

Abstract

Nicotinamide N-methyltransferase (NNMT) catalyzes the methylation of nicotinamide to form N-methylnicotinamide. Overexpression of NNMT is associated with a variety of diseases, including a number of cancers and metabolic disorders, suggesting a role for NNMT as a potential therapeutic target. By structural modification of a lead NNMT inhibitor previously developed in our group, we prepared a diverse library of inhibitors to probe the different regions of the enzyme’s active site. This investigation revealed that incorporation of a naphthalene moiety, intended to bind the hydrophobic nicotinamide binding pocket via π–π stacking interactions, significantly increases the activity of bisubstrate-like NNMT inhibitors (half-maximal inhibitory concentration 1.41 μM). These findings are further supported by isothermal titration calorimetry binding assays as well as modeling studies. The most active NNMT inhibitor identified in the present study demonstrated a dose-dependent inhibitory effect on the cell proliferation of the HSC-2 human oral cancer cell line.
Introduction
Nicotinamide N-methyltransferase (NNMT) is an important metabolic enzyme that catalyzes the transfer of a methyl group from the cofactor, S-adenosyl-l-methionine (SAM), onto its various substrates, most notably nicotinamide (NA) and other pyridines, to form 1-methyl-nicotinamide (MNA) or the corresponding pyridinium ions.1−3 The past decade has seen a renewed interest in the biological function of NNMT in a range of human diseases. While it was previously assumed that NNMT’s primary roles were limited to nicotinamide metabolism and xenobiotic detoxification of endogenous metabolites, broader roles of NNMT in human health and disease are becoming clearer.4 NNMT has been found to be overexpressed in a variety of diseases, including metabolic disorders,5−7 cardiovascular disease,8,9 cancer,10−14 and Parkinson’s disease.15,16 In general, overexpression of NNMT has been linked to disease progression in the aforementioned afflictions, with the exception of its role in Parkinson’s disease where NNMT seems to be neuroprotective.17,18 Collectively, NNMT appears to play a unique role in the regulation of post-translational modifications and signal transduction, making it an attractive and viable therapeutic target.
Despite the growing interest, few small-molecule NNMT inhibitors have been described to date. Among these structures, the product of the enzymatic reaction, MNA, is a known inhibitor of NNMT and has generally been used in biochemical activity assays.19 Recently, Cravatt and co-workers reported chloroacetamide-based covalent NNMT inhibitors that react with cysteine C165 in the SAM-binding pocket of the enzyme.20 Notably, Sanofi researchers have also recently reported a series of nicotinamide analogues that inhibit NNMT activity, leading to decreased MNA production, stabilization of insulin levels, glucose regulation, and weight loss in mouse models of metabolic disorders.21,22 In another approach, the group of Watowich focused on the development of inhibitors based on NNMT’s alternative substrate, quinoline. Their compounds showed improvement of symptoms in diet-induced obese mice.23 Previous work in our group has focused on bisubstrate inhibitors designed to mimic the transition state of the methylation reaction catalyzed by NNMT with compound 1 (Figure 1) showing activity on par with the known general methyltransferase inhibitor, sinefungin.24
Figure 1.

Schematic overview of the design strategy of the second generation of inhibitors based on trivalent bisubstrate compounds 124 and 2.31
Designing bisubstrate analogues as inhibitors is an established and effective strategy that has been applied to a range of methyltransferase enzymes, including catechol O-methyltransferase,25,26 histone lysine methyltransferases,27 arginine methyltransferases,28−30 and more recently nicotinamide N-methyltransferase.24,31 A recently published co-crystal structure of a bisubstrate inhibitor bound to NNMT [Protein Data Bank (PDB) ID: 6CHH] clearly delineates key interactions with residues in the enzyme active site, providing valuable information for further optimization of improved bisubstrate-like inhibitors.31 The work here described builds on our previous findings for “trivalent” inhibitor 1, which is assumed to simultaneously bind in the adenosine, amino acid, and nicotinamide binding pockets of the NNMT active site. Based on insights provided by recent NNMT crystal structures, we have designed new inhibitors, wherein the nicotinamide moiety is replaced by other aromatic substituents accompanied by variation in the length of the linker connecting the amino acid moiety. Based on the high conservation of the residues in the adenosine binding pocket, no changes were made to the adenosine group. A schematic overview of the design strategy is presented in Figure 1.
Results and Discussion
Design
The ternary crystal structure of NNMT (PDB ID: 3ROD) reveals the interactions of nicotinamide and S-adenosyl-l-homocysteine (SAH) with the active site residues.32 The active site can be roughly divided into three binding regions for the adenosine group, the amino acid moiety, and the nicotinamide unit. The starting point was a trivalent bisubstrate compound, 1, which was designed to bind all three binding regions. To find the optimal substitutions, a systematic approach was applied, where variations were made to the nicotinamide mimic on the one hand and the amino acid moiety on the other. The benzamide group, representing nicotinamide, was also replaced by methyl benzoate or benzoic acid moieties. Notably, the crystal structure of the NNMT–nicotinamide–SAH ternary complex reveals π–π stacking between tyrosine (Tyr) residue Y204 and the nicotinamide substrate.32 We, therefore, also prepared an analogue bearing a naphthalene unit in the presumed nicotinamide position with the aim of introducing stronger π–π stacking with the tyrosine residues of the NNMT active site. We also explored variation of the amino acid moiety as part of our design strategy: in some analogues the amine of the amino acid unit was omitted to reduce charge and in others the carboxylic acid was replaced by the corresponding primary amide. In addition, variation in the length of the carbon chain linking the amino acid moiety was examined. Furthermore, inspired by the structure of histone methyltransferase DOTL1 inhibitor pinometostat,33 we also investigated the incorporation of an isopropyl group to replace the amino acid moiety entirely.
Synthesis
Key aldehyde intermediates (compounds 6, 8, 9, 16, 17, 22, 23, 27, and 28) required for the synthesis of the various bisubstrate analogues pursued were prepared from commercially available materials, in good overall yields, as summarized in scheme 1–3. The trivalent inhibitors were then prepared via a convenient double-reductive amination strategy starting from the commercially available 2′-3′-O-isopropylidene-6-aminomethyl-adenosine starting material and the corresponding aldehydes (Schemes 4 and 5).
Scheme 1. Synthetic Route for Aldehydes 6, 8, and 9.

Reagents and conditions: (a) NaOH, MeOH, room temperature (rt), 16 h (95%); (b) (i) SOCl2, reflux, 2 h, (ii) tritylamine, CH2Cl2, 0 °C to rt, 2 h (72%); (c) diisobutylaluminum hydride (DIBAL-H), −78 °C to rt, 2 h (85%); (d) pyridinium dichromate (PDC), CH2Cl2, rt, 2 h (53–64%); (e) NaBH4, BF3.Et2O, tetrahydrofuran (THF), 0 °C to rt, 2 h (89%); (f) LiOH, THF/H2O (2:1); (g) 2-tert-butyl-1,3-diisopropylisourea, CH2Cl2, tert-butanol (39% over two steps).
Scheme 3. Synthetic Route for Aldehydes 27 and 28.

Reagents and conditions: (a) CH3NHOCH3·HCl, BOP, Et3N, CH2Cl2, rt, 2 h (85–88%); (b) (Boc)2O, Et3N, DMAP, CH2Cl2 (94%); (c) DIBAL-H in hexanes (1 M), THF, −78 °C, assumed quant.
Scheme 4. Synthesis of Intermediate Compounds 29–32.

Reagents and conditions: (a) NaBH(OAc)3, AcOH, 1,2-dichloroethane (DCE), rt, overnight (50–74%).
Scheme 5. Representative Scheme for the Synthesis of the Final Compounds; Shown for Compounds 1 and 57–61.

The same procedure was used starting from aldehydes 30–32 to form intermediate compounds 39–56 and 80 and final compounds 62–79 and 81 as detailed in the Experimental Section. Reagents and conditions: (a) aldehyde, NaBH(OAc)3, AcOH, DCE, rt, overnight (49–77%); (b) (i) trifluoroacetyl (TFA), CH2Cl2, rt, 2 h, (ii) H2O, rt, 30 min (47–73%).
The preparation of aromatic aldehydes 6, 8, and 9 began with the selective mono-deprotection of dimethyl isophthalate using sodium hydroxide (Scheme 1).34 Monomethyl isophthalate (3) was subsequently transformed into trityl-protected amide 4 using tritylamine via its acid chloride intermediate and reduced by diisobutylaluminum hydride (DIBAL-H) to give alcohol 5. The alcohol was oxidized to aldehyde 6 using pyridinium dichromate (PDC). For aldehydes 8 and 9, the carboxylic acid of 3 was selectively reduced using a mixture of sodium borohydride and boron trifluoride diethyl etherate.35 The resulting alcohol (7) was oxidized using PDC to yield the corresponding aldehyde (8). Following hydrolysis of the methyl ester in 8 and subsequent conversion to the tert-butyl ester, aldehyde 9 was obtained.36
Aliphatic aldehydes 16 and 17 containing trityl-protected amide functionalities were prepared from succinimide and glutarimide, respectively (Scheme 2). The cyclic amides were first trityl-protected and subsequently ring-opened using potassium hydroxide. Reduction to the corresponding alcohols and oxidization using PDC gave aldehydes 16 and 17.37,38 In an analogous fashion, aldehydes 22 and 23, both containing tert-butyl ester moieties, were prepared by ring opening of succinic or glutaric anhydride with tert-butyl alcohol to obtain mono-esters 18 and 19.39,40 The carboxylic acid functionalities were reduced to alcohols 20 and 21 and then oxidized using PDC to yield aldehydes 22 and 23.
Scheme 2. Synthetic Route for Aldehydes 16, 17, 22, and 23.

Reagents and conditions: (a) TrtCl, CH3CN, K2CO3, rt, 48 h (20–28%); (b) KOH, EtOH, reflux, overnight (37–93%) (c) NaBH4, BF3·Et2O, THF, 0 °C to rt, 2 h (64–81%); (d) PDC, CH2Cl2, rt, 2 h (65–78%); (e) tert-butanol, 4-dimethylaminopyridine (DMAP), N-hydroxysuccinimide, Et3N, toluene, overnight (25–93%).
Aldehydes 27 and 28, both containing protected amino acid functionalities, were prepared starting from the appropriately protected aspartic acid and glutamic acid building blocks (Scheme 3). Conversion of the side chain carboxylates to their corresponding Weinreb amides yielded intermediates 24 and 25. Reduction of aspartate-derived 24 with DIBAL-H gave amino acid aldehyde 27 in high yield. For the preparation of aldehyde 28, a similar route was followed with the addition of a second Boc-protection of intermediate 25 to avoid an intramolecular cyclization side reaction.24,41
With the necessary aldehyde building blocks in hand, assembly of the bisubstrate inhibitors was performed in each case starting from commercially available 2′-3′-O-isopropylidene-6-aminomethyl-adenosine (Scheme 4). Using a reliable reductive amination approach, aromatic aldehydes 6, 8, and 9, and commercially available 2-naphthaldehyde were each coupled to the protected adenosine species to yield intermediates 29–32. These intermediates were next connected with aliphatic aldehydes 16, 17, 22, 23, 27, and 28 or acetone via a second reductive amination step to give the corresponding protected tertiary amine intermediates 33–56 (Scheme 5). Global deprotection of the acid-labile protecting groups was carried out in CH2Cl2/TFA (1:1) with isopropylidene group cleavage facilitated by subsequent addition of water. The crude products were purified by preparative high-performance liquid chromatography (HPLC) to yield bisubstrate analogues 1 and 57–60.
Inhibition Studies
The bisubstrate analogues were next tested for their NNMT inhibitory activity using a method recently developed in our group.2 This assay employs ultra-high-performance (UHP) hydrophilic liquid interaction chromatography (HILIC) coupled to quadrupole time-of-flight mass spectrometry (Q-TOF-MS) to rapidly and efficiently assess NNMT inhibition by analysis of the formation of MNA. The NNMT inhibition of all compounds was initially screened at a fixed concentration of 250 μM for all of the compounds. In cases where at least 50% inhibition was detected at this concentration, full inhibition curves were measured in triplicate to determine the corresponding half-maximal inhibitory concentration (IC50) values. As reference compounds, we included the well-established and general methyltransferase inhibitors sinefungin and SAH. In addition, we also synthesized two recently described NNMT inhibitors, compound 2 and 6-(methylamino)-nicotinamide, following the procedures described in the corresponding publications.21,31 The structures of these reference compounds are provided in Figure 2.
Figure 2.

Chemical structures of the reference compounds used in NNMT inhibition studies.
