

Abstract
Recent developments in high-resolution mass spectrometry (HRMS) technology enabled ultrasensitive detection of proteins, peptides, and metabolites in limited amounts of samples, even single cells. However, extraction of trace-abundance signals from complex datasets (m/z value, separation time, signal abundance) that result from ultrasensitive studies requires improved data processing algorithms. To bridge this gap, we here developed “Trace,” a software framework that incorporates machine learning (ML) to automate feature selection and optimization for the extraction of trace-level signals from HRMS data. The method was validated using primary (raw) and manually curated datasets from single-cell metabolomic studies of the South African clawed frog (Xenopus laevis) embryo using capillary electrophoresis electrospray ionization HRMS. We demonstrated that Trace combines sensitivity, accuracy, robustness with high data processing throughput to recognize signals, including those previously identified as metabolites in single-cell capillary electrophoresis HRMS measurements that we conducted over several months. These performance metrics combined with a compatibility with MS data in open-source (mzML) format make Trace an attractive software resource to facilitate data analysis for studies employing ultrasensitive high-resolution MS.
Graphical Abstract

INTRODUCTION
Recent advances in high-resolution mass spectrometry (HRMS) extended this powerful technology to the detection of important biomolecules in limited amounts of samples, even single cells (reviewed in references 1–10). However, broader adoption of ultrasensitive HRMS is hampered by a limited number of software tools capable of extracting trace-level signals from complex datasets. Trace amounts of materials from tissue biopsies, small populations of cells, or single cells yield signals with lower signal-to-noise ratios (SNRs) than expected from classical cell-pooling studies. Exacerbating challenges in sensitivity, most successful software packages assume chromatographic peak shapes, potentially leaving transient peaks undetected from trace-level analyses by emerging separation technologies such as fast nano-flow liquid chromatography (nanoLC), capillary electrophoresis (CE), and ion mobility separation. Although semi-manual data analysis alleviates some of these challenges,11 manual inspection of hundreds of gigabytes of data is labor-intensive, has low throughput, and is practically unscalable to system-wide studies generating hundreds of files for analysis. For ultrasensitive HRMS to gain broader throughput and adaptation in bioanalysis, including in single-cell studies, software packages are needed with a capability to recognize trace-level signals from complex HRMS datasets.
Many successful software packages designed for HRMS laid the groundwork to detect peaks and perform metabolomics data analysis. Representative successful toolboxes for LC- and gas chromatography (GC) MS datasets include but are not limited to XCMS,12,13 Metabox,14 and MetaboAnalyst.15 Noise reduction with traditional signal processing techniques is usually the first step of data processing in the m/z or chromatographic dimension. Typically, molecular features are surveyed based on m/z-selected chromatograms (also known as extracted ion chromatograms, EICs) by moving average windows,16 median filters,17 or a wavelet transform.18 The resulting molecular features are credentialed using various algorithms. For example, VIPER,19 OpenMS,20 MZmine,21 vectorized peak detection,17 and XCMS22 examine the m/z or time dimension to find chromatographic peaks with satisfactory shape (e.g., Gaussian) and signal-to-noise ratio (SNR). Other software tools, such as LCMS-2D23 and MapQuand,24 simultaneously survey the m/z and time spaces for peak detection. However, to enhance robustness, thresholding by SNR often needs to be augmented with additional peak finding features, such as evaluation of Gaussian similarity, peak width, and shape (e.g., noisy or zigzag).
Signal detection for trace-sensitive HRMS calls for advances in data analysis. Although computationally cost-efficient, binning along the m/z dimension risks the recognition of trace-level signals by potentially splitting them or merging them with other signals, including those due to chemical background or electronic noise. EICs successfully recover abundant signals by taking into consideration the time of separation (t) for a given m/z value. However, limiting data analysis to signals with predefined m/z and separation time (molecular features) constitutes an essentially rule-based framework, which although aids the selection of targeted signals, still inefficiently discriminates signals with relatively low SNR. For example, we observed that six typically used features (zigzag, Gaussian similarity, SNR, significance, TPAR, and sharpness)25 form broad and overlapping distributions for both true and false signals. These engineered features thus fail to discriminate true signals from false ones for border-case SNRs. Recently, semi-automated approaches of targeted data analysis successfully recognized trace-sensitive metabolite signals during single-cell HRMS,11,26–28 albeit at the expense of low data analysis throughput, high manual labor, and requirement for significant expertise in computation. To enhance trace-level studies using HRMS, software tools capable of automated and high-fidelity inspection are needed.
Here we developed Trace, a software framework that uses deep machine learning (ML) techniques for signal detection for HRMS (Fig. 1). We first treated the naturally three-dimensional information from HRMS—these are the m/z, time of separation (t), and signal abundance (peak area or peak height)—as two-dimensional images (m/z, t). Next, we applied deep convolution neural networks for pattern recognition. After training the model on a reference dataset, we validated our neural network model for accuracy. Comparison with popular data processing strategies (XCMS and MetaboAnalyst) suggests that the neural network model presented here achieves consistent prediction of high accuracy and low false positive rates, which we demonstrated using data from single-cell metabolomic analysis of identified Xenopus laevis embryonic cells. Trace was found to be sufficiently robust, reproducible, and sensitive to facilitate large-scale trace-sensitive studies by HRMS.
