

Summary
One of the primary goals of genomic medicine, utilizing genomics in clinical care, is to improve diagnosis through identification of genomic conditions to improve clinical management, prevent complications, and promote health. In this paper we explore how genomic medicine is being used to obtain molecular diagnoses for patients with previously undiagnosed diseases in the prenatal, pediatric, and adult clinical settings. We focus on the role of clinical genomic sequencing (exome and genome) in aiding patients with undiagnosed conditions despite extensive clinical evaluation and prior testing. In particular, we explore the impact of combining genomic and phenotypic data and integrating across multiple data types to improve diagnoses for patients with undiagnosed diseases, along with how these genomic sequencing diagnoses change clinical management.
Introduction
Genomic medicine is defined by the National Human Genome Research Institute (NHGRI) as “an emerging medical discipline that involves using genomic information about an individual as part of their clinical care (e.g., for diagnostic or therapeutic decision-making) and the health outcomes and policy implications of that clinical use.1” An introduction to genomic medicine can be found in “Opportunities, Resources, and Techniques for Implementing Genomics in Clinical Care” in this series and key definitions in Panel 1. With the continuing decreases in the cost of DNA sequencing,2 clinical exome and genome sequencing are being used across diverse clinical settings with the goal of increasing diagnostic rates and improving clinical management. In exome sequencing, the protein-coding regions (or exons) of the genome are sequenced, while genome sequencing includes both protein-coding and non-protein-coding regions of the genome. For more information on the clinical utility of exome and genome sequencing for genomic medicine see “Building Evidence and Measuring Clinical Outcomes for Genomic Medicine” in this series. In this paper, we use clinical genomic sequencing to refer to the clinical use of exome or genome DNA sequencing and diagnosis to refer to an etiological molecular diagnosis as a step beyond a descriptive diagnostic name for a condition with unknown cause.
Panel 1: Key genomic medicine definitions.
Exome sequencing – seqeuncing the protein-coding regions (or exons) of the genome
Genome sequencing – seqeuncing both protein-coding and non-protein-coding regions of the genome
Clinical genomic sequencing – clinical use of exome or genome DNA sequencing
Monogenic inheritance – a single gene is causative of the disease
Oligogenic inheritance – variants in more than one gene influence disease
Polygenic risk score – calculates the cumulative risk of many genetic variants that all have a small effect on disease risk by using a weighted sum
Allelic heterogeneity – different variants in the same gene leading to variable phenotypes and a spectrum of disease severity
Clinical utility – results can be used to inform clinical decisions and management
Personal utility – results have benefits to an individual or family beyond clinical care
One rapidly emerging area of genomic medicine involves establishing a diagnosis for patients with complex phenotypes (or combinations of phenotypes) that have defied conventional medical evaluation. Initial successes were reported from the NIH Undiagnosed Diseases Program3 and more recently from the Undiagnosed Diseases Network;4 in turn, this has led to the global Undiagnosed Diseases Network International (UDNI) effort including programs in 16 countries.5 A common element in all the global undiagnosed patient programs that make up the UDNI is the utilization of genomics as an important component of the diagnostic process. The International Rare Diseases Research Consortium (IRDiRC) has also recognized the importance of diagnosis in their global rare disease goals for 2017–2027 with goal 1 seeking to have “all patients coming to medical attention with a suspected rare disease…diagnosed within one year if their disorder is known in the medical literature; [and] all currently undiagnosable individuals…enter[ing] a globally coordinated diagnostic and research pipeline.”6 In this paper, we consider a patient to have an undiagnosed disease if the individual has received an appropriate, extensive clinical evaluation based on their presenting symptoms and signs yet remain without an etiologic diagnosis. Such individuals may also have received targeted genetic testing or low-resolution chromosomal copy number analyses (e.g., chromosomal microarray) based on their clinical presentation and/or may have a suspected diagnosis, but no genomic-based diagnosis of disease has been made.
Many patients with undiagnosed diseases are eventually found to have rare diseases. In the United States, the Orphan Drug Act of 1983 and Rare Disease Act of 2002 define rare diseases as conditions that affect fewer than 200,000 people in the United States.7,8 However, while rare diseases are individually rare, there are so many of them (estimated at ~7,000) that, altogether, they affect 25–30 million people in the United States, amounting to nearly one in ten Americans.9 Based on patient surveys,10 rare disease patients in the United States spend an average of 7.6 years on their diagnostic odyssey to reach a diagnosis, and 5.6 years in the United Kingdom. Reflecting several years spent without a diagnosis, the NIH Undiagnosed Diseases Program reported that pediatric applications peaked in children ages 4–6 with congenital onset of disease and teenagers age 16–18 with onset of symptoms at early school age11. Living with an undiagnosed disease also presents a significant burden on patients and their families. Patients visit an average of four primary care physicians and four specialists before reaching their diagnosis, repeat testing with an average of two to three misdiagnoses, have difficulty locating specialists, receive conflicting treatment guidance, and have difficulty coordinating care amongst clinicians.10 Beyond the lengthy diagnostic odyssey, adult patients with rare diseases report lower health-related quality of life compared with the population at large and patients with common chronic diseases.12 While some parents of undiagnosed children have been shown to be tolerant of uncertainty (remaining actively engaged in health care, and having confidence in performing coping behaviors when faced with life challenges), 35–40% also experience anxiety and depression.13
With a desire to bring an end to the diagnostic odyssey as soon as possible, genomic sequencing has been investigated as a potential key diagnostic modality for patients with undiagnosed diseases. Given that at most 46% of patients presenting to medical genetics specialists and suspected of having a genetic disorder are currently diagnosed using traditional genetic diagnostic evaluations, comprehensive clinical evaluations, targeted genetic testing, and chromosomal copy number analyses,14 approaches for improving diagnostic rates are still needed. In this paper, we will explore the impact of combining genomic and phenotypic data and integrating across multiple data types to improve diagnoses in undiagnosed patients, along with how these genomic sequencing diagnoses change clinical management. Figure 1 illustrates a vision for implementing genomic medicine for patients with undiagnosed diseases integrating these points starting from the undiagnosed patient in box #1.