The results of the NNMT inhibition studies are summarized in Table 1 and clearly show that only minor adjustments to the functional groups found in the enzyme’s natural substrates are tolerated. Among the compounds studied, the most potent inhibition was observed when the aliphatic moiety corresponded to the same length in the amino acid side chain as present in the methyl donor SAM. Notably, the preferred aromatic moiety was found to be the naphthalene group, an apparent confirmation of our hypothesis that increased π–π stacking can lead to enhanced binding in the nicotinamide pocket. The bisubstrate analogue containing both of these elements (compound 78), displayed the highest inhibitory activity against NNMT with an IC50 of 1.41 μM. Interestingly, the amino acid and naphthyl moieties were also found to independently enhance the activity of the other inhibitors prepared. In this way, a suboptimal moiety at one position can be compensated for—to an extent—by including either the SAM amino acid motif or the naphthalene unit at the other position. For example, bisubstrate analogues containing the benzamide, benzoic acid, or methyl benzoate groups only show inhibitory activity if they also contain the amino acid motif (compounds 1, 2, 66, and 72) with IC50 values of 4.36–23.4 μM, respectively. On the other hand, among the bisubstrate analogues lacking the amino acid motif, inclusion of the naphthalene moiety (compounds 74–79) enhances NNMT inhibition, albeit with moderate IC50 values in the range of 52.6–129.9 μM.
Table 1. Tabulated Overview of the Chemical Structures and Inhibition Results of the Final Compounds and Reference Compounds.

Assays performed in triplicate on at least six different inhibitor concentrations. Standard errors of the mean reported.
Other notable findings were the results obtained with the reference compounds. The general methyltransferase inhibitors, sinefungin and SAH, showed inhibitory activities in line with those previously reported.24 Interestingly, the 6-methylamino-NA compound, recently described by Sanofi to be a submicromolar inhibitor,21 gave an IC50 of 19.8 μM in our assay. The recently published bisubstrate analogue 2 exhibited good activity (IC50 4.4 μM) on par with published values.31 Given the potent inhibition measured for both compounds 2 and 78, we also prepared and tested compound 81, an analogue of 78 bearing the same naphthyl moiety but with the amino acid motif containing an additional methylene unit as in 2. Somewhat surprisingly, this linker elongation resulted in a complete loss of inhibitory activity (IC50 > 250 μM).
To gain insight into the selectivity of compound 78, we also tested its activity against representative members of both the arginine and lysine families of methyltransferases, PRMT1 and NSD2, respectively. In both cases, compound 78 was tested at a concentration of 50 μM and showed no significant inhibition (>50% of the enzyme’s activity remained), see Table S1.
Isothermal Titration Calorimetry (ITC) Binding Studies
To further evaluate the binding interactions of the most active bisubstrate analogues with NNMT, isothermal titration calorimetry (ITC) studies were performed. Compounds 1, 66, 72, and 78, all containing the amino acid moiety but with varying aromatic substituents, were investigated. As illustrated in Figure 3, the dissociation constants (Kd) measured for these compounds track very well with the IC50 values measured in the in vitro assay. Compounds 1 and 66 display similar binding to NNMT with Kd values of 36 and 25 μM, respectively, whereas compound 72 binds less tightly with a Kd of 124 μM. In good agreement with the results of the inhibition assay, the most active inhibitor, compound 78, also displayed the highest binding affinity for NNMT with a Kd of 5.6 μM. As expected, the inhibitors were each found to bind the enzyme with a 1:1 stoichiometry.
Figure 3.

ITC isotherms and thermograms including thermodynamic binding parameters measured for compounds 1, 66, 72, and 78 with human NNMT.
Modeling Studies
To further investigate the way in which the inhibitors bind within the NNMT active site, modeling studies were performed. Working from the available crystal structure of the NNMT protein bounded to nicotinamide and SAH (PDB ID: 3ROD),32 compounds 1, 2, 78, and 81 were modeled in the binding pocket. In an attempt to explain the significant difference in the activity of 78 and 81, additional molecular dynamic simulations were also performed for compounds 1, 2, 78, and 81. Although these simulations suggest differences in the binding interaction of the compounds (Figure S1, Supporting Information), the calculated binding energies for each are all very similar (Table S2, Supporting Information). In terms of their active site orientations, compounds 1, 2, 78, and 81 are all predicted to position their three branches roughly in the same regions of the active site; however, their orientations and interactions are quite different.
From the modeling data, two distinct features are apparent. First, when the chain linking the amino acid moiety is shorter (as in compounds 1 and 78), the formation of an intramolecular hydrogen bond interaction was observed between the carboxylate of the amino acid moiety and the protonated tertiary amine (see Figure 4). This intramolecular interaction is highly stable for compound 78 and less stable for compound 1. This additional interaction reduces the entropic energy of the ligand, thereby potentially stabilizing its binding, and re-orients the amino acid part in the pocket, preventing the polar interactions with neighboring residues (e.g., Y25, D61, Y69, and T163) observed when the chain is longer (as present in compounds 2 and 81). This intramolecular hydrogen bond may explain the difference in activity observed between compounds 78 and 81. The second distinct feature is the tyrosine-rich environment around the naphthalene moiety of 78 compared to the nicotinamide unit of 1. The orientation of the tyrosine residues surrounding this part of the molecule leads to π–π stacking interactions with the naphthalene and hints at an explanation for the strong inhibition and high affinity of compound 78 with the NNMT protein (Figure 4).
Figure 4.
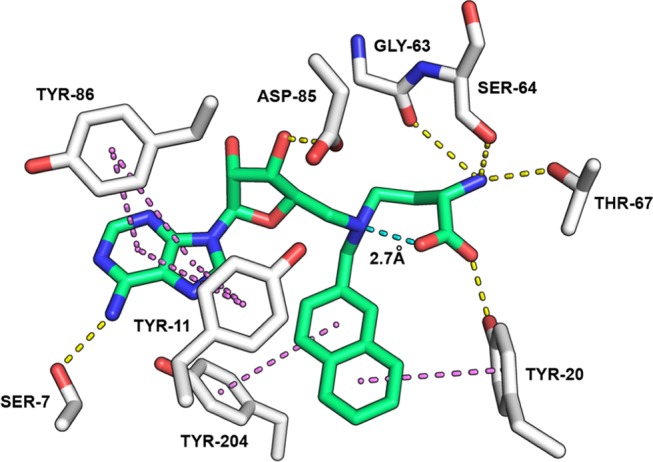
Modeling results for compound 78 in the NNMT active site (PDB ID: 3ROD). Molecular dynamics simulation indicates the presence of an intramolecular hydrogen bond (2.7 Å, shown in cyan) specific to compound 78 (in green) that would be expected to reduce the entropic energy of the ligand and potentially stabilize binding to NNMT (in white). Proposed intermolecular hydrogen bond network (in yellow) and π–π stacking interactions with Tyr residues (in purple) stabilize compound 78 in the NNMT active site (hydrogens omitted for clarity).
Cell-Based Assays
To evaluate the cellular activity of the bisubstrate inhibitors, the compounds were tested for their effect on cell proliferation in the human oral cancer cell line, HSC-2. We recently found that NNMT expression levels are high in this particular cell line and may contribute to its proliferation and tumorigenic capacity.42 As shown in Figure 5, there were no significant differences in the cell proliferation rate between HSC-2 cells treated with dimethyl sulfoxide (DMSO) at 0.1% concentration and cells grown with only the culture medium, at any time of each performed assay. Upon treatment with the NNMT inhibitors, cell proliferation was not significantly inhibited by compounds 1, 2, and 81 (Figure 5). In contrast, relative to the DMSO control, treatment with compound 78 led to a notable decrease in cell proliferation. In particular, cell proliferation was significantly (p < 0.05) inhibited by compound 78 at 10 μM (20% reduction), 50 μM (21% reduction), and 100 μM (27% reduction) concentrations, 48 h after treatment. Interestingly, at the longest 72 h time point taken, treatment with compound 78 leads to an even greater and significant (p < 0.01) decrease in cell proliferation (44% reduction), at the highest concentration (100 μM) (Figure 5).
Figure 5.

Results of the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) cell viability assay on HSC-2 human oral cancer cells. Only compound 78 showed a significant effect on cell proliferation after 48 and 72 h.
We next investigated the effect of compound 78 on cellular NNMT activity by assessing its impact on MNA production in the same HSC-2 cell line. Cells were treated with 100 μM of 78, and MNA levels were determined after 0, 1, 2, and 3 days. Cells treated with compound 78 show a significant (p < 0.01) decrease in the levels of MNA (50% reduction) compared to controls after 48 h. Interestingly, at 72 h an increase in cellular MNA production was detected; however, the same effect was also observed in the DMSO control (but not in the untreated control), suggesting an effect attributable to longer term DMSO exposure. The results of the cellular MNA analysis are presented in Figure S2, Supporting Information.
Conclusions
Building from our earlier findings with first reported ternary bisubstrate NNMT inhibitor 1,24 we designed and prepared a focused library of novel inhibitors to provide new structure–activity insights. In doing so, various structural motifs were investigated for their ability to enhance inhibitor activity and binding within the NNMT active site. By probing the SAM and NA binding pockets with different spacers and functional groups, we found that the optimal ligands are the endogenous amino acid side chain and the naphthalene moiety. Among the naphthalene-containing bisubstrate analogues prepared, compound 78 showed the most potent NNMT inhibition. In this way, the activity of our initial NNMT inhibitor 1 (IC50 14.9 μM) was improved 10-fold with compound 78, displaying an IC50 value of 1.41 μM. Notably, using an assay designed to directly measure NNMT product formation, compound 78 was shown to be more potent than most other NNMT inhibitors reported to date. ITC-based binding studies provided additional insights into the affinity of the inhibitors for the enzyme with the measured Kd value following a trend similar to that observed for the IC50 data obtained in the in vitro inhibition assays. From modeling studies, the improved activity of compound 78 can be rationalized by the apparent presence of an intramolecular hydrogen bonding interaction predisposing the compound to an active conformation with lower entropic cost. In addition, the modeling indicates that the naphthalene group in 78 is properly oriented so as to benefit from additional π–π stacking interactions with several tyrosine residues in the nicotinamide binding pocket of the enzyme. The cellular data obtained for compound 78 show a significant inhibitory effect on cell proliferation in HSC-2 oral cancer cells. These promising results provide important new insights for the design and further optimization of potent NNMT inhibitors.
Experimental Procedures
General Procedures
All reagents employed were of American Chemical Society grade or finer and were used without further purification unless otherwise stated. For compound characterization, 1H NMR spectra were recorded at 400 MHz with chemical shifts reported in parts per million downfield relative to tetramethylsilane, H2O (δ 4.79), CHCl3 (7.26), or DMSO (δ 2.50). 1H NMR data are reported in the following order: multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; and m, multiplet), coupling constant (J) in hertz (Hz) and the number of protons. Where appropriate, the multiplicity is preceded by br, indicating that the signal was broad. 13C NMR spectra were recorded at 101 MHz with chemical shifts reported relative to CDCl3 (δ 77.16), methanol (δ 49.00), or DMSO (δ 39.52). The 13C NMR spectra of the compounds recorded in D2O could not be referenced. High-resolution mass spectrometry (HRMS) analysis was performed using a Q-TOF instrument. Compounds 1,242,313,347,348,369,3710,4312,3814,3816,4418,4019,4020,4521,4622,4723,4024,4125,4126,4827,41 and 28(48) were prepared as previously described and had NMR spectra and mass spectra consistent with the assigned structures. Purity was confirmed to be ≥95% by analytical reversed-phase HPLC using a Phenomenex Kinetex C18 column (5 μm, 250 × 4.6 mm2) eluted with a water–acetonitrile gradient moving from 0 to 100% CH3CN (0.1% TFA) in 30 min. The compounds were purified via preparative HPLC using a ReproSil-Pur C18-AQ column (10 μm, 250 × 22 mm2) eluted with a water–acetonitrile gradient moving from 0 to 50% CH3CN (0.1% TFA) over 60 min at a flow rate of 12.0 mL/min with UV detection at 214 and 254 nm.
Methyl 3-(Tritylcarbamoyl)benzoate (4)
Monomethyl isophthalate 3 (0.98 g, 5.4 mmol) was refluxed in 10 mL of SOCl2 at 90 °C for about 1 h (until the reaction mixture was a clear solution). SOCl2 was removed under reduced pressure and the acid chloride intermediate was redissolved in 15 mL of dry CH2Cl2 and transferred to a cooled (ice bath) solution of tritylamine (1.41 g, 5.4 mmol) and 2 mL of triethylamine in 30 mL of CH2Cl2. The reaction was stirred overnight under a N2 atmosphere, allowing the mixture to warm to room temperature. After the reaction was completed [monitored by thin-layer chromatography (TLC) (petroleum ether/CH2Cl2 = 1:1)], the reaction mixture was washed with water and brine, and the organic phase was dried over Na2SO4 and concentrated. The crude product was purified by column chromatography (petroleum ether/CH2Cl2 = 2:1) to give compound 4 as a white powder (1.64 g, 72% yield). 1H NMR (400 MHz, CDCl3) δ 8.45 (t, J = 1.6 Hz, 1H), 8.18 (m, 1H), 8.03 (m, 1H), 7.53 (t, J = 7.8 Hz, 1H), 7.41–7.26 (m, 15H), 3.94 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 166.3, 165.4, 144.5, 135.6, 132.5, 131.7, 130.6, 128.9, 128.7, 128.1, 128.1, 127.6, 127.2, 71.0, 52.4. HRMS [electrospray ionization (ESI)]: calcd for C28H23NO3 [M + Na]+ 444.1576, found 444.1581.
