

Abstract
Aims
The study objective was to evaluate the pharmacokinetics of the selective progesterone receptor modulator vilaprisan in participants with hepatic impairment. Additionally, the safety and tolerability of vilaprisan were investigated.
Methods
In this phase 1, open‐label, nonrandomised, parallel‐group, pharmacokinetic study, men and women with mild or moderate hepatic impairment (Child–Pugh grade A or B) and control participants with normal hepatic function matched by age, weight and sex received a single oral 2 mg dose of vilaprisan. Key pharmacokinetic parameters, relationships between parameters and safety outcomes were measured.
Results
Thirty‐six participants completed the study: 9 with mild hepatic impairment, 9 with moderate hepatic impairment and 18 matched control participants with normal hepatic function. Vilaprisan reached maximum plasma concentrations after 1–2 hours. Unbound vilaprisan exposure was 1.44‐fold higher for participants with mild hepatic impairment vs controls (90% confidence interval: 0.91–2.26), and 1.74‐fold higher for participants with moderate impairment vs controls (90% confidence interval: 1.09–2.78). The maximum observed unbound peak concentrations were similar for participants with hepatic impairment and matched controls. Vilaprisan 2 mg was well tolerated and the incidence of treatment‐emergent adverse events was similar across cohorts.
Conclusion
Only mild increases of <1.75‐fold in exposure were observed in participants with mild or moderate hepatic impairment compared with control participants. No safety concern was identified. These data, alongside the excellent safety profile observed in phase 1 and 2 studies, do not indicate that a dose adjustment would be required in patients with mild or moderate hepatic impairment.
Keywords: clinical pharmacology, drug development, gynaecology/obstetrics, pharmacokinetics, phase 1
What is already known about this subject
Vilaprisan is predominantly metabolised by CYP3A4 in the liver, and excreted in faeces
Systemic exposure may be increased in patients with mild/moderate hepatic impairment due to reduced metabolic activity
There is a need to understand the impact of hepatic impairment on the pharmacokinetics of vilaprisan to support dosing recommendations
What this study adds
Only mild increases in vilaprisan exposure were observed in participants with mild or moderate hepatic impairment compared with controls
No safety concern was identified in patients with mild or moderate hepatic impairment
There is no indication that dose adjustment would be required for patients with mild or moderate hepatic impairment
1. INTRODUCTION
Vilaprisan is a highly potent selective progesterone receptor modulator under investigation for the treatment of symptomatic uterine fibroids (UF) and endometriosis. UF (leiomyomas or myomas) are the most common benign smooth muscle tumours of the myometrium and are clinically relevant in up to 40% of premenopausal women aged 35–40 years.1 Symptoms include heavy menstrual bleeding, which can lead to anaemia, fatigue, painful periods, abdominal protuberance, painful intercourse or pelvic pressure, and bladder or bowel dysfunction. UF have also been linked with reproductive issues that can negatively impact on fertility and pregnancy.2, 3
In 2 phase 2 clinical studies in women with UF, treatment with vilaprisan resulted in effective bleeding control and reductions in fibroid volume, and demonstrated a favourable safety profile.4, 5 Based on these promising results, vilaprisan is currently being investigated in phase 3 studies in patients for long‐term treatment of UF.6
Previous clinical drug–drug interaction studies with the potent cytochrome P450 3A4 (CYP3A4) inhibitor itraconazole and the potent CYP3A4 inducer rifampicin showed a mean increase by 6.2‐fold and a reduction by 96% of the vilaprisan exposure, respectively, confirming preclinical investigations that vilaprisan is extensively metabolised by CYP3A4.7, 8 Biotransformation occurs mainly by oxidation reactions at the steroid skeleton, as well as reductions of the 3‐keto group, leading to a complex metabolite pattern with rather low exposure (<10%) to single metabolites in plasma. The 2 main metabolites elicit only marginal pharmacological activity at the progesterone receptor, not contributing to clinical efficacy. Vilaprisan is highly bound to plasma proteins with an unbound fraction of 5.29%. Furthermore, based on a physiologically based pharmacokinetic model, the estimated fraction metabolised by CYP3A4 was >0.9 for vilaprisan.8, 9 The fraction escaping first‐pass metabolism in the gut (FG) was estimated to be about 0.65 and the hepatic bioavailability was ~0.93 (FH), consistent with an oral bioavailability of 61%.7 Clinical results from an intravenous microtracer study showed that vilaprisan is extensively distributed into tissues, as reflected by a large volume of distribution at steady state of ~360 L, and has a moderate plasma clearance of ~7 L/h with negligible contribution of renal clearance to total elimination. The terminal plasma elimination half‐life of ~40 hours is similar after intravenous and oral administration.7 In vitro interaction studies suggested no clinically relevant interaction of vilaprisan with the main CYP enzymes or transporters (i.e. as a perpetrator). The metabolic activity of CYP3A4 may be lower in patients with mild or moderate hepatic impairment than in patients with normal hepatic function, which could potentially result in decreased clearance and therefore higher systemic exposure.10, 11, 12 Hence, the pharmacokinetics of vilaprisan may be altered in patients with reduced liver function. It is anticipated that vilaprisan could be used for patients with comorbidities, including those with mild or moderate hepatic impairment. Therefore, it is important to gain a clear understanding of the impact of hepatic impairment on the pharmacokinetics of vilaprisan, to guide labelling and dosing recommendations for this subgroup of the target patient population.
The aim of this phase 1 study was to investigate the pharmacokinetics of vilaprisan in participants of both sexes with mild or moderate hepatic impairment, as defined by Child–Pugh criteria in comparison with matched control participants with normal hepatic function. In addition, the safety and tolerability of vilaprisan were evaluated.13
2. METHODS
2.1. Study design and treatments
This was a phase 1, multicentre, open‐label, nonrandomised, parallel‐group, single‐dose, pharmacokinetic study (clinicaltrials.gov number: NCT03092999; EudraCT: 2015–005232‐18). The primary objective was to compare the pharmacokinetics of vilaprisan following a single oral dose between participants with mild or moderate hepatic impairment, defined according to the Child–Pugh classification grade A (CP‐A) or B (CP‐B), and sex‐, age‐ and weight‐matched control participants with normal hepatic function. The secondary objective was to evaluate the safety and tolerability of vilaprisan in the study population.