Figure 1.
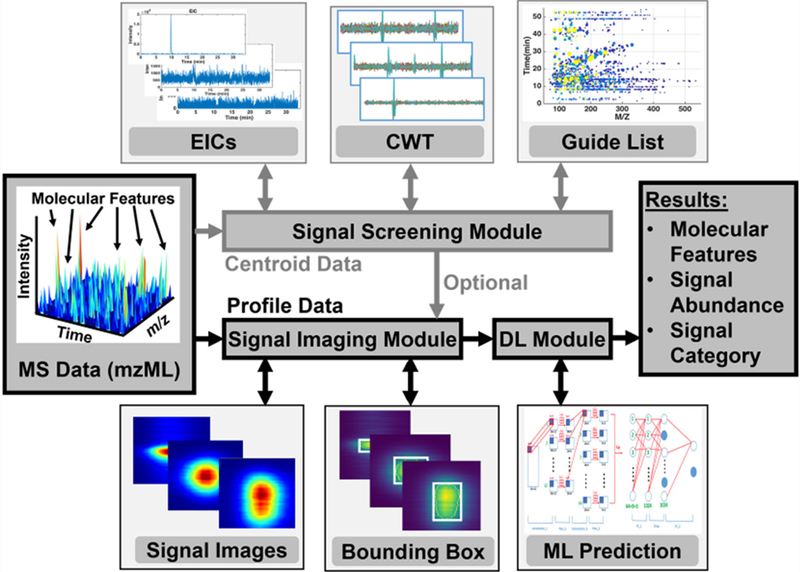
Flowchart of signal detection highlighting major data processing modules in Trace. (Top panel) Optionally, the “signal screening module” is executed to construct a series of extracted ion chromatograms (EICs) for inspection by a continuous wavelet transform (CWT), thus generating a “guide list” of signal candidates (m/z and separation time values) for follow-up data analysis. (Bottom panel) A trained convolution neural network validates each potential signal (true or false) by generating the corresponding “signal image”, which is then rescaled to a bounding box to be fed to a convolution network tabulating predicted true signals. The results are a list of molecular features (m/z and separation time) and their corresponding signal abundance (peak height or area).
EXPERIMENTAL SECTION
Study Design.
This study was designed to develop and validate Trace and to test its utility for high-throughput metabolomics. To approximate high biological variability and varying experimental conditions, which often result from large-scale metabolomic studies, we pooled CE-ESI-HRMS data that were recorded on N = 5 different D11 cells in this study or acquired during our earlier experiments. Each cell was measured from a different embryo from a different set of parents and analyzed in technical replicates. A unique identifier assigned to each resulting file is described in Supplementary Information Table 1 (Table S1). For example, “D11 cellE1T1” corresponds to the first technical replicate analysis of the D11 cell that was sampled from the first embryo in this study. For a bias-free evaluation of software performance, the identity of each file was blinded during development and testing of Trace; this information was only revealed after data analysis to aid results interpretation.
Single-cell HRMS of Identified Embryonic Cells.
Experimental details are provided in the SI document. All protocols related to the humane care and treatment of Xenopus laevis were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Maryland, College Park (IACUC # R-DEC-17-57) or the George Washington University (IACUC # A311). D11 cells were identified in 16-cell X. laevis embryos, whence 10 nL were aspirated in situ using a pulled capillary following our recent protocol.27 Metabolites were extracted from the aspirate and analyzed using a laboratory-built CE-ESI platform that was coupled to a quadrupole time-of-flight mass spectrometer (Impact HD, Bruker Daltonics, Billerica, MA). This platform and its operation were described in detail elsewhere.26,27
Data Analysis.
Raw MS data were processed following our established protocols.26,27 Trace software codes were developed in Python (version 2.7) and are provided with a User Manual in the SI document.
Safety Considerations.
Chemicals and biological samples were handled following standard safety procedures. CE capillaries and ES emitters, which pose a potential needle-stick hazard, were handled with gloves and safety goggles. To prevent users from exposure to high voltage, which presents electrical shock hazard, all connective parts of the CE-ESI setup were earth-grounded or isolated in a Plexiglass enclosure equipped with a safety interlock-enabled door.
RESULTS AND DISCUSSION
Trace: Software Design and Implementation.
Trace was designed to integrate several interconnected modules with each carrying out a specific task (Fig. 1). Optionally, the “signal screening” module may initially be utilized to speed up downstream data processing. Extracted ion chromatograms (EICs) were obtained by projecting the intensity profile I (m/z, t) of centroid data from the (m/z, t) space to time by binning m/z into a series of discrete values with a specific bin size, thus reducing dimensionality and data size. For example, with a mass accuracy of <5 ppm and a typical signal width of 0.010 Da in our FIRMS dataset, a bin width of ± 0.005 m/z unit was chosen for this study. For each centroid (m/z)0, the maximal ion signal intensity within a range ((m/z)0 − Δw, (m/z)0 + Δw) was assigned as the intensity of (m/z)0. A continuous wavelet transform (CWT) approach (reviewed in reference 18) was adopted to rapidly screen peaks along each monitored m/z channel (EICs). For each EIC, the CWT was applied using the Python package “SciPy.signal.” A complete list of user-controlled parameters, including cutoff values that require optimization in a given study, is provided in the SI (see “Trace User Manual”). Last, by screening the EICs corresponding to each discrete m/z value with increasingly more relaxed cutoffs, we obtained larger datasets of potential signals that we expected to reflect most of the true signals. The center location ((m/z)0, t0) of each potential signal was recorded for further analysis.
