

Abstract
The degeneration in the locus coeruleus associated with Alzheimer’s disease suggests an involvement of the noradrenergic system in the disease pathogenesis. The role of depleted norepinephrine was tested in adult and aged rhesus macaques to develop a potential model for testing Alzheimer’s disease interventions. Monkeys were injected with the noradrenergic neurotoxin N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP4) or vehicle at 0, 3, and 6 months; brains were harvested at 9 months. Reduced norepinephrine in the locus coeruleus was accompanied by decreased dopamine β-hydroxylase staining and increased amyloid-β load in the aged group, and the proportion of potentially toxic amyloid-β42 peptide was increased. Immunohistochemistry revealed no effects on microglia or astrocytes. DSP4 treatment altered amyloid processing, but these changes were not associated with the induction of chronic neuroinflammation. These findings suggest norepinephrine deregulation is an essential component of a nonhuman primate model of Alzheimer’s disease, but further refinement is necessary.
Keywords: Alzheimer’s disease, DSP4, locus coeruleus, neurotoxin, nonhuman primate, norepinephrine
INTRODUCTION
Alzheimer’s disease (AD) currently afflicts 5.7 million Americans and 18 million people worldwide; without successful intervention, this number is estimated to triple by 2050 [1]. Cognitive neural systems seem preferentially impacted by AD, with severe impairment of memory and attention occurring with amyloid-β (Aβ) accumulation, tangle formation, neuroinflammation, and neuronal loss [2]. Biochemical alterations and onset of pathology may begin decades before observable dementia. Parenchymal deposits isolated from clinical AD cases contain aggregates of Aβ40 and Aβ42, including soluble monomers and oligomers [3]. Several factors control Aβ generation from the amyloid-p protein precursor (AβPP), which is itself regulated epigenetically and post-transcriptionally [4–6]. Likewise, regulating AβPP processing enzymes, such as β-site AβPP cleaving enzyme-1 (BACE1), is important for Aβ generation [7]. Furthermore, differential activity in production, clearance, and Aβ degradation pathways contribute to AD development [8–10]. Environmental insults and toxicants may also contribute to AD, exemplified by exposure to metallic lead causing overexpression of AβPP and Aβ [11], oxidative stress [12], and AD-like pathology in rodents and primates [13].
The forebrain receives noradrenergic input from the locus coeruleus (LC) and utilizes norepinephrine (NE) in a variety of receptor-mediated functions, ranging from modulating attentive behavior to mediating inflammatory responses [14–16]. As such, the LC has been an anatomical area of great interest in gaining an understanding of AD pathogenesis and for development of potential therapies for AD and other neurodegenerative processes [17–21]. Further, a recent report demonstrates a potential key role of tyrosine-hydroxylase expressing neurons projecting to the hippocampus for post-encoding memory enhancement, in synergy with dopaminergic systems [22]. NE suppresses inflammatory genes that promote cytokine production, thereby mitigating inflammatory responses [23, 24]; an action suggesting a neuroprotective role for the NE neural system. Moreover, LC degeneration and NE loss have been documented in AD [25, 26], and other dementias [27,28], with neuronal dysfunction occurring early in disease pathogenesis [29]. Increased cell loss within the LC of AD patients suggests a potential linkage between LC degeneration and exacerbation of pathology through these axonal connections [28,29]. Taken together, there appears to be a clear linkage between the LC and NE neural systems in the pathogenesis of neurodegenerative disease [25, 30].
Administration of the neurotoxin N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP4) to AD-transgenic mice resulted in increased Aβ pathology and produced LC impairment [29, 31]. Moreover, targeted LC degeneration induced activated glia and inflammatory pathways, and reduced neprilysin expression with elevated Aβ deposition in AD rodent models [31, 32]. An understanding of the cross-talk between catecholamines and AβPP remains in its early stages of discovery. Notably, amine oxidase activity of AβPP modulates systemic and local catecholamine levels, such as dopamine and NE [33]. Mechanisms responsible for increased AD pathology due to LC impairment and, more specifically, the role of the NE system remain unclear. Recent work suggests that integrity of LC plays an important role in the broader context of dementia. For example, the initial stages of AD include deficits in egocentric (subject-to-object) recognition and allocentric (object-to-object) spatial representations. Both AD and amnestic mild cognitive impairment displayed allocentric deficits, which might result from concurrent dysregulations in the locus coeruleus-noradrenaline system or pre-frontal cortex [34]. Although Aβ peptides in nonhuman primates (NHPs) are structurally identical to those in humans, few studies have investigated NE depletion in an animal model with natural Aβ accumulation [35]. However, even with this natural age-related Aβ accumulation, NHPs do not develop all AD-like symptoms spontaneously [35]. This reiterates the need for an enhanced NHP model to examine mechanisms of idiopathic development. For this reason, our goal was to utilize DSP4 to induce NE depletion and to examine pathways contributing to the exacerbation of age-related development of amyloid pathology. Taken together, the present work of developing an AD-like pathology in an NHP model with reduced levels of NE and increased levels of AβPP would be a significant step toward discovering and testing novel drug targets for the human neurodegenerative diseases, such as AD, Down’s syndrome, frontotemporal dementia, and Lewy body dementia.
METHODS
Study subjects
Animals included eleven adult (14–17 years old) and six aged (19–25 years old) female rhesus macaques (Macaca mulatta) randomized into a treatment (nadult = 6 and naged = 3) or control (nadult = 5 and naged = 3) group. The study focused on females because AD incidence is significantly higher in women [2]. Monkeys were maintained at the National Institutes of Health Animal Center (Poolesville, MD) and housed in standard NHP caging on a 12:12 h light cycle with ad libitum access to water and fed a standard NHP diet at approximately ad libitum levels. Monkeys were observed daily for food consumption and overall well-being. Animal husbandry and all experimental procedures complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were conducted under approved protocols by the NIA Institutional Animal Care and Use Committee.
Experimental design
At each procedure, monkeys were fasted overnight and anesthetized with ketamine (7–10 mg/kg, IM). Initial restraint was supplemented with partial doses of ketamine, as needed, to maintain anesthesia. Monkeys in the treatment group received an initial injection of N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP4; Sigma-Aldrich, St. Louis, MO) at a 40 mg/kg (IP) dose. To prevent incidental serotonergic depletion, each experimental monkey was injected with zimelidine (Sigma-Aldrich; 10 mg/kg, IP) 45min prior to DSP4. To maintain NE depletion, subsequent dosing of DSP4 (10 mg/kg, IP) occurred at 3 and 6 months again preceded by zimelidine (10 mg/kg, IP). Controls received saline vehicle (IP) under identical conditions and at the same timepoints as the experimental group. Animals were euthanized for tissue harvest 9 months following the initial treatment. The timing and the treatment protocol were confirmed in a preliminary experiment in which this dose resulted in significant depression of norepinephrine with minimal distress and side effects.
