaction of total template in this case (Fig. 4B). In contrast, the linear range of rAAV8RSM is much narrower than that of rAAV2RSM (data not shown), demonstrating that proper detection ranges must be established for each system when using the ratio of data of each group as an indicator for evaluating the quality of rAAV vector products.
As already described, we could discriminate the intact rAAV genome (SV40+/ITR+ group) from incomplete genomes (SV40+/ITR− or SV40−/ITR+ groups) using 2D ddPCR. We, therefore, hypothesized that rAAV activity is correlated with the quantity of intact genome determined by 2D ddPCR. To confirm this hypothesis, rAAV2RSM stock was incubated at 37°C for 0, 1, 3, or 7 days, and analyzed by 2D ddPCR and qPCR. The activity of treated rAAV2RSM was determined by FACS analysis of GFP-expressed Vero cells 48 h postinfection. As expected, the activity of rAAV2RSM gradually decreased as the incubation time increased (Fig. 5A). Correlations between rAAV2RSM activity and the values obtained using qPCR or 2D ddPCR are plotted in Fig. 5B. The ddPCR titers of the SV40+/ITR+ group were highly correlated with rAAV2RSM activity with an R2 of 0.9847. However, the qPCR titer of the same samples determined using SV40 showed no relationship with AAV activity (Fig. 5B). The ratio of incomplete vg increased as the incubation time increased (Fig. 5C). The relationship between amount of the double positive population and the activity needs to be further established, our data revealed the potential of the 2D ddPCR assay to measure stability as a surrogate of the true activity assay.
Figure 5.

Correlation between the integrity of rAAV2RSM evaluated by ddPCR and transfection activity. (A) rAAV2RSM was incubated at 37°C for 0, 1, 3, or 7 days and activity was detected as TU toward Vero cell by FACS analysis after 48 h of infection. The activity of rAAV was demonstrated as the ratio to day 0. (B) Correlation between ddPCR or qPCR titer and TU activity. The vector genome titers of rAAV2RSM were simultaneously detected by qPCR (SV40 titer using ScaI-digested pTR-UF11 as standard) and ddPCR (ITR+/SV40+ group). Values are indicated as mean ± SD (activity n = 3, titer n = 4). (C) The percentage of incomplete vector genome of treated rAAV2RSM. TU, transduction activity.
2D ddPCR can be used to evaluate impurity of the plasmid DNA used in the manufacture of rAAV. To demonstrate this, we choose two primer probe sets against ITR (within the rAAV vg region) and a replication origin (ori, within the plasmid backbone), labeled them with HEX and FAM, respectively, and conducted 2D ddPCR analysis (ori and ITR 2D ddPCR) (Fig. 6A). Similar to the droplet profiles of the SV40 and ITR primer/probe sets, the droplets with FAM and HEX signals are distributed into four groups: the ori+/ITR+ group (vector plasmid), the ori+/ITR− group (plasmid backbone impurity), the ori−/ITR+ group (rAAV candidates), and the ori−/ITR− group (Fig. 6B). After converting the number of drops in each group to concentration according to the Poisson distribution, we determined that 5.76%, 13.9%, and 80.34% of the droplets were ori+/ITR+, ori+/ITR−, and ori−/ITR+, respectively, for rAAV2RSM (Fig. 6B), and 0.06%, 1.27%, and 98.14% for rAAV8RSM (Fig. 6C). This revealed that 20% of the rAAV2RSM and 1.3% of the rAAV8RSM templates contained impurity of plasmid backbone.
Figure 6.

Plasmid impurity of the rAAV vector products detected by 2D ddPCR. (A) Gene structure of positive strand of rAAV, dashed line is the plasmid backbone linked with the 5′ end and 3′ end of the ITR sequence. Narrow and thick arrows are indicated as the position of ITR and ori (replication origin) primer probe target site in this research, respectively. (B, C) Dot plot profiles of FAM-labeled ori primer probe (channel 1, amplitude) and HEX-labeled ITR primer probe (channel 2, amplitude). Droplets of 2D signals were separated into four groups. Ratio of each group is indicated in percentage. (B) rAAV2RSM. (C) rAAV8RSM. rAAV8RSM, rAAV reference standard material for serotype 8.
Discussion
qPCR is routinely utilized in rAAV titration due to its sensitivity, robustness, and accuracy, and thus qPCR was established by the international AAVRSM Working Group as an SOP for the vg titration of rAAVRSM. In this study, we analyzed the effect of the secondary structure of the target genome on qPCR titer by comparing the amplification of the rAAV2RSM genome, SmaI-digested plasmid, linearized plasmid, and circular plasmid DNA. As shown in Fig. 2A, the amplification efficiency of qPCR was impeded by the structure of the plasmid tail, especially when the amplification target was near the ITR structure. Therefore, a protocol for determining the absolute number of copies of the rAAV vector shouldo be established.