The “signal imaging” module was designed to probe the local spectral environment for each signal. We used the gridded profile data, viz. I(m/z, t), to extract signal intensity around each detected signal centered at ((m/z)0, t0). The imaging window was set at 12 bins (0.002 Da for each) in the m/z dimension and 60 bins (30 s /60 = 0.5 s for each) in the temporal dimension. The generated 12 × 60 pixel2 images, which are referred to as “signal images” in this study, essentially enquire the chemical space around each molecular feature. To outline the contour of an actual signal and the minimal rectangle encompassing it, these signal images were segmented using the OpenCV package as follows: for each image, pixel values were normalized to the range of 0–255, and a threshold of 130 was set for identifying the image contour. Abounding box was defined as the minimal rectangle that contains the contour associated with the image center and has a size larger than 1/60 of the image size. The bounding box was resized to 40 × 20 pixel2 with interpolation. Therefore, each image contains only one bounding box as a result of this step. Finally, if an image contained no bounding box, it was labeled as a false signal and eliminated from future processing. Trace also provides users with the option to adjust the size of these settings as needed for a particular study.
The “machine learning” (ML) module was tasked to categorize signal images as true or false signals. To automate signal assessment, we employed deep learning (DL) models, which efficiently leverage an example dataset or past experience to solve/predict an outcome,29 namely true vs. false in this work. Encouraged by successful image recognition using ML/DL across wide applications, such as medical image analysis30, pathology31, astrophysics32, biology33, and FIRMS34, we adopted a convolutional neural network (CNN) for signal shape evaluation. CNN represents a class of deep neural networks that require no prior feature engineering. Instead, CNN learns a set of weight-sharing filters to represent features at different scales. This scheme of ab initio feature selection presents a major advantage of CNN.35 The well-known LeNet5 CNN has achieved a remarkable precision in handwritten digit recognition for MNIST dataset.36
We utilized a similar network architecture as LeNet5 and implemented it with Google’s DL framework TensorFlow37. The input to our network was the signal image of size 60×12 pixel2 (or 40×20 pixel2 if the bounding box detection is included). The first hidden layer of the network was composed of 32 convolutional kernels of size 4 × 4 pixel2 followed by a 2 × 2 pixel2 max pooling process. These convolutional kernels served as filters to extract local features of images, while the max pooling process further abstracts the local features. The convolutions were zero padded with stride of 1 so that the output has the same size as the input. The second layer was similar to the first layer, except it featured 64 instead of 32 convolutional kernels. The third layer contained 256 densely connected neurons. To reduce possible overfitting, dropout with a rate of 0.5 was applied before the output layer for the training process. For the testing process and subsequent prediction, the dropout rate was set to 0 (dropout turned off). Rectified Linear Unit (ReLU) was used as the activation function. The readout layer was a softmax regression that outputs the probability of being a true signal for a given image. This probability was designated as the prediction confidence score. For comparison, a simple neural network (SNN) containing only one hidden layer of 64 neurons was also tested below, which yielded only slightly inferior results.
A large dataset is necessary to train most basic machine learning and more advanced deep learning (DL) models, and our study is no exception. The ML model was developed based on manually curated data that we obtained on single dorsal-animal (D11) cells fromX. laevis embryos using single-cell CE-ESI-HRMS. The CE-ESI-HRMS files that resulted from the analysis of the identified cells are described in Table S1 and are provided open-access (see Metabolomics Workbench). To perform supervised learning of our models, we manually curated a total of 4,546 potential signal images from 3 different samples: 1,464 features were marked as true metabolic signals [value “1” assigned for (m/z, t) values yielding a peak in the corresponding MS data] and 3,082 features were labeled as false metabolic signals [value “0” assigned for (m/z, t) values yielding no peak in the corresponding MS data]. Select examples are shown in Supplementary Figure 1 (Fig. S1). The complete set of data that were used for training are tabulated in Table S2. These data provided a rich baseline to train the neural network model for the analysis of other single cells in this study.
The neural network was trained by adjusting the weights using back propagation and gradient descent algorithms. We used the cross entropy as our loss function for minimization to adjust the weights. The loss function can be expressed as: where yn and stands for the actual and predicted probability for the nth image to be a true signal, respectively. N is the number of training images at each step. In our case, the batch size for each training step was set to 64. The training optimizer that we used is ADAM in TensorFlow37 with a learning rate of 1 × 10−4 Other parameters were set as default in TensorFlow. The model was trained until the final training accuracy converged with little fluctuation. From our hype-parameter tuning experimentation, 5,000 or so training steps were found to be sufficient, and the process took ~5 min on a PC equipped with a NVIDIA K20 Graphics Processing Unit (GPU) in our laboratory. After the neutral network machine was trained to high accuracy (>99%), the bounding box of the signal images to be evaluated were fed to the machine probability score assignment (Fig. 1). This probability was used as the prediction confidence score: the higher the probability is, the more likely the correspondence is to a true signal (e.g., a valid metabolite in our study).