The study was conducted from March 2017 to July 2017 at 2 investigational sites in Germany. Vilaprisan 2 mg immediate‐release tablet was administered as a single oral dose under fasting conditions. An overview of the study design is shown in Figure 1.
Figure 1.
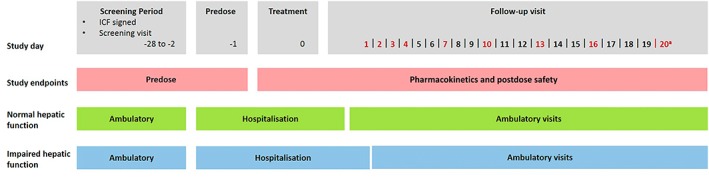
Study design overview. aLast follow‐up visit. ICF, informed consent form. Red text denotes the days of the follow‐up visits. Participants with impaired hepatic function were hospitalized for two days after treatment, controls for one day after treatment
All local legal and regulatory requirements were met and the study was conducted in accordance with the ethical principles of the Declaration of Helsinki and the International Conference on Harmonisation guideline E6: Good Clinical Practice. The study was approved by the relevant independent ethics committee (Ethics Committee of the Ärztekammer Schleswig‐Holstein). Written informed consent was obtained directly from all participants prior to any study procedures being undertaken.
2.2. Participants
Although the target population for vilaprisan is women with UF or endometriosis, the study was conducted in both male and female participants, as physiological differences between male and female participants were not expected to influence the main study outcomes. Three parallel treatment groups were included: participants with either (i) mild (CP‐A) or (ii) moderate (CP‐B) hepatic impairment (Table 1), or (iii) participants with normal hepatic function (control participants) matched with regard to sex, age (±15%), and body weight (±15%).
Table 1.
Child–Pugh classification for participants with hepatic impairment13
| Measurement | 1 point | 2 points | 3 points |
|---|---|---|---|
| Albumin (g L−1) | >35 | 28–35 | <28 |
| Ascites | Absent | Slight quantity | Moderate quantity |
| Bilirubin (mg dL−1) | <2 mg dL−1 | 2–3 mg dL−1 | >3 mg dL−1 |
| Hepatic encephalopathya | None | I–II | III–IV |
| Prothrombin time (Quick's test value; %) | >60 | 40–60 | <40 |
The degree of hepatic impairment was classified as CP‐A (5–6 points) or CP‐B (7–9 points). Participants with CP‐C (10–15 points) were not eligible for study enrolment. CP‐A, Child–Pugh classification grade A; CP‐B, Child–Pugh classification grade B.
Encephalopathy Grading System:
Grade I: restless, sleep disturbed, irritable/agitated, tremor, impaired handwriting
Grade II: lethargic, time‐disoriented, inappropriate, asterixis, ataxia
Grade III: somnolent, stuporous, place‐disoriented, hyperactive reflexes, rigidity
Grade IV: unrousable coma, no personality/behaviour, decerebrate.
White/Caucasian men and women aged 18–79 years with a body mass index of 18–34 kg m-2 were included in the study. Participants were required to use adequate contraception. For participants with hepatic impairment, documentation of liver cirrhosis was required by either histopathology, laparoscopy, Fibroscan or ultrasound. Impaired liver function was classified as mild or moderate according to the Child–Pugh criteria provided in Table 1.13 Liver function had to be stable with the same Child–Pugh class in the last 2 months prior to screening.
Participants were excluded from study participation if they had any gastrointestinal, cerebrovascular, cardiovascular, renal, pulmonary or other hepatic disease or malignancy, or clinically relevant deviations from the normal range in physical or gynaecological examinations, clinical chemistry, haematology, or urinalysis. Full subject selection criteria are provided in the Supporting Information.
2.3. Assessment and variables
Plasma (blood samples taken 1.0 hour pre‐vilaprisan dosing and at 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 4.0, 6.0, 8.0, 12.0, and 16.0 hours postdosing and also on days 1, 2, 3, 4, 7, 10, 13, 16, and 20 post‐dosing) and urine concentrations (urine samples taken 1.0 hour pre‐vilaprisan dosing and continuously until 6 hours postdosing, then continuously until Day 1 postdosing) of vilaprisan were determined from the pharmacokinetic samples taken. Vilaprisan was determined in plasma and urine after addition of [13C6]vilaprisan as an internal standard followed by separation employing liquid chromatography and tandem mass spectrometric using an Agilent system 1200.
Vilaprisan was determined in individual urine and plasma samples using a 150 × 4.6 mm Prodigy ODS3, 5‐μm analytical column (Phenomenex, 00F‐4097‐E0) following elution with an acetonitrile/ammonium acetate buffer gradient system. All methods were validated according to the relevant European and US guidelines.14, 15 The fraction of unbound (fu) vilaprisan was determined at 2, 24, and 48 hours post‐dosing in plasma ex vivo using the Transil method.16 Transil is a widely used rapid dialysis method using immobilised plasma proteins on membrane beads. Plasma samples were spiked with carbon 14‐labelled vilaprisan at a nominal concentration of 750 μg L−1, enabling reliable detection of vilaprisan in plasma. The detection limits were within the concentration range up to 2537 μg L−1, for which linear protein binding has been demonstrated for vilaprisan. For details and an overview of the performance of the bioanalytical methods, see Supporting Information.