We attempted to use ddPCR to determine the number of genome copies of rAAV because this technique does not require a calibration standard, as previously mentioned by Lock et al.15 ddPCR was found to be compatible with a broad range of concentrations and provided reproducible measurements of rAAV2RSM genome, regardless of the amplification target selected, for example, the presence or absence of ITR secondary conformation. In contrast to the study of Lock et al.,15 we exploited 2D ddPCR by using two different primers and probes labeled with FAM and HEX, which provided information about the integrity of the rAAV vector whereas qPCR could not. In this article, analyses were conducted using two sets of primer/probes targeting the SV40 and ITR sequences, which are distantly positioned in the rAAV vg, and revealed that rAAV2RSM contains ∼40% incomplete genomes. These incomplete genomes might be degradation products formed during purification and genome extraction, due to impurity of the vector plasmid used for vector synthesis, or result from packaging of prematurely terminated defective interfering genomes,21 or packaged in a vector production process using HEK-293 cells.22
Most importantly, we revealed that the complete genome titer (SV40+/ITR+ group) detected using 2D ddPCR measurement highly correlated with infectivity (transduction activity). 2D ddPCR is the only evaluation method that simultaneously reflects integrity and activity. In this context, an accelerated (37°C) stability study instead of a long-term stability study revealed that loss of infectivity activity by rAAV may, in part, be due to the instability of the genome structure as the ratio of SV40−/ITR+ group and SV40+/ITR− group increased (Fig. 5C).
2D ddPCR can also be used to evaluate impurity of the plasmid DNA used in the manufacture of rAAV by choosing two primer probe sets against ITR (within the rAAV vg region) and a replication origin (ori, within the plasmid backbone), labeled them with HEX and FAM, respectively. We revealed that the ratio of impurities (from the vector plasmid or plasmid backbone) to the rAAV candidate of rAAV2RSM and rAAV8RSM was 20% and 1.3%, respectively (Fig. 6B, C). The qPCR ratio of plasmid impurity was also determined routinely by comparing the titer of the plasmid backbone (antibiotic-resistant gene or replication origin) with the rAAV genomes from separate qPCR detections. The values obtained were 1.87%, 1–8%, and 26.1% for the final bulk purified product of good manufacturing practice-produced self-complementary serotype 8 AAV vector for a hemophilia B clinical trial,23 a laboratory-made rAAV2 containing a single-stranded vg, and a laboratory-made self-complementary vector,24,25 respectively. In our experience, ratio of plasmid impurity in rAAVRSM determined by qPCR was less than that by 2D ddPCR (data not shown). Besides that, when we detected the ori titer by qPCR, the standard curve of ori was 2 Ct. shifted down than that of ITR. Therefore, the effect of the bias between the qPCR results of plasmid backbone amplicon and rAAV amplicon should not be ignored when using qPCR to evaluate the plasmid impurity of rAAV vector product.
In this study, we attempted to clarify the usefulness of ddPCR and 2D ddPCR for characterizing the single-stranded rAAV vector. The use of ddPCR provided the absolute number of copies of rAAV, and the use of optimized condition might show a correlation between the infection activity of rAAV and genome integrity. Nevertheless, additional inspection is necessary for the application of 2D ddPCR in self-complementary rAAV vector.
Overall, our study demonstrated the reliable measurement of rAAV vector titration by ddPCR without the need for a calibration standard, in contrast to qPCR. Moreover, the titer of the rAAV vector includes the intact viral genome detected by 2D ddPCR with two different primers and probes for the same virus genome. In addition, the titer of the intact viral genome was highly correlated to the transfection activity of the rAAV vector. The combination of different primers and probes allows quantification of, for example, residual vector plasmid DNA or cell host genome DNA, and helper virus used for vector production. Therefore, the detailed information regarding the composition of rAAV vectors provided by 2D ddPCR can be a powerful tool in titration, evaluating impurities, and stability of rAAV vectors for clinical trials.
Acknowledgment
This study was supported by Research on Regulatory Science of Pharmaceuticals and Medical Devices from the Japan Agency for Medical Research and Development (AMED).
Author Disclosure
All authors report no conflicts of interest and no personal financial interest directly relevant to the content of this article.