3-(Hydroxymethyl)-N-tritylbenzamide (5)
Methyl 3-(tritylcarbamoyl)benzoate 4 (0.56 g, 1.33 mmol) was dissolved in dry CH2Cl2 (20 mL) under a N2 atmosphere, the reaction solution was cooled down to −78 °C, and then diisobutylaluminum hydride (DIBAL-H) (5.5 mL, 1.0 M hexane solution) was added slowly. The reaction mixture was stirred at −78 °C for 2 h. Saturated (sat.) aqueous (aq) NH4Cl (50 mL) was added slowly to quench the reaction at −78 °C, followed by the addition of a saturated Rochelle salt solution (100 mL). The mixture was stirred at room temperature overnight, extracted with CH2Cl2, and the organic layers were dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by column chromatography (CH2Cl2/EtOAc = 9:1) to obtain 5 as a white powder (0.44 g, 85% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.92 (s, 1H), 7.78 (s, 1H), 7.75–7.71 (m, 1H), 7.47 (d, J = 7.8 Hz, 1H), 7.40 (t, J = 7.6 Hz, 1H), 7.36–7.18 (m, 15H), 5.26 (br, 1H), 4.54 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 167.0, 145.3, 143.0, 135.5, 129.6, 128.9, 128.3, 127.9, 126.7, 126.5, 126.2, 79.6, 69.9, 69.9, 63.0. HRMS (ESI): calcd for C27H23NO2 [2M + Na]+ 809.3355, found 809.3359.
3-Formyl-N-tritylbenzamide (6)
3-(Hydroxymethyl)-N-tritylbenzamide 5 (0.20 g, 0.51 mmol) and pyridinium dichromate (PDC) (0.23 g, 0.61 mmol) were placed in a 50 mL round bottom flask and 10 mL of dry CH2Cl2 was added under a N2 atmosphere at room temperature. The reaction was stirred till completion, as monitored by TLC (petroleum ether/CH2Cl2 = 5:1). The mixture was filtered and the organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The resulting crude product was purified by column chromatography (petroleum ether/CH2Cl2 = 9:1) to obtain 6 as a white powder (0.13 g, yield 64%). 1H NMR (400 MHz, DMSO-d6) δ 10.09 (s, 1H), 9.31 (s, 1H), 8.39 (s, 1H), 8.17 (d, J = 7.7 Hz, 1H), 8.06 (d, J = 7.7 Hz, 1H), 7.68 (t, J = 7.7 Hz, 1H), 7.41–7.17 (m, 15H). 13C NMR (101 MHz, CDCl3) δ 191.5, 165.1, 144.4, 136.5, 136.2, 133.0, 132.5, 129.5, 128.6, 128.5, 128.1, 127.7, 127.3, 77.2, 71.1. HRMS (ESI): calcd for C27H21NO2 [2M + Na]+ 805.3042, found 805.3047.
N-(Triphenylmethyl)glutarimide (11)
Glutarimide (2.8 g, 25 mmol), triphenylchloromethane (7.4 g, 25 mmol), and potassium carbonate (3.7 g, 25 mmol) were added to 100 mL of acetonitrile, and the mixture was stirred at room temperature overnight. Saturated aqueous NaHCO3 (50 mL) was added, and the mixture was extracted with EtOAc. The combined organic layers were dried with anhydrous Na2SO4, and the solvent was removed under reduced pressure. The crude product was purified by column chromatography (petroleum ether/EtOAc = 4:1) to obtain 11 as a white powder (1.8 g, yield 20%). 1H NMR (400 MHz, DMSO-d6) δ 7.45–7.35 (m, 6H), 7.20 (t, J = 7.8 Hz, 6H), 7.08 (t, J = 7.3 Hz, 3H), 2.66 (t, J = 6.4 Hz, 4H), 2.01 (p, J = 6.5 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 172.4, 143.4, 128.5, 127.3, 125.9, 35.5, 16.7. HRMS (ESI): calcd for C24H21NO2 [M + Na]+ 378.1470, found 378.1493.
5-Oxo-5-(tritylamino)pentanoic Acid (13)
To 2.80 g of KOH dissolved in 50 mL of ethanol was added N-tritylglutarimide 11 (1.00 g, 2.8 mmol), and the mixture was refluxed for 48 h. The mixture was then concentrated to dryness and redissolved in H2O. Acidification of the basic solution with conc. HCl to pH = 2 and filtration of the product gave compound 13 as a white powder (0.96 g, yield 91%). 1H NMR (400 MHz, CD3OD) δ 7.30–7.17 (m, 15H), 2.37 (t, J = 7.4 Hz, 2H), 2.25 (t, J = 7.4 Hz, 2H), 1.79–1.87 (m 2H). 13C NMR (101 MHz, CD3OD) δ 175.5, 173.3, 144.6, 128.6, 127.3, 127.2, 126.7, 126.3, 35.2, 32.6, 20.7. HRMS (ESI): calcd for C24H23NO3 [M + Na]+ 396.1576, found 396.1573.
5-Hydroxy-N-tritylpentanamide (15)
To a solution of 13 (2.60 g, 6.96 mmol) in dry THF (60 mL) cooled to 0 °C was added NaBH(OAc)3 (0.28 g, 7.3 mmol). The solution was stirred until evolution of H2 stopped, and BF3.OEt2 (1.1 mL, 8.8 mmol) was added dropwise. The reaction was stirred at room temperature for 4 h. The reaction was quenched by adding 50 mL of H2O at 0 °C. The mixture was extracted with EtOAc, and the combined organic layers were washed with sat. aq Na2CO3 and brine and dried over Na2SO4. The crude product was purified by column chromatography (100% EtOAc) to give compound 15 as a white powder (1.60 g, 64% yield). 1H NMR (400 MHz, CDCl3) δ 7.22–6.74 (m, 15H), 6.36 (br, 1H), 3.29–3.19 (br, 2H), 2.01 (t, J = 7.2 Hz, 2H), 1.46–1.36 (m, 2H), 1.24 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 171.9, 144.7, 128.6, 127.9, 127.0, 62.0, 37.0, 32.0, 21.4. HRMS (ESI): calcd for C24H25NO2 [M + Na]+ 382.1783, found 382.1783.
5-Oxo-N-tritylpentanamide (17)
5-Hydroxy-N-tritylpentanamide 15 (1.30 g, 3.6 mmol) and PDC (2.00 g, 5.4 mmol) were dissolved in 50 mL of dry CH2Cl2 and stirred for 2 h under a N2 atmosphere at room temperature. The mixture was filtered, and the organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The crude product was purified by column chromatography (100% CH2Cl2) to give compound 17 as an off-white powder (0.84 g, 65% yield). 1H NMR (400 MHz, CDCl3) δ 9.71 (s, 1H), 7.36–7.10 (m, 15H), 6.59 (s, 1H), 2.44 (t, J = 7.0 Hz, 2H), 2.32 (t, J = 7.2 Hz, 2H), 1.97–1.88 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 202.0, 170.8, 144.6, 128.6, 127.9, 127.0, 70.5, 42.9, 36.1, 17.9. HRMS (ESI): calcd for C24H23NO2 [M + Na]+ 380.1626, found 380.1629.
3-(((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]-dioxol-4-yl)methyl)amino)methyl)-N-tritylbenzamide (29)
3-Formyl-N-tritylbenzamide 6 (1.22 g, 3.12 mmol), 2′-3′-O-isopropylidene-6-aminomethyl-adenosine (1.00 g, 3.43 mmol), and acetic acid (0.45 mL, 8 mmol) were dissolved in 1,2-dichloroethane (DCE, 50 mL) and stirred at room temperature under a N2 atmosphere. After 3 h, NaBH(OAc)3 (1.09 g, 5.15 mmol) was added, and the reaction mixture was stirred overnight at room temperature. The reaction was quenched by adding 1 N NaOH solution (50 mL), and the product was extracted with CH2Cl2. The combined organic layers were washed with brine and dried over Na2SO4. The solvent was evaporated, and the crude product was purified by column chromatography (10% MeOH in CH2Cl2) to give compound 29 as a white powder (1.25 g, 59% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.89 (s, 1H), 8.34 (s, 1H), 8.06 (s, 1H), 7.79 (s, 1H), 7.71 (d, J = 7.7 Hz, 1H), 7.43 (d, J = 7.7 Hz, 1H), 7.39–7.24 (m, 15H), 7.20 (m, 3H), 6.09 (d, J = 3.1 Hz, 1H), 5.76 (s, 1H), 5.46 (M, 1H), 5.00 (m, 1H), 4.28–4.23 (m, 1H), 3.73 (s, 2H), 2.75–2.66 (m, 2H), 1.54 (s, 3H), 1.31 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.9, 156.5, 153.1, 149.3, 145.3, 140.4, 135.6, 128.9, 128.3, 127.9, 127.6, 126.7, 126.5, 119.7, 113.7, 89.7, 85.3, 83.1, 82.6, 69.9, 55.3, 53.0, 50.8, 27.5, 25.7. HRMS (ESI): calcd for C40H39N7O4 [M + Na]+ 704.2961, found 704.2975.
Methyl 3-((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)amino)benzoate (30)
Following the procedure described for compound 29, coupling methyl 3-formylbenzoate 8 (0.51 g, 3.12 mmol) and 2′-3′-O-isopropylidene-6-aminomethyl-adenosine (1.00 g, 3.43 mmol) afforded compound 30 as a white powder (0.92 g, 65% yield). 1H NMR (400 MHz, CDCl3) δ 8.08 (s, 1H), 7.92 (s, 1H), 7.90–7.83 (m, 2H), 7.44 (d, J = 7.6 Hz, 1H), 7.32 (t, J = 7.6 Hz, 1H), 6.37 (d, J = 5.7 Hz, 2H), 5.95 (d, J = 3.1 Hz, 1H), 5.45 (m, 1H), 5.04 (m, 1H), 4.40–4.34 (m, 1H), 3.86 (s, 3H), 3.79 (s, 2H), 2.90–2.83 (m, 2H), 1.58 (s, 3H), 1.35 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.1, 155.8, 155.8, 153.0, 149.2, 140.4, 140.4, 139.8, 132.6, 132.6, 130.1, 129.1, 129.1, 128.4, 128.4, 128.2, 120.2, 114.4, 91.0, 85.5, 83.2, 83.2, 82.2, 82.2, 53.3, 52.1, 52.1, 50.6, 27.3, 27.2, 25.4, 25.3. HRMS (ESI): calcd for C22H26N6O5 [M + H]+ 455.2043, found 455.2050.
tert-Butyl 3-(((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)amino)methyl)benzoate (31)
Following the procedure described for compound 29, coupling tert-butyl 3-formylbenzoate 9 (0.64 g, 3.12 mmol) and 2′-3′-O-isopropylidene-6-aminomethyl-adenosine (1.00 g, 3.43 mmol) afforded compound 31 as a white powder (0.77 g, 50% yield). 1H NMR (400 MHz, CDCl3) δ 8.11 (s, 1H), 7.89 (s, 1H), 7.86–7.83 (m, 2H), 7.43 (d, J = 7.7 Hz, 1H), 7.31 (t, J = 7.6 Hz, 1H), 6.36–6.27 (m, 2H), 5.96 (d, J = 3.3 Hz, 1H), 5.46 (m, 1H), 5.04 (m, 1H), 4.38 (m, 1H), 3.80 (s, 2H), 2.94–2.81 (m, 2H), 1.58 (s, 3H), 1.55 (s, 9H), 1.36 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 165.7, 155.8, 155.8, 153.0, 149.3, 140.2, 139.8, 132.0, 132.0, 129.0, 128.2, 128.1, 120.3, 114.5, 91.0, 85.4, 83.2, 82.2, 80.9, 53.4, 50.6, 28.1, 27.3, 25.4. HRMS (ESI): calcd for C25H32N6O5 [M + H]+ 497.2512, found 497.2511.