Pharmacokinetic parameters were calculated based on the actual concentration–time data by a noncompartmental approach using WinNonlin, version 5.3. Two main pharmacokinetic parameters were defined for this study: area under the concentration vs time curve in plasma from zero to infinity for unbound drug (AUCu), and maximum observed unbound drug concentration (Cmax,u). Further pharmacokinetic parameters specified for this study were: area under the concentration vs time curve from zero to infinity after a single dose (AUC); maximum observed drug concentration (Cmax); time to reach Cmax; terminal half‐life; total and unbound apparent oral vilaprisan clearance; apparent total and unbound volume of distribution during the terminal phase; urinary excretion of vilaprisan during 24 hours post dose; renal clearance from plasma; and fraction of unbound vilaprisan in plasma (fu).
Safety assessments included recording of adverse events (AEs), serious AEs, reasons for study discontinuation, vital signs, electrocardiogram, and clinical laboratory tests (blood and urine; haematology and biochemistry). AEs were recorded continuously. Vital signs were measured 1 day and 1 hour pre‐vilaprisan dosing, at 2.0 hours postdosing, and also on days 1, 2, 3 and 13 postdosing. Electrocardiogram measurements were conducted 1 day and 1 hour pre‐vilaprisan dosing, at 2 hours postdosing, and also on days 1 and 13 postdosing. Clinical laboratory tests were performed 1 day pre‐vilaprisan dosing, and 1, 3 and 13 days postdosing.
2.4. Statistics
All participants treated with vilaprisan were included in the pharmacokinetic analyses. Analyses of variance (ANOVAs), including fixed hepatic group effects, were used for the comparison of the pharmacokinetic parameters AUCu and Cmax,u between study groups. Log‐transformed parameter values were used, as the parameters were assumed to be log‐normally distributed. The point estimates and 90% confidence intervals (CIs) of the ratios of the groups were derived by retransforming the point estimates (least squares means) and the 90% CIs of the corresponding models to the original scale.
Relationships between the primary variables AUCu and Cmax,u of vilaprisan, and the baseline hepatic function parameters (prothrombin time [Quick's test], albumin, bilirubin, Child–Pugh score) were explored using pairwise scatter plots (figures not shown), Spearman's rank correlation coefficients or Pearson's correlation coefficients with corresponding 95% CIs. The pharmacokinetic parameters of vilaprisan in urine were summarised using descriptive statistics.
No formal statistical sample size estimation had been performed for this study due to the exploratory character of the study. Sample size was based on recommendations from the Food and Drug Administration Guidance for Industry on hepatic impairment studies.17 Based on experience in previous pharmacokinetic studies with vilaprisan at Bayer, the chosen sample size of n = 36 subjects, i.e. 9 subjects with mild (CP‐A) hepatic impairment, 9 subjects with moderate (CP‐B) hepatic impairment, and 18 sex‐, age‐ and weight‐ matched subjects, was considered to be sufficient to detect a moderate, at least 2‐fold increase in AUC.
2.5. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY,18 and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/2018.19
3. RESULTS
3.1. Disposition of participants
There were 4 (10%) screening failures out of the 40 participants screened. A total of 36 participants were assigned to treatment and completed the study: 9 participants with mild hepatic impairment (CP‐A), 9 participants with moderate hepatic impairment (CP‐B) and 18 matched control participants (Figure 2).
Figure 2.
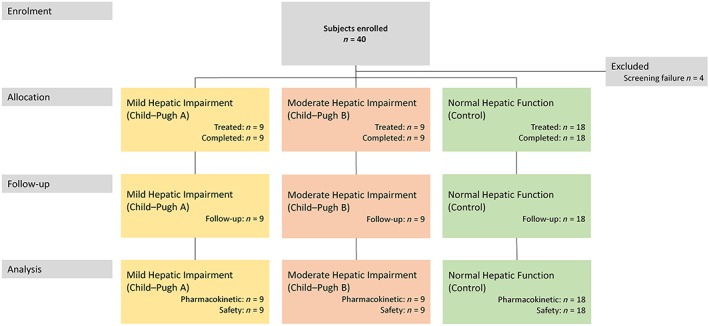
Subject disposition
Baseline characteristics for all treated participants (n = 36) are summarised in Table 2. Two‐thirds (66.7%) of the participants were male, the mean age was 62.3 years (standard deviation 8.5 years; range: 48–78 years) and mean baseline body mass index was 27.18 kg m−2 (standard deviation 3.99 kg m−2).
Table 2.
Demographic and baseline characteristics
| Matched control mild hepatic impairment n = 9 (100%) | Mild hepatic impairment n = 9 (100%) | Matched control moderate hepatic impairment n = 9 (100%) | Moderate hepatic impairment n = 9 (100%) | Total n = 36 (100%) | |
|---|---|---|---|---|---|
| Gender | |||||
| Male, n (%) | 6 (66.7) | 6 (66.7) | 6 (66.7) | 6 (66.7) | 24 (66.7) |
| Female, n (%) | 3 (33.3) | 3 (33.3) | 3 (33.3) | 3 (33.3) | 12 (33.3) |
| Race | |||||
| White, n (%) | 9 (100.0) | 9 (100.0) | 9 (100.0) | 9 (100.0) | 36 (100.0) |
| Age (y), mean ± SD | 58.7 ± 6.7 | 60.2 ± 6.0 | 63.6 ± 9.8 | 66.9 ± 9.7 | 62.3 ± 8.5 |
| Weighta (kg), mean ± SD | 82.20 ± 18.33 | 82.86 ± 18.03 | 81.91 ± 15.37 | 85.17 ± 17.74 | 83.03 ± 16.70 |
| Heighta (cm), mean ± SD | 174.67 ± 11.01 | 174.00 ± 7.86 | 174.89 ± 9.97 | 172.78 ± 6.51 | 174.08 ± 8.66 |
| BMIa (kg m-2), mean ± SD | 26.69 ± 3.92 | 27.12 ± 4.53 | 26.58 ± 3.11 | 28.32 ± 4.67 | 27.18 ± 3.99 |
| Smoking history | |||||
| Never | 5 (55.6%) | 1 (11.1%) | 6 (66.7%) | 3 (33.3%) | 15 (41.7%) |
| Former | 1 (11.1%) | 3 (33.3%) | 1 (11.1%) | 3 (33.3%) | 8 (22.2%) |
| Current | 3 (33.3%) | 5 (55.6%) | 2 (22.2%) | 3 (33.3%) | 13 (36.1%) |
| Other tobacco | |||||
| Never | 8 (88.9%) | 8 (88.9%) | 9 (100.0%) | 9 (100.0%) | 34 (94.4%) |
| Former | 0 | 1 (11.1%) | 0 | 0 | 1 (2.8%) |
| Current | 1 (11.1%) | 0 | 0 | 0 | 1 (2.8%) |
| Alcohol use | |||||
| Abstinent | 6 (66.7%) | 7 (77.8%) | 5 (55.6%) | 8 (88.9%) | 26 (72.2%) |
| Light | 3 (33.3%) | 2 (22.2%) | 4 (44.4%) | 1 (11.1%) | 10 (27.8%) |
| Laboratory parameters, mean ± SDb | |||||
| Albumin (g dL−1) | 4.31 ± 0.23 | 4.17 ± 0.30 | 4.33 ± 0.23 | 3.88 ± 0.52 | ‐ |
| Bilirubin (mg dL−1) | 0.533 ± 0.390 | 0.600 ± 0.492 | 0.411 ± 0.127 | 0.867 ± 0.730 | ‐ |
| Child–Pugh score, n | |||||
| 5 | ‐ | 5 | ‐ | ‐ | ‐ |
| 6 | ‐ | 4 | ‐ | ‐ | ‐ |
| 7 | ‐ | ‐ | ‐ | 5 | ‐ |
| 8 | ‐ | ‐ | ‐ | 1 | ‐ |
| 9 | ‐ | ‐ | ‐ | 3 | ‐ |
BMI, body mass index; SD, standard deviation.
