

Abstract
Selective immunoglobulin M deficiency (SIGMD) is an uncommon primary immunodeficiency disorder. We herein report an SIGMD patient with autoimmune hepatitis. A 21-year-old Japanese man was transferred to our hospital because of acute liver dysfunction. His serum IgM level was low, whereas those of IgG and IgA were normal, indicating that he had SIGMD. We diagnosed him with acute-onset autoimmune hepatitis, and his liver function test findings gradually recovered with corticosteroid administration. Although SIGMD with autoimmune diseases has been reported, the clinical features and pathogenesis have not yet been clarified. We have summarized previous reports on SIGMD patients with autoimmune diseases.
Keywords: autoimmune hepatitis, selective immunoglobulin M deficiency
Introduction
Selective immunoglobulin M deficiency (SIGMD) is a rare primary immunodeficiency disease associated with serious infectious diseases, autoimmune diseases and malignancies (1). In SIGMD patients, the serum immunoglobulin M (IgM) level is below two standard deviations of the mean, and the IgG and IgA levels are normal (1). Although a number of patients with SIGMD have been reported after Hobbs et al. first described young patients with SIGMD in 1967 (2), few large-scale studies have explored the prevalence of SIGMD.
Autoimmune hepatitis (AIH), which is predominant in middle-age and elderly women, is a liver disease caused by the loss of tolerance to autoantigens (3). Although several studies have shown that autoimmune diseases are frequently present in adults with SIGMD, few SIGMD patients with AIH have been reported.
We herein report a young patient with SIGMD who experienced acute-onset AIH and review previous reports on SIGMD patients with autoimmune diseases.
Case Report
A 21-year-old Japanese man presented to a local hospital with fatigue and nausea. He had frequently experienced symptoms of a common cold for one year before visiting the hospital. Regarding his medical history, he had atopic dermatitis. He had no history of regular alcohol intake or smoking. He was not regularly taking any medicines, supplements, or illicit drugs.
He was admitted to the hospital because of liver dysfunction with high levels of serum aspartate aminotransferase (AST, 977 U/L) and alanine aminotransferase (ALT, 2,191 U/L) (Table 1). He was negative for hepatitis B surface antigen, hepatitis C virus antibody, anti-nuclear antibody (ANA), and anti-liver kidney microsome type 1 (anti-LKM-1) antibody. His serum level of IgM [reference range (RR) in the hospital: 33-190 mg/dL] was low (2 mg/dL), whereas those of IgG (RR: 870-1,700 mg/dL) and IgA (RR: 110-410 mg/dL) were normal (1,095 and 175 mg/dL, respectively). Abdominal computed tomography (CT) revealed periportal collar signs and edematous thickening of the gallbladder wall, indicating the presence of acute hepatitis (Fig. 1). CT did not show any signs of malignant disease. A percutaneous liver biopsy was performed nine days after his admission, and the histological examination did not show lobular/portal plasma cell infiltration, fibrosis, or centrilobular inflammation/necrosis, but moderate lobular inflammation and interface hepatitis in the portal areas was found (Fig. 2). No emperipolesis, cobblestone appearance, or rosette formation of hepatocytes was noted.
Table 1.
Laboratory Data on Admission to the Previous Hospital.