9-((3aR,4R,6R,6aR)-2,2-Dimethyl-6-(((naphthalen-2-ylmethyl)amino)methyl)tetrahydrofuro-[3,4-d][1,3]dioxol-4-yl)-9H-purin-6-amine (32)
Following the procedure described for compound 29, coupling 2-naphthaldehyde (0.49 g, 3.12 mmol) and 2′-3′-O-isopropylidene-6-aminomethyl-adenosine (1.00 g, 3.43 mmol) afforded compound 32 as a white powder (1.03 g, 74% yield). 1H NMR (400 MHz, CDCl3) δ 8.11 (s, 1H), 7.88 (s, 1H), 7.78 (m, 3H), 7.70 (s, 1H), 7.48–7.38 (m, 3H), 6.05 (s, 2H), 5.99 (d, J = 3.3 Hz, 1H), 5.48 (m, 1H), 5.06 (m, 1H), 4.45–4.39 (m, 1H), 3.95 (s, 2H), 3.01–2.87 (m, 2H), 2.33 (br, 2H), 1.61 (s, 3H), 1.38 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 155.7, 153.0, 149.3, 139.9, 137.4, 133.3, 132.6, 128.0, 127.6 126.4, 126.4, 126.0, 125.5, 120.3, 114.5, 91.0, 85.6, 83.3, 82.3, 53.9, 50.7, 27.3, 25.4. HRMS (ESI): calcd for C24H26N6O3 [M + H]+ 447.2145, found 447.2167.
3-(((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]-dioxol-4-yl)methyl)(4-oxo-4-(tritylamino)butyl)amino)methyl)-N-tritylbenzamide (33)
Oxo-N-tritylbutanamide 16 (62 mg, 0.18 mmol), compound 29 (100 mg, 0.15 mmol), and AcOH (one drop) were dissolved in 1,2-dichloroethane (DCE, 10 mL) and stirred at room temperature under a N2 atmosphere. After 3 h, NaBH4 (49 mg, 0.23 mmol) was added, and the reaction mixture was stirred overnight at room temperature. The reaction was quenched by adding 1 N NaOH (10 mL), and the product was extracted with CH2Cl2. The combined organic layers were washed with brine and dried over Na2SO4. The solvent was evaporated, and the crude product was purified by column chromatography (10% MeOH in CH2Cl2) to give compound 33 as a white powder (83 mg, 55% yield). 1H NMR (400 MHz, CDCl3) δ 8.15 (s, 1H), 7.69 (s, 1H), 7.67 (s, 1H), 7.53 (d, J = 7.1 Hz, 2H), 7.39–7.09 (m, 32H), 6.61 (s, 1H), 5.95 (d, J = 1.9 Hz, 1H), 5.65 (s, 2H), 5.36 (m, 1H), 4.89 (m, 1H), 4.40–4.34 (m, 1H), 3.56 (d, J = 3.4 Hz, 2H), 2.68 (d, J = 6.8 Hz, 2H), 2.46 (m, 2H), 2.26 (m, 2H), 1.81–1.69 (m, 2H), 1.52 (s, 3H), 1.30 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.5, 166.7, 155.4, 152.9, 149.0, 144.8, 144.7, 140.0, 139.9, 135.3, 131.5, 128.8, 128.7, 128.0, 127.9, 127.0, 126.9, 125.3, 114.1, 90.8, 85.7, 83.8, 83.4, 70.7, 70.4, 58.6, 56.0, 53.5, 34.9, 27.0, 25.3, 22.7. HRMS (ESI): calcd for C63H60N8O5 [M + H]+ 1009.4765, found 1009.4765.
3-(((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(5-oxo-5-(tritylamino)pentyl)amino)methyl)-N-tritylbenzamide (34)
Following the procedure described for compound 33, coupling compound 29 (100 mg, 0.15 mmol) with 5-oxo-N-tritylpentanamide 17 (64 mg, 0.18 mmol) afforded compound 34 as a white powder (88 mg, 57% yield). 1H NMR (400 MHz, CDCl3) δ 8.16 (s, 1H), 7.67 (s, 2H), 7.57 (br, 1H), 7.52 (s, 1H), 7.41–7.13 (m, 32H), 6.62 (s, 1H), 5.96 (d, J = 1.8 Hz, 1H), 5.83 (br, 2H), 5.38 (m, 1H), 4.92 (m, 1H), 4.40–4.34 (m, 1H), 3.54 (s, 2H), 2.65 (d, J = 6.9 Hz, 2H), 2.46–2.38 (m, 2H), 2.13 (m, 2H), 1.56 (s, 3H), 1.42–1.33 (m, 2H), 1.30 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.6, 166.6, 155.5, 152.9, 149.1, 144.8, 144.8, 140.3, 140.0, 135.2, 131.6, 128.8, 128.7, 128.2, 128.0, 127.9, 127.5, 127.0, 127.0, 125.4, 114.1, 90.9, 85.9, 83.8, 83.4, 77.3, 70.7, 70.4, 58.6, 56.1, 53.9, 37.1, 27.1, 26.3, 25.4, 23.1. HRMS (ESI): calcd for C64H62N8O5 [M + H]+ 1023.4921, found 1023.4918.
tert-Butyl 4-((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro-[3,4-d][1,3]dioxol-4-yl)methyl)(3-(tritylcarbamoyl)benzyl)amino)butanoate (35)
Following the procedure described for compound 33, coupling tert-butyl 4-oxobutanoate 22 (29 mg, 0.18 mmol) and compound 29 (100 mg, 0.15 mmol) afforded compound 35 as a white powder (61 mg, 49% yield). 1H NMR (400 MHz, CDCl3) δ 8.16 (s, 1H), 7.70 (d, J = 5.8 Hz, 2H), 7.58 (d, J = 7.6 Hz, 2H), 7.38–7.15 (m, 17H), 5.97 (d, J = 2.0 Hz, 3H), 5.36 (m, 1H), 4.93 (m, 1H), 4.35 (m, 1H), 3.63–3.52 (m, 2H), 2.76–2.63 (m, 2H), 2.47 (t, J = 7.1 Hz, 2H), 2.23–2.12 (m, 2H), 1.75–1.65 (m, 2H), 1.55 (s, 3H), 1.36 (s, 9H), 1.29 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 172.7, 166.7, 155.5, 152.9, 149.0, 144.8, 144.8, 140.0, 139.9, 131.6, 128.8, 128.7, 128.0, 128.0, 127.0, 125.5, 120.1, 90.8, 85.8, 83.9, 83.4, 80.1, 70.7, 61.9, 58.8, 55.9, 53.4, 33.0, 32.4, 28.0, 27.1, 25.3, 22.4. HRMS (ESI): calcd for C48H53N7O6 [M + H]+ 824.4136, found 824.4142.
tert-Butyl 5-((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro-[3,4-d][1,3]dioxol-4-yl)methyl)(3-(tritylcarbamoyl)benzyl)amino)pentanoate (36)
Following the procedure described for compound 33, coupling tert-butyl 5-oxopentanoate 23 (31 mg, 0.18 mmol) and compound 29 (100 mg, 0.15 mmol) afforded compound 36 as a white powder (67 mg, 53% yield). 1H NMR (400 MHz, CDCl3) δ 8.17 (s, 1H), 7.70 (d, J = 8.7 Hz, 2H), 7.58 (d, J = 7.8 Hz, 1H), 7.51 (s, 1H), 7.42–7.27 (m, 14H), 7.19 (t, J = 7.7 Hz, 1H), 5.98 (s, 1H), 5.72 (s, 2H), 5.38 (m, 1H), 4.94 (s, 1H), 4.40–4.33 (m, 1H), 3.57 (s, 2H), 2.70–2.65 (m, 2H), 2.47 (t, J = 7.0 Hz, 2H), 2.13 (t, J = 7.2 Hz, 2H), 1.57 (s, 3H), 1.52–1.45 (m, 2H), 1.41 (s, 9H), 1.32 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 172.9, 166.6, 155.4, 152.9, 149.1, 144.8, 140.2, 139.9, 135.2, 131.6, 128.7, 128.2, 128.0, 127.4, 127.0, 125.4, 120.2, 114.1, 90.8, 85.8, 83.9, 83.4, 80.0, 70.7, 58.7, 56.0, 54.0, 35.2, 28.1, 27.1, 26.3, 25.3, 22.7. HRMS (ESI): calcd for C49H55N7O5 [M + H]+ 838.4292, found 838.4298.
tert-Butyl (S)-4-((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(3-(tritylcarbamoyl)benzyl)amino)-2-((tert-butoxycarbonyl)amino)butanoate (37)
Following the procedure described for compound 33, coupling tert-butyl (R)-2-((tert-butoxycarbonyl)amino)-4-oxobutanoate 27 (49 mg, 0.18 mmol) and compound 29 (100 mg, 0.15 mmol) afforded compound 37 as a white powder (94 mg, 67% yield). 1H NMR (400 MHz, CDCl3) δ 8.15 (s, 1H), 7.70 (m, 2H), 7.57 (d, J = 9.9 Hz, 2H), 7.41–7.14 (m, 15H), 5.98 (s, 1H), 5.59 (s, 2H), 5.37 (m, 2H), 4.91 (s, 1H), 4.36 (s, 1H), 4.17 (s, 1H), 3.62 (d, J = 13.8 Hz, 1H), 3.54 (d, J = 13.8 Hz, 1H), 2.76–2.48 (m, 4H), 1.99 (d, J = 6.2 Hz, 1H), 1.76 (br, 1H), 1.57 (s, 3H), 1.39 (m, 15H), 1.32 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.6, 166.6, 155.4, 152.9, 149.0, 144.8, 140.0, 139.5, 135.4, 131.6, 128.8, 128.0, 127.0, 125.5, 114.2, 90.7, 85.6, 83.9, 83.4, 81.7, 79.4, 70.7, 58.9, 58.2, 55.9, 52.7, 50.9, 50.6, 36.5, 29.7, 28.3, 28.2, 28.0, 27.9, 27.1, 25.3. HRMS (ESI): calcd for C53H61N8O8 [M + H]+ 939.4749, found 939.4784.
3-(((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(isopropyl)amino)methyl)-N-tritylbenzamide (38)
Following the procedure described for compound 33, coupling 5 mL of dry acetone (large excess) and compound 29 (100 mg, 0.15 mmol) afforded compound 38 as a white powder (68 mg, 63% yield). 1H NMR (400 MHz, CDCl3) δ 8.22 (s, 1H), 7.75 (s, 1H), 7.60 (d, J = 10.4 Hz, 2H), 7.49–7.40 (m, 2H), 7.33–7.21 (m, 15H), 5.91 (s, 1H), 5.34 (m, 2H), 4.92–4.87 (m, 1H), 4.23 (d, J = 3.2 Hz, 1H), 3.57 (s, 2H), 2.92–2.83 (m, 1H), 2.76–2.68 (m, 1H), 2.59 (m, 1H), 1.49 (s, 3H), 1.25 (s, 3H), 1.01 (d, J = 6.6 Hz, 3H), 0.90 (d, J = 6.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 166.7, 155.7, 152.9, 149.0, 144.8, 141.4, 139.9, 135.2, 131.4, 128.7, 128.3, 128.0, 127.1, 125.4, 120.2, 114.0, 90.9, 86.5, 83.7, 83.3, 70.7, 54.4, 51.8, 50.5, 27.1, 25.4, 18.7, 17.2. HRMS (ESI): calcd for C43H45N7O4 [M + H]+ 724.3611, found 724.3618.
Methyl 3-(((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro-[3,4-d][1,3]dioxol-4-yl)methyl)(4-oxo-4-(tritylamino)butyl)amino)methyl)benzoate (39)
Following the procedure described for compound 33, coupling 4-oxo-N-tritylbutanamide 16 (62 mg, 0.18 mmol) and compound 30 (68 mg, 0.15 mmol) afforded compound 39 as a white powder (63 mg, 54% yield). 1H NMR (400 MHz, CDCl3) δ 8.17 (s, 1H), 7.90 (s, 1H), 7.84–7.79 (m, 1H), 7.77 (s, 1H), 7.40 (d, J = 7.7 Hz, 1H), 7.31–7.08 (m, 17H), 6.60 (s, 1H), 5.97 (d, J = 2.2 Hz, 1H), 5.67 (s, 2H), 5.34 (m, 1H), 4.86 (m, 1H), 4.34 (m, 1H), 3.85 (s, 3H), 3.57 (m, 2H), 2.74–2.62 (m, 2H), 2.45 (t, J = 7.0 Hz, 2H), 2.31–2.16 (m, 2H), 1.75 (m, 2H), 1.53 (s, 3H), 1.32 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.5, 167.0, 155.4, 153.0, 149.1, 144.7, 139.9, 139.6, 133.3, 129.9, 129.7, 128.7, 128.2, 128.1, 127.9, 127.0, 126.9, 120.2, 114.2, 90.8, 85.4, 83.7, 83.4, 70.4, 58.5, 55.7, 53.6, 52.1, 34.9, 27.0, 25.2, 22.7. HRMS (ESI): calcd for C45H48N7O6 [M + H]+ 782.3666, found 782.3666.