At screening.
Values calculated for all control participants (pooled matched control participants with mild and moderate hepatic impairment, n=18)
In line with the matching of control participants, sex, age and body weight were similar across cohorts. There were 5 and 4 participants with Child–Pugh score 5 or 6, respectively, in the mild hepatic impairment group, and 5, 1 and 3 participants with Child–Pugh score of 7, 8 or 9, respectively, in the moderate hepatic impairment group.
The study selection criteria excluded participants using CYP3A4 or P‐glycoprotein inhibitors. Based on the current knowledge of the pharmacokinetic and metabolic characteristics of vilaprisan, none of the concomitant medications received by any of the study participants was expected to have any interaction with vilaprisan, and thereby influence interpretation of the data. A table with the concomitant medication administered is provided in the Supporting Information.
3.2. Pharmacokinetic results
All pharmacokinetic data are discussed with reference to the geometric mean. Following single oral dose administration of vilaprisan 2 mg, geometric mean maximum concentrations were observed at 2.5, 2.0, 1.5 and 2.0 hours postadministration in the matched control cohorts for mild and moderate hepatic impairment, mild hepatic impairment and moderate hepatic impairment cohorts, respectively (Figure 3A).
Figure 3.
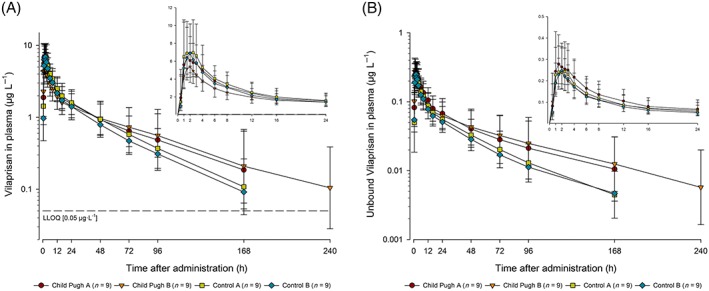
Total (A) and unbound (B) vilaprisan plasma concentrations (μg L−1) over time after a single oral 2 mg dose administered under fasting conditions in participants with mild (Child–Pugh‐A) or moderate hepatic impairment (Child–Pugh‐B) and matched control participants with normal hepatic function (control mild hepatic impairment, control moderate hepatic impairment). Semi‐logarithmic scale (error bars are standard deviations): Inset: linear scale for the first 24 h postdose. Planned sampling times used. Predose sample was set to 0 hours. lower limit of quantification = 0.05 μg L−1
Thereafter, plasma concentrations declined in a biphasic pattern in all 4 cohorts, with an initial rapid distribution phase followed by a second slower elimination phase that became apparent at lower vilaprisan concentrations. Plasma concentrations of vilaprisan during the elimination phase were higher in the participants with hepatic impairment compared with participants with normal hepatic function (moderate hepatic impairment > mild hepatic impairment > matched control participants for mild hepatic impairment cohort > matched control participants for moderate hepatic impairment cohort). Pharmacokinetic parameters with associated variability for vilaprisan by study group are summarised in Table 3.
Table 3.