| WBC | 7,100 | /µL | TSH | 0.77 | µU/dL | |||
| Neut | 60.8 | % | free T3 | 2.65 | pg/mL | |||
| Eosi | 20.1 | % | free T4 | 1.16 | ng/dL | |||
| Baso | 0.6 | % | Na | 141 | mmol/L | |||
| Mono | 9.1 | % | K | 4 | mmol/L | |||
| Lymp | 9.4 | % | Cl | 103 | mmol/L | |||
| RBC | 4.87 | ×106/µL | Glucose | 105 | mg/dL | |||
| Hb | 14.5 | g/dL | HbA1c | 5.6 | % | |||
| Hct | 43.8 | % | IgG | 1,095 | mg/dL | |||
| PLT | 17.7 | ×104/µL | IgA | 175 | mg/dL | |||
| PT% | 96.5 | % | IgM | 2 | mg/dL | |||
| APTT | 31.9 | sec | ANA | <40 | × | |||
| Fibrinogen | 207 | mg/dL | Anti-LKM-1 Ab | <5.0 | index | |||
| T-Bil | 1.6 | U/L | Anti-ds DNA Ab | 2.4 | U/mL | |||
| D-Bil | 0.8 | U/L | Anti-Sm Ab | <1.0 | U/mL | |||
| AST | 977 | U/L | AMA | <20 | × | |||
| ALT | 2,191 | U/L | P-ANCA | <1.0 | U/mL | |||
| LDH | 435 | U/L | HBsAg | (-) | ||||
| ALP | 828 | U/L | HBsAb | (-) | ||||
| ChE | 208 | U/L | HBcAb | (-) | ||||
| Amylase | 114 | U/L | HBV DNA | (-) | ||||
| BUN | 13.7 | mg/dL | HCV Ab | (-) | ||||
| Creatinine | 0.69 | mg/dL | HCV RNA | (-) | ||||
| UA | 6.6 | mg/dL | HAV IgM | (-) | ||||
| TP | 6.8 | mg/dL | HEV IgA | (-) | ||||
| Albumin | 4.4 | mg/dL | EBV EA IgG | (-) | ||||
| TG | 135 | mg/dL | EBV VCA IgM | (-) | ||||
| T-Cho | 157 | mg/dL | EB VCA IgG | (+) | ||||
| Ferritin | 991 | ng/mL | EB EBNA | (-) | ||||
| NH3 | 39 | µg/dL | HSV IgM | (-) | ||||
| AFP | 2.6 | ng/mL | VZV IgM | (-) | ||||
| CEA | 2.0 | ng/mL | VZV IgG | (+) | ||||
| CA19-9 | 9.7 | U/mL | B19 IgM | (-) | ||||
| CMV IgM | (-) |
WBC: white blood cell, Neut: neutrophil, Eosi: eosinophil, Baso: basophil, Mono: monocyte, Lymp: lymphocyte, RBC: red blood cell, Hb: hemoglobin, Hct: hematocrit, PLT: platelet, PT: prothrombin time, APTT: activated partial thromboplastin time, T-Bil: total bilirubin, D-Bil: direct bilirubin, AST: aspartate aminotransferase, ALT: alanine aminotransferase, LDH: lactate dehydrogenase, ALP: alkaline phosphatase, ChE: cholinesterase, BUN: blood urea nitrogen, UA: uric acid, TP: total protein, TG: triglycerides, T-Cho: total cholesterol, AFP: alpha-fetoprotein, CEA: carcinoembryonic antigen, CA19-9: carbohydrate antigen 19-9, TSH: thyroid-stimulating hormone, Ig: immunoglobulin, ANA: anti-nuclear antibody, Anti-LKM-1 Ab: anti-liver kidney microsome type 1 antibody, Anti-ds DNA Ab: anti-double stranded DNA IgG antibody, Anti-Sm Ab: anti-Smith antibody, AMA: anti-mitochondrial antibody, p-ANCA: myeloperoxidase-anti-neutrophil cytoplasmic antibody, HBs Ag: hepatitis B surface antigen, HBs Ab: hepatitis B surface antibody, HBc: hepatitis B core, HCV: hepatitis C virus, HAV: hepatitis A virus, HEV: hepatitis E virus, EBV: Epstein-Barr virus, EA: early antigen, VCA: virus capsid antigen, EBNA: EBV nuclear antigen, HSV: herpes simplex virus, VZV: varicella zoster virus, B19: Parvovirus B19, CMV: cytomegalovirus
Figure 1.

Abdominal contrast enhanced computed tomography scans. (A) Areas of low attenuation around the portal vein and its branches (periportal collar signs) were recognized. (B) Edematous thickening of the gallbladder wall appeared.
Figure 2.
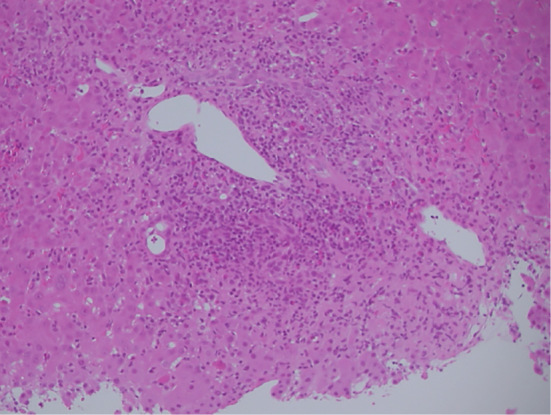
An image of a liver biopsy specimen (Hematoxylin and Eosin staining, original magnification ×200). The presence of interface hepatitis with dense portal lymphocyte infiltrate disrupting the limiting plate is shown.