Figure 1.
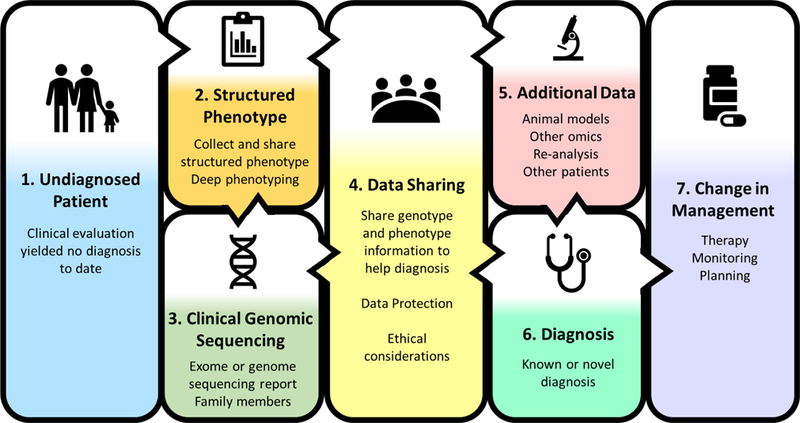
A vision for implementing genomic medicine for patients with undiagnosed diseases
Combining Genomic and Phenotypic Data to Improve Diagnoses
One significant barrier to reaching a diagnosis in undiagnosed patients is the variable quality and quantity of phenotypic data available to the clinical sequencing laboratory searching for a causative genomic variant. Laboratories performing clinical genomic sequencing use such phenotype data while interpreting the sequence variants they encounter to determine pathogenicity and prioritization of variants in their clinical reports. However, many laboratories report receiving only limited and highly variable phenotypic information from the referring provider (Table 1). In addition, the benefits of data sharing, discussed further below, cannot be realized unless phenotypic data are collected in a structured and readily shareable fashion. One clinical diagnostic laboratory reported receiving variable phenotype information ranging from International Classification of Diseases–version 9 (ICD-9) codes, to completion of a phenotype checklist on the genomic sequencing submission form, to submission of a clinical summary, to multiple clinical notes and laboratory test results.15 Another laboratory reported receiving case summaries of two to five pages in free text.16 While many groups generated standardized Human Phenotype Ontology (HPO)17 terms to capture the primary clinical indication and sometimes other symptoms when comparing diagnostic rates (Table 1), only a few utilized application solutions to produce HPO terms rather than relying on manual curation.18,19 Phenotype application solutions can aid in the collection of HPO terms and currently include Phenomizer20to rank diseases using signs and symptoms producing a phenotype driven differential diagnosis (http://compbio.charite.de/phenomizer/) and BioLark21 a concept recognition tool used to produce HPO terms from clinical notes. Another tool, Phenolyzer22 uses phenotypes combined with prior biological knowledge to implicate genes and disease-associated genomic variants. In a family with multiple conditions, including Prader–Willi Syndrome, Hereditary Hemochromatosis, dysautonomia-like symptoms, Tourette Syndrome, and other conditions, 21 HPO candidate terms were identified.23 Increasing the number of HPO terms analyzed with Phenolyzer22 in combinations of 1 to 6 of the 21 candidate HPO terms increased the chance of a known variant being prioritized as “high confidence”.23 This increase in confidence highlights the need for more complete and systematic collection of structured phenotype information, such as HPO terms, along with improved tools for collecting phenotype data rapidly and in an automated fashion, to facilitate interpretation of genomic sequencing for undiagnosed diseases. In Figure 1 this is shown with the collection of structured phenotype data in box #2 before the interpretation of genomic sequencing results in box #3.