Summary of vilaprisan pharmacokinetic parameters
| Parameter geometric mean (CV%) | Matched control mild hepatic impairment n = 9 (CV%) | Mild hepatic impairment n = 9 (CV%) | Matched control moderate hepatic impairment n = 9 (CV%) | Moderate hepatic impairment n = 9 (CV%) |
|---|---|---|---|---|
| Total vilaprisan concentration | ||||
| Cmax (μg L−1) | 7.95 (43.6) | 7.05 (34.1) | 8.70 (23.0) | 6.85 (34.5) |
| tmax (h)a | 2.00 (1.00–3.00) | 1.50 (1.00–4.00) | 1.00 (1.00–4.00) | 2.00 (0.50–3.00) |
| AUC (μg·h L−1) | 162 (48.8) | 187 (74.4) | 142 (41.1) | 195 (66.8) |
| CL/F (L h−1) | 12.4 (48.8) | 10.7 (74.4) | 14.1 (41.1) | 10.2 (66.8) |
| Vz/F (L) | 696 (46.3) | 824 (39.3) | 801 (26.2) | 1040 (38.4) |
| t1/2 (h) | 39.0 (41.9) | 53.3 (69.8) | 39.4 (42.5) | 70.4 (49.2) |
| CLR(0–24) L h−1 | 0.0443 (90.2) | 0.0684 (61.9) | 0.0486 (52.4) | 0.0671 (81.5) |
| Unbound vilaprisan concentration | ||||
| fu (%) | 3.50 | 4.36 | 3.61 | 4.44 |
| Cmax,u (μg L−1) | 0.278 (43.4) | 0.307 (32.9) | 0.314 (26.7) | 0.304 (39.1) |
| Cmax,u (μg L−1) ANOVA, LS mean (90% CI) | 1.1043 (0.8360–1.4588) | 0.9685 (0.7331–1.2793) | ||
| AUCu (μg·h L−1) | 5.65 (52.9) | 8.13 (79.6) | 5.13 (45.7) | 8.93 (66.0) |
| AUCu (μg·h L−1) ANOVA, LS mean (90% CI) | 1.4379 (0.9143–2.2615) | 1.7422 (1.0924–2.7785) | ||
| CLu/F (L h−1) | 354 (52.9) | 246 (79.6) | 390 (45.7) | 224 (66.0) |
| Vz,u/F (L) | 19 900 (54.0) | 18 900 (41.0) | 22 200 (33.9) | 22 700 (45.3) |
AUC, area under the concentration vs time curve; AUCu, area under the concentration vs time curve in plasma from zero to infinity for unbound drug; CI, confidence interval; CL/F, total apparent oral vilaprisan clearance; CLu/F, unbound oral vilaprisan clearance; CLR, renal clearance from plasma; Cmax, maximum observed drug concentration; Cmax,u, maximum observed unbound drug concentration; CV, coefficient of variation; LS, least‐square; tmax, time to reach Cmax; t1/2, terminal half‐life; Vz/F, apparent volume of distribution during the terminal phase; Vz,u/F, apparent volume of distribution during the terminal phase for unbound drug.
Median values (range).
Vilaprisan Cmax was slightly lower in participants with hepatic impairment compared with their matched control participants; geometric mean values of 7.95, 8.70, 7.05 and 6.85 μg L−1 were observed in the matched control cohorts for mild hepatic impairment and moderate hepatic impairment, mild hepatic impairment and moderate hepatic impairment cohorts, respectively (Figure 4, bottom). No pattern was observed for geometric mean time to reach Cmax between participants with mild or moderate hepatic impairment and their matched control participants. Overall, vilaprisan exposure was higher in participants with hepatic impairment compared with the matched control participants; geometric mean AUC values of 162, 142, 187 and 195 μg·h L−1 were observed in the control mild hepatic impairment, control moderate hepatic impairment, mild hepatic impairment and moderate hepatic impairment cohorts, respectively (Table 3).
Figure 4.
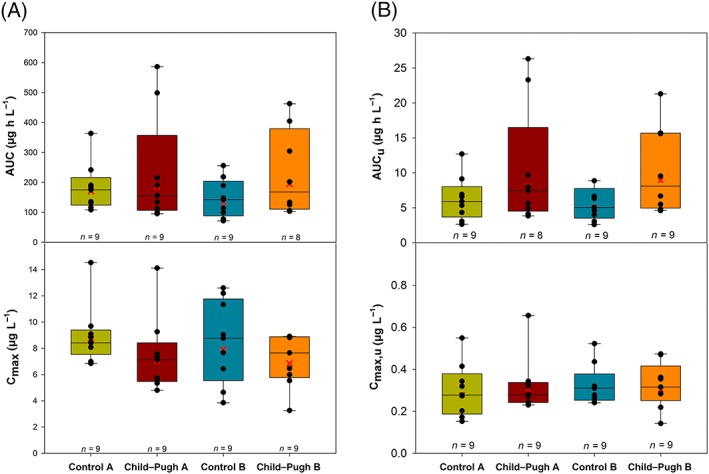
Vilaprisan total (A) and unbound (B) area under the concentration–time curve (AUC; top) and total and unbound maximum observed plasma concentration (Cmax; bottom) after a single oral 2 mg dose administered under fasting conditions in participants with mild (Child–Pugh‐A) or moderate hepatic impairment (Child–Pugh‐B) and matched control participants with normal hepatic function (control mild hepatic impairment, control moderate hepatic impairment). Vertical line in box represents median, box: 25–75 percentile, vertical lines extend from the box as far as the data extend to, at most, 1.5 times the interquartile range, symbols: individual data
The CL/F of vilaprisan was similar between the mild and moderate hepatic impairment cohorts (10.7 and 10.2 L h−1, respectively), and slightly higher in the control mild hepatic impairment and control moderate hepatic impairment cohorts (12.4 and 14.1 L h−1, respectively).
The apparent volume of distribution was higher in participants with hepatic impairment compared with their respective control groups and increased from mild to moderate hepatic impairment groups (mild hepatic impairment vs control mild hepatic impairment: 824 vs 696 L; moderate hepatic impairment vs control moderate hepatic impairment: 1040 vs 801 L).
The vilaprisan terminal half‐life appeared to be longer in participants with moderate hepatic impairment than in participants with mild hepatic impairment (70.4 vs 53.3 hours), and longer than in participants in the control cohorts (control mild hepatic impairment, 39.0 hours; control moderate hepatic impairment, 39.4 hours).
Consistent with previous data, vilaprisan had only marginal renal excretion in all treatment groups.7 Geometric mean values of renal clearance up to 24 hours of 0.0684, 0.0671, 0.0443 and 0.0486 L h−1 were determined in mild hepatic impairment, moderate hepatic impairment, control mild hepatic impairment and control moderate hepatic impairment cohorts.