He was diagnosed with acute-onset AIH based on the pathological findings, and steroid therapy with prednisolone (PSL, 50 mg/day) was started, with the dose later tapered (Fig. 3). However, while his liver function gradually recovered, the serum aminotransferase levels were still abnormal despite the patient taking 30 mg/day PSL. At 36 days after his admission, he was transferred to our hospital for further examinations and treatment.
Figure 3.

The clinical course of the present case after admission to the previous hospital. ALT: alanine aminotransferase, AST: aspartate aminotransferase, IgG: Immunoglobulin G, UDCA: ursodeoxycholic acid
On admission to our hospital, he was 170 cm in height and 53.8 kg in weight with a blood pressure of 103/67 mmHg, pulse 62 beats per minute and regular, and body temperature 36.4℃. His consciousness was lucid. He did not have icterus or abdominal symptoms.
Regarding laboratory data, his serum IgM level (RR in our hospital: 33-183 mg/dL) was extremely low (<4 mg/dL) although the serum IgG (RR: 861-1,747 mg/dL) and IgA (RR: 93-393 mg/dL) levels were almost normal (867 mg/dL and 150 mg/dL, respectively), as the laboratory data from the previous hospital had shown. A flow cytometric analysis of the peripheral blood revealed normal rates of CD4+ and CD8+ T lymphocytes (36.8% and 26.3%, respectively) and CD19+ B lymphocytes (28.1%). He was positive for anti-Epstein Barr virus capsid antigen IgG, and anti-Varicella Zoster Virus IgG, indicating that his specific IgG producibility was normal. Lymphocyte blastoid transformation by process hazard analysis (PHA) did not show dysfunction of T cells (PHA+: 16,604 cpm, control: 113 cpm). These results indicated that he had SIGMD as the underlying disease.
The laboratory data also showed high levels of serum AST (66 U/L) and ALT (188 U/L). According to the revised scoring system of the International Autoimmune Hepatitis Group (IAIHG) (4), he was scored as probable AIH (score 12). Because some human leukocyte antigen (HLA) types have been reported to be risk factors for AIH, we determined his HLA haplotype, and it was revealed that he had DRB1*04:05 and DQB1*04:01, both of which have been reported to be associated with AIH (5). Considering these laboratory data and pathological findings along with the evidence from previous reports that adults with SIGMD frequently develop autoimmune diseases, he was diagnosed as having acute-onset AIH with SIGMD.
We continued steroid therapy (PSL 30 mg/day) and tapered the dose. During the tapering of the corticosteroid dose, ursodeoxycholic acid (UDCA, 600 mg/day) was added, and the liver function gradually recovered. During the treatment period, the serum level of IgM was not changed, while that of IgG was slightly reduced. On the 26th day after the admission to our hospital, he was discharged with PSL (15 mg/day) and UDCA (600 mg/day). Eight days after discharge, his serum AST and ALT levels increased once again (177 and 485 U/L, respectively). The dose of PSL was therefore increased to 30 mg/day and tapered again more slowly. His serum aminotransferase levels subsequently decreased gradually, and six months after the start of PSL, they had normalized.
Discussion
SIGMD is a disorder characterized by a low level of serum IgM and normal IgG and IgA levels with a normal T cell function. Some patients with SIGMD may be asymptomatic, but more commonly, the frequency of infections with microbes is reported to be higher than in healthy people (6). There have been few large-scale studies concerning the prevalence of patients with SIGMD. Entezari et al. reported that the prevalence of patients with SIGMD was 0.37% in the screening of a blood bank of more than 3,000 healthy adults (7). Another cohort study obtained from a retrospective analysis based on a PubMed database reported that the prevalence of SIGMD was 0.26% (8). A complete absence (<4 mg/dL) of serum IgM is relatively uncommon; indeed, in a cohort of 55 patients with SIGMD, only 4 patients had complete absence of serum IgM (1).