Tissue collection
Animals were restrained with ketamine (7–10 mg/kg, IM) and deeply anesthetized using B-Euthanasia-D (80 mg/kg, IV) for transcardial perfusion with cold 0.9% saline. Following tissue harvest, each brain was divided along the medial longitudinal fissure and blocked in 1 cm increments using a coronal brain matrix for use in biochemical and immunohistochemical assays. The right hemisphere was immediately frozen in isopentane and stored at –80°C for biochemical analyses; the left hemisphere was immersion-fixed in 4% paraformaldehyde for 48 h, placed through a series of graded sucrose solutions until the blocks sank in 30% sucrose, and then frozen at –80°C.
Enzyme linked immunosorbent assay (ELISA)
Tissue punches from specific brain regions were taken from fresh frozen tissue blocks for analysis of catecholamines and Aβ levels. Levels in brain homogenate were standardized by sample protein levels measured using the BCA (bicinchoninic acid) Protein Assay (Pierce-Thermo Scientific, Rockford, IL). Punches for catecholamine measurements were homogenized (Fisher Scientific PowerGen125) in 0.1 N HCL, centrifuged (at 15,000 g) for 15min at 4°C, and the supernatant was collected for ELISA. Levels of NE were measured in duplicate from punches taken from prefrontal cortex, caudate nucleus of the striatum, hippocampus, cingulate cortex, temporal cortex, and LC using specific ELISAs and run per the manufacturer’s instructions (Rocky Mountain Diagnostics, Inc. Colorado Springs, CO). Average optical density values (Bio-Rad 480 microplate reader-450 nm filter) for each region were interpolated on 4-PL standard curves to determine NE concentrations and standardized per individual protein content (BCA; Pierce-Thermo Scientific).
Additional punches from prefrontal and temporal cortex were homogenized in five volumes of non-detergent homogenization buffer (50 mM NaCl with protease inhibitor cocktail), and then centrifuged at 30,000 g for 4h (equivalent to a centrifugation of 100,0000 g for 1 h) at 4°C to generate a soluble fraction supernatant. Levels of the Aβ peptide with 40 amino acid fragment (Aβ40) and with 42 amino acid fragment (Aβ42) were measured independently in each region by sensitive, specific sandwich ELISA per the manufacturer’s instructions (Invitrogen, Camarillo CA), with all samples run in duplicate. Average optical density values (Bio-Rad 480 microplate reader-540 nm filter) for each region were interpolated on 4-PL standard curves to determine isoform concentrations and standardized for each sample per protein content (BCA; Pierce-Thermo Scientific).
Immunohistochemistry
Fixed blocks were sectioned (50 μm) on a freezing stage sliding microtome and placed into cryopreservation buffer for storage at –20°C until immunohistochemical (IHC) staining. Serial sections were collected from blocks throughout the brain to examine prefrontal, temporal, parietal, and entorhinal cortex. Regions of interest were delineated based on landmarks from a rhesus monkey stereotaxic atlas and used to identify standardized regions across individuals [36]. Sections were stained with the following primary antibodies: Anti-Aβ 6E10 (1:1000, Covance, Emeryville, CA), Anti-dopamine β-hydroxylase (DBH) (1:1000 Millipore, Temecula, CA), and Anti-IBA1 (1:3000, Wako Chemicals, Richmond, VA) to examine Aβ, NE, and microglia. Stained sections were mounted onto PLUS slides and air-dried. Sections were counterstained with 2% cresyl violet, dehydrated, and then cover-slipped using permanent mounting medium. After drying, Aβ in neocortical regions, NE containing cell bodies located in the LC, and microglia were examined. Images were quantified using ImageJ (NIH) to analyze specific staining patterns for each antibody. Thresholding and exclusion criteria were adjusted to isolate positively stained regions from background controls to determine percentage of area stained within the region of interest [37]. All samples were blinded and codes were revealed following the completion of image analysis.
Due to the rarity of 6E10 immunostaining (6E10 IR) in rhesus macaques under the age of 25 years [38], each observation of Aβ was counted using a 10x objective on three sections spaced 750 μm apart from frontal, entorhinal/temporal, and parietal regions. Within each region, counts of Aβ occurrences were summed for each region of interest, and total neocortical load was determined by adding regional counts for each monkey and then averaged for aged control and DSP4 treated groups. DBH-IR was used to identify NE synthesizing neurons in the LC. The LC was imaged using a 4x objective; % area was calculated and averaged for two sections per animal spaced 100 μm apart. For microglia (IBA1), % area stained on three non-overlapping adjacent 10x images from cortex were calculated on sections spaced 500 μm apart from each animal and averaged to obtain semi-quantitative measurements.
Western blot analyses
Sample lysates (intracellular fractions) from prefrontal and temporal cortex were prepared using M-PER (Pierce) buffer, supplemented with protease inhibitor for analysis of AβPP and BACE1. Equal amounts of lysate protein were loaded onto 10% polyacrylamide ‘Criterion’ gels (BioRad, Hercules, CA) and subjected to electrophoresis for 1.5 h at 180 volts at room temperature (PAGE). Proteins were transferred electrophoretically onto PVDF membranes at 4°C. Membranes were blocked for 1 h in nonfat milk dissolved in TBST and incubated overnight serially with primary antibodies against AβPP-CT (Calbiochem, Billerica, MA), BACE1 (3D5, generously provided by Dr. Robert Vasser) and β-actin (Sigma-Aldrich). Membranes were incubated with secondary antibody for 1 h followed by an ECL detection step. Bands were visualized and normalized to β-actin using NIH “Image J” software.
Statistics
Data were analyzed by one-way ANOVA (JMP9, SAS Institute, Cary, NC), and directional contrasts were used to examine a priori comparisons of interest and tested at a significance level of p <0.05. The a priori comparisons tested in this study were the following: 1) DSP4 will exert deleterious effects in adult animals compared to age-matched animals injected with vehicle; 2) aged animals injected with DSP4 will exhibit increased pathology compared to age-matched controls; 3) aged animals would show significantly more pathology than adults, as tested in the control groups. These comparisons tested levels of NE through ELISA and IHC to measure regional reductions and identify neurons synthesizing NE, levels of Aβ pathology using IHC, ELISA and western Blot to measure increased amyloid deposition, pathological AβPP processing indicated by Aβ42:40 ratio and BACE1, and finally measured microglia using IHC to determine phenotypes indicative of chronic neuroinflammation. The number of planned comparisons for each dependent measure was restricted (k–1) to test the three above hypotheses to minimize family wise type I error.