3.3. Protein binding
Impaired hepatocyte function in patients with hepatic impairment could lead to decreased synthesis and dysfunction of albumin, resulting in decreased plasma protein binding and altered drug disposition.20 As vilaprisan binds primarily to albumin, plasma protein binding was assessed in the different groups. The geometric mean fraction of unbound vilaprisan in plasma was slightly higher in participants with hepatic impairment (mild: 0.0436; moderate: 0.0444) than in control subjects (control mild hepatic impairment: 0.0350; control moderate hepatic impairment: 0.0361). The fraction of unbound vilaprisan was similar at all 3 time points (2, 24 and 48 hours postdose), showing that protein binding was independent of drug concentration within that range. The concentration of unbound vilaprisan in the plasma over time is shown in Figure 3B. The observed pharmacokinetic parameters for unbound vilaprisan in participants with hepatic impairment and their matched control cohorts are summarised in Table 3 .
Overall, unbound vilaprisan exposure was higher in participants with hepatic impairment than in the matched control cohorts (Figure 4, top). Geometric mean AUCu was 8.13 μg·h L−1 in participants with mild impairment compared with 5.65 μg·h L−1 in healthy mild hepatic impairment—an increase of 1.44‐fold. Similarly, participants with moderate impairment had an increase of 1.74‐fold compared with their matched control cohorts. Geometric mean AUCu was 8.93 μg·h L−1 in participants with moderate hepatic impairment compared with 5.13 μg·h L−1 in the matched control moderate hepatic impairment cohort. There was relatively high variability for AUCu measurements of 52.9, 45.7, 79.6 and 66.0% coefficient of variation in the control mild hepatic impairment, control moderate hepatic impairment, mild hepatic impairment, and moderate hepatic impairment cohorts, respectively. The Cmax,u was approximately equal for the participants with hepatic impairment and the control cohorts, with a moderate degree of variability in measurements ranging from 26.7–43.4% coefficient of variation (Table 3).
3.4. Relationship between pharmacokinetic parameters and hepatic function parameters
The exploratory relationships between the primary pharmacokinetic parameters AUCu and Cmax,u and the baseline hepatic function parameters albumin, bilirubin and prothrombin time were investigated. No correlation was observed between Cmax,u and prothrombin time (r = 0.0681) nor between Cmax,u and bilirubin (r = −0.0554). Weak negative correlations were observed between AUCu and prothrombin time (r = −0.2418), and Cmax,u and albumin levels (r = −0.3385). There was a moderate positive correlation between AUCu and bilirubin level (r = 0.5593) and a moderate negative correlation between AUCu and albumin (r = −0.4568).
3.5. Relationship between pharmacokinetic parameters and Child–Pugh scores
The relationships between the pharmacokinetic parameters for vilaprisan and the individual Child–Pugh scores for the hepatic impaired participants were explored by plotting the Cmax,u and AUCu of vilaprisan as a function of Child–Pugh scores (Figure 5). For Cmax,u no correlation was observed. For AUCu, the subjects with a Child–Pugh score of 6 and 9 had the highest geometric mean exposure compared with subjects with Child–Pugh scores of 5, 7 and 8, indicating no clear correlation.
Figure 5.
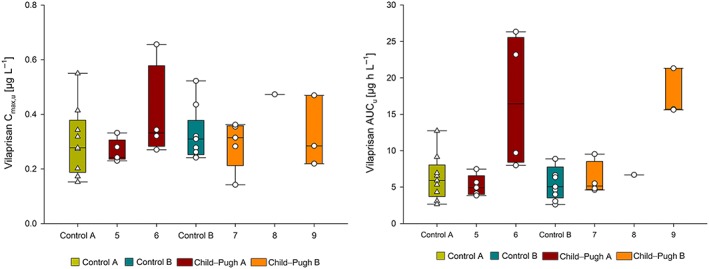
Vilaprisan unbound maximum observed plasma concentration (Cmax,u; A) and area under the concentration‐time curve (AUCu; B) by Child–Pugh score (CP‐A: 5–6 points, CP‐B: 7–9 points). Vertical line in box represents median, box: 25–75 percentile, vertical lines extend from the box as far as the data extend to, at most, 1.5 times the interquartile range, symbols: individual data
3.6. Safety
All 36 participants (100%) who received vilaprisan were included in the safety analysis. Overall, 8 participants (22.2%) reported at least 1 treatment‐emergent adverse event (TEAE), and 4 (11.1%) reported >1 vilaprisan‐related TEAE. The groups had similar incidences of TEAEs and vilaprisan‐related TEAEs. TEAEs were generally mild (5/36 participants, 13.9%) or moderate (2/36 participants, 5.6%) in intensity. The most frequent TEAEs were headache (11.1%, n = 4) and nausea (5.6%, n = 2). No participant experienced an SAE or death, and there were no study discontinuations due to AEs. All TEAEs are detailed in the Supporting Information.
4. DISCUSSION
Vilaprisan, a highly potent selective progesterone receptor modulator, is under clinical development for the treatment of symptomatic UF and endometriosis. Since it is largely metabolised by the liver through oxidative and reductive metabolism, an increase in vilaprisan exposure was expected in patients with hepatic impairment. This phase 1, single‐dose, open‐label, parallel‐group study evaluated the impact of mild (CP‐A) or moderate (CP‐B) hepatic impairment on the pharmacokinetics of vilaprisan in adult White/Caucasian participants. A single oral vilaprisan 2 mg dose was given, in line with the dose that was selected for the phase 3 trials in UF.4 Participants with severe hepatic impairment were not included in this study as they are not part of the target patient population for vilaprisan. Additionally, for such patients the clinical management of the severity of hepatic impairment may be the primary focus.
Single‐dose oral administration of vilaprisan 2 mg resulted in mild increases of <1.75‐fold in total and unbound systemic exposure of vilaprisan in participants with mild or moderate hepatic impairment compared with participants with normal hepatic function. The largest effect was seen in participants with moderate hepatic impairment, with an increased AUCu of approximately 1.74‐fold compared with their matched control cohorts.