Methyl 3-(((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro-[3,4-d][1,3]dioxol-4-yl)methyl)(5-oxo-5-(tritylamino)pentyl)amino)methyl)benzoate (40)
Following the procedure described for compound 33, coupling 5-oxo-N-tritylpentanamide 17 (64 mg, 0.18 mmol) and compound 30 (68 mg, 0.15 mmol) afforded compound 40 as a white powder (67 mg, 53% yield). 1H NMR (400 MHz, CDCl3) δ 8.17 (s, 1H), 7.89 (s, 1H), 7.83 (d, J = 7.8 Hz, 1H), 7.76 (s, 1H), 7.44 (d, J = 7.6 Hz, 1H), 7.26–7.17 (m, 14H), 6.69 (s, 1H), 6.35 (br, 2H), 5.99 (d, J = 1.8 Hz, 1H), 5.39 (m, 1H), 4.90 (m, 1H), 4.36 (m, 1H), 3.83 (s, 3H), 3.62–3.48 (m, 2H), 2.65 (m, 2H), 2.41 (t, J = 6.9 Hz, 2H), 2.14 (p, J = 8.2 Hz, 2H), 1.56 (s, 3H), 1.33 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.7, 167.1, 155.8, 152.9, 149.1, 144.8, 139.9, 139.8, 133.4, 129.9, 129.8, 128.7, 128.2, 128.2, 127.9, 126.9, 114.1, 90.8, 85.6, 83.7, 83.4, 70.4, 58.5, 55.8, 53.8, 52.1, 37.1, 27.1, 26.3, 25.3, 23.1. HRMS (ESI): calcd for C46H49N7O6 [M + H]+ 796.3823, found 796.3814.
Methyl 3-(((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro-[3,4-d][1,3]dioxol-4-yl)methyl)(4-(tert-butoxy)-4-oxobutyl)amino)methyl)benzoate (41)
Following the procedure described for compound 33, coupling tert-butyl 4-oxobutanoate 22 (29 mg, 0.18 mmol) and compound 30 (68 mg, 0.15 mmol) afforded compound 41 as a white powder (65 mg, 73% yield). 1H NMR (400 MHz, CDCl3) δ 8.13 (s, 1H), 7.85 (s, 1H), 7.80 (d, J = 8.1 Hz, 2H), 7.41 (d, J = 7.6 Hz, 1H), 7.23 (t, J = 7.6 Hz, 1H), 6.47 (s, 2H), 5.98 (d, J = 1.9 Hz, 1H), 5.33 (d, J = 6.4 Hz, 1H), 4.87 (m, 1H), 4.30 (m, 1H), 3.84 (s, 3H), 3.61–3.48 (m, 2H), 2.75–2.69 (m, 2H), 2.43 (m, 2H), 2.16–2.10 (m, 2H), 1.53 (s, 3H), 1.39–1.30 (m, 15H). 13C NMR (101 MHz, CDCl3) δ 172.8, 167.0, 155.8, 152.9, 148.9, 139.7, 139.6, 133.3, 129.9, 129.7, 128.2, 128.1, 120.0, 114.1, 114.1, 90.7, 85.4, 83.7, 83.3, 80.0, 61.6, 61.1, 58.6, 55.6, 53.4, 52.0, 32.9, 32.3, 28.0, 27.5, 27.0, 25.3, 22.3. HRMS (ESI): calcd for C30H40N6O7 [M + H]+ 597.3037, found 597.3037.
Methyl 3-(((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro-[3,4-d][1,3]dioxol-4-yl)methyl)(5-(tert-butoxy)-5-oxopentyl)amino)methyl)benzoate (42)
Following the procedure described for compound 33, coupling tert-butyl 5-oxopentanoate 23 (31 mg, 0.18 mmol) and compound 30 (68 mg, 0.15 mmol) afforded compound 42 as a white powder (56 mg, 61% yield). 1H NMR (400 MHz, CDCl3) δ 8.12 (s, 1H), 7.84 (s, 1H), 7.79 (d, J = 7.8 Hz, 2H), 7.39 (d, J = 7.5 Hz, 1H), 7.21 (t, J = 7.7 Hz, 1H), 6.52 (s, 2H), 5.97 (s, 1H), 5.36–5.30 (m, 1H), 4.86 (m, 1H), 4.33–4.26 (m, 1H), 3.82 (s, 3H), 3.58 (d, J = 13.8 Hz, 1H), 3.47 (d, J = 13.7 Hz, 1H), 2.69–2.56 (m, 2H), 2.43–2.35 (m, 2H), 2.07 (t, J = 6.8 Hz, 2H), 1.52 (s, 3H), 1.48–1.42 (m, 2H), 1.34 (s, 9H), 1.30 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 172.8, 167.0, 155.8, 152.9, 149.0, 139.7, 139.7, 133.3, 129.9, 129.7, 128.2, 128.1, 128.1, 120.1, 114.1, 90.8, 85.5, 83.6, 83.5, 83.3, 79.9, 58.6, 55.6, 53.9, 52.0, 35.2, 28.0, 27.1, 26.2, 25.3, 22.7. HRMS (ESI): calcd for C31H42N6O7 [M + H]+ 611.3193, found 611.3182.
Methyl 3-(((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro-[3,4-d][1,3]dioxol-4-yl)methyl)((S)-4-(tert-butoxy)-2-((tert-butoxycarbonyl)amino)-4-oxobutyl)amino)methyl)benzoate (43)
Following the procedure described for compound 33, coupling tert-butyl (R)-2-((tert-butoxycarbonyl)amino)-4-oxobutanoate 27 (49 mg, 0.18 mmol) and compound 30 (68 mg, 0.15 mmol) afforded compound 43 as a white powder (62 mg, 58% yield). 1H NMR (400 MHz, CDCl3) δ 8.19 (s, 1H), 7.87 (d, J = 6.5 Hz, 2H), 7.82 (s, 1H), 7.48 (d, J = 7.7 Hz, 1H), 7.30 (t, J = 7.9 Hz, 1H), 6.01 (s, 1H), 5.73 (s, 2H), 5.38 (m, 2H), 4.89 (m, 1H), 4.35 (m, 1H), 4.20–4.11 (m, 1H), 3.90 (s, 3H), 3.71–3.52 (m, 2H), 2.78 (m, 1H), 2.65 (m, 2H), 2.51 (m, 1H), 1.96 (s, 2H), 1.76 (m, 1H), 1.59 (s, 3H), 1.40 (m, 18H), 1.37 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.6, 167.0, 155.4, 155.4, 153.0, 149.1, 139.9, 139.2, 133.4, 130.0, 129.8, 128.4, 128.3, 120.3, 114.3, 90.7, 85.3, 83.8, 83.3, 81.7, 79.4, 58.6, 55.7, 52.7, 52.1, 50.5, 29.5, 28.3, 27.9, 27.1, 25.3. HRMS (ESI): calcd for C35H49N7O9 [M + H]+ 712.3670, found 712.3682.
Methyl 3-(((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro-[3,4-d][1,3]dioxol-4-yl)methyl)(isopropyl)amino)methyl)benzoate (44)
Following the procedure described for compound 33, coupling dry acetone (5 mL, large excess) and compound 30 (68 mg, 0.15 mmol) afforded compound 44 as a white powder (42 mg, 57% yield). 1H NMR (400 MHz, CDCl3) δ 8.22 (s, 1H), 7.94 (s, 1H), 7.84 (d, J = 7.7 Hz, 1H), 7.77 (s, 1H), 7.52 (d, J = 7.6 Hz, 1H), 7.29 (t, J = 7.7 Hz, 1H), 5.96 (d, J = 2.4 Hz, 2H), 5.36 (dd, J = 6.4, 2.4 Hz, 1H), 4.87 (dd, J = 6.4, 3.0 Hz, 1H), 4.26–4.20 (m, 1H), 3.88 (s, 3H), 3.62 (d, J = 14.2 Hz, 1H), 3.54 (d, J = 14.2 Hz, 1H), 2.88 (p, J = 6.6 Hz, 1H), 2.73–2.59 (m, 2H), 1.53 (s, 3H), 1.33 (s, 3H). 13C NMR (101 MHz, chloroform-d) δ 167.1, 155.6, 152.9, 149.2, 141.0, 139.9, 133.2, 129.9, 129.6, 128.1, 128.1, 120.2, 114.0, 91.0, 86.1, 83.5, 83.2, 60.3, 54.3, 52.0, 51.4, 50.3, 27.1, 25.3, 21.0, 18.9, 16.8, 14.2. HRMS (ESI): calcd for C35H32N6O5 [M + H]+ 497.2512, found 497.2510.
tert-Butyl 3-(((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro-[3,4-d][1,3]dioxol-4-yl)methyl)(4-oxo-4-(tritylamino)butyl)amino)methyl)benzoate (45)
Following the procedure described for compound 33, coupling 4-oxo-N-tritylbutanamide 16 (62 mg, 0.18 mmol) and compound 31 (75 mg, 0.15 mmol) afforded compound 45 as a white powder (93 mg, 75% yield). 1H NMR (400 MHz, CDCl3) δ 8.23 (s, 1H), 7.90 (s, 1H), 7.84–7.79 (m, 2H), 7.43 (d, J = 7.6 Hz, 1H), 7.31–7.16 (m, 17H), 6.68 (s, 1H), 6.07 (s, 2H), 6.01 (d, J = 2.2 Hz, 1H), 5.39 (m, 1H), 4.89 (m, 1H), 4.37 (m, 1H), 3.66 (d, J = 13.9 Hz, 1H), 3.55 (d, J = 13.9 Hz, 1H), 2.76 (m, 1H), 2.66 (m, 1H), 2.47 (t, J = 6.8 Hz, 2H), 2.28 (m, 2H), 1.77 (m, 2H), 1.59 (s, 9H), 1.56 (s, 3H), 1.35 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.6, 165.7, 155.6, 153.0, 149.1, 144.8, 139.7, 139.4, 132.8, 131.9, 129.6, 128.7, 128.1, 128.0, 127.9, 126.9, 120.2, 114.2, 90.8, 85.4, 83.6, 83.4, 80.9, 70.4, 58.6, 55.7, 53.6, 34.9, 28.2, 27.1, 25.3, 22.7. HRMS (ESI): calcd for C49H53N7O6 [M + H]+ 824.4136, found 824.4123.
tert-Butyl 3-(((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro-[3,4-d][1,3]dioxol-4-yl)methyl)(5-oxo-5-(tritylamino)pentyl)amino)methyl)benzoate (46)
Following the procedure described for compound 33, coupling 5-oxo-N-tritylpentanamide 17 (64 mg, 0.18 mmol) and compound 31 (75 mg, 0.15 mmol) afforded compound 46 as a white powder (97 mg, 77% yield). 1H NMR (400 MHz, CDCl3) δ 8.23 (d, J = 7.6 Hz, 1H), 7.90 (d, J = 9.6 Hz, 1H), 7.85–7.79 (m, 2H), 7.47 (d, J = 7.7 Hz, 1H), 7.32–7.15 (m, 16H), 6.71 (d, J = 8.4 Hz, 1H), 6.35 (d, J = 14.9 Hz, 2H), 6.03 (d, J = 2.1 Hz, 1H), 5.43 (m, 1H), 4.93 (m, 1H), 4.41–4.37 (m, 1H), 3.65 (d, J = 13.8 Hz, 1H), 3.54 (d, J = 13.8 Hz, 1H), 2.75–2.62 (m, 1H), 2.48–2.39 (m, 2H), 2.16 (t, J = 7.2 Hz, 2H), 1.59 (s, 12H), 1.45–1.37 (m, 2H), 1.36 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.7, 165.8, 155.8, 153.0, 149.1, 144.8, 144.8, 139.9, 139.6, 132.9, 131.8, 129.7, 128.7, 128.2, 128.1, 128.0, 128.0, 127.9, 126.9, 120.2, 114.1, 90.9, 90.8, 85.6, 83.7, 83.6, 83.4, 80.9, 70.4, 58.6, 58.6, 55.7, 53.8, 53.6, 37.1, 34.9, 28.2, 27.1, 27.1, 26.3, 25.3, 25.3, 23.1, 22.7. HRMS (ESI): calcd for C49H55N7O6 [M + H]+ 838.4292, found 838.4314.
tert-Butyl 3-(((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro-[3,4-d][1,3]dioxol-4-yl)methyl)(4-(tert-butoxy)-4-oxobutyl)amino)methyl)benzoate (47)
Following the procedure described for compound 33, coupling tert-butyl 4-oxobutanoate 22 (29 mg, 0.18 mmol) and compound 31 (75 mg, 0.15 mmol) afforded compound 47 as a white powder (64 mg, 67% yield). 1H NMR (400 MHz, CDCl3) δ 8.21 (s, 1H), 7.86–7.77 (m, 3H), 7.42 (d, J = 7.6 Hz, 1H), 7.26 (d, J = 7.7 Hz, 1H), 6.15 (s, 2H), 5.99 (d, J = 2.2 Hz, 1H), 5.36 (m, 1H), 4.88 (m, 1H), 4.32 (m, 1H), 3.65 (d, J = 13.8 Hz, 1H), 3.50 (d, J = 13.8 Hz, 1H), 2.78–2.73 (m, 1H), 2.64–2.59 (m,1H), 2.42 (m, 2H), 2.22–2.09 (m, 2H), 1.55 (s, 12H), 1.36 (s, 9H), 1.33 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 172.8, 165.7, 155.7, 153.0, 149.1, 139.8, 139.5, 132.8, 131.8, 129.6, 128.1, 128.0, 120.2, 114.2, 90.8, 85.5, 83.7, 83.3, 80.8, 80.0, 58.7, 55.6, 53.4, 33.0, 28.2, 28.0, 27.1, 25.3, 22.4. HRMS (ESI): calcd for C33H46N6O7 [M + H]+ 639.3506, found 639.3506.