Although familial cases of SIGMD have been reported (9), no inheritance patterns or responsible genes have yet been identified. The European Society for Immunodeficiencies Registry defines SIGMD as a serum IgM level repeatedly below two standard deviations of normal with normal levels of serum IgA, IgG, and IgG subclasses, an absence of T cell defects, and an absence of causative external factors (http://www.esid.org). Our case showed a complete absence of serum IgM repeatedly and almost normal IgG and IgA. In addition, a flow cytometric analysis of peripheral blood revealed almost normal rates of CD4+ and CD8+ T lymphocytes and CD19+ B lymphocytes, and lymphocyte blastoid transformation by PHA showed no dysfunction of T cells. External factors causing SIGMD were not found. Based on these findings, we diagnosed the present patient with SIGMD.
AIH is a disease of the hepatic parenchyma that can present in acute or chronic forms, in common with many autoimmune diseases. AIH is associated with non-organ-specific antibodies in the context of hepatic autoimmunity (10). In Japan, a study in a certain region revealed that the incidence and prevalence of AIH were 2.23 and 23.4 per 100,000, respectively (age-standardized to the Japanese population) (11). AIH often presents as a chronic disease; however, it can also have an acute presentation (12). A nationwide survey in Japan revealed that acute hepatitis was seen in 11.7% of patients with AIH (13). Acute AIH has some characteristics: (a) recent onset (<30 days), (b) significant clinical symptoms, (c) remarkable biochemical elevations (serum bilirubin level and/or aminotransferase levels) (12). Although serum IgG or autoantibody levels are frequently elevated in chronic AIH, they may not be elevated in patients with acute-onset AIH (12,14-16). Therefore, these laboratory data are sometimes not reliable. The histological features are also varied, and previous studies have reported that lobular/portal inflammation and interface hepatitis were frequently observed in patients with acute-onset AIH (17,18). The IAIHG criteria have been often used for the diagnosis of AIH (4), but the scoring system may be also unreliable in patients with acute-onset AIH. A previous study reported that the frequency of an overall diagnosis of AIH by the IAIHG system among 70 patients with fulminant liver failure caused by acute-onset AIH was 40% (19). Therefore, the diagnosis of acute-onset AIH needs to be made comprehensively from various viewpoints.
The present patient showed acute-onset clinical symptoms of fatigue and nausea and markedly elevated serum aminotransferase levels, suggesting he had acute hepatitis. Evidence of a viral infection, alcoholic hepatitis, drug-induced liver injury, or other liver diseases was absent. Although he showed atypical findings of AIH, with the absence of elevated levels of serum IgG and autoantibodies, he was defined as probable AIH according to the revised IAIHG scoring system, and the histological findings were consistent with the features of AIH. Furthermore, he had HLA-DR/DQ types, which have been reported to be associated with AIH in Japan (5). In addition, autoimmune diseases are more frequent in patients with SIGMD than in the general population (1), with an estimated rate of 8% according to a long-term follow-up study (6). Given the above, we diagnosed him with acute-onset AIH and started corticosteroid therapy. A previous study noted that, in acute-onset AIH, the response rate to PSL therapy ranges from 36% to 100% (12). In our case, the reaction to corticosteroid treatment was relatively good, and his serum aminotransferase levels have remained adequate with oral low-dose of PSL. This good response to steroid therapy also supports the diagnosis of AIH.
Previous reports of autoimmune diseases associated with SIGMD on PubMed are presented in Table 2 (20-30). Although collagen diseases or thyroid diseases with SIGMD have been reported relatively frequently, there is only one case report of AIH with SIGMD. No other cases of AIH accompanied by the “complete” absence of serum IgM have been reported.
Table 2.