Table 1:
Genomic sequencing and phenotyping methods from studies of undiagnosed patients unselected for a specific phenotype published in 2014–2018
| Study PMID | First author | Year | # Patients | Age group | Type of sequencing | Individuals studied | Phenotyping method |
|---|---|---|---|---|---|---|---|
| 30304647 | Splinter K | 2018 | 382 | pediatric and adult | exome and genome | families when available | cross-disciplinary team selects HPO terms |
| 25326635 | Yang Y | 2014 | 2000 | prenatal to adult | exome | proband with parent Sanger confirmation when available | clinical data provided by referring physician |
| 25326637 | Lee H | 2014 | 814 | pediatric and adult | exome | proband, trio, and other family members | referring physician reported primary clinical indication and differential diagnosis |
| 28496993 | Bick D | 2017 | 22 | pediatric | genome | proband with parent Sanger confirmation when available | referring physician case summary |
| 26633542 | Retterer K | 2016 | 3040 | pediatric and adult | exome | proband and up to 4 family members when available | referring physician provided primary clinical diagnosis and ICD-9 code, used to select HPO terms |
| 26938784 | Stark Z | 2016 | 80 | infants | exome | proband | diagnostic investigations from referring clinicians and medical records, HPO terms collected at enrollment |
| 28567303 | Stavropoulos D | 2016 | 100 | pediatric | genome | proband | PhenoTips collection after clinical geneticist exam using HPO terms |
| 26633545 | Posey J | 2016 | 486 | adults | exome | proband with parent Sanger confirmation when available | available clinical information used to generate HPO terms |
| 25356970 | Farwell K | 2015 | 500 | prenatal to adult | exome | trios when available | clinical and test history from referring provider summarized by molecular geneticist or genetic counselor |
| 30266093 | Normand E | 2018 | 146 | prenatal | exome | proband or trio | fetal phenotype converted to HPO categories using Phenomizer |
| 27848944 | Trujillano D | 2017 | 1000 | prenatal to adult | exome | trios when available | referring provider clinical data used to generate HPO terms |
| 28973083 | Meng L | 2017 | 278 | infants | exome | proband or trio | BioLark and manual review of clinical notes used to generate HPO terms |
One setting in which combining genomic approaches with structured phenotypic information has been adopted to accelerate the diagnostic process is in neonatal medicine for critically ill infants. There is a particularly acute need for accurate phenotyping to support rapid diagnoses in the neonatal intensive care unit (NICU) setting to reduce infant morbidity and mortality.24 The critical impact of early diagnosis and effective treatment on neonatal development has helped drive the adoption of natural-language-processing and machine-learning algorithms to extract structured phenotypes from electronic medical records.24 Combining detailed genomic sequencing and structured phenotypic data can help clinicians direct their diagnostic searches towards suspected genes and diseases (many of which can be pre-defined based on past experience), allowing more rapid diagnoses that can change clinical management. By extracting phenotypes directly from the medical record of infants in the NICU, Farnaes et al. demonstrated derivation of rich phenotypic data to enhance the interpretation of genome sequences leading to provisional diagnoses in as little as 26–48 hours from the time of blood sample receipt.24 Such computational approaches can provide more complete phenotypes as structured data that, when coupled with differential diagnosis gene lists tied to commonly encountered patterns of phenotypes, allow for the creation of computer simulated gene panels, thereby speeding up genomic sequence interpretation in the clinical setting. While such techniques may speed up diagnosis through the use of automation, it should be noted that some novel findings may be missed by such an approach that may be picked up using more traditional clinical genomic sequencing approaches. Implemented as a sequential process, automation can enhance the rapid diagnosis of sick neonates who have a disorder that is familiar to neonatologists, while moving those neonates for whom a diagnosis is not apparent into the more time intensive clinical discovery interpretation pipeline. Sharing data utilizing structured phenotypes also allows for knowledge generation about disease mechanisms through research studies combining genomic data and standardized phenotype terms. Research studies have used multiple HPO terms to describe complex clinical disease phenotypes in a structured fashion allowing for the identification of novel genomic variants associated with specific phenotypes.25 HPO terms can also be used to align human clinical phenotypes to model organism phenotypes to aid understanding of disease mechanisms.25
However, when analyses are limited by observed phenotypes, determining a priori what phenotypes have a genetic etiology in an undiagnosed patient may be difficult. A patient’s clinical presentation may reflect multiple diagnoses (2 or more), yielding a blended phenotype that does not fit exactly with a single condition.4,26–29 Multiple diagnoses were encountered in 3–7% of diagnosed cases in 5 studies of patients unselected for phenotype.15,26–29 While the genes implicated in these diagnoses may be related to distinct phenotypes seen in the patient that combine to account for some or all of a patient’s clinical findings, related or overlapping phenotypes may also be caused by genes interacting in the same pathway. Analyses also often assume a monogenic inheritance model in which a single gene is causative of the disease; however, undiagnosed disease may be caused by oligogenic inheritance in which variants in more than one gene influence disease. Many common variants can also be associated with rare diseases, as has been demonstrated for rare severe neurodevelopmental disorders using polygenic risk scores.30 A polygenic risk score calculates the cumulative risk of many genetic variants that all have a small effect on disease risk by using a weighted sum. Additionally, novel diagnoses may come from newly described findings or phenotypic expansions of known conditions31,32 that may not fit with phenotypic expectations. Therefore, deep phenotyping, that systematically catalogues signs and symptoms of disease rather than focusing on a single primary diagnosis, may assist with disentangling the genetic contributions of undiagnosed diseases. In Figure 1, deep phenotyping is also included in box #2 with an emphasis on the importance of data sharing with box #4 to interpret clinical genomic sequencing in box #3.
Phenotyping can also be a challenge due to the timing of clinical genomic sequencing, as is the case in the prenatal setting. Prenatal imaging, including fetal computed tomography scanning, echocardiography, magnetic resonance imaging, and ultrasonography, can be utilized to detect prenatal phenotypes and help confirm suspected diagnoses. Current practice guidelines recommend chromosomal microarray analysis and karyotyping for fetal anomalies detected by fetal imaging, identifying aneuploidy, chromosomal rearrangements, and copy number variants (deletions and duplications) responsible for these detected anomalies in 30–40% of pregnancies.18,33,34 However, such testing leaves approximately 60% of fetuses with detected anomalies undiagnosed.18 Such undiagnosed cases may benefit from rapid prenatal clinical genomic sequencing. Normand et al. demonstrated a 35% diagnosis rate (22/62 fetuses with at least one structural anomaly detected by fetal imaging) using prenatal exome sequencing of trios for ongoing pregnancies often after negative karyotype and microarray.18 Further studies to enhance our knowledge of prenatal phenotypes and structured phenotype sharing will continue to improve our ability to link genomic variation to these early onset conditions.