RESULTS
Effects on noradrenergic system
Three DSP4 injections over 9 months reduced mean levels of NE in the LC, compared to age-matched controls, in adult (p = 0.008, Fig. 1A) and aged monkeys (p = 0.037). Age alone was not significantly related to NE levels as adult and aged controls had similar levels of NE (p = 0.373). DSP4 did not significantly alter NE content in the cortical projection areas, and only the temporal cortex region was significantly lower in the aged control compared to adult (p = 0.046, Fig. 1B). The small sample size likely limited the ability to detect differences. Additionally, in both the hippocampus and the non-projecting striatum, NE content was not altered by DSP4 treatment or with age (Fig. 1C).
Fig. 1.
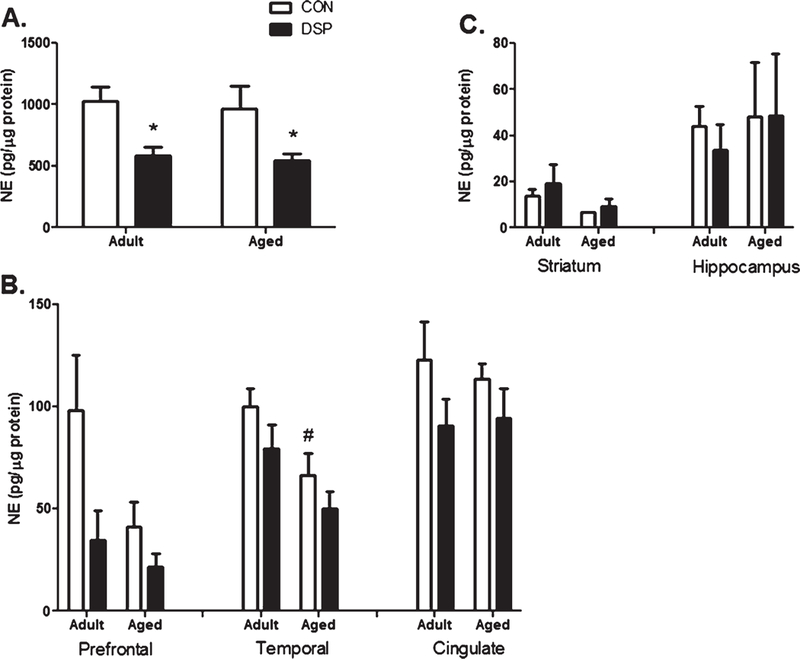
NE content in LC. Presented as mean ± SEM for treatment (nadult = 6 and naged = 3) or control (nadult = 5 and naged = 3) groups: A) Locus Coeruleus: NE concentrations decreased following DSP4 injections in both adult and aged monkeys (*p <0.05) compared to age- matched CON. B) An effect on NE content due to DSP4 was not detected in the three cortical projection regions (Prefrontal- adult: p = 0.141, aged: p = 0.287; Temporal-adult: p = 0.103, aged: 0.224; Cingulate- adult: 0.085, aged: 0.268); however, aged CON monkeys had lower NE concentrations in the temporal region than adult CON (#p < 0.05); C) NE content of the striatum and hippocampus were not affected by DSP4 or age.
Serial brain sections in the aged DSP4 treated monkeys showed reduced DBH staining compared to the age-matched controls (p = 0.04, Fig. 2). However, DSP4 treatment in the adult group had no effect, nor did age alone.
Fig. 2.
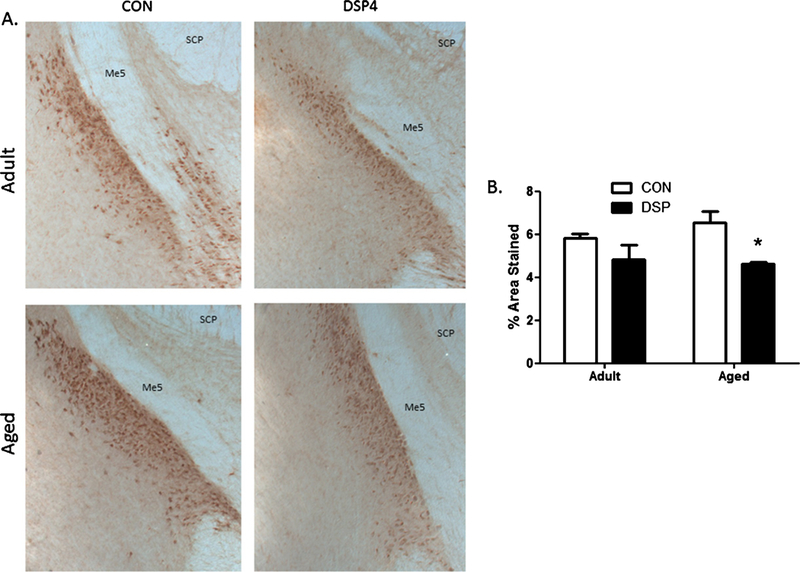
NE neurons in the LC. A) Representative photomicrographs (4x) of DBH-immunoreactivity identifying LC neurons (brown punctate staining) and location verified with anatomical landmarks for superior cerebellar peduncle (SCP) and mesencephalic trigeminal nucleus (Me5). B) Semi-quantitative determination of % area stained in serial sections through the LC. *p <0.05 DSP4 treated compared to age-matched CON (treatment (nadult = 6 and naged = 3) or control (nadult = 5 and naged = 3) groups).
Amyloid-β
Serial brain sections analyzed for extracellular deposits of Aβ in frontal (Fig. 3A) and temporal cortex, and hippocampus using 6E10-IR and Congo red histological staining showed Aβ staining in isolated regions in aged monkeys (Fig. 3B). Diffuse and compact staining was observed in neocortex and preferentially localized to deep layers of aged rhesus monkeys (Fig. 3A). Total Aβ count was significantly elevated with age (p = 0.0001) and following DSP4 in aged animals (p = 0.0001, Fig. 3C). Subcortical areas were devoid of Aβ stained by 6E10, and all monkeys were Congo red negative, with vascular deposition nearly non-existent across individuals.
Fig. 3.