Reported Cases of Autoimmune Diseases with SIGMD.
| Age (years) |
Gender | Associated disease | IgM (mg/dL) |
IgG (mg/dL) |
Reference | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 51 | Female | SLE | 10 | 2,340 | (21) | |||||
| 43 | Female | SLE | 5 | 1,426 | (22) | |||||
| 37 | Female | SLE | 15 | 1,506 | (22) | |||||
| 33 | Female | SLE | 17 | 1,532 | (22) | |||||
| 47 | Female | SLE | 23 | 1,567 | (22) | |||||
| 41 | Female | SLE | 25 | 1,900 | (22) | |||||
| 42 | Female | SLE | 28 | 1,316 | (22) | |||||
| 36 | Male | SLE | 30 | 1,267 | (22) | |||||
| 59 | Female | SLE | 32 | 2,546 | (22) | |||||
| 30 | Female | SLE | 37 | 1,109 | (22) | |||||
| 63 | Female | SLE | 43 | 1,319 | (22) | |||||
| 36 | Female | SLE | 44 | 1,226 | (22) | |||||
| 55 | Female | SLE | 44 | 1,441 | (22) | |||||
| 29 | Female | SLE | 25 | 1,597 | (23) | |||||
| 30 | Male | SLE | 6 | 936 | (23) | |||||
| 73 | Female | SLE | 12 | 1,180 | (24) | |||||
| 77 | Female | SLE | 18 | 1,120 | (24) | |||||
| 23 | Male | Celiac disease | 48 | 2,100 | (25) | |||||
| 26 | Male | Celiac disease | 27 | 1,700 | (25) | |||||
| 75 | Male | Celiac disease | 48 | 1,700 | (25) | |||||
| 52 | Male | Celiac disease | <32 | NA | (20) | |||||
| 63 | Female | Sjögren’s syndrome | <5 | 3,030 | (24) | |||||
| 53 | Female | Sjögren’s syndrome | 16 | 1,500 | (24) | |||||
| 70 | Male | Hashimoto’s disease | <3 | 1,972 | (25) | |||||
| 50 | Female | Hashimoto’s disease | 34 | 921 | (26) | |||||
| 39 | Female | Hypothyroidism | 16 | 729 | (26) | |||||
| 65 | Male | Hypothyroidism | <5 | 1,350 | (24) | |||||
| 67 | Male | Hypothyroidism | 27 | NA | (27) | |||||
| 53 | Female | Rheumatoid arthritis | 17 | 2,159 | (23) | |||||
| 46 | Female | Rheumatoid arthritis | 39 | 985 | (26) | |||||
| 68 | Female | CREST syndrome | 37 | NA | (27) | |||||
| 57 | Male | Autoimmune glomerulonephritis | 34 | 1,500 | (28) | |||||
| 34 | Male | Psoriasis pustulosa | 1 | 1,314 | (29) | |||||
| 64 | Female | Autoimmune hepatitis | 11 | 2,942 | (30) | |||||
| 21 | Male | Autoimmune hepatitis | 2 | 1,095 | Present case |
SIGMD: selective immunoglobulin M deficiency, IgM: immunoglobulin M, IgG: immunoglobulin G, SLE: systematic lupus erythematosus, NA: not available
The mechanisms underlying autoimmunity with SIGMD are unknown. One suggested possibility is cross-linking of IgM Fc receptor (FcμR) and B-cell receptor (BCR) by IgM autoantibody-self-antigen complexes, causing the induction of anergy (20). Mouse mutants for FcμR showed impairment in specific responses of IgG antibodies against T-dependent and T-independent antigens, resulting in the development of autoimmunity (31). In addition, the deficiency of CXCR3+ B-cells in SIGMD patients and a relationship between CXCR3+ cells and autoimmunity have been reported (32,33). Other potential mechanisms reported in the literature include the loss of central tolerance as a result of BCR editing, loss of peripheral tolerance due to a deficiency of isotype-specific regulatory lymphocytes, and an impaired clearance of apoptotic cells (1). In the present AIH case, high levels of serum IgG or autoantibodies were not seen, probably because AIH was caused by the loss of tolerance or an impairment of anergy resulting from a deficiency in IgM by the mechanisms described above, which differed from those in patients with typical AIH.
We herein report a rare case of a young man who developed acute-onset AIH with SIGMD. Corticosteroid therapy has been effective, and the state of AIH has been well-controlled. Although the risk of serious infections is regarded as smaller in adult patients with SIGMD than in pediatric patients (1), an adult patient with a life-threatening infection has been reported (34). We should carefully treat the complications of infectious diseases because of the need for low-dose long-term corticosteroid administration. Because cases of autoimmune diseases, especially AIH, accompanied by SIGMD have seldom been reported, further studies are needed to reveal the etiology, pathogenesis, and clinical features.