Evaluation of Model Performance.
The performance of our CNN was benchmarked against successful ML models, specifically decision tree, random forest, and SNN. The training dataset was randomly split to assign 80% of the samples for training the model and the remaining 20% for performance testing. SNN and CNN was 10-times cross-validated by randomly splitting the training dataset each time. Test accuracy was quantified using mean and standard deviation as follows (see Fig. S2): 78.1 ± 0.3% for decision tree, 86.6 ± 0.1% for random forest, 90.5 ± 0.4% for SNN, and 91.9 ± 0.4% for CNN. The receiver operating characteristic (ROC) curve is shown in Figure S3 with the area-under-curve (AUC) of 0.97. These results suggest that our trained CNN model achieved adequate accuracy and stability to perform metabolic analysis of other samples.
The trained CNN model was tested for a randomly chosen file. The file “D11 cellE2T2” was manually curated following our established protocols26,27 to extract molecular features as a reference. Trace was employed to search signals with “soft,” “normal,” and “strict” settings, which respectively correspond to increasingly conservative conditions for peak finding. The signal images and the bounding boxes were constructed for each potential signal, which were then evaluated by the neural network already trained to produce the final list of predicted signals. The results are presented in Figure 2A. The number of predicted signals converged despite increasingly more relaxed criteria used for initial signal screening. The initial screening produced 1096, 2780, and 7898 potential signal images, respectively (Fig. 2A, see “Initial Signal”), of which 649, 1321, and 2734 had features that were detected by bounding boxes, respectively (Figure 2A, see “Signal with BB”). The final evaluation by the model resulted in 324, 462, and 492 true signals, respectively (Figure 2A, see “Final Signal”). Table S3 tabulates the signals that were found from these experiments. Compared to the normal condition, the soft criterion only produced 30 additional true signals by the neural network model even though more than 5,000 additional potential signals were examined. Therefore, the list of true signals was exhausted under the soft condition. Detection of ~500 signals agrees with results from our manual curation of the data, supporting the accuracy of molecular feature finding by Trace.
Figure 2.
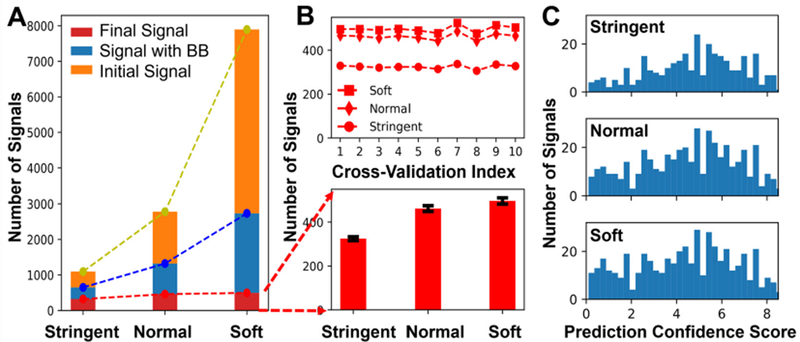
Performance evaluation of Trace. (A) The numbers of predicted true signals gradually converged for initial screening of three increasingly more relaxed criteria (strict, normal, and soft). (B) Ten-fold cross-validation of the neural network model for the three initial screenings showed a consistent total number of true signals, reflecting the robustness of the model. (C) Histogram of prediction confidence scores. The high end of confidence score distribution hardly changed from normal to soft condition, suggesting that the model effectively identified most of true signals.
The consistency of the model was evaluated by cross-validation. We independently trained and tested the network 10 different times, each time using randomly chosen signal images from the initial training set (4,546 images). As shown in Figure 2B, the predicted number of true signals was comparable between the individual models. Combined, these results show that the model was robust. Furthermore, the consistency and robustness of the model was also validated by the histogram of prediction confidence score of the true signals by the neural network, as shown in Figure 2C. The higher the prediction confidence score of a signal is, the more likely it was classified as a true signal. Figure 2C also revealed little change in the histograms as the initial screening criteria were relaxed (e.g., compare with “soft” conditions): Most of the newly predicted signals exhibited relatively low scores and were likely around the border between true and false signal. This means that the model effectively identified signals of good quality, namely those with high confidence scores. The neural network model thus achieved the desired consistency and sensitivity to actively remove the need for fine-tuning initial screening parameters. By simply choosing lax enough parameters, the true signals were not missed from the set of images to be examined by the model.