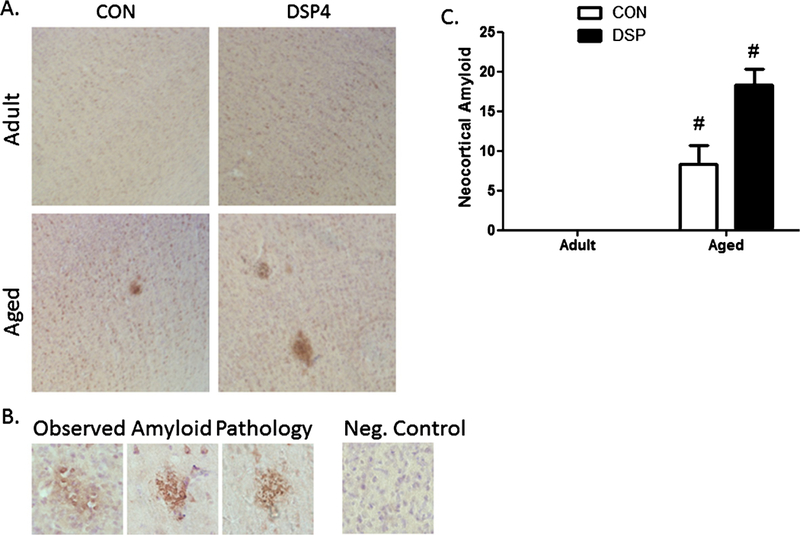
Observed amyloid pathology. A) Photomicrographs (4x) of 6E10 immunoreactivity (brown) and cresyl violet counterstain (purple) in frontal cortex showing extracellular amyloid deposition in deeper cortical layers in aged animals and following DSP4; B) Higher magnification (40x) observation of Aβ morphology in aged control and DSP4 treated animals; Negative control (no 6E10) showed no non-specific staining and only cell bodies stained with cresyl violet; C) Quantification of area stained for amyloid for each treatment group. Amyloid was not detected in CON and DSP4 treated adults; aged monkeys were significantly different than treatment-matched adults (treatment (nadult = 6 and naged = 3) or control (nadult = 5 and naged = 3) groups) (#p < 0.05), but there was no difference between the two aged cohorts.
The ratio of soluble Aβ42:Aβ40 increased after DSP4 in the aged cohort but not in adults (Fig. 4), with a significantly elevated ratio in both prefrontal (p = 0.025) and temporal cortex (p = 0.028) in the aged cohort. There was no significant age-related difference overall when comparing the untreated adult and aged individuals.
Fig. 4.
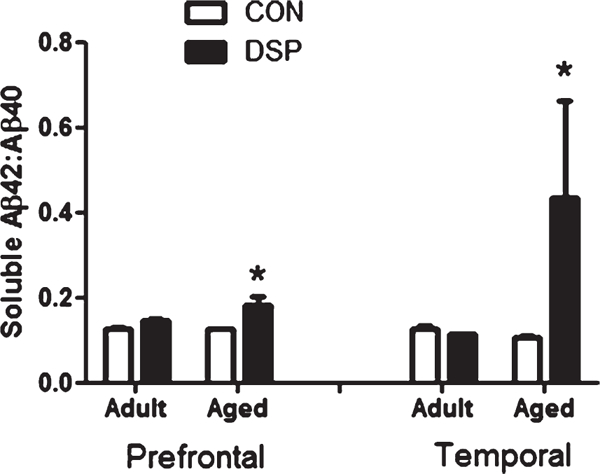
Ratio of soluble amyloid isoforms. Prefrontal and temporal cortex of aged monkeys, showed a shift toward a more pathological ratio of soluble isoforms in DSP4 treated compared to age-matched CON (treatment (nadult = 6 and naged = 3) or control (nadult = 5 and naged = 3) groups). (*p < 0.05).
AβPP processing
The rate limiting step for Aβ generation is cleavage of AβPP by BACE1 enzyme. Levels of intracellular AβPP showed a strong age-related increase of AβPP in temporal cortex (p = 0.0004); and DSP4 treatment dramatically increased AβPP in adult temporal cortex (p = 0.00006; Fig. 5A). Conversely, the aged group showed a reduction in AβPP Increased BACE1 was observed following DSP4 treatment in temporal cortex of the adult group (p = 0.0009) and the frontal cortex of the aged (p = 0.007; Fig. 5B).
Fig. 5.
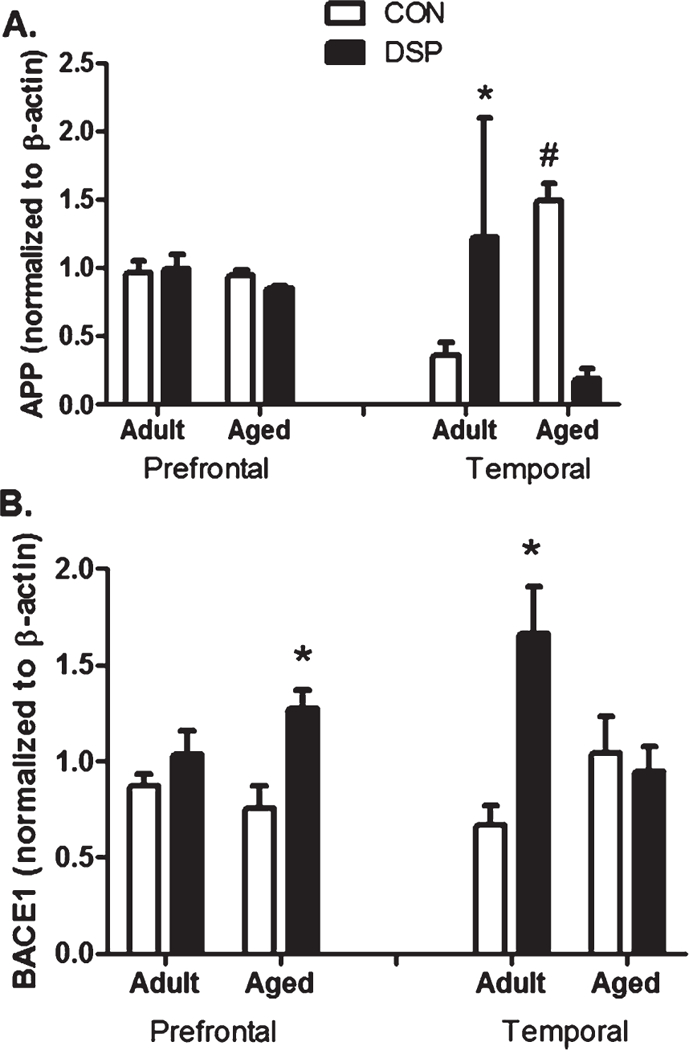
AβPP processing. A) Total AβPP and B) BACE1 in prefrontal and temporal cortex samples normalized to B-actin. *p < 0.05 DSP4 treated compared to age-matched CON; #p < 0.05 aged CON compared to adult CON (treatment (nadult = 6 and naged = 3) or control (nadult = 5 and naged = 3) groups).
Microglia
Resting and activated microglia [39] were visualized using IBA1-IR in serial cortical sections. Microglia morphology was examined to distinguish an activated phenotype and resting populations of microglia were found widely distributed throughout the brain. Activated morphology was rarely observed in control or DSP4 treated brains and no difference in immunoreactivity was detected in resting populations (p = 0.776, Fig. 6).
Fig. 6.