Phenotypes can also change over time and have variable intensities in their presentation. For example, some conditions exhibit allelic heterogeneity, i.e., different variants in the same gene leading to variable phenotypes and a spectrum of disease severity. Lysosomal storage disorders (e.g., Fabry, Gaucher, and Pompe disease) exhibit allelic heterogeneity leading to variable age at onset from the newborn period through childhood and into adulthood, highlighting that conditions may not always be easily classified into newborn, childhood, or adult onset. This points to the critical importance of linking longitudinal phenotypic and genotypic data. In addition to collecting up-front phenotypic data, further phenotyping based on the results of genomic sequence data can improve our understanding of how conditions progress and develop from the prenatal to pediatric and adult time periods, and the associated changes in phenotypes. Such data will also allow for determination if a variant explains all of a phenotype improving our biological understanding to help achieve diagnoses and connecting these diagnoses to therapeutic strategies across the lifespan. Up to date information will therefore be critical, requiring that databases of phenotypic and genetic variant information be updated regularly and shared broadly. Figure 1 demonstrates this link between box #2 structured phenotype data and box #3 clinical genomic sequencing interpretation both feeding into box #4 data sharing to generate diagnoses box #6 across the lifetime.
Improving Diagnoses by Integrating Multiple Data Types
Genomic sequencing data can also be integrated with additional data types (such as model organism, metabolome, and transcriptome data) to improve diagnoses as shown in Figure 1 box #5. In the first 20 months of the Undiagnosed Diseases Network, combining clinical exome and genome sequencing data with functional information from studying drosophila and zebrafish animal models led to diagnoses in eight patients, while metabolomics data contributed to diagnoses in three others out of 132 diagnoses.4 Clinical assessment by a medical geneticist to obtain additional targeted phenotypic and molecular data, based on information derived from genomic sequence data, has also been shown to lead to more accurate diagnoses and a net increase in diagnoses from 36% to 43% (16 diagnoses promoted to definitive and 5 demoted from definitive to possible or unlikely yielding a net 67 diagnoses out of 155 cases).35 The inclusion of transcriptome (RNA sequencing) data when combined with genomic sequencing information has also improved diagnostic yield, including in cases where the disease specific tissue may be inaccessible resulting in the use of more accessible cell sources such as fibroblasts or peripheral blood mononuclear cells.36–38 The sequencing of RNA in muscle biopsies from 50 undiagnosed patients with muscle disease yielded 17 new diagnoses in families where prior DNA sequencing had yielded no genomic diagnosis.36 Additionally, sequencing of RNA from peripheral blood mononuclear cells supported the first long-read genomic sequencing diagnosis of a novel pathogenic deletion in PRKAR1A in a patient with Carney complex.38 In Carney complex variation in PRKAR1A leads to degradation of a subunit of protein kinase A, thereby triggering increased activation leading to uncontrolled cell growth, and an increased risk of benign tumors.38 The sequencing of fibroblast RNA additionally provided support for the pathogenicity of a novel, de novo heterozygous IGF2 splice site variant in an Australian Aboriginal, leading to a diagnosis of Silver-Russell syndrome (a syndrome characterized by poor growth before and after birth).37 This finding represents the first Australian Aboriginal family known to be diagnosed with IGF2-related Silver-Russell syndrome and highlights the importance of genomic testing in diverse populations to clarify phenotypic features within and across populations with different ethnic backgrounds. Utilizing additional data (such as those from animal models, metabolomics studies, clinical re-assessment, and RNA sequencing) can thus add to the clinical genomic sequencing results to improve diagnoses.
For rare disease diagnoses, data sharing (Figure 1, box #4) is also critical for identifying additional patients with the same or similar phenotypes, strengthening evidence that variant in a gene is associated, especially when a gene has not previously been associated with any human disease. Matchmaker Exchange (https://www.matchmakerexchange.org/) is an excellent example of a federated data sharing model with multiple connected databases, or nodes, that is designed to allow patients, clinicians, and researchers to share information.39 With 7 databases currently sharing data, Matchmaker Exchange matches patients based on genes and phenotypes. Patients can access Matchmaker Exchange through GeneMatcher (https://genematcher.org/) and MyGene2 (https://www.mygene2.org/), while all nodes support data entry and matching by clinicians and researchers. Sharing phenotypic and genomic data has also been critical to the success of multiple global initiatives to diagnose previously undiagnosed patients in the Undiagnosed Disease Network International (http://www.udninternational.org/).4,5,40–43 Gainotti et al. highlighted the importance of patient and family participation in undiagnosed research, recommending that patients’ active participation should be maximized. This allows for patients to describe their own phenotype, choose how much information they share in matchmaking databases, and express preferences for what genomic results they find relevant to return to them.43 Such active participation in data sharing drives patient matching and clinical diagnosis and furthers discovery related to the etiology of these previously undiagnosed conditions. Databases, such as the Genome Aggregation Database (gnomAD, http://gnomad.broadinstitute.org/) that share standardized information about allele frequencies seen in the general population and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) that share relationships between variants and phenotypes, are also important resources for determining if a suspected genomic variant is seen in the general population and for reporting new variant-phenotype assertions.