The authors state that they have no Conflict of Interest (COI).
References
- 1. Gupta S, Gupta A. Selective IgM deficiency-an underestimated primary immunodeficiency. Front Immunol 8: 1-7, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hobbs JR, Milner RDG, Watt PJ. Gamma-M deficiency predisposing to meningococcal septicaemia. Br Med J 4: 583-586, 1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sahebjam F, Vierling JM. Autoimmune hepatitis. Front Med 9: 187-219, 2015. [DOI] [PubMed] [Google Scholar]
- 4. Alvarez F, Berg PA, Bianchi FB, et al. . International Autoimmune Hepatitis Group Report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol 31: 929-938, 1999. [DOI] [PubMed] [Google Scholar]
- 5. Umemura T, Katsuyama Y, Yoshizawa K, et al. . Human leukocyte antigen class II haplotypes affect clinical characteristics and progression of type 1 autoimmune hepatitis in Japan. PLoS ONE 9: e100565, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Louis AG, Gupta S. Primary selective IgM deficiency: an ignored immunodeficiency. Clin Rev Allergy Immunol 46: 104-111, 2014. [DOI] [PubMed] [Google Scholar]
- 7. Entezari N, Adab Z, Zeydi M, et al. . The prevalence of selective immunoglobulin M deficiency (SIgMD) in Iranian volunteer blood donors. Hum Immunol 77: 7-11, 2016. [DOI] [PubMed] [Google Scholar]
- 8. Goldstein MF, Goldstein AL, Dunsky EH, Dvorin DJ, Belecanech GA, Shamir K. Selective IgM immunodeficiency: retrospective analysis of 36 adult patients with review of the literature. Ann Allergy Asthma Immunol 97: 717-730, 2006. [DOI] [PubMed] [Google Scholar]
- 9. Jones DM, Tobin BM, Butterworth A. Three cases of meningococcal infection in a family, associated with a deficient immune response. Arch Dis Child 48: 742-743, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heneghan MA, Yeoman AD, Verma S, Smith AD, Longhi MS. Autoimmune hepatitis. Lancet 382: 1433-1444, 2013. [DOI] [PubMed] [Google Scholar]
- 11. Yoshizawa K, Joshita S, Matsumoto A, et al. . Incidence and prevalence of autoimmune hepatitis in the Ueda area, Japan. Hepatol Res 46: 878-883, 2016. [DOI] [PubMed] [Google Scholar]
- 12. Wang Q, Yang F, Miao Q, Krawitt EL, Gershwin ME, Ma X. The clinical phenotypes of autoimmune hepatitis: a comprehensive review. J Autoimmun 66: 98-107, 2016. [DOI] [PubMed] [Google Scholar]
- 13. Takahashi A, Arinaga-Hino T, Ohira H, et al. . Autoimmune hepatitis in Japan: trends in a nationwide survey. J Gastroenterol 52: 631-640, 2017. [DOI] [PubMed] [Google Scholar]
- 14. Ohira H, Abe K, Takahashi A, Watanabe H. Autoimmune hepatitis: recent advances in the pathogenesis and new diagnostic guidelines in Japan. Intern Med 54: 1323-1328, 2015. [DOI] [PubMed] [Google Scholar]
- 15. Ohira H. Current status and issues of autoimmune hepatitis in Japan. Kanzo 56: 167-178, 2015(in Japanese). [Google Scholar]
- 16. Joshita S, Yoshizawa K, Umemura T, et al. . Clinical features of autoimmune hepatitis with acute presentation: a Japanese nationwide survey. J Gastroenterol 53: 1079-1088, 2018. [DOI] [PubMed] [Google Scholar]
- 17. Okano N, Yamamoto K, Sakaguchi K, et al. . Clinicopathological features of acute-onset autoimmune hepatitis. Hepatol Res 25: 263-270, 2003. [DOI] [PubMed] [Google Scholar]
- 18. Nguyen Canh H, Harada K, Ouchi H, et al. . Acute presentation of autoimmune hepatitis: a multicentre study with detailed histological evaluation in a large cohort of patients. J Clin Pathol 70: 961-969, 2017. [DOI] [PubMed] [Google Scholar]