tert-Butyl 3-(((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro-[3,4-d][1,3]dioxol-4-yl)methyl)(5-(tert-butoxy)-5-oxopentyl)amino)methyl)benzoate (48)
Following the procedure described for compound 33, coupling tert-butyl 5-oxopentanoate 23 (31 mg, 0.18 mmol) and compound 31 (75 mg, 0.15 mmol) afforded compound 48 as a white powder (72 mg, 73% yield). 1H NMR (400 MHz, CDCl3) δ 8.20 (s, 1H), 7.84–7.77 (m, 3H), 7.42 (d, J = 7.6 Hz, 1H), 7.24 (t, J = 7.6 Hz, 1H), 6.19 (s, 2H), 5.99 (d, J = 2.2 Hz, 1H), 5.37 (m, 1H), 4.88 (m, 1H), 4.35–4.30 (m, 1H), 3.65–3.48 (1H), 2.71–2.59 (m, 1H), 2.46–2.38 (m, 2H), 2.10 (t, J = 7.1 Hz, 2H), 1.55 (s, 12H), 1.44 (m, 2H), 1.37 (s, 9H), 1.33 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 172.9, 165.7, 155.7, 153.0, 149.1, 139.7, 139.5, 132.9, 131.8, 129.6, 128.0, 128.0, 120.2, 114.1, 90.8, 85.5, 83.6, 83.3, 80.8, 79.9, 58.7, 55.6, 53.9, 35.2, 28.2, 28.1, 27.1, 26.2, 25.3, 22.7. HRMS (ESI): calcd for C34H48N6O7 [M + H]+ 653.3663, found 653.3669.
tert-Butyl 3-(((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro-[3,4-d][1,3]dioxol-4-yl)methyl)((S)-4-(tert-butoxy)-3-((tert-butoxycarbonyl)amino)-4-oxobutyl)amino)methyl)benzoate (49)
Following the procedure described for compound 33, coupling tert-butyl (R)-2-((tert-butoxycarbonyl)amino)-4-oxobutanoate 27 (49 mg, 0.18 mmol) and compound 31 (75 mg, 0.15 mmol) afforded compound 49 as a white powder (85 mg, 75% yield). 1H NMR (400 MHz, CDCl3) δ 8.18 (s, 1H), 7.79 (d, J = 6.7 Hz, 3H), 7.44 (s, 1H), 7.28–7.23 (m, 1H), 6.20 (s, 2H), 5.99 (s, 1H), 5.50–5.43 (m, 1H), 5.34 (d, J = 5.6 Hz, 1H), 4.86 (m, 1H), 4.31 (m, 1H), 4.15–4.07 (m, 1H), 3.67 (br, 1H), 3.47 (br, 1H), 2.76 (br, 2H), 2.59 (m, 2H), 2.44 (m, 2H), 1.93 (m, 1H), 1.73 (m, 1H), 1.54 (s, 12H), 1.35 (m, 21H). 13C NMR (101 MHz, CDCl3) δ 171.6, 165.6, 155.7, 155.7, 155.3, 153.0, 149.1, 139.8, 139.0, 132.9, 131.8, 129.6, 128.2, 128.2, 120.2, 114.3, 90.6, 85.3, 83.7, 83.3, 81.6, 80.9, 79.3, 58.8, 55.7, 52.7, 50.5, 29.4, 28.3, 28.2, 27.9, 27.1, 25.4. HRMS (ESI): calcd for C38H55N7O9 [M + H]+ 754.4140, found 754.4129.
tert-Butyl 3-(((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(isopropyl)amino)methyl)benzoate (50)
Following the procedure described for compound 33, coupling 5 mL of dry acetone (large excess) and compound 31 (75 mg, 0.15 mmol) afforded compound 50 as a white powder (85 mg, 79% yield). 1H NMR (400 MHz, CDCl3) δ 8.26 (s, 1H), 7.90 (s, 1H), 7.80 (m, 2H), 7.51 (d, J = 7.6 Hz, 1H), 7.28 (t, J = 7.7 Hz, 1H), 5.97 (d, J = 2.4 Hz, 1H), 5.92 (s, 2H), 5.37 (m, 1H), 4.86 (m, 1H), 4.26–4.20 (m, 1H), 3.64 (br, 1H), 3.54 (br, 1H), 2.87 (m, 1H), 2.73–2.56 (br, 2H), 1.57 (s, 9H), 1.53 (s, 3H), 1.33 (s, 3H), 1.03 (d, J = 6.6 Hz, 3H), 0.89 (d, J = 6.5 Hz, 3H). 13C NMR (101 MHz, CDCl3-d) δ 165.8, 155.5, 153.0, 149.2, 140.8, 139.9, 132.7, 131.8, 129.4, 128.0, 127.9, 120.2, 114.0, 91.0, 86.1, 83.4, 83.2, 80.8, 54.5, 51.3, 50.4, 28.2, 27.1, 25.3, 19.0, 16.7. HRMS (ESI): calcd for C28H38N6O5 [M + H]+ 539.2982, found 539.2982.
4-((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(naphthalen-2-ylmethyl)amino)-N-tritylbutanamide (51)
Following the procedure described for compound 33, coupling 4-oxo-N-tritylbutanamide 16 (62 mg, 0.18 mmol) and compound 32 (67 mg, 0.15 mmol) afforded compound 51 as a white powder (85 mg, 73% yield). 1H NMR (400 MHz, CDCl3) δ 8.10 (s, 1H), 7.79–7.73 (m, 2H), 7.72–7.66 (m, 2H), 7.61 (s, 1H), 7.46–7.38 (m, 3H), 7.28–7.12 (m, 15H), 6.59 (s, 1H), 5.97 (d, J = 2.2 Hz, 1H), 5.81 (s, 2H), 5.30 (m, 1H), 4.83 (m, 1H), 4.38 (s, 1H), 3.77 (d, J = 13.7 Hz, 1H), 3.63 (d, J = 13.7 Hz, 1H), 2.78–2.64 (m, 2H), 2.51 (t, J = 6.9 Hz, 2H), 2.30–2.20 (m, 2H), 1.79 (m, 2H), 1.52 (s, 3H), 1.31 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.6, 155.5, 153.0, 144.8, 139.7, 136.6, 133.2, 132.7, 128.7, 127.9, 127.8, 127.6, 127.6, 127.4, 127.2, 126.9, 126.0, 125.6, 114.2, 90.8, 85.4, 83.7, 83.4, 70.4, 59.1, 55.8, 53.8, 35.0, 27.0, 25.3, 22.8. HRMS (ESI): calcd for C47H47N7O4 [M + H]+ 774.3768, found 774.3769.
5-((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(naphthalen-2-ylmethyl)amino)-N-tritylpentanamide (52)
Following the procedure described for compound 33, coupling 5-oxo-N-tritylpentanamide 17 (64 mg, 0.18 mmol) and compound 32 (67 mg, 0.15 mmol) afforded compound 52 as a white powder (85 mg, 64% yield). 1H NMR (400 MHz, CDCl3) δ 8.12 (s, 1H), 7.84–7.55 (m, 5H), 7.42 (m, 3H), 7.28–7.16 (m, 14H), 6.57 (s, 1H), 5.98 (s, 1H), 5.76 (s, 2H), 5.35 (d, J = 6.3 Hz, 1H), 4.88 (d, J = 6.2 Hz, 1H), 4.39 (s, 1H), 3.75 (d, J = 13.6 Hz, 1H), 3.62 (d, J = 13.6 Hz, 1H), 2.77–2.62 (m, 2H), 2.52–2.37 (m, 2H), 2.16–2.09 (m, 2H), 1.56 (s, 3H), 1.45–1.38 (m, 2H), 1.32 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.7, 155.4, 152.9, 149.2, 144.8, 139.9, 136.8, 133.2, 132.7, 128.7, 128.4, 127.9, 127.8, 127.6, 127.4, 127.2, 127.0, 125.9, 125.5, 120.2, 114.1, 90.9, 85.7, 83.7, 83.5, 70.4, 59.1, 55.8, 53.8, 37.2, 27.1, 26.3, 25.3, 23.2. HRMS (ESI): calcd for C48H49N7O4 [M + H]+ 788.3924, found 788.3932.
tert-Butyl 4-((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro-[3,4-d][1,3]dioxol-4-yl)methyl)(naphthalen-2-ylmethyl)amino)butanoate (53)
Following the procedure described for compound 33, coupling tert-butyl 4-oxobutanoate 22 (29 mg, 0.18 mmol) and compound 32 (67 mg, 0.15 mmol) afforded compound 53 as a white powder (67 mg, 76% yield). 1H NMR (400 MHz, CDCl3) δ 8.12 (s, 1H), 7.82 (s, 1H), 7.78–7.60 (m, 4H), 7.48–7.35 (m, 3H), 6.74 (s, 2H), 6.02 (s, 1H), 5.34 (m, 1H), 4.89 (m, 1H), 4.38 (m, 1H), 3.79 (d, J = 13.6 Hz, 1H), 3.60 (d, J = 13.6 Hz, 1H), 2.80 (m, 1H), 2.65 (m, 1H), 2.57–2.44 (m, 2H), 2.29–2.13 (m, 2H), 1.75 (p, J = 7.3 Hz, 2H), 1.57 (s, 3H), 1.40–1.08 (m, 12H). 13C NMR (101 MHz, CDCl3) δ 172.9, 156.0, 152.9, 152.9, 149.0, 139.6, 136.7, 133.2, 132.7, 127.8, 127.6, 127.3, 127.2, 125.9, 125.5, 120.1, 114.1, 90.8, 85.5, 83.7, 83.5, 80.0, 59.2, 55.7, 53.6, 33.0, 28.0, 27.5, 27.1, 25.4, 22.4. HRMS (ESI): calcd for C32H40N6O5 [M + H]+ 589.3138, found 589.3143.
tert-Butyl 5-((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro-[3,4-d][1,3]dioxol-4-yl)methyl)(naphthalen-2-ylmethyl)amino)pentanoate (54)
Following the procedure described for compound 33, coupling tert-butyl 5-oxopentanoate 23 (31 mg, 0.18 mmol) and compound 32 (67 mg, 0.15 mmol) afforded compound 54 as a white powder (62 mg, 69% yield). 1H NMR (400 MHz, CDCl3) δ 8.11 (s, 1H), 7.80 (d, J = 24.4 Hz, 2H), 7.73 (d, J = 8.4 Hz, 2H), 7.64 (s, 1H), 7.47–7.38 (m, 3H), 6.42 (s, 2H), 6.02 (s, 1H), 5.34 (d, J = 6.3 Hz, 1H), 4.90 (m, 1H), 4.42–4.36 (m, 1H), 3.80 (d, J = 13.6 Hz, 1H), 3.62 (d, J = 13.6 Hz, 1H), 2.77 (m, 1H), 2.70–2.62 (m, 1H), 2.54 (s, 2H), 2.15 (t, J = 6.7 Hz, 2H), 1.58 (s, 3H), 1.48 (d, J = 9.8 Hz, 2H), 1.41 (s, 9H), 1.35 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 172.9, 155.8, 152.9, 149.1, 139.7, 136.8, 133.2, 132.7, 127.7, 127.6, 127.5, 127.3, 127.2, 125.9, 125.5, 120.2, 114.1, 90.9, 85.5, 83.7, 83.6, 83.4, 79.9, 59.1, 55.7, 54.1, 35.3, 28.1, 28.0, 27.1, 26.3, 25.3, 22.8. HRMS (ESI): calcd for C33H42N6O5 [M + H]+ 603.3295, found 603.3311.
tert-Butyl (R)-4-((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro-[3,4-d][1,3]dioxol-4-yl)methyl)(naphthalen-2-ylmethyl)amino)-2-((tert-butoxycarbonyl)amino)butanoate (55)
Following the procedure described for compound 33, coupling tert-butyl (R)-2-((tert-butoxycarbonyl)amino)-4-oxobutanoate 27 (49 mg, 0.18 mmol) and compound 32 (67 mg, 0.15 mmol) afforded compound 55 as a white powder (72 mg, 68% yield). 1H NMR (400 MHz, CDCl3) δ 8.08 (s, 1H), 7.87–7.67 (m, 4H), 7.61 (s, 1H), 7.54–7.39 (m, 3H), 6.27 (d, J = 11.3 Hz, 2H), 6.00 (s, 1H), 5.71–5.61 (m, 1H), 5.30 (d, J = 5.1 Hz, 1H), 4.84 (m, 1H), 4.39–4.34 (m, 1H), 4.23–4.14 (m, 1H), 3.84 (d, J = 13.5 Hz, 1H), 3.59 (d, J = 13.5 Hz, 1H), 2.82 (br, 2H), 2.65 (br, 2H), 2.57–2.51 (m, 1H), 2.06–1.94 (m, 1H), 1.86–1.78 (m, 1H), 1.57 (s, 3H), 1.50–1.18 (m, 21H). 13C NMR (101 MHz, CDCl3) δ 171.7, 155.4, 152.9, 149.0, 139.7, 136.2, 133.2, 132.7, 127.9, 127.6, 127.6, 127.5, 127.2, 127.1, 125.9, 125.6, 120.1, 114.3, 90.7, 85.3, 83.7, 83.4, 81.6, 79.3, 59.2, 55.7, 52.9, 50.8, 29.4, 28.3, 27.9, 27.9, 27.1, 25.4. HRMS (ESI): calcd for C33H42N6O5 [M + H]+ 704.3772, found 704.3777.