Changing Clinical Management Based on Genomic Sequencing Diagnoses
Utilizing collected phenotypic information, clinical genomic sequencing (exome or genome) aims to diagnose patients and change clinical management (Figure 1, box #7). Diagnostic rates for clinical exome and genome sequencing vary dramatically depending upon the patient population (Table 2); however, multiple studies have shown overall diagnostic rates of 25–35% for pediatric and adult undiagnosed diseases.4,15,16,18,19,26,27,44–46 In general, diagnostic rates tend to be higher in children and lower in adults. Using proband-only exome sequencing, Stark et al. reported one of the highest diagnostic rates in infants suspected of monogenic disorders at 58% (46/80).47 On the other end of the age spectrum, Posey et al. saw one of the lowest diagnostic rates in adults at 18% (85/486).28 The diagnostic rate in adults over age 30 (10%, 24/231) was lower than in adults age 18–30 (24%, 61/285), potentially due in part to the lower availability of parental samples for analysis in older adults. This lack of parental samples limits the ability to detect de novo variants (i.e. variants not inherited from either parent) as part of the clinical report (box #3).28 Environmental effects may also have a greater impact on adult conditions leading to increased non-genetic variation contributing to the phenotype, making it more difficult to identify causative genetic variants. Fewer recurrent molecular diagnoses of different variants in the same gene were seen in adult (11%, 9/85) versus pediatric patients (57%, 266/463) p-value < 0.0001 suggesting that greater genomic diversity may underlie adult disorders.26,28 Additionally, phenotypes seen in infants may be more severe than those seen in adults due to survivability.
Table 2:
Diagnostic rates across different ages from studies of undiagnosed patients unselected for a specific phenotype published in 2014–2018
| Study PMID | First author | Year | Age group | Diagnostic rate |
|---|---|---|---|---|
| 30304647 | Splinter K | 2018 | pediatric and adult | 132/382 (35%) |
| 25326635 | Yang Y | 2014 | fetus | 6/11 (55%) |
| <5 years | 247/900 (27%) | |||
| 5–18 years | 210/845 (25%) | |||
| >18 years | 41/244 (17%) | |||
| overall | 504/2000 (25%) | |||
| 25326637 | Lee H | 2014 | pediatric and adult | 213/814 (26%) |
| 28496993 | Bick D | 2017 | pediatric | 8/22 (36%) |
| 26633542 | Retterer K | 2016 | pediatric and adult | 876/3040 (29%) |
| 26938784 | Stark Z | 2016 | 0–2 years | 46/80 (58%) |
| 28567303 | Stavropoulos D | 2016 | <1 month – 18 years | 34/100 (34%) |
| 26633545 | Posey J | 2016 | adults | 85/486 (18%) |
| 25356970 | Farwell K | 2015 | prenatal | 2/2 (100%) |
| 0–3 months | 6/12 (50%) | |||
| <1 year | 7/36 (19%) | |||
| 1–5 years | 67/194 (35%) | |||
| 5–12 years | 30/117 (26%) | |||
| 12–18 years | 19/58 (33%) | |||
| 18–40 years | 14/45 (31%) | |||
| >40 years | 5/36 (14%) | |||
| overall | 152/500 (30%) | |||
| 30266093 | Normand E | 2018 | prenatal | 46/146 (32%) |
| 27848944 | Trujillano D | 2017 | prenatal | 4/23 (17%) |
| <1 year | 42/141 (30%) | |||
| 1–5 years | 128/394 (32%) | |||
| 5–15 years | 73/285 (26%) | |||
| 15–30 years | 23/81 (28%) | |||
| >30 years | 10/38 (26%) | |||
| unknown age | 27/38 (71%) | |||
| overall | 307/1000 (31%) | |||
| 28973083 | Meng L | 2017 | infants (<100 days) | 102/278 (37%) |
A single phenotype category, as may be defined by HPO terms, can also exhibit variability in diagnostic rate.15,18,19,26,28,44–47 Neurological phenotypes are one of the most frequent primary indications for referral, with diagnostic rates from 27% to 42% in six studies.15,18,19,26,45,46 Additionally, there are differences in diagnostic rates by phenotype. 15,18,19,26,28,44–47 Variability in the phenotype yielding the highest diagnosis rate can also be seen across studies, with 8 studies reporting 8 different phenotypes: hearing (55%)15, craniofacial (46%)18, abnormalities of blood (65%)19, retinal disorders (48%)44, obstetric (43%)45, connective tissue (44%)46, neurometabolic disorder (74%)47, and neurodevelopmental (28%)28. It is also important to consider the denominator used when comparing diagnosis statistics between different phenotypes and studies, as illustrated by Trujillano et al., whose study of 1000 families included 771 (77%) with neurological abnormalities, of which 229 received a diagnosis.46 Thus, while only 30% (229/771) of the families with neurological abnormalities received a diagnosis, 75% (229/307) of the 307 families diagnosed had neurological abnormalities.46
Moreover, multiple studies reported changes in management for 33–94% of patients diagnosed including changes in therapy from receiving a diagnosis (Table 3).4,15,16,18,19,27,47 In the prenatal setting, having a diagnosis before birth provides the opportunity for direct intervention, including surgical procedures before or immediately after birth. For example, Deprest et al. noted the opportunity for rapid clinical genomic sequencing to provide diagnostic information about whether congenital diaphragmatic hernia is isolated or associated with other fetal abnormalities, helping to decide if fetal endoluminal tracheal occlusion would be beneficial as a prenatal intervention.48 Currently, abnormal genomic findings are used as exclusion criteria for fetal endoluminal tracheal occlusion trials due to a worse prognosis. Diagnostic information can also be used by families to prepare for when and where delivery will occur and to give insights about condition specific care challenges. In Normand et al., prenatal detection of a pathogenic COL1A1 variant allowed the parents time to learn about osteogenesis imperfecta, a condition characterized by bones that break easily, and to connect with other families about strategies to prevent such breaks.18 Delivery strategies to minimize trauma can also be employed. Diagnostic information may additionally be useful for family planning purposes and have a positive psychosocial impact for parents.18 In the unfortunate situation where an inevitably fatal condition is identified, care planning includes avoidance of unnecessary and futile intensive care, with development of plans for palliative comfort care that can reduce suffering and financial stress. Rapid genomic sequencing of critically ill infants in the NICU has been shown by multiple groups to change clinical management, ranging from 33–72% in three