The pharmacokinetic profile of vilaprisan in the control groups was very similar to that observed in previous studies conducted in postmenopausal women.20 Furthermore, as expected, no relevant differences in the pharmacokinetics were seen between male and female subjects. Although vilaprisan is intended to be used for long‐term treatment, investigating a single dose of 2 mg in this study was justified considering that vilaprisan shows linear pharmacokinetics upon multiple dosing, thereby allowing us to extrapolate these results to a multiple dosing scenario.22
The expected vilaprisan exposure at steady state for participants with moderate hepatic impairment, given the anticipated accumulation due to the apparent slight prolongation of the terminal elimination half‐life, is within the ranges included in a previous multiple‐dose study.22 In this previously conducted study, no safety concerns were identified with vilaprisan at doses up to 30 mg day−1 over 28 days.22 No safety concerns are expected upon multiple daily dosing in patients with mild or moderate hepatic impairment, based on these pharmacokinetic observations.
The unbound fractions of vilaprisan measured in this study (~0.04) were similar to those previously observed in the plasma of healthy volunteers in vitro (0.053, data not shown). There was no major difference in protein binding between the different sampling time points in all 4 cohorts. Plasma protein binding was slightly lower in participants with mild and moderate hepatic impairment, resulting in a higher fu of vilaprisan than in the matched controls. The higher fu may be caused, at least in part, by the lower albumin concentrations in hepatic impaired participants, given that it is a major binding protein for vilaprisan. The slightly higher systemic exposure of vilaprisan in mild and moderately impaired participants in this study is consistent with low hepatic extraction compounds such as vilaprisan. For such drugs, elimination is primarily related to the intrinsic metabolic clearance and unbound fraction in plasma. The hepatic intrinsic clearance tends to decrease with increasing degree of hepatic impairment due to the reduced metabolic capacity and/or due to bypassing the perfusion of functional hepatic tissue through the formation of shunts. By contrast, the unbound fraction in the participants with hepatic impairment was only slightly higher in participants with hepatic impairment, suggesting that the change in protein binding has no relevant effect on the clearance of vilaprisan. In accordance with this understanding, a prolonged terminal elimination half‐life was observed in participants with mild and moderate hepatic impairment. Of note, even in the cohort with moderate hepatic impairment, the increase in exposure was only mild (i.e. <1.75‐fold), indicating that the metabolic activity of CYP3A4, the main enzyme responsible for the clearance of vilaprisan,7 was only slightly reduced in this participant group. This could reliably be shown with vilaprisan where no compensating alternative elimination route such as involvement of extrahepatic eliminations is in place, for example renal excretion, which was marginal in all cohorts. Of interest, the maximum observed total and unbound vilaprisan concentrations were not different between the control and hepatic impairment cohorts, indicating that the absorption from the gastrointestinal tract into systemic circulation is not different in participants with mild and moderate hepatic impairment.
With respect to the investigation of the relationship between the primary pharmacokinetic and hepatic function parameters, only a weak negative correlation was observed between Cmax,u and albumin. This correlation is consistent with the assumption that the lower albumin concentration in participants with hepatic impairment results in higher fu and therefore higher Cmax,u. There was a moderate negative correlation between AUCu and albumin and a moderate positive correlation with bilirubin, as expected, that reflect the extent of hepatic impairment resulting in an increase in systemic vilaprisan exposure.
In this study, a single oral dose of vilaprisan 2 mg was well tolerated by participants of both sexes with mild or moderate hepatic impairment, and in participants with normal hepatic function. Safety findings in this study were comparable with observations from previous phase 1 and phase 2 studies in which vilaprisan was well tolerated, with no drug‐related serious AEs even at the maximal daily doses used in females.4, 5, 7, 21, 23 Here, the most frequently observed TEAEs were headache and nausea, which were also among the most common drug‐related AEs reported alongside ovarian and cervical cyst (identified at ultrasound), fatigue, abdominal or pelvic pain, other gastrointestinal disorders (flatulence, constipation), hot flushes, and dizziness in previous studies.4, 5, 7, 21, 23
A limitation of this study, which is inherent in the primary objective to investigate the pharmacokinetics of vilaprisan, is that safety data were only collected in a small participant population and after single oral dosing (n = 36). Nevertheless, the observed favourable safety profile supports more extensive safety evaluations of vilaprisan during the phase 3 clinical studies in women with UF with/without mild or moderate hepatic impairment. A single oral dose administration of 2 mg vilaprisan to participants with mild or moderate hepatic impairment resulted in a mild increase in systemic exposure of vilaprisan to <2‐fold compared with their matched control participants. Vilaprisan was well tolerated in participants with hepatic impairment and participants with normal hepatic function, and no safety concerns were identified. Considering the excellent safety profile of vilaprisan as demonstrated in several phase 1 and 2 studies, the pharmacokinetic results from this study do not indicate a need for dose adjustment in women with UF or endometriosis who also have mild or moderate hepatic impairment.
COMPETING INTERESTS
All authors except A.H. are employees of Bayer AG. A.H.'s employer received funding from Bayer AG for conducting the study. The authors declare that they have no other real or potential competing interests.
CONTRIBUTORS
All authors participated in research design, contributed to the analysis and interpretation of the data and the writing of the manuscript. All authors have approved the manuscript as written.
5.
Supporting information
Data SI
Supporting information
ACKNOWLEDGEMENTS
This study was funded by Bayer AG, Berlin, Germany, including collection, analysis, and interpretation of the data. Medical writing support was provided by Katherine Sutherland, PhD, of Huntsworth Health Ltd, with funding from Bayer AG.
The authors sincerely thank the participants who took part in this study as well as the staff who assisted with the study at each site. The clinical part of the study was conducted at CRS Clinical Research Services Kiel GmbH, Kiel and Lübeck, Germany. A.H. was the principal investigator of the study. Measurements of vilaprisan were carried out at Bayer AG, Wuppertal, Germany. Safety laboratory analyses were performed at Synlab Pharma Institute, a division of Synlab Umweltinstitut GmbH, Berlin, Germany.