9-((3aR,4R,6R,6aR)-6-((Isopropyl(naphthalen-2-ylmethyl)amino)methyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-9H-purin-6-amine (56)
Following the procedure described for compound 33, coupling 5 mL of dry acetone (large excess) and compound 32 (67 mg, 0.15 mmol) afforded compound 56 as a white powder (35 mg, 48% yield). 1H NMR (400 MHz, CDCl3) δ 8.21 (s, 1H), 7.83–7.68 (m, 5H), 7.53 (m, 1H), 7.47–7.39 (m, 2H), 6.26 (s, 2H), 5.98 (d, J = 2.3 Hz, 1H), 5.32 (m, 1H), 4.85 (m, 1H), 4.32–4.27 (m, 1H), 3.79 (d, J = 13.9 Hz, 1H), 3.64 (d, J = 14.0 Hz, 1H), 3.00–2.93 (m, 1H), 2.78 (m 1H), 2.64 (m, 1H), 1.53 (s, 3H), 1.31 (s, 3H), 1.09 (d, J = 6.6 Hz, 3H), 0.95 (d, J = 6.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 155.7, 152.9, 149.1, 139.7, 138.0, 133.2, 132.7, 127.7, 127.6, 127.5, 127.2, 127.0, 125.9, 125.4, 120.2, 113.9, 91.1, 86.2, 83.5, 83.3, 77.3, 54.9, 51.4, 50.4, 27.0, 25.3, 19.2, 16.6. HRMS (ESI): calcd for C27H32N6O3 [M + H]+ 489.2614, found 489.2611.
3-(((4-Amino-4-oxobutyl)(((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)amino)methyl)benzamide (57)
To a solution of compound 33 (100 mg, 0.098 mmol) in 5 mL of CH2Cl2 was added 5 mL of TFA, and the mixture was stirred at room temperature. After 2 h, 2 mL of H2O was added, and the mixture was stirred for 1 h at room temperature. The mixture was concentrated, and the crude product was purified by preparative HPLC affording compound 57 as a white powder. 1H NMR (400 MHz, D2O) δ 8.46–8.06 (m, 2H), 7.87–7.26 (m, 4H), 6.08 (br, 1H), 4.75–4.36 (m, 4H), 4.27 (br, 1H), 3.84–3.27 (m, 4H), 2.38 (br, 2H), 2.10 (br, 2H). 13C NMR (101 MHz, D2O) δ 177.5, 162.8, 162.5, 149.6, 143.8, 134.8, 134.1, 132.7, 129.6, 129.1, 128.3, 118.9, 117.6, 114.7, 90.4, 77.7, 73.6, 71.5, 57.9, 54.8, 31.8, 19.0. HRMS (ESI): calcd for C22H28N8O5 [M + H]+ 485.2261, found 485.2265.
3-(((5-Amino-5-oxopentyl)(((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxy-tetrahydrofuran-2-yl)methyl)amino)methyl)benzamide (58)
Following the procedure described for compound 57, compound 34 (50 mg, 0.049 mmol) was deprotected to obtain compound 58 as a white powder (16 mg, 56% yield). 1H NMR (400 MHz, D2O) δ 8.43–8.12 (m, 2H), 7.84–7.26 (m, 4H), 6.08 (br, 1H), 4.65–4.21 (m, 5H), 3.63–3.48 (m, 2H), 3.34 (br, 2H), 2.35 (br, 2H), 1.84 (br, 2H), 1.58 (br, 2H). 13C NMR (101 MHz, D2O) δ 177.6, 170.6, 162.8, 149.6, 147.3, 143.8, 134.8, 129.7, 129.6, 129.4, 129.1, 128.3, 117.6, 114.7, 90.5, 77.8, 77.4, 71.6, 71.4, 57.8, 54.6, 32.6, 22.4, 21.0. HRMS (ESI): calcd for C23H30N8O5 [M + H]+ 499.2417, found 499.2420.
4-((((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(3-carbamoylbenzyl)amino)butanoic Acid (59)
Following the procedure described for compound 57, compound 35 (50 mg, 0.060 mmol) was deprotected to obtain compound 59 as a white powder (21 mg, 60% yield). 1H NMR (400 MHz, D2O) δ 8.38–8.06 (m, 2H), 7.71–7.26 (m, 4H), 6.05 (br, 1H), 4.64–4.21 (m, 5H), 3.53 (br, 2H), 3.35 (s, 2H), 2.41 (br, 2H), 2.02 (br, 2H). 13C NMR (101 MHz, D2O) δ 176.4, 170.5, 149.6, 147.3, 143.8, 134.8, 132.7, 129.6, 129.5, 128.3, 117.5, 114.6, 90.4, 77.7, 73.5, 71.4, 57.8, 52.8, 38.6, 30.2, 18.4. HRMS (ESI): calcd for C22H28N7O6 [M + H]+ 486.2101, found 486.2103.
5-((((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(3-carbamoylbenzyl)amino)pentanoic Acid (60)
Following the procedure described for compound 57, compound 36 (50 mg, 0.059 mmol) was deprotected to obtain compound 60 as a white powder (17 mg, 50% yield). 1H NMR (400 MHz, D2O) δ 8.29 (br, 2H), 7.84–7.58 (m, 3H), 7.46 (br, 1H), 6.13 (br, 1H), 4.70–4.33 (m, 5H), 3.66 (br, 2H), 3.48–3.31 (m, 2H), 2.42 (br, 2H), 1.88 (s, 2H), 1.65 (br, 2H). 13C NMR (101 MHz, D2O) δ 177.7, 170.8, 163.0, 150.5, 147.6, 145.1, 143.1, 134.5, 132.9, 129.7, 129.3, 128.4, 119.0, 117.7, 90.3, 77.7, 73.3, 32.8, 22.4, 21.0. HRMS (ESI): calcd for C23H30N7O6 [M + H]+ 500.2258, found 500.2267.
3-(((((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(isopropyl)amino)methyl)benzamide (61)
Following the procedure described for compound 57, compound 38 (50 mg, 0.069 mmol) was deprotected to obtain compound 61 as a white powder (22 mg, 60% yield). 1H NMR (400 MHz, acetone-d6) δ 8.48–8.39 (m, 2H), 8.26 (br, 1H), 7.94 (d, J = 7.8 Hz, 1H), 7.9–7.73 (m, 2H), 7.46 (m, 1H), 6.81 (br, 1H), 6.13 (d, J = 3.4 Hz, 1H), 4.74 (br, 2H), 4.65 (s, 1H), 4.53 (br, 1H), 4.46 (br, 1H), 3.93–3.69 (m, 3H), 3.31 (s, 1H), 1.49–1.45 (m, 6H). 13C NMR (101 MHz, acetone-d6) δ 152.7, 148.4, 146.1, 142.5, 135.0, 134.0, 130.5, 130.2, 119.9, 90.6, 79.3, 73.5, 72.5, 55.6, 54.3, 54.0, 51.4, 16.6, 15.0. HRMS (ESI): calcd for C21H28N7O4 [M + H]+ 442.2203, found 442.2203.
Methyl 3-(((4-Amino-4-oxobutyl)(((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)amino)methyl)benzoate (62)
Following the procedure described for compound 57, compound 39 (50 mg, 0.064 mmol) was deprotected to obtain compound 62 as a white powder (20 mg, 53% yield). 1H NMR (400 MHz, D2O) δ 8.38–7.98 (m, 2H), 7.88–7.50 (m, 3H), 7.35 (br, 1H), 6.05 (br, 1H), 4.64–4.32 (m, 4H), 4.20 (br, 1H), 3.78 (s, 3H), 3.55 (br, 1H), 3.47–3.30 (m, 2H), 2.39 (br, 2H), 2.08 (br, 2H). 13C NMR (101 MHz, D2O) δ 177.5, 167.3, 149.5, 147.2, 143.7, 143.6, 135.8, 134.6, 131.3, 130.7, 129.9, 129.8, 129.4, 129.1, 118.8, 90.6, 77.8, 77.4, 73.8, 73.1, 71.7, 71.4, 57.6, 56.9, 55.2, 54.8, 53.6, 52.7, 31.8, 19.0. HRMS (ESI): calcd for C23H29N7O6 [M + H]+ 500.2258, found 500.2265.
Methyl 3-(((5-Amino-5-oxopentyl)(((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)amino)methyl)benzoate (63)
Following the procedure described for compound 57, compound 40 (50 mg, 0.063 mmol) was deprotected to obtain compound 63 as a white powder (21 mg, 55% yield). 1H NMR (400 MHz, D2O) δ 8.44–8.07 (m, 2H), 7.96–7.34 (m, 4H), 6.12 (br, 1H), 4.50 (br, 4H), 4.32 (s, 1H), 3.86 (s, 3H), 3.62 (br, 1H), 3.52–3.34 (m, 2H), 2.32 (br, 2H), 1.89 (br, 2H), 1.68 (br, 2H). 13C NMR (101 MHz, D2O) δ 178.7, 167.5, 162.6, 149.6, 143.8, 143.6, 135.9, 130.9, 130.0, 129.9, 129.5, 129.1, 117.6, 114.7, 111.8, 71.4, 52.7, 33.9, 22.4, 21.9. HRMS (ESI): calcd for C24H31N7O6 [M + H]+ 514.2414, found 514.2415.
4-((((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(3-(methoxycarbonyl)benzyl)amino)butanoic Acid (64)
Following the procedure described for compound 57, compound 41 (50 mg, 0.084 mmol) was deprotected to obtain compound 64 as a white powder (24 mg, 49% yield). 1H NMR (400 MHz, D2O) δ 8.29 (s, 1H), 8.11 (s, 1H), 7.94–7.64 (m, 4H), 7.45 (t, J = 7.9 Hz, 1H), 6.09 (s, 1H), 4.62 (br, 4H), 4.49 (br, 1H), 3.88 (s, 3H), 3.67 (br, 2H), 3.55–3.41 (m, 6.9 Hz, 3H), 2.53 (t, J = 6.4 Hz, 2H), 2.18–2.09 (m, 2H). 13C NMR (101 MHz, D2O) δ 176.8, 167.6, 150.3, 147.4, 144.8, 143.3, 130.1, 129.8, 129.5, 129.2, 90.5, 77.7, 71.6, 57.7, 52.8, 30.6, 18.5. HRMS (ESI): calcd for C23H28N6O7 [M + H]+ 501.2098, found 501.2097.
5-((((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(3-(methoxycarbonyl)benzyl)amino)pentanoic Acid (65)
Following the procedure described for compound 57, compound 42 (50 mg, 0.082 mmol) was deprotected to obtain compound 65 as a white powder (30 mg, 59% yield). 1H NMR (400 MHz, D2O) δ 8.22 (br, 2H), 7.86 (br, 2H), 7.67 (br, 1H), 7.46 (br, 1H), 6.08 (br, 1H), 4.55 (br, 4H), 4.35 (br, 1H), 3.88 (s, 3H), 3.64 (br, 1H), 3.43 (br, 2H), 2.45 (br, 2H), 1.90 (br, 2H), 1.67 (br, 2H). 13C NMR (101 MHz, D2O) δ 178.1, 168.0, 150.3, 144.5, 143.8, 130.5, 130.2, 129.9, 129.6, 118.0, 115.1, 72.0, 53.1, 33.1, 23.6, 22.8, 21.4. HRMS (ESI): calcd for C24H30N6O7 [M + H]+ 515.2254, found 515.2257.