studies.19,24,47 Of note, some of the management changes are to palliative care (19/53 or 36% of management changes in Meng et al. and 1/18 or 6% in Farnaes et al.); however, even this change can be of personal utility to the family, as it brings an end to the diagnostic odyssey and clinical utility as it allows for a peaceful withdrawal of invasive interventions that would prove ineffective.19,24 A test has clinical utility if the results can be used to inform clinical decisions and management, while a test with personal utility has benefits to an individual or family beyond clinical care. Management changes, such as changes in medications and surgical procedures, also led to avoided morbidity in 26% (11/42) of infants in Farnaes et al. including seizure control with a change in medication in an infant with Early Infantile Epileptic Encephalopathy type seven and avoidance of a surgical Kasai procedure in an infant with Alagille syndrome.24 In the Undiagnosed Diseases Network, diagnosis of pediatric and adult patients led to a change in therapy in 28 patients (21% of the 132 individuals diagnosed), a change in care other than therapy (such as changes in diagnostic strategy) in 49 patients (37%), and variant-specific genetic counseling in 48 patient (36%).4 Having a diagnosis can affect care from the prenatal to the adult setting with changes in medical management (including starting or stopping therapies or other interventions based on the diagnostic results) and modified genetic counseling based on recurrence risk, which may influence reproductive planning.4,15,16,18,19,27,47
Table 3:
Examples of management changes after diagnosis for studies of undiagnosed patients unselected for a specific phenotype published in 2014–2018
| Study PMID | First author | Year | Age group | Management changes |
|---|---|---|---|---|
| 30304647 | Splinter K | 2018 | pediatric and adult | 21% (28/132) change in therapy, 37% (49/132) change in care other than therapy, 36% (48/132) variant-specific genetic counseling |
| 28496993 | Bick D | 2017 | pediatric | 75% (6/8) impact on medical management or surveillance, 4 changes in medication, 6 medical surveillance |
| 26633542 | Retterer K | 2016 | pediatric and adult | 5 reported with suggested intervention or treatment |
| 26938784 | Stark Z | 2016 | 0–2 years | 33% (15/46) clinical management changed (3 additional treatment started, 5 treatments stopped or modified) |
| 28567303 | Stavropoulos D | 2016 | <1 month – 18 years | 94% (32/34) change in clinical management |
| 30266093 | Normand E | 2018 | prenatal | 4 medical management changes, 15 reproductive planning, 10 recurrence risk of 19 cases with information |
| 28973083 | Meng L | 2017 | infants (<100 days) | 52 infants affected medical management |
There are also ethical considerations (Figure 1, box #4) regarding the use of clinical genomic sequencing for diagnosis of previously undiagnosed conditions. The American College of Genetics and Genomics (ACMG) recommends a minimal gene list for analysis as secondary findings when clinical genomic sequencing is conducted across the age spectrum.49 Genes on this list as well as additional medically actionable findings including incidental, secondary, and carrier-status findings as determined by the clinical genomics laboratories, are provided on clinical reports.15,16,18,19,26–28,44 In the pediatric and prenatal setting, genomic sequencing results returned to parents may have implications for their own health or family planning decisions. Which incidental or secondary results should be returned in the clinical context is also a matter of debate, with professionals having different opinions about what results should be returned in the pediatric setting.49–55 For adults, carrier results may have direct relevance for reproductive planning, and incidental or secondary findings may have broader implications for other family members. Furthermore, even if a condition is not currently medically actionable, having a diagnosis may still have clinical and personal utility for the family, including an end to the economic and psychological cost of the diagnostic odyssey, the possibility of family member testing, information for reproductive decision-making, and social support related to the diagnosis.56 Data protection (Figure 1, box #4) is also critical to ensure that trust in data sharing is maintained while not preventing access to information that may help lead to diagnoses and changes in management.
Conclusion
Genomic medicine can help undiagnosed patients and their families end their diagnostic odysseys. While adult patients may currently exhibit lower diagnostic rates, structured longitudinal phenotyping (Figure 1, box #2) may improve our understanding of the connection between genomic variants and phenotypes across the lifespan. Challenges also remain for improving structured phenotype collection methods, getting laboratories access to the latest data on genes and phenotypes globally, improving methods to integrate additional datatypes with clinical genomic sequencing, and making information on diagnoses sharable in a way that protects patients. Figure 1 also illustrates the central role that data sharing plays in genomic medicine to link phenotypes with clinical genomic sequencing report, additional data types, and diagnoses, leading to changes in clinical management.
Such diagnoses matter, not just for the potential treatments they may lead to, but also for the closure and the peace of mind provided by finally receiving a diagnosis. Diagnoses derived from genomic sequencing findings provide a variety of benefits to patients and their families, including new opportunities for therapeutic interventions and condition-specific management that can lead to improved outcomes and quality of life. The diagnostic results can be used for family planning, cascade testing of other family members, justifying social and educational services, and connecting to other families and condition-specific support groups. Data sharing and data standards are critical to the future success of genomic medicine, since clinical data can inform research discoveries that, in turn, can return new knowledge to inform clinical care. This virtuous cycle is a core component of genomic medicine and will only be enhanced by the new technologies and innovations being brought to bear for the population of patients affected with rare and undiagnosed diseases.