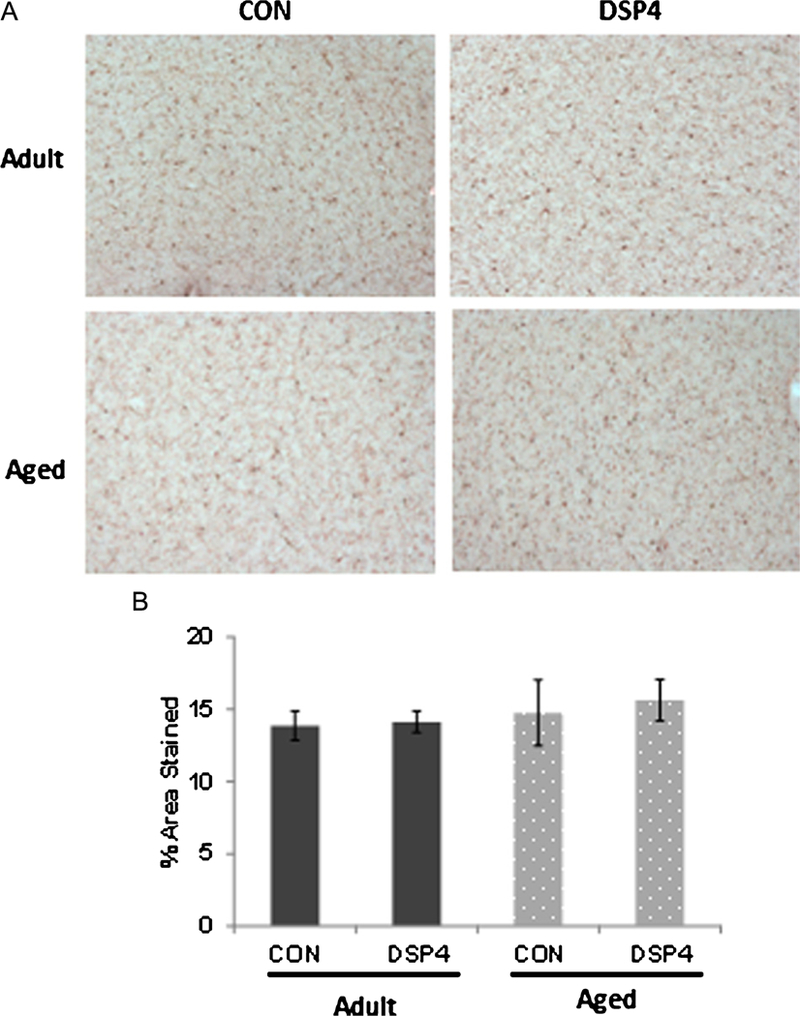
Microglia distribution in cortex. A) Microglia were widely distributed throughout the brain; staining pattern was similar across adult and aged monkeys regardless of treatment group. B) Semi-quantitative determination of % area stained in serial sections from frontal cortex (p > 0.05). DSP4: nadult = 6 and naged = 3 Control: nadult = 5 and naged = 3.
DISCUSSION
This study aimed to determine if DSP4, a noradrenergic neurotoxin, could elevate cerebral Aβ pathology in a NHP with 95% genetic homology to humans, and thus, establish a potential model for idiopathic AD and a means toward unraveling mechanisms that contribute to disease progression in animals that develop Aβ deposits naturally. A key role of LC impairment in AD development was established as DSP4 injections accelerated the onset and incidence of AD-like pathology. NE loss exacerbated the progression of AD pathology. In this study, the reduction of NE contributed to increased Aβ pathology via altered AβPP processing. These data support other observations linking the noradrenergic system and delay of the progression of the AD neuropathology. Furthermore, these long-lasting modifications were likely produced in the absence of chronic neuroinflammation, as there were no observable differences in distributions or morphology of microglia between DSP4 and control brains.
DSP4 treatment (50 mg/kg) in rodents caused exacerbation of AD-like neuropathology with NE depletion [29, 33]. DSP4 in AD transgenic mice often utilized multiple high dose injections; however, to minimize transient peripheral side effects noted in our pilot study (unpublished observations), 40 mg/kg for the high dose injection and 10 mg/kg was used for subsequent dosing in our experimental design. We observed that aging females and long-term NE depletion in the LC resulted in increased Aβ pathology in cortical regions of rhesus monkeys and thus revealed potential mechanisms and pathways impacted by DSP4 that influence pathology.
Aβ load almost doubled in NE-depleted aged macaques. Previous studies with AD-transgenic animals and post-mortem examination of AD-human brains showed significant loss of NE neurons in the LC [20, 40]. Markers of neuroinflammation and the AβPP pathway in AD relevant brain regions elucidated mechanisms influencing Aβ accumulation when challenged with NE depletion using DSP4. Furthermore, altered LC function caused by DSP4 appeared to exacerbate Aβ load and the coalescing of plaques in cortical areas receiving noradrenergic projections. Here we also demonstrated that NE depletion in the LC associated with cortical increases in BACE1, the secretase responsible for generating pathogenic Aβ, and influenced AβPP in the absence of chronic neuroinflammation.
The long-lasting reduction of NE suggests that LC function was impaired by DSP4. Our findings also support that the primary action of DSP4 was not restricted to LC terminals and that DSP4 effects also occurred at LC cell bodies possibly acting via the NE transporter [41, 42]. Equivalent levels of NE in cortical projection regions coupled with reduced NE content in the LC suggest that increased activity could compensate for a subset of impaired neurons [43], although our data demonstrate that LC dysfunction still permitted increased Aβ deposition.
There appeared to be noradrenergic system suppression following DSP4 as evidenced by a reduction in DBH at necropsy, suggesting an age-dependent capacity for recovery, with diminished response in aged individuals. Documented recovery occurred in young rodents within weeks to months following single DSP4 injection, and NE content in LC and projection regions recovered within 3 months [44, 45]. Long-lasting reduction of DBH-IR and decreased NE after DSP4 observed in the aging female rhesus monkey support the hypothesis of greater sensitivity and reduced plasticity associated with long-term effects of DSP4.
Similar to humans, an age-related increase of Aβ occurred in rhesus females. Our findings were comparable to other reports of cerebral Aβ in NHPs, with diffuse and compact extracellular Aβ pathology in frontal and temporal cortex; however, we observed more isolated staining in prefrontal, temporal, and parietal cortices. Aβ increased following DSP4 injection, demonstrating that NE depletion exacerbated deposition and accumulation of neocortical Aβ in female aged rhesus macaques. Elevated cortical Aβ deposition was not observed in the adult group after DSP4, possibly due to differences in natural onset and pattern of Aβ deposition during aging or due to CSF clearance [46]. LC degeneration has been reported in cases of mild cognitive impairment and may herald early stages of AD [28]. Loss of NE altered the ratio of soluble Aβ in cortical regions from aged animals; soluble monomers represent an earlier stage of pathology suggesting NE depletion also influences the generation and aggregation of Aβ isoforms. Elevated levels of these monomers and oligomers correlate with impaired cognition in AD patients, and soluble oligomers impair synaptic function in animal models [47]. The pathology associated with oligomer accumulation was the basis for another NHP model of AD. Here, Aβ oligomers were injected into the intracerebro-ventricles of NHPs leading to tau pathology, an observation that does not generally occur spontaneously in macaques [48].