These performance metrics were compared with software packages that are widely used for the analysis of nanoLC-HRMS type data. As an example, the single-cell CE-ESI-MS file that we recorded for the D11 cellE2T2 was analyzed using XCMS22, the CWT-based18 python function scipy.signal.find_peaks_cwt, and Trace. Results of this comparison are presented in Figure 3A. The XCMS online server (version 3.7.1) using the default parameters recognized 2,805 molecular features, whereas cipy.signal.find_peaks_cwt using an EIC bin width of 0.010 Da returned thousands of signals: the number of molecular features were 7,898 for soft, 2,780 for normal, and 1,096 for strict setting. A limited number of features were recognized by both XCMS and CWT (see overlap for ~960 signals), suggesting that the rest of signals were too variable or false. Indeed, follow-up manual inspection of EICs found most of the signals returned by XCMS or CWT as false positives. In comparison, Trace uncovered ~500 molecular features, most of which were also found by XCMS and CWT. Semi-manual curation revealed a similar number of molecular features to those found by Trace. It is worth noting that Trace also returned signals that were not detected by XCMS or CWT (with normal criteria). Follow-up manual curation revealed these extra features as true signals with relatively low SNR or ion count intensity. Therefore, the neural network approach by Trace was able to extract true signals with low false-positive rates from trace-level CE-ESI-HRMS studies.
Figure 3.

Benchmarking of molecular feature detection. (A) Overlap of reported molecular features from the cell “D11 cellE2T2” by XCMS (default settings), CWT (normal criteria), and Trace. (B) Distribution of quality measures for true and false signals in Trace, revealing the use of advanced deep machine learning to categorize signals.
We asked whether Trace used any simple feature(s) for distinguishing true from false signals. The following 6 features of signal quality25 were calculated for the signal images of known false and known true signals (by manual validation): Zigzag evaluates the smoothness of the EIC; Gaussian Similarity describes how similar the target shape is to a standard Gaussian curve; SNR describes the abundance of a signal with respect to the surrounding background noise; significance evaluates the ratio of peak intensity and peak baseline; triangle peak area similarity ratio (TPAR) describes the similarity of the observed peak shape to a triangle; and sharpness evaluates the dynamics of the signal how fast the signal peak emerges and vanishes. The distribution of these 6 features are shown for true and false signals in Figure 3B. The Gaussian Similarity and SNR presented with a slightly separated distribution with enhanced values for true signals. Similarly, the distribution of both Zigzag and Sharpness for true signal group formed peaks at small values. Nevertheless, the feature distributions were broad with considerable overlaps between true and false signals. Thus, some of these measures are helpful but not discriminative enough for accurate signal prediction. This finding highlights the necessity of using more advanced ML and DL approaches for signal categorization (true vs. false), such as those implemented in Trace.
Case Study: Trace for Single-cell Metabolomics.
We tested the performance of Trace on single-cell CE-ESI-MS data that we obtained on identified dorsal-animal (D11) cells in the 16-cell X. laevis embryo. Briefly, a pulled microcapillary was used to aspirate an ~10 nL portion of the D11 cell from N = 5 different embryos, each from a different set of parents. The metabolites from each aspirate were extracted in 4 μL of aqueous mixture of 40% acetonitrile and 40% methanol, and an ~10 nL portion of each single-cell extract was individually analyzed in one-to-three technical replicates by CE-ESI-HRMS following our established protocols27. Each resulting file were assigned a unique identifier (Table S1), although this information was only revealed to facilitate the interpretation of results.
By design, this study accounted for significant technical and biological variability to allow us to test Trace for large-scale metabolomics projects. Our dataset contained several sources of variability, including the following: differences in sample processing due to multiple users (2 users here); shifts in CE separation time due to the use of different CE capillaries and separation conditions over multiple experiments (4 capillaries and ~2 separation conditions used here); varying detection sensitivity resulting from different ionization efficiencies due to differences in custom-built CE-ESI ion sources (4 different CE-ESI interfaces built here); and differences in m/z detection sensitivity due to user-defined MS tuning parameters (2 independent tunings performed here). Therefore, these conditions provided an excellent opportunity to establish the performance of Trace for studies spanning variable experimental, instrumental, and biological conditions.
Reproducibility and robustness were determined for the DL model. Figure 4 presents the signal images that Trace generated for representative metabolites that were identified in N = 5 different D11 cells (see Methods). A consequence of our study design was variable reproducibility in separation times (refer to Study Design in Methods). Experiments that were performed under similar conditions yielded <3% error in migration time (e.g., same-day analysis of E3T1 and E3T3, see Table S1)), which is indicative of the optimal reproducibility of our CE platform.26,27 Non-linear time time-alignment using our established approach11,26 can further reduce variance to −0.1% error, facilitating metabolite identification and quantification. In stark contrast, migration times between D11 cellE2T2 us. D11 cellE5T, analyzed 3 months apart, varied by up to ~30–40% for argininosuccinate (Fig. 4A), S-adenosylmethionine (Fig. 4B), hypoxanthine (Fig. 4C), and creatine (Fig. 4D). Despite these inherent variances in separation and detection, the signal images displayed a remarkable similarity for the different metabolites. For example, the signal images appeared reproducibly circular for creatine, circular with a broader spread for argininosuccinate, oval with horizontal spreading for S-adenosinemethionine, and vertically oval for hypoxanthine. In theory, these image consistencies that Trace recovered across the different samples, even under suboptimal experimental conditions, may be utilized to perform feature selection by machine learning and deep learning models across different samples. It does not escape our attention that a signal-image driven classification of metabolite signals raises an opportunity to complement classical metabolomics with a potentially metabolite-dependent piece of information: the shape of the image signal.