- 19. Yeoman AD, Westbrook RH, Al-Chalabi T, et al. . Diagnostic value and utility of the simplified International Autoimmune Hepatitis Group (IAIHG) criteria in acute and chronic liver disease. Hepatology 50: 538-545, 2009. [DOI] [PubMed] [Google Scholar]
- 20. Chen S, Shamriz O, Toker O, Fridlender ZG, Tal Y. Recurrent eosinophilic pneumonia in a patient with isolated immunoglobulin M deficiency and celiac disease. Isr Med Assoc J 17: 526-527, 2015. [PubMed] [Google Scholar]
- 21. Ohno T, Inaba M, Kuribayashi K, Masuda T, Kanoh T, Uchino H. Selective IgM deficiency in adults: phenotypically and functionally altered profiles of peripheral blood lymphocytes. Clin Exp Immunol 68: 630-637, 1987. [PMC free article] [PubMed] [Google Scholar]
- 22. Saiki O, Saeki Y, Tanaka T, et al. . Development of selective IgM deficiency in systemic lupus erythematosus patients with disease of long duration. Arthritis Rheum 30: 1289-1292, 1987. [DOI] [PubMed] [Google Scholar]
- 23. Inoue T, Okumura Y, Shirahama M, et al. . Selective partial IgM deficiency: functional assessment of T and B lymphocytes in vitro. J Clin Immunol 6: 130-135, 1986. [DOI] [PubMed] [Google Scholar]
- 24. Chovancova Z, Kralickova P, Pejchalova A, et al. . Selective IgM deficiency: clinical and laboratory features of 17 patients and a review of the literature. J Clin Immunol 37: 559-574, 2017. [DOI] [PubMed] [Google Scholar]
- 25. Blecher TE, Brzechwa-Ajdukiewicz A, McCarthy CF, Read AE. Serum immunoglobulins and lymphocyte transformation studies in coeliac disease. Gut 10: 57-62, 1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yel L, Ramanuja S, Gupta S. Clinical and immunological features in IgM deficiency. Int Arch Allergy Immunol 150: 291-298, 2009. [DOI] [PubMed] [Google Scholar]
- 27. Janssen LMA, Macken T, Creemers MCW, Pruijt JFM, Eijk JJJ, Vries E. Truly selective primary IgM deficiency is probably very rare. Clin Exp Immunol 191: 203-211, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Antar M, Lamarche J, Peguero A, Reiss A, Cole S. A case of selective immunoglobulin M deficiency and autoimmune glomerulonephritis. Clin Exp Nephrol 12: 300-304, 2008. [DOI] [PubMed] [Google Scholar]
- 29. Yamasaki T. Selective IgM deficiency: functional assessment of peripheral blood lymphocytes in vitro. Intern Med 31: 866-870, 1992. [DOI] [PubMed] [Google Scholar]
- 30. Arahata M, Tajiri K, Nomoto K, Tsuneyama K, Minami S, Shimizu Y. A novel type of selective immunoglobulin M deficiency in a patient with autoimmune liver cirrhosis with recurrent hepatocellular carcinoma: a case report and review of the literature. Int Arch Allergy Immunol 161: 91-96, 2013. [DOI] [PubMed] [Google Scholar]
- 31. Gupta S, Agrawal S, Gollapudi S, Kubagawa H. FcμR in human B cell subsets in primary selective IgM deficiency, and regulation of FcμR and production of natural IgM antibodies by IGIV. Hum Immunol 77: 1194-1201, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Louis AG, Agrawal S, Gupta S. Analysis of subsets of B cells, Breg, CD4Treg and CD8Treg cells in adult patients with primary selective IgM deficiency. Am J Clin Exp Immunol 5: 21-32, 2016. [PMC free article] [PubMed] [Google Scholar]
- 33. Henneken M, Dörner T, Burmester GR, Berek C. Differential expression of chemokine receptors on peripheral blood B cells from patients with rheumatoid arthritis and systemic lupus erythematosus. Arthritis Res Ther 7: R1001-R1013, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hong R, Gupta S. Selective immunoglobulin M deficiency in an adult with Streptococcus pneumoniae sepsis and invasive aspergillosis. J Investig Allergol Clin Immunol 18: 214-218, 2008. [PubMed] [Google Scholar]