Neuroglial cells are critical transducers of neuroimmune inflammatory responses; activated microglia phagocytize damaged cells, clear Aβ, and become chronically activated with progressive neural disease [49]. DSP4 caused sustained activation in AD-transgenic mouse studies with increased secretion of proinflammatory cytokines [23,29]. However, we did not see evidence of chronic neuroinflammation in our monkey study, as assessed by IBA1 staining to assess morphology. It is possible that the dosing schedule allowed sufficient time between injections to restore a resting state morphology for neuroglia populations, including microglia and astrocytes. Further, the presence of increased Aβ without coexisting activation of microglia or astrocytes suggests that the observed Aβ deposits alone were inadequate to cause a sustained inflammatory response in NHPs.
Accumulation of Aβ isoforms and plaque formation occurs through imbalances between Aβ production and clearance. Pro-inflammatory cytokines modulate AβPP processing, increasing its generation and p-secretase cleavage [50, 51]; while neuroglia and proteases actively degrade Aβ [52, 53]. DSP4 increased Aβ deposition in neocortex and sustained elevations in AβPP and BACE1, suggest altered AβPP processing contributed to increased Aβ deposition rather than decreases by neuroglia since these populations did not display phenotypes associated with Aβ degradation.
Finally, the effects of DSP4 treatment in decreasing NE levels in both adult and aged rhesus and increasing AβPP levels in temporal cortex is interesting. This reciprocal activity may not be coincidental. Duce et al. [33] recently reported that AβPP acts as a ferroxidase in neurons and other cells, the iron-export activity that ceruloplasmin mediates in glia. AβPP also oxidizes synthetic amines and catecholamines catalytically via a site encompassing its ferroxidase motif. Accordingly, AβPP knockout mice have significantly higher levels of dopamine and NE in brain, plasma, and select tissues. These findings support a role for AβPP in extracellular catecholaminergic clearance, which is consistent with our data (high AβPP and low NE). Down’s syndrome has similar attributes with a trisomy of chromosome 21 that carries AβPP, and AβPP amine oxidase activity at clinically increased levels. Interestingly, these patients have reduced brain catecholamine levels and blunted sympathetic response [33]. High AβPP and low NE levels are also observed in our NHP model. Likewise, cognitive defects in a Down’s syndrome mouse model appear to be in part due to adrenergic deficiency; elevating NE levels can partially restore this deficit.
Altogether, our results of modulating neural catecholamine levels in the NHP model, which is closer to human than rodent, assumes increased significance. This could be utilized to further study the important NE effect for several major classes of psychotropic drugs. Further, our present work coupled with the recent finding that AβPP has modifiable activity which influences NE levels, has significant consequences for clinical pharmacology.
In conclusion, we report elevated Aβ pathology following DSP4 induced LC impairment using a modified dosing schedule to minimize peripheral adverse effects of long term DSP4 treatment in an NHP model. Although we did not monitor potential transient inflammatory effects, treatment resulted in long-term reduction of NE in the LC and clearly influenced Aβ deposition in macaques. These data highlight the importance of an NHP model for AD; a model that shows similar spontaneous initiation and progression of the neural disease. Notably, our present work has utilized primates which are expensive and challenging concerning their access, handling, and experimentation, yet provide significant advantages over the more commonly used and readily available rodents, including wild type/transgenic strains. The sequence of AβPP and Aβ peptide is identical between NHPs and humans. Further, these genes/proteins are all driven by the native regulatory element (e.g., endogenous promoter) present in the host (NHP), unlike that in transgenic rodent animals. In short, the low number used in our study would nevertheless provide results that are very meaningful and translatable for a human disease, AD.
It is critical to consider the stage of aging; it would have been interesting to include an older postmenopausal group to assess plasticity. Compensatory actions and recovery mechanisms are important mediators to overall dysfunction that require study to elucidate their contribution in NHPs. Clearance of Aβ isoforms across the blood-brain barrier mediated by low density lipoprotein receptors and the receptor for advanced glycation end products (RAGE) also warrants further investigation. These investigations should shed additional light on mechanisms involved in the DSP4 mediated enhancement of pathology associated with AD. Finally, relatively few studies have compared these aging processes across NHPs and addressed the chronology of amyloid burden and other age-related neurodegenerative diseases [54]. Studies addressing age-related deposition of amyloid across multiple NHPs will provide additional insights into parallel degenerative disease and distinguish parallel but independent components of neurodegenerative disease processes.
ACKNOWLEDGMENTS
This research was supported in part by the National Institute on Aging Intramural Research Program of the NIH; NIH Grants AG18379, AG18884, and AG42804 to DKL; AG031387 to MAO; the Alzheimer’s Association (Zenith award and IIRG-206418) to DKL and BR; and the University of Maryland Department of Animal and Avian Sciences. The authors thank Kelli Vaughan for editorial assistance. Research conducted in partial fulfillment of the requirements for the Ph.D. degree for the University of Maryland (KBD).
Footnotes
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0487r2).