The authors would like to thank the following colleagues from Bayer AG, Berlin, Germany, for their contributions to this manuscript: Antonia Kohnke for support with data analysis and some of the figures. Medical writing support was provided by Katherine Sutherland, PhD, Huntsworth Health Ltd, London, UK, with funding from Bayer AG, Berlin, Germany.
Chattopadhyay N, Riecke K, Ligges S, Zimmermann T, Halabi A, Schultze‐Mosgau M‐H. Effect of hepatic impairment on the pharmacokinetics of vilaprisan: An open‐label, single‐dose, parallel‐group study. Br J Clin Pharmacol. 2019;85:2011–2021. 10.1111/bcp.13992
Data Availability Statement:The data that support the findings of this study are available from the corresponding author upon reasonable request.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Baird DD, Dunson DB, Hill MC, Cousins D, Schectman JM. High cumulative incidence of uterine leiomyoma in black and white women: ultrasound evidence. Am J Obstet Gynecol. 2003;188(1):100–107. [DOI] [PubMed] [Google Scholar]
- 2. Sohn GS, Cho S, Kim YM, et al. Current medical treatment of uterine fibroids. Obstet Gynecol Sci. 2018;61(2):192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stewart EA, Cookson CL, Gandolfo RA, Schulze‐Rath R. Epidemiology of uterine fibroids: a systematic review. BJOG. 2017;124(10):1501–1512. [DOI] [PubMed] [Google Scholar]
- 4. Bradley L, Singh S, Simon J, et al. Vilaprisan in women with uterine fibroids: randomized phase 2b ASTEROID 1 study. Fertil Steril. 2019;111(2):240–248. [DOI] [PubMed] [Google Scholar]
- 5. Gemzell‐Danielsson K, Ramirez K, Petersdorf K, Faustmann T, Seitz C. Efficacy and safety of the selective progesterone receptor modulator (PRM) vilaprisan – data from the phase 2 ASTEROID 2 study. In: ESG Congress, 2017.
- 6. Seitz C, Al‐Hendy A, Bradley L, et al. The clinical development of vilaprisan: the ASTEROID phase 3 program. Florence: SEUD; 2018. [Google Scholar]
- 7. Schultze‐Mosgau MH, Hoechel J, Prien O, et al. Characterization of the pharmacokinetics of vilaprisan: bioavailability, excretion, biotransformation, and drug‐drug interaction potential. Clin Pharmacokinet. 2018;57(8):1001–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chattopadhyay N, Kanacher T, Casjens M, et al. CYP3A4‐mediated effects of rifampicin on the pharmacokinetics of vilaprisan and its UGT1A1‐mediated effects on bilirubin glucuronidation in humans. Br J Clin Pharmacol. 2018;84(12):2857–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kanacher T, Frechen S, Wendl T, Block M, Rottmann A, Chattopadhyay N. Towards a detailed understanding of the impact of drug–drug interactions on vilaprisan exposure by PBPK modeling and simulation. DDI Workshop Öhningen, Germany: Marbach Castle; 2017. [Google Scholar]
- 10. Verbeeck RK. Pharmacokinetics and dosage adjustment in patients with hepatic dysfunction. Eur J Clin Pharmacol. 2008;64(12):1147–1161. [DOI] [PubMed] [Google Scholar]
- 11. Pentikainen PJ, Valisalmi L, Himberg JJ, Crevoisier C. Pharmacokinetics of midazolam following intravenous and oral administration in patients with chronic liver disease and in healthy subjects. J Clin Pharmacol. 1989;29(3):272–277. [DOI] [PubMed] [Google Scholar]
- 12. Kleinbloesem CH, van Harten J, Wilson JP, Danhof M, van Brummelen P, Breimer DD. Nifedipine: kinetics and hemodynamic effects in patients with liver cirrhosis after intravenous and oral administration. Clin Pharmacol Ther. 1986;40(1):21–28. [DOI] [PubMed] [Google Scholar]
- 13. Pugh RN, Murray‐Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60(8):646–649. [DOI] [PubMed] [Google Scholar]
- 14. EMA . Guideline on bioanalytical method validation. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf, 2011. Accessed June 4, 2018.
- 15. FDA . Guidance for Industry: Bioanalytical Method Validation 2001. https://www.fda.gov/downloads/drugs/guidances/ucm070107.pdf, 2001. (Accessed June 4, 2018).
- 16. Schuhmacher J, Kohlsdorfer C, Buhner K, Brandenburger T, Kruk R. High‐throughput determination of the free fraction of drugs strongly bound to plasma proteins. J Pharm Sci. 2004;93(4):816–830. [DOI] [PubMed] [Google Scholar]
- 17. FDA . Guidance for Industry: Pharmacokinetics inpatients with impaired hepatic function: study design, data analysis, and impact on dosing and labeling. 2003. https://www.fda.gov/media/71311/download (Accessed July 02, 2019)
- 18. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res. 2018;46(D1):D1091‐D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alexander SPH, Cidlowski JA, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol. 2017;174(Suppl 1):S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bernardi M, Ricci CS, Zaccherini G. Role of human albumin in the management of complications of liver cirrhosis. J Clin Exp Hepatol. 2014;4(4):302–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schuett B, Kaiser A, Schultze‐Mosgau MH, et al. Pharmacodynamics and safety of the novel selective progesterone receptor modulator vilaprisan: a double‐blind, randomized, placebo‐controlled phase 1 trial in healthy women. Hum Reprod. 2016;31(8):1703–1712. [DOI] [PubMed] [Google Scholar]
- 22. Schultze‐Mosgau MH, Schuett B, Hafner FT, et al. Pharmacokinetics and safety of the selective progesterone receptor modulator vilaprisan in healthy postmenopausal women. Int J Clin Pharmacol Ther. 2017;55(01):16–24. [DOI] [PubMed] [Google Scholar]
- 23. Schuett B, Schultze‐Mosgau MH, Draeger C, et al. Effect of the novel selective progesterone receptor modulator vilaprisan on ovarian activity in healthy women. J Clin Pharmacol. 2018;58(2):228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data SI
Supporting information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.