Acknowledgements
The authors would like to thank William A. Gahl, MD, PhD for review of the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Human Genome Research Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Genomic Medicine and Health Care https://www.genome.gov/27552451/what-is-genomic-medicine/. Accessed: October 14, 2018.
- 2.The Cost of Sequencing a Human Genome https://www.genome.gov/sequencingcosts/. Accessed: October 14, 2018.
- 3.Gahl WA, Markello TC, Toro C, et al. The National Institutes of Health Undiagnosed Diseases Program: insights into rare diseases. Genet Med 2012. January; 14(1):51–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Splinter K, Adams DR, Bacino CA, et al. Effect of Genetic Diagnosis on Patients with Previously Undiagnosed Disease. N Engl J Med 2018. October 10. [DOI] [PMC free article] [PubMed]
- 5.Taruscio D, Groft SC, Cederroth H, et al. Undiagnosed Diseases Network International (UDNI): White paper for global actions to meet patient needs. Mol Genet Metab 2015. December; 116(4):223–5. [DOI] [PubMed] [Google Scholar]
- 6.International Rare Diseases Research Consortium: Vision & Goals http://www.irdirc.org/about-us/vision-goals/. Accessed: March 18, 201.
- 7.Orphan Drug Act of 1983 https://www.govinfo.gov/content/pkg/STATUTE-96/pdf/STATUTE-96-Pg2049.pdf. Accessed: October 14, 2018.
- 8.Rare Diseases Act of 2002 https://www.govinfo.gov/content/pkg/CRPT-107hrpt543/pdf/CRPT-107hrpt543.pdf. Accessed: October 14, 2018.
- 9.Genetic and Rare Diseases Information Center FAQs About Rare Diseases https://rarediseases.info.nih.gov/diseases/pages/31/faqs-about-rare-diseases. Accessed: October 14, 2018.
- 10.Rare Disease Impact Report: Insights from patients and the medical community http://www.journalofraredisorders.com/pub/IssuePDFs/RareDiseaseImpactReportforWeb.pdf. Accessed: October 14, 2018.
- 11.Tifft CJ, Adams DR. The National Institutes of Health undiagnosed diseases program. Curr Opin Pediatr 2014. December; 26(6):626–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bogart KR, Irvin VL. Health-related quality of life among adults with diverse rare disorders. Orphanet J Rare Dis 2017. December 7; 12(1):177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McConkie-Rosell A, Hooper SR, Pena LDM, et al. Psychosocial Profiles of Parents of Children with Undiagnosed Diseases: Managing Well or Just Managing? J Genet Couns 2018. August; 27(4):935–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shashi V, McConkie-Rosell A, Rosell B, et al. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet Med 2014. February; 16(2):176–82. [DOI] [PubMed] [Google Scholar]
- 15.Retterer K, Juusola J, Cho MT, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med 2016. July; 18(7):696–704. [DOI] [PubMed] [Google Scholar]
- 16.Bick D, Fraser PC, Gutzeit MF, et al. Successful Application of Whole Genome Sequencing in a Medical Genetics Clinic. J Pediatr Genet 2017. June; 6(2):61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Köhler S, Vasilevsky NA, Engelstad M, et al. The Human Phenotype Ontology in 2017. Nucleic Acids Res 2017. January 4; 45(D1):D865–D876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Normand EA, Braxton A, Nassef S, et al. Clinical exome sequencing for fetuses with ultrasound abnormalities and a suspected Mendelian disorder. Genome Med 2018. September 28; 10(1):74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meng L, Pammi M, Saronwala A, et al. Use of Exome Sequencing for Infants in Intensive Care Units: Ascertainment of Severe Single-Gene Disorders and Effect on Medical Management. JAMA Pediatr 2017. December 4; 171(12):e173438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Köhler S, Schulz MH, Krawitz P, et al. Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am J Hum Genet 2009. October; 85(4):457–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Groza T, Köhler S, Doelken S, et al. Automatic concept recognition using the human phenotype ontology reference and test suite corpora. Database (Oxford) 2015. February 27; 2015. pii: bav005. [DOI] [PMC free article] [PubMed]
- 22.Yang H, Robinson PN, Wang K. Phenolyzer: phenotype-based prioritization of candidate genes for human diseases. Nat Methods 2015. September; 12(9):841–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fang H, Wu Y, Yang H, et al. Whole genome sequencing of one complex pedigree illustrates challenges with genomic medicine. BMC Med Genomics 2017. February 23; 10(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farnaes L, Hildreth A, Sweeney NM, et al. Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. NPJ Genom Med 2018. April 4; 3:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Köhler S, Vasilevsky NA, Engelstad M, et al. The Human Phenotype Ontology in 2017. Nucleic Acids Res 2017. January 4;45(D1):D865–D876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 2014. November 12; 312(18):1870–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stavropoulos DJ, Merico D, Jobling R, et al. Whole Genome Sequencing Expands Diagnostic Utility and Improves Clinical Management in Pediatric Medicine. NPJ Genom Med 2016. January 13; 1 pii: 15012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Posey JE, Rosenfeld JA, James RA, et al. Molecular diagnostic experience of whole-exome sequencing in adult patients. Genet Med 2016. July; 18(7):678–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Posey JE, Harel T, Liu P, et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N Engl J Med 2017. January 5; 376(1):21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niemi MEK, Martin HC, Rice DL, et al. Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature 2018. October;562(7726):268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Posey JE, Rosenfeld JA, et al. Phenotypic expansion in DDX3X - a common cause of intellectual disability in females. Ann Clin Transl Neurol 2018. September 15; 5(10):1277–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan QK, Cope H, Spillmann RC, et al. Further evidence for the involvement of EFL1 in a Shwachman-Diamond-like syndrome and expansion of the phenotypic features. Cold Spring Harb Mol Case Stud 2018. October 1; 4(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.American College of Obstetricians and Gynecologists’ Committee on Practice Bulletins—Obstetrics, Committee on Genetics, Society for Maternal–Fetal Medicine. Practice bulletin no. 162: prenatal diagnostic testing for genetic disorders. Obstet Gynecol 2016: e108–22. [DOI] [PubMed]