REFERENCES
- [1].Alzhiemer’s Association (2018) 2018 Alzheimer’s disease facts and figures. Alzheimers Dement 14, 367–429. [Google Scholar]
- [2].Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Petzold GC, Town T, Morgan D, Shinohara ML, Perry VH, Holmes C, Bazan NG, Brooks DJ, Hunot S, Joseph B, Deigendesch N, Garaschuk O, Boddeke E, Dinarello CA, Breitner JC, Cole GM, Golen-bock DT, Kummer MP (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14, 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. [DOI] [PubMed] [Google Scholar]
- [4].Shaw KT, Utsuki T, Rogers J, Yu QS, Sambamurti K, Brossi A, Ge YW, Lahiri DK, Greig NH (2001) Phenser- ine regulates translation of beta-amyloid precursor protein mRNA by a putative interleukin-1 responsive element, a target for drug development. Proc Natl Acad Sci USA 98, 7605–7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Long JM, Ray B, Lahiri DK (2012) MicroRNA-153 physiologically inhibits expression of amyloid-beta precursor protein in cultured human fetal brain cells and is dysregulated in a subset of Alzheimer disease patients. J Biol Chem 287, 31298–31310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lahiri DK, Maloney B, Zawia NH (2009) The LEARn model: An epigenetic explanation for idiopathic neurobiological diseases. Mol Psychiatry 14, 992–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Long JM, Ray B, Lahiri DK (2014) MicroRNA-339–5p down-regulates protein expression of beta-site amyloid precursor protein-cleaving enzyme 1 (BACE1) in human primary brain cultures and is reduced in brain tissue specimens of Alzheimer disease subjects. J Biol Chem 289, 5184–5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wang S, Wang R, Chen L, Bennett DA, Dickson DW, Wang DS (2010) Expression and functional profiling of neprilysin, insulin-degrading enzyme, and endothelin-converting enzyme in prospectively studied elderly and Alzheimer’s brain. J Neurochem 115, 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Miners JS, Baig S, Palmer J, Palmer LE, Kehoe PG, Love S (2008) Abeta-degrading enzymes in Alzheimer’s disease. Brain Pathol 18, 240–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Atwood CS, Martins RN, Smith MA, Perry G (2002) Senile plaque composition and posttranslational modification of amyloid-beta peptide and associated proteins. Peptides 23, 1343–1350. [DOI] [PubMed] [Google Scholar]
- [11].Basha MR, Wei W, Bakheet SA, Benitez N, Siddiqi HK, Ge YW, Lahiri DK, Zawia NH (2005) The fetal basis of amyloidogenesis: Exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. J Neurosci 25, 823–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bolin CM, Basha R, Cox D, Zawia NH, Maloney B,Lahiri DK, Cardozo-Pelaez F (2006) Exposure to lead and the developmental origin of oxidative DNA damage in the aging brain. FASEB J 20, 788–790. [DOI] [PubMed] [Google Scholar]
- [13].Wu J, Basha MR, Brock B, Cox DP, Cardozo-Pelaez F, McPherson CA, Harry J, Rice DC, Maloney B, Chen D, Lahiri DK, Zawia NH (2008) Alzheimer’s disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): Evidence for a developmental origin and environmental link for AD. J Neurosci 28, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Feinstein DL, Heneka MT, Gavrilyuk V, Dello Russo C, Weinberg G, Galea E (2002) Noradrenergic regulation of inflammatory gene expression in brain. Neurochem Int 41, 357–365. [DOI] [PubMed] [Google Scholar]
- [15].Marien MR, Colpaert FC, Rosenquist AC (2004) Noradrenergic mechanisms in neurodegenerative diseases: A theory. Brain Res Brain Res Rev 45, 38–78. [DOI] [PubMed] [Google Scholar]
- [16].Sara SJ (2009) The locus coeruleus and noradrenergic modulation of cognition. Nat Rev Neurosci 10, 211–223. [DOI] [PubMed] [Google Scholar]
- [17].Feinstein DL, Kalinin S, Braun D (2016) Causes, consequences, and cures for neuroinflammation mediated via the locus coeruleus: Noradrenergic signaling system. J Neurochem 139, 154–178. [DOI] [PubMed] [Google Scholar]
- [18].Mravec B, Lejavova K, Cubinkova V (2014) Locus (coeruleus) minoris resistentiae in pathogenesis of Alzheimer’s disease. Curr Alzheimer Res 11, 992–1001. [DOI] [PubMed] [Google Scholar]
- [19].German DC, Manaye KF, White CL 3rd, Woodward DJ, Mclntire DD, Smith WK, Kalaria RN, Mann DM (1992) Disease-specific patterns oflocus coeruleus cell loss. Ann Neurol 32, 667–676. [DOI] [PubMed] [Google Scholar]
- [20].Zarow C, Lyness SA, Mortimer JA, Chui HC (2003) Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol 60, 337–341. [DOI] [PubMed] [Google Scholar]
- [21].Rüb U, Stratmann K, Heinsen H, Turco DD, Seidel K, Dunnen Wd, Korf H-W (2016) The brainstem tau cytoskeletal pathology of Alzheimer’s disease: A brief historical overview and description of its anatomical distribution pattern, evolutional features, pathogenetic and clinical relevance. Curr Alzheimer Res 13, 1178–1197. [DOI] [PubMed] [Google Scholar]
- [22].Takeuchi T, Duszkiewicz AJ, Sonneborn A, Spooner PA, Yamasaki M, Watanabe M, Smith CC, Fernandez G, Deisseroth K, Greene RW, Morris RGM (2016) Locus coeruleus and dopaminergic consolidation of everyday memory. Nature 537, 357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Heneka MT, Nadrigny F, Regen T, Martinez-Hernandez A, Dumitrescu-Ozimek L, Terwel D, Jardanhazi-Kurutz D, Walter J, Kirchhoff F, Hanisch UK, Kummer MP (2010) Locus ceruleus controls Alzheimer’s disease pathology by modulating microglial functions through norepinephrine. Proc Natl Acad Sci USA 107, 6058–6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Madrigal JL, Feinstein DL, Dello Russo C (2005) Norepinephrine protects cortical neurons against microglial-induced cell death. J Neurosci Res 81, 390–396. [DOI] [PubMed] [Google Scholar]
- [25].Mann DM, Yates PO, Hawkes J (1982) The noradrenergic system in Alzheimer and multi-infarct dementias. J Neurol Neurosurg Psychiatry 45, 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Phillips C, Fahimi A, Das D, Mojabi FS, Ponnusamy R, Salehi A (2016) Noradrenergic system in Down syndrome and Alzheimer’s disease: A target for therapy. Curr Alzheimer Res 13, 68–83. [DOI] [PubMed] [Google Scholar]
- [27].Brunnstrom H, Friberg N, Lindberg E, Englund E (2011) Differential degeneration of the locus coeruleus in dementia subtypes. Clin Neuropathol 30, 104–110. [DOI] [PubMed] [Google Scholar]
- [28].Grudzien A, Shaw P, Weintraub S, Bigio E, Mash DC, Mesulam MM (2007) Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer’s disease. Neurobiol Aging 28, 327–335. [DOI] [PubMed] [Google Scholar]