References
- 1. Maguire AM, Simonelli F, Pierce EA, et al. . Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med 2008;358:2240–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Scallan CD, Lillicrap D, Jiang H, et al. . Sustained phenotypic correction of canine hemophilia A using an adeno-associated viral vector. Blood 2003;102:2031–2037 [DOI] [PubMed] [Google Scholar]
- 3. George LA, Sullivan SK, Giermasz A, et al. . Hemophilia B gene therapy with a high-specific-activity factor IX variant. N Engl J Med 2017;377:2215–2227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gaudet D, Methot J, Kastelein J. Gene therapy for lipoprotein lipase deficiency. Curr Opin Lipidol 2012;23:310–320 [DOI] [PubMed] [Google Scholar]
- 5. Ferreira V, Twisk J, Kwikkers K, et al. . Immune responses to intramuscular administration of alipogene tiparvovec (AAV1-LPL(S447X)) in a phase II clinical trial of lipoprotein lipase deficiency gene therapy. Hum Gene Ther 2014;25:180–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. LoDuca PA, Hoffman BE, Herzog RW. Hepatic gene transfer as a means of tolerance induction to transgene products. Curr Gene Ther 2009;9:104–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Clark KR, Liu X, McGrath JP, et al. . Highly purified recombinant adeno-associated virus vectors are biologically active and free of detectable helper and wild-type viruses. Hum Gene Ther 1999;10:1031–1039 [DOI] [PubMed] [Google Scholar]
- 8. Moulay G, Scherman D, Kichler A. Fasting increases the in vivo gene delivery of AAV vectors. Clin Transl Sci 2010;3:333–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gonin P, Gaillard C. Gene transfer vector biodistribution: Pivotal safety studies in clinical gene therapy development. Gene Ther 2004;11(Suppl. 1):S98–S108 [DOI] [PubMed] [Google Scholar]
- 10. Lock M, McGorray S, Auricchio A, et al. . Characterization of a recombinant adeno-associated virus type 2 reference standard material. Hum Gene Ther 2010;21:1273–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moullier P, Snyder RO. International efforts for recombinant adeno-associated viral vector reference standards. Mol Ther 2008;16:1185–1188 [DOI] [PubMed] [Google Scholar]
- 12. Hou Y, Zhang H, Miranda L, et al. . Serious overestimation in quantitative PCR by circular (supercoiled) plasmid standard: Microalgal pcna as the model gene. PLoS One 2010;5:e9545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang F, Cui X, Wang M, et al. . A reliable and feasible qPCR strategy for titrating AAV vectors. Med Sci Monit Basic Res 2013;19:187–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hindson BJ, Ness KD, Masquelier DA, et al. . High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem 2011;83:8604–8610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lock M, Alvira MR, Chen SJ, et al. . Absolute determination of single-stranded and self-complementary adeno-associated viral vector genome titers by droplet digital PCR. Hum Gene Ther Methods 2014;25:115–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aurnhammer C, Haase M, Muether N, et al. . Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Hum Gene Ther Methods 2012;23:18–28 [DOI] [PubMed] [Google Scholar]
- 17. Pinheiro LB, Coleman VA, Hindson CM, et al. . Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal Chem 2012;84:1003–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vassalli G, Bueler H, Dudler J, et al. . Adeno-associated virus (AAV) vectors achieve prolonged transgene expression in mouse myocardium and arteries in vivo: A comparative study with adenovirus vectors. Int J Cardiol 2003;90:229–238 [DOI] [PubMed] [Google Scholar]
- 19. Slanina H, Weger S, Stow ND, et al. . Role of the herpes simplex virus helicase-primase complex during adeno-associated virus DNA replication. J Virol 2006;80:5241–5250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Werling NJ, Satkunanathan S, Thorpe R, et al. . Systematic comparison and validation of quantitative real-time PCR methods for the quantitation of adeno-associated viral products. Hum Gene Ther Methods 2015;26:82–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kapranov P, Chen L, Dederich D, et al. . Native molecular state of adeno-associated viral vectors revealed by single-molecule sequencing. Hum Gene Ther 2012;23:46–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burnham B, Nass S, Kong E, et al. . Analytical ultracentrifugation as an approach to characterize recombinant adeno-associated viral vectors. Hum Gene Ther Methods 2015;26:228–242 [DOI] [PubMed] [Google Scholar]
- 23. Allay JA, Sleep S, Long S, et al. . Good manufacturing practice production of self-complementary serotype 8 adeno-associated viral vector for a hemophilia B clinical trial. Hum Gene Ther 2011;22:595–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wright JF. Product-related impurities in clinical-grade recombinant AAV vectors: characterization and risk assessment. Biomedicines 2014;2:80–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schnodt M, Buning H. Improving the quality of adeno-associated viral vector preparations: the challenge of product-related impurities. Hum Gene Ther Methods 2017;28:101–108 [DOI] [PubMed] [Google Scholar]