- 34.Committee on Genetics and the Society for Maternal-Fetal Medicine. Committee Opinion No.682: Microarrays and next-generation sequencing technology: the use of advanced genetic diagnostic tools in obstetrics and gynecology. Obstet Gynecol 2016: e262–8. [DOI] [PubMed]
- 35.Baldridge D, Heeley J, Vineyard M, et al. The Exome Clinic and the role of medical genetics expertise in the interpretation of exome sequencing results. Genet Med 2017. September; 19(9):1040–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cummings BB, Marshall JL, Tukiainen T, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med 2017. April 19; 9(386). pii: eaal5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poulton C, Azmanov D, Atkinson V, et al. Silver Russel syndrome in an aboriginal patient from Australia. Am J Med Genet A 2018. August 27. [DOI] [PubMed]
- 38.Merker JD, Wenger AM, Sneddon T, et al. Long-read genome sequencing identifies causal structural variation in a Mendelian disease. Genet Med 2018. January; 20(1):159–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Philippakis AA, Azzariti DR, Beltran S, et al. The Matchmaker Exchange: a platform for rare disease gene discovery. Hum Mutat 2015. October; 36(10):915–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.López-Martín E, Martínez-Delgado B, Bermejo-Sánchez E, Alonso J; SpainUDP Network, Posada M. SpainUDP: The Spanish Undiagnosed Rare Diseases Program. Int J Environ Res Public Health 2018. August 14;15(8). pii: E1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baynam G, Bowman F, Lister K, et al. Improved Diagnosis and Care for Rare Diseases through Implementation of Precision Public Health Framework. Adv Exp Med Biol 2017; 1031:55–94. [DOI] [PubMed] [Google Scholar]
- 42.Adachi T, Kawamura K, Furusawa Y, et al. Japan’s initiative on rare and undiagnosed diseases (IRUD): towards an end to the diagnostic odyssey. Eur J Hum Genet 2017. September; 25(9):1025–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gainotti S, Mascalzoni D, Bros-Facer V, et al. Meeting Patients’ Right to the Correct Diagnosis: Ongoing International Initiatives on Undiagnosed Rare Diseases and Ethical and Social Issues. Int J Environ Res Public Health 2018. September 21; 15(10). pii: E2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee H, Deignan JL, Dorrani N, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 2014. November 12; 312(18):1880–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Farwell KD, Shahmirzadi L, El-Khechen D, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med 2015. July; 17(7):578–86. [DOI] [PubMed] [Google Scholar]
- 46.Trujillano D, Bertoli-Avella AM, Kumar Kandaswamy K, et al. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur J Hum Genet 2017. February; 25(2):176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stark Z, Tan TY, Chong B, et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med 2016. November; 18(11):1090–1096. [DOI] [PubMed] [Google Scholar]
- 48.Deprest J, Brady P, Nicolaides K, et al. Prenatal management of the fetus with isolated congenital diaphragmatic hernia in the era of the TOTAL trial. Semin Fetal Neonatal Med 2014. December; 19(6):338–48. [DOI] [PubMed] [Google Scholar]
- 49.Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 2017. February; 19(2):249–255 [DOI] [PubMed] [Google Scholar]
- 50.American Academy of Pediatrics, Committee on Bioethics, Committee on Genetics; and American College of Medical Genetics and Genomics, Social, Ethical, and Legal Issues Committee. Ethical and policy issues in genetic testing and screening of children. Pediatrics 2013. March; 131(3):620–2. [DOI] [PubMed] [Google Scholar]
- 51.Ross LF, Saal HM, David KL, et al. Technical report: Ethical and policy issues in genetic testing and screening of children. Genet Med 2013. March; 15(3):234–45. [DOI] [PubMed] [Google Scholar]
- 52.Botkin J Ethical Issues in Pediatric Genetic Testing and Screening for Current Opinion in Pediatrics. Curr Opin Pediatr 2016. December; 28(6):700–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wilfond BS, Fernandez CV, Green RC. Disclosing Secondary Findings from Pediatric Sequencing to Families: Considering the “Benefit to Families”. J Law Med Ethics 2015. Fall; 43(3):552–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kleiderman E, Knoppers BM, Fernandez CV, et al. Returning incidental findings from genetic research to children: views of parents of children affected by rare diseases. J Med Ethics 2014. October; 40(10):691–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barajas M, Ross LF. Pediatric Professionals’ Attitudes about Secondary Findings in Genomic Sequencing of Children. J Pediatr 2015. May; 166(5):1276–1282.e7. [DOI] [PubMed] [Google Scholar]
- 56.ACMG Board of Directors. Clinical utility of genetic and genomic services: a position statement of the American College of Medical Genetics and Genomics. Genet Med 2015. June; 17(6):505–7 [DOI] [PubMed] [Google Scholar]