- [29].Heneka MT, Ramanathan M, Jacobs AH, Dumitrescu-Ozimek L, Bilkei-Gorzo A, Debeir T, Sastre M, Galldiks N, Zimmer A, Hoehn M, Heiss WD, Klockgether T, Staufenbiel M (2006) Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J Neurosci 26, 1343–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Heneka MT, Galea E, Gavriluyk V, Dumitrescu-Ozimek L, Daeschner J, O’Banion MK, Weinberg G, Klockgether T, Feinstein DL (2002) Noradrenergic depletion potentiates beta-amyloid-induced cortical inflammation: Implications for Alzheimer’s disease. J Neurosci 22, 2434–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kalinin S, Gavrilyuk V, Polak PE, Vasser R, Zhao J, Heneka MT, Feinstein DL (2007) Noradrenaline deficiency in brain increases beta-amyloid plaque burden in an animal model of Alzheimer’s disease. Neuro boil Aging 28, 1206–1214. [DOI] [PubMed] [Google Scholar]
- [32].Pugh PL, Vidgeon-Hart MP, Ashmeade T, Culbert AA, Seymour Z, Perren MJ, Joyce F, Bate ST, Babin A, Virley DJ, Richardson JC, Upton N, Sunter D (2007) Repeated administration of the noradrenergic neurotoxin N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP-4) modulates neuroinflammation and amyloid plaque load in mice bearing amyloid precursor protein and presenilin-1 mutant transgenes. J Neuroinflammation 4, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Duce JA, Ayton S, Miller AA, Tsatsanis A, Lam LQ, Leone L, Corbin JE, Butzkueven H, Kilpatrick TJ, Rogers JT, Barnham KJ, Finkelstein DI, Bush AI (2013) Amine oxidase activity of beta-amyloid precursor protein modulates systemic and local catecholamine levels. Mol Psychiatry 18, 245–254. [DOI] [PubMed] [Google Scholar]
- [34].Ruggiero G, Iavarone A, Iachini T (2018) Allocentric to egocentric spatial switching: Impairment in aMCI and Alzheimer’s disease patients? Curr Alzheimer Res 15,229–236. [DOI] [PubMed] [Google Scholar]
- [35].Heuer E, Rosen RF, Cintron A, Walker LC (2012) Nonhuman primate models of Alzheimer-like cerebral proteopathy. Curr Pharm Des 18, 1159–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Paxinos G, Huang H-F, Petrides M, Toga AW (2009) The Rhesus Monkey Brain in Stereotaxic Coordinates, Elsevier, London, UK. [Google Scholar]
- [37].Tynan RJ, Naicker S, Hinwood M, Nalivaiko E, Buller KM, Pow DV, Day TA, Walker FR (2010) Chronic stress alters the density and morphology of microglia in a subset of stress-responsive brain regions. Brain Behav Immun 24, 1058–1068. [DOI] [PubMed] [Google Scholar]
- [38].Uno H, Alsum PB, Dong S, Richardson R, Zimbric ML, Thieme CS, Houser WD (1996) Cerebral amyloid angiopathy and plaques, and visceral amyloidosis in aged macaques. Neurobiol Aging 17, 275–281. [DOI] [PubMed] [Google Scholar]
- [39].Streit WJ, Braak H, Xue QS, Bechmann I (2009) Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol 118, 475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].O’Neil JN, Mouton PR, Tizabi Y, Ottinger MA, Lei DL, Ingram DK, Manaye KF (2007) Catecholaminergic neuronal loss in locus coeruleus of aged female dtg APP/PS1 mice. J Chem Neuroanat 34, 102–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ordway GA, Stockmeier CA, Cason GW, Klimek V (1997) Pharmacology and distribution of norepinephrine transporters in the human locus coeruleus and raphe nuclei. J Neurosci 17, 1710–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sanders JD, Happe HK, Bylund DB, Murrin LC (2005) Development of the norepinephrine transporter in the rat CNS. Neuroscience 130, 107–117. [DOI] [PubMed] [Google Scholar]
- [43].Szot P, White SS, Greenup JL, Leverenz JB, Peskind ER, Raskind MA (2006) Compensatory changes in the noradrenergic nervous system in the locus ceruleus and hippocampus of postmortem subjects with Alzheimer’s disease and dementia with Lewy bodies. J Neurosci 26, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fornai F, Bassi L, Torracca MT, Alessandri MG, Scalori V, Corsini GU (1996) Region- and neurotransmitter-dependent species and strain differences in DSP-4-induced monoamine depletion in rodents. Neurodegeneration 5, 241–249. [DOI] [PubMed] [Google Scholar]
- [45].Szot P, Miguelez C, White SS, Franklin A, Sikkema C, Wilkinson CW, Ugedo L, Raskind MA (2010) A comprehensive analysis of the effect of DSP4 on the locus coeruleus noradrenergic system in the rat. Neuroscience 166, 279–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lahiri DK, Ray B (2012) Abnormal cerebrospinal fluid (CSF) dynamics in Alzheimer’s disease and normal pressure hydrocephalus: CSF-amyloid beta precursor protein metabolites as possible biomarkers. Eur J Neurol 20, 211–213. [DOI] [PubMed] [Google Scholar]
- [47].Krafft GA, Klein WL (2010) ADDLs and the signaling web that leads to Alzheimer’s disease. Neuropharmacology 59, 230–242. [DOI] [PubMed] [Google Scholar]
- [48].Forny-Germano L, Lyra e Silva NM, Batista AF, Brito-Moreira J, Gralle M, Boehnke SE, Coe BC, Lablans A, Marques SA, Martinez AM, Klein WL, Houzel JC, Ferreira ST, Munoz DP, De Felice FG (2014) Alzheimer’s disease-like pathology induced by amyloidbeta oligomers in nonhuman primates. J Neurosci 34, 13629–13643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Streit WJ, Mrak RE, Griffin WS (2004) Microglia and neuroinflammation: A pathological perspective. J Neuro inflammation 1, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bourne KZ, Ferrari DC, Lange-Dohna C, Rossner S, Wood TG, Perez-Polo JR (2007) Differential regulation of BACE1 promoter activity by nuclear factor-kappaB in neurons and glia upon exposure to beta-amyloid peptides. J Neurosci Res 85, 1194–1204. [DOI] [PubMed] [Google Scholar]
- [51].Carrero I, Gonzalo MR, Martin B, Sanz-Anquela JM, Arevalo-Serrano J, Gonzalo-Ruiz A (2012) Oligomers of beta-amyloid protein (Abeta1–42) induce the activation of cyclooxygenase-2 in astrocytes via an interaction with interleukin-1beta, tumour necrosis factor-alpha, and a nuclear factor kappa-B mechanism in the rat brain. Exp Neurol 236, 215–227. [DOI] [PubMed] [Google Scholar]
- [52].Lee CY, Landreth GE (2010) Therole of microgliain amyloid clearance from the AD brain. J Neural Transm 117, 949–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Pihlaja R, Koistinaho J, Kauppinen R, Sandholm J, Tanila H, Koistinaho M (2011) Multiple cellular and molecular mechanisms are involved in human Abeta clearance by transplanted adult astrocytes. Glia 59, 1643–1657. [DOI] [PubMed] [Google Scholar]
- [54].Kalinin S, Willard SL, Shively CA, Kaplan JR, Register TC, Jorgensen MJ, Polak PE, Rubinstein I, Feinstein DL (2013) Development of amyloid burden in African Green monkeys. Neurobiol Aging 34, 2361–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]