Figure 4.
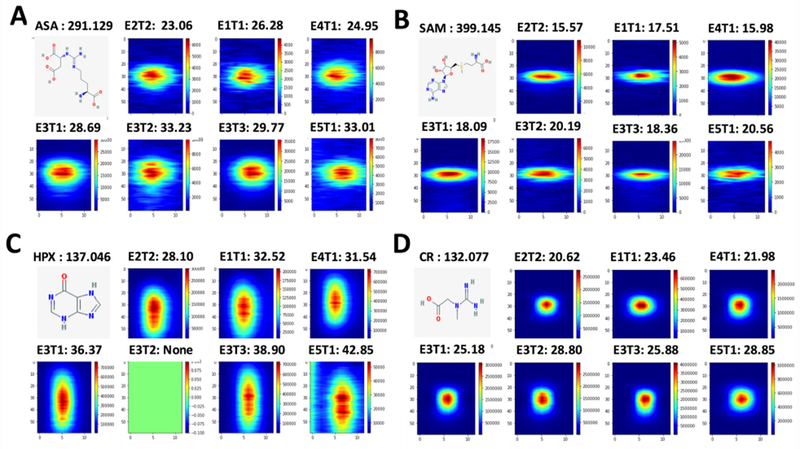
Signal images of representative identified metabolites among N = 4 different D11 cells, each from a different embryo from a different set of parents: (A) argininosuccinate (ASA); (B) S-adenosylmethionine (SAM); (C) hypoxanthine (HPX); and (D) creatine (CR). For each metabolite, the accurate m/z is provided with the corresponding signal image labeling the time of separation as was detected (not aligned data). The current machine learning model did not detect HPX in the cell D11 cellE3T2. The high similarity of signal images across the different samples confirms robust and sensitive operation by Trace, suggesting a utility for large-scale trace-sensitive studies.
The reproducibility of finding molecular features was determined between the biological replicates. The CE-ESI-HRMS data were processed for a subset of identified metabolites (Table S4) following our semi-manual method (see protocols in reference 11, 26) and the neural network model that was trained earlier (Table S2). Migration times were aligned based on 5 confidently identified metabolites following a third-order correction function (see protocols in references 11, 26). The average separation time of the compounds was chosen as reference for calculating separation time deviations. The corrected time was calculated by adjusting the original separation time with the corresponding correction function from a third-order fitting, as is standard in our single-cell CE-MS protocols.11 These data, tabulated in Table S5, allowed us to quantify the reproducibility of data analysis.
Common features were compared. Figure 5A plots the relative error (standard deviation) of corrected separation times for 36 metabolites that we identified from the D11 cell labeled “D11 cellE2T2.” A less than 1% (relative standard deviation) in corrected migration time revealed robust feature finding by Trace, even under high technical and biological variability that resulted from our experimental design. Out of the possible 216 signals (36 signals for each of the 6 samples), 9 were not experimentally detected by CE-ESI-MS in select cells, likely due to biological variation. Another set of 13 signals out of the experimentally detected 207 signals were not detected by Trace in some specific cells: 5 were removed by the initial CWT scanning and the remaining 8 were not recognized as true signals by the ML model. The corresponding false negative rate was 6.28% (13/207) for Trace and 3.96% (8/202) for ML. These rates are comparable with the test error (~8%), indicating that these misclassifications largely fall within the expected performance of our current model. Future revisions of the software may enhance the fidelity of signal finding, e.g., by collecting more comprehensive training sets and employing deeper neural network architectures. For most metabolites, close alignment between the corrected migration times with robust and accurate signal classification validates that the neural network model, once trained on a reference sample, can efficiently and accurately detect true signals in trace-sensitive MS datasets.
Figure 5.
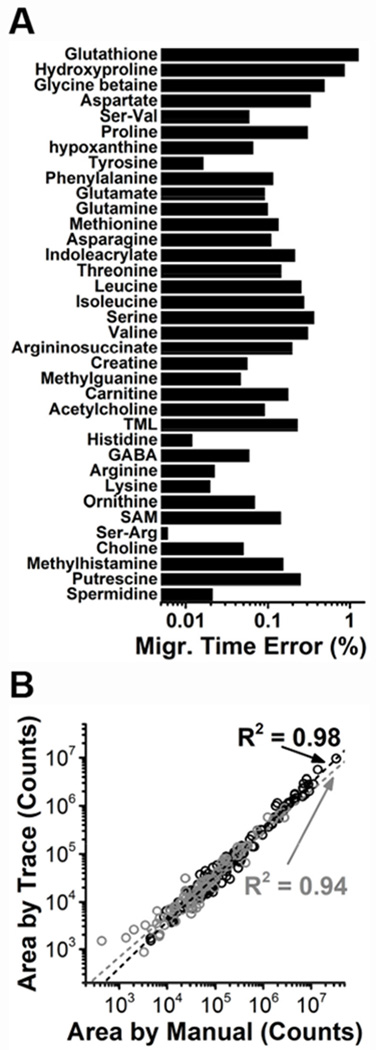
A case study of Trace for single-cell CE-ESI-MS of metabolites in single X. laevis cells. (A) Evaluation of molecular feature recognition in the temporal domain (migration times). Trace detected signals between N = 5 different D11 cells with <1% separation time error (data from only one experiment shown). (B) Assessment of quantitative reproducibility. Peak areas from manual data integration correlated with those returned by Trace for data from this study (black data points) and two independent measurements on the same cell type from 2014 (grey data points). Linear regression coefficients (R2) are shown.
The reproducibility of signal quantification was also tested. In metabolomics, under-the-curve peak areas or peak heights are often used as a proxy for metabolite concentration. We and others leveraged this information from single-cell CE-ESI-MS to compare the metabolic state of identified single cells in A. californica28,38,39, rat ganglia11,40, and X laevis embryos26,27. To validate quantification by Trace, peak areas and peak heights that were calculated by Trace (see Table S5) were compared to peak areas that we manually integrated for 100 manually curated molecular features. Figure 5B presents the results for data from a current study on a D11 cell from X. laevis as well as a previous study26. Linear regression coefficients (R2) were > 0.9 for peak areas over a 3-log-order dynamic range, revealing excellent correlations between data obtained by manual curation and Trace in spite of the long duration of the study. In addition, peak areas from Trace correlated with peak heights from Trace (R2 = 0.93), suggesting that peak heights from the software can also be used for quantification, particularly for peaks with limited SNRs. Intriguingly, the model that was trained in the current study successfully recognized molecular features from data collected 4 years ago, thus demonstrating robust, quantitative data processing by Trace to aid trace-sensitive MS.
CONCLUSIONS
Trace complements currently available metabolomic software packages with robust performance to extract signals from trace-sensitive metabolomics studies. Once trained on a curated dataset, our neural network model consistently predicts molecular features with high accuracy and low false positive rates. Automated data processing provides a significant gain in data analysis throughput compared to semi-manual inspection of MS data. This case study also demonstrated Trace to deliver sufficient robustness to analyze large-scale metabolomics data from trace-sensitive studies, including those spanning substantial duration (e.g., 4 years tested here). These performance metrics are encouraging to meet increasing data processing demands that result from trace-sensitive HRMS, including single-cell investigations that are rapidly gaining throughput.
Trace also presents developmental opportunities for future studies. An open-access code facilitates future updates for add-on features. For example, separation times may be automatically aligned between samples based on known (identified) molecular features (reference standards), thus aiding identifications, such as isobaric (identical m/z) or closely separating/overlapping species (e.g., leucine and isoleucine). Accurate alignment of m/z vs. time domains between metadata can help identify molecular features using reference standards. Overlapping signals that challenge CWT-based initial screening in the time dimension may be distinguished by evaluating the entire 2D image via a grid search. The software modules may also be integrated into other successful platforms to expand data analysis capabilities. Trace can be efficiently used to process data from large-scale studies with exciting developmental potentials to support studies using trace-sensitive HRMS.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Sally A. Moody (The George Washington University) for providing a portion of the embryos that were analyzed for this study. This work was supported by the National Institutes of Health awards 7R03CA211635 (to PN. and C.Z.) and 1R35GM124755 (to P.N.).
Footnotes
Supporting Information. The electronic Supplementary Information document contains a user manual for Trace as well as supplementary figures and tabular data. The MS-MS/MS data from this study is available in mzML format at the NIH Common Fund’s Metabolomics Data Repository and Coordinating Center (supported by NIH grant, U01-DK097430) website, the Metabolomics Workbench, http://www.metabolomicsworkbench.org, under Project ID PR000686. The data can be accessed directly via the following Project DOI: https://doi.org/10.21228/M80Q2W.
REFERENCES
- 1.Chen X; Love JC; Navin NE; Pachter L; Stubbington MJT; Svensson V; Sweedler JV; Teichmann SA, Nat. Biotechnol 2016, 34, 1111–1118. [DOI] [PubMed] [Google Scholar]
- 2.Acunha T; Simo C; Ibanez C; Gallardo A; Cifuentes A, J. Chromatogr., A 2016, 1428, 326–335. [DOI] [PubMed] [Google Scholar]
- 3.Rubakhin SS; Romanova EY; Nemes P; Sweedler JV, Nat. Methods 2011, 8, S20–S29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rubakhin SS; Lanni EJ; Sweedler JV, Curr. Opin. Biotechnol 2013, 24, 95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang YY; Huang YY; Wu JH; Liu N; Deng JW; Luan TG, Trends Anal. Chem 2017, 90, 14–26. [Google Scholar]
- 6.Comi TJ; Do TD; Rubakhin SS; Sweedler JV, J. Am. Chem. Soc 2017, 139, 3920–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zenobi R, Science 2013, 342, 1243259. [DOI] [PubMed] [Google Scholar]
- 8.Yin L; Zhang Z; Liu Y; Gao Y; Gu J, Analyst 2018, In print, DOI: 10.1039/C8AN01190G. [DOI] [PubMed] [Google Scholar]
- 9.Passarelli MK; Ewing AG, Curr. Opin. Chem. Biol 2013, 17, 854–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang LW; Vertes A, Angew. Chem. Int. Ed 2018, 57, 4466–4477. [DOI] [PubMed] [Google Scholar]
- 11.Nemes P; Rubakhin SS; Aerts JT; Sweedler JV, Nat. Protoc 2013, 8, 783–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benton HP; Wong DM; Trauger SA; Siuzdak G, Anal. Chem 2008, 80, 6382–6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forsberg EM; Huan T; Rinehart D; Benton HP; Warth B; Hilmers B; Siuzdak G, Nat. Protoc 2018, 13, 633–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wanichthanarak K; Fan S; Grapov D; Barupal DK; Fiehn O, PLoS One 2017, 12, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chong J; Soufan O; Li C; Caraus I; Li SZ; Bourque G; Wishart DS; Xia JG, Nucleic Acids Res 2018, 46, W486–W494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Radulovic D; Jelveh S; Ryu S; Hamilton TG; Foss E; Mao Y; Emili A, Mol. Cell. Proteomics 2004, 3, 984–997. [DOI] [PubMed] [Google Scholar]
- 17.Hastings CA; Norton SM; Roy S, Rapid Commun. Mass Spectrom 2002, 16, 462–467. [DOI] [PubMed] [Google Scholar]
- 18.Du P; Kibbe WA; Lin SM, Bioinformatics 2006, 22, 2059–2065. [DOI] [PubMed] [Google Scholar]
- 19.Monroe ME; Tolić N; Jaitly N; Shaw JL; Adkins JN; Smith RD, Bioinformatics 2007, 23, 2021–2023. [DOI] [PubMed] [Google Scholar]
- 20.Sturm M; Bertsch A; Gröpl C; Hildebrandt A; Hussong R; Lange E; Pfeifer N; Schulz-Trieglaff O; Zerck A; Reinert K; Kohlbacher O, BMC Bioinformatics 2008, 9, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katajamaa M; Miettinen J; Orešič M, Bioinformatics 2006, 22, 634–636. [DOI] [PubMed] [Google Scholar]
- 22.Gowda H; Ivanisevic J; Johnson CH; Kurczy ME; Benton HP; Rinehart D; Nguyen T; Ray J; Kuehl J; Arevalo B; Westenskow PD; Wang JH; Arkin AP; Deutschbauer AM; Patti GJ; Siuzdak G, Anal. Chem 2014, 86, 6931–6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Du P; Sudha R; Prystowsky MB; Angeletti RH, Bioinformatics 2007, 23, 1394–1400. [DOI] [PubMed] [Google Scholar]
- 24.Leptos KC; Sarracino DA; Jaffe JD; Krastins B; Church GM, Proteomics 2006, 6, 1770–1782. [DOI] [PubMed] [Google Scholar]
- 25.Zhang W; Zhao PX, BMC Bioinformatics 2014, 15, S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Onjiko RM; Moody SA; Nemes R, Proc. Natl. Acad. Sci. U. S. A 2015, 772, 6545–6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Onjiko RM; Portero EP; Moody SA; Nemes P, Anal. Chem 2017, 89, 7069–7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nemes P; Knolhoff AM; Rubakhin SS; Sweedler JV, Anal. Chem 2011, 83, 6810–6817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alpaydin E, Introduction to machine learning. MIT press: 2014. [Google Scholar]
- 30.Lu L; Zheng Y; Carneiro G; Yang L, Deep Learning and Convolutional Neural Networks for Medical Image Computing. Springer: 2017. [Google Scholar]
- 31.Janowczyk A; Madabhushi A, J. Pathol. Inform 2016, 7, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kremer J; Stensbo-Smidt K; Gieseke R; Pedersen KS; Igel C, IEEE Intelligent Systems 2017, 32, 16–22. [Google Scholar]
- 33.Angermueller C; Pärnamaa X; Parts L; Stegle O, Mol. Syst. Biol 2016, 12, 878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woldegebriel M; Derks E, Anal. Chem 2016, 89, 1212–1221. [DOI] [PubMed] [Google Scholar]
- 35.LeCun Y; Bengio Y; Hinton G, Nature 2015, 521, 436–444. [DOI] [PubMed] [Google Scholar]
- 36.LeCun Y; Bottou L; Bengio Y; Haffner R, Proceedings of the IEEE 1998, 86, 2278–2324. [Google Scholar]
- 37.Abadi M; Agarwal A; Barham P; Brevdo E; Chen Z; Citro C; Corrado GS; Davis A; Dean J; Devin M, arXiv preprint arXiv: 1603.04467 2016, [Google Scholar]
- 38.Lapainis T; Rubakhin SS; Sweedler JV, Anal. Chem 2009, 81, 5858–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu JX; Aerts JT;Rubakhin SS; Zhang XX; Sweedler JV, Analyst 2014, 139, 5835–5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aerts JT; Louis KR; Crandall SR; Govindaiah G; Cox CL; Sweedler JV, Anal. Chem 2014, 86, 3203–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.