(S)-2-Amino-4-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(3-(methoxycarbonyl)benzyl)amino)butanoic Acid (66)
Following the procedure described for compound 57, compound 43 (50 mg, 0.070 mmol) was deprotected to obtain compound 66 as a white powder (26 mg, 60% yield). 1H NMR (400 MHz, D2O) δ 8.24 (s, 1H), 8.01 (s, 1H), 7.72 (br, 1H), 7.64 (d, J = 7.8 Hz, 1H), 7.54 (d, J = 7.8 Hz, 1H), 7.31 (t, J = 7.8 Hz, 1H), 6.03 (s, 1H), 4.56–4.52 (m, 1H), 4.50–4.35 (m, 4H), 4.07 (m, 1H), 3.74 (s, 3H), 3.69–3.54 (m, 4H), 2.55–2.45 (m, 1H), 2.41–2.33 (m, 1H). 13C NMR (101 MHz, D2O) δ 170.8, 167.2, 162.7, 162.3, 149.5, 147.2, 143.6, 143.6, 131.0, 130.0, 129.5, 129.3, 129.1, 118.8, 117.6, 114.7, 90.8, 77.5, 73.5, 71.4, 53.9, 50.7, 24.4. HRMS (ESI): calcd for C23H29N7O7 [M + H]+ 516.2207, found 516.2206.
Methyl 3-(((((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(isopropyl)amino)methyl)benzoate (67)
Following the procedure described for compound 57, compound 44 (50 mg, 0.101 mmol) was deprotected to obtain compound 67 as a white powder (33 mg, 59% yield). 1H NMR (400 MHz, D2O) δ 8.19 (d, J = 3.9 Hz, 1H), 7.99–7.54 (m, 4H), 7.29 (t, J = 7.6 Hz, 1H), 5.91 (s, 1H), 4.62–4.46 (m, 3H), 4.33–4.25 (m, 1H), 4.20 (br, 1H), 3.87 (s, 3H), 3.79–3.62 (m, 2H), 3.39–3.28 (m, 1H), 1.54–1.37 (m, 6H). 13C NMR (101 MHz, D2O) δ 167.6, 149.7, 147.3, 144.1, 143.3, 136.1, 131.3, 130.1, 129.7, 128.7, 118.7, 114.8, 90.6, 78.6, 73.6, 71.4, 58.7, 55.2, 52.7, 50.5, 16.2, 15.7. HRMS (ESI): calcd for C22H28N6O5 [M + H]+ 457.2199, found 457.2196.
3-(((4-Amino-4-oxobutyl)(((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)amino)methyl)benzoic Acid (68)
Following the procedure described for compound 57, compound 45 (50 mg, 0.061 mmol) was deprotected to obtain compound 68 as a white powder (15 mg, 42% yield). 1H NMR (400 MHz, D2O) δ 8.21 (br, 2H), 7.86 (br, 2H), 7.65 (d, J = 7.6 Hz, 1H), 7.43 (br, 1H), 6.08 (br, 1H), 4.70–4.24 (m, 5H), 3.63 (br, 1H), 3.52–3.38 (m, 2H), 2.48 (br, 2H), 2.12 (br, 2H). 13C NMR (101 MHz, D2O) δ 177.5, 149.8, 147.4, 144.0, 143.6, 131.2, 130.3, 129.9, 129.1, 117.7, 114.8, 90.5, 77.7, 71.5, 31.8, 19.1. HRMS (ESI): calcd for C22H26N7O6 [M + H]+ 486.2101, found 486.2089.
3-(((5-Amino-5-oxopentyl)(((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)amino)methyl)benzoic Acid (69)
Following the procedure described for compound 57, compound 46 (50 mg, 0.059 mmol) was deprotected to obtain compound 69 as a white powder (19 mg, 65% yield). 1H NMR (400 MHz, D2O) δ 8.19 (br, 2H), 7.83 (br, 2H), 7.63 (br, 1H), 7.40 (br, 1H), 6.03 (br, 1H), 4.58–4.41 (m, 4H), 4.31 (br, 1H), 3.63 (br, 1H), 3.42 (d, J = 7.8 Hz, 2H), 2.34 (br, 2H), 1.90 (br, 2H), 1.68 (br, 2H). 13C NMR (101 MHz, D2O) δ 178.7, 149.7, 143.9, 143.5, 130.3, 129.8, 129.8, 129.1, 120.6, 117.7, 114.8, 105.0, 77.9, 71.5, 33.9, 21.9. HRMS (ESI): calcd for C23H30N7O6 [M + H]+ 500.2258, found 500.2253.
3-(((((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(3-carboxypropyl)amino)methyl)benzoic Acid (70)
Following the procedure described for compound 57, compound 47 (50 mg, 0.078 mmol) was deprotected to obtain compound 70 as a white powder (21 mg, 46% yield). 1H NMR (400 MHz, D2O) δ 8.27 (s, 1H), 8.14 (s, 1H), 7.85 (br, 2H), 7.64 (d, J = 7.7 Hz, 1H), 7.49–7.35 (m, 1H), 6.08 (br, 1H), 4.55 (br, 5H), 3.65 (br, 1H), 3.48–3.43 (m, 2H), 2.52 (br, 2H), 2.13 (br, 2H). 13C NMR (101 MHz, D2O) δ 176.6, 168.8, 149.8, 147.4, 144.0, 143.5, 131.4, 130.4, 129.7, 129.1, 117.7, 114.8, 90.5, 77.7, 71.6, 30.4, 22.1, 18.5. HRMS (ESI): calcd for C22H27N6O7 [M + H]+ 487.1941, found 487.1945.
3-(((((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(4-carboxybutyl)amino)methyl)benzoic Acid (71)
Following the procedure described for compound 57, compound 48 (50 mg, 0.076 mmol) was deprotected to obtain compound 71 as a white powder (24 mg, 52% yield). 1H NMR (400 MHz, D2O) δ 8.20 (br, 2H), 7.83 (br, 2H), 7.63 (d, J = 7.5 Hz, 1H), 7.42 (br, 1H), 6.05 (br, 1H), 4.53 (br, 4H), 4.31 (br, 1H), 3.63 (br, 1H), 3.49–3.32 (m, 2H), 2.42 (br, 2H), 1.88 (br, 2H), 1.66 (br, 2H). 13C NMR (101 MHz, D2O) δ 177.7, 150.0, 144.3, 143.4, 131.4, 130.4, 130.1, 129.7, 129.1, 117.7, 114.8, 90.4, 77.7, 71.6, 32.7, 22.4, 21.0. HRMS (ESI): calcd for C23H29N6O7 [M + H]+ 501.2098, found 501.2096.
3-((((S)-3-Amino-3-carboxypropyl)(((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)amino)methyl)benzoic Acid (72)
Following the procedure described for compound 57, compound 49 (50 mg, 0.066 mmol) was deprotected to obtain compound 72 as a white powder (24 mg, 61% yield). 1H NMR (400 MHz, D2O) δ 8.23 (bs, 1H), 8.06 (s, 1H), 7.76 (s, 1H), 7.69 (d, J = 7.6 Hz, 1H), 7.56 (d, J = 7.8 Hz, 1H), 7.33 (t, J = 7.8 Hz, 1H), 6.06–5.98 (m, 1H), 4.59–4.34 (m, 5H), 4.06 (m, 1H), 3.72–3.51 (m, 4H), 2.55–2.4 (m, 1H), 2.40–2.32 (m, 1H). 13C NMR (101 MHz, D2O) δ 171.0, 168.4, 162.8, 162.4, 149.5, 147.3, 143.7, 143.5, 131.3, 130.3, 129.7, 129.5, 129.1, 118.9, 117.6, 114.7, 90.6, 77.5, 73.5, 71.5, 50.9, 24.4. HRMS (ESI): calcd for C22H27N7O7 [M + H]+ 502.2050, found 502.2048.
3-(((((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(isopropyl)amino)methyl)benzoic Acid (73)
Following the procedure described for compound 57, compound 50 (50 mg, 0.093 mmol) was deprotected to obtain compound 73 as a white powder (27 mg, 54% yield). 1H NMR (400 MHz, D2O) δ 8.21 (s, 1H), 8.16 (s, 1H), 7.90 (s, 1H), 7.58 (m, 2H), 7.27 (t, J = 7.3 Hz, 1H), 5.89 (s, 1H), 4.55 (t, J = 10.4 Hz, 2H), 4.31–4.15 (m, 2H), 3.98–3.90 (m, 1H), 3.78 (br, 1H), 3.32 (br, 1H), 1.50 (m, 6H). 13C NMR (101 MHz, D2O) δ 215.3, 168.7, 149.9, 147.3, 144.4, 143.1, 136.1, 131.7, 129.9, 128.7, 118.7, 117.7, 114.8, 89.7, 78.5, 73.5, 71.4, 58.8, 55.3, 51.7, 30.1, 16.2, 15.7. HRMS (ESI): calcd for C21H26N6O5 [M + H]+ 443.2043, found 443.2040.
4-((((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(naphthalen-2-ylmethyl)amino)butanamide (74)
Following the procedure described for compound 57, compound 51 (50 mg, 0.064 mmol) was deprotected to obtain compound 74 as a white powder (22 mg, 58% yield). 1H NMR (400 MHz, D2O) δ 8.11 (br, 1H), 7.57–7.25 (m, 4H), 7.09 (d, J = 8.0 Hz, 1H), 5.89 (br, 1H), 4.35 (br, 4H), 3.94–3.63 (m, 1H), 3.49–3.22 (m, 3H), 2.44 (br, 2H), 2.13–1.82 (br, 2H). 13C NMR (101 MHz, D2O) δ 177.5, 148.4, 146.0, 143.3, 142.3, 132.1, 131.6, 129.5, 128.0, 127.7, 127.2, 127.0, 126.8, 126.7, 125.1, 118.0, 90.6, 77.8, 74.1, 71.2, 58.0, 55.9, 54.9, 32.0, 19.0. HRMS (ESI): calcd for C25H29N7O4 [M + H]+ 492.2359, found 492.2363.
5-((((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(naphthalen-2-ylmethyl)amino)pentanamide (75)
Following the procedure described for compound 57, compound 52 (50 mg, 0.063 mmol) was deprotected to obtain compound 75 as a white powder (23 mg, 60% yield). 1H NMR (400 MHz, D2O) δ 8.11 (s, 1H), 7.72–7.61 (m, 2H), 7.55–7.48 (m, 2H), 7.46–7.39 (m, 1H), 7.37–7.15 (m, 2H), 5.89 (br, 1H), 4.66–4.48 (m, 2H), 4.42–4.11 (m, 3H), 3.61–3.33 (m, 4H), 2.43–2.27 (m, 2H), 1.89 (d, J = 9.8 Hz, 2H), 1.77–1.61 (m, 2H). 13C NMR (101 MHz, D2O) δ 178.8, 148.6, 146.3, 143.8, 143.4, 142.7, 142.2, 132.4, 131.8, 129.9, 128.2, 127.8, 127.4, 126.8, 125.1, 90.7, 78.1, 74.2, 71.3, 58.6, 57.8, 55.8, 54.6, 34.0, 22.5, 22.0. HRMS (ESI): calcd for C26H31N7O4 [M + H]+ 506.2516, found 506.2520.
4-((((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(naphthalen-2-ylmethyl)amino)butanoic Acid (76)
Following the procedure described for compound 57, compound 53 (50 mg, 0.085 mmol) was deprotected to obtain compound 76 as a white powder (30 mg, 60% yield). 1H NMR (400 MHz, D2O) δ 8.14 (br, 1H), 7.75–7.23 (m, 8H), 5.95 (br, 1H), 4.62–4.54 (m, 1H), 4.43–4.26 (m, 4H), 3.69–3.53 (m, 2H), 3.45 (m, 2H), 2.54 (t, J = 6.5 Hz, 2H), 2.19–2.04 (m, 2H). 13C NMR (101 MHz, D2O) δ 179.8, 179.8, 152.0, 149.1, 146.3, 145.7, 134.9, 134.4, 132.5, 130.6, 129.9, 129.8, 129.3, 121.0, 120.2, 117.3, 93.3, 80.2, 74.2, 60.8, 33.6, 21.4. HRMS (ESI): calcd for C23H30N8O5 [M + H]+ 493.2199, found 493.2199.
5-((((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(naphthalen-2-ylmethyl)amino)pentanoic Acid (77)
Following the procedure described for compound 57, compound 54 (50 mg, 0.083 mmol) was deprotected to obtain compound 77 as a white powder (31 mg, 61% yield). 1H NMR (400 MHz, D2O) δ 8.27 (br, 2H), 7.86–7.55 (m, 5H), 7.52–7.47 (m, 1H), 7.40 (d, J = 7.7 Hz, 1H), 5.95 (s, 1H), 4.66–4.40 (m, 4H), 4.31 (br, 1H), 3.70–3.43 (m, 4H), 2.50 (br, 2H), 1.97 (br, 2H), 1.80–1.67 (br, 2H). 13C NMR (101 MHz, D2O) δ 178.0, 163.0, 162.7, 149.5, 146.6, 143.8, 143.1, 132.4, 131.9, 130.0, 128.0, 127.3, 127.2, 126.8, 117.7, 38.6, 32.9, 21.2. HRMS (ESI): calcd for C26H28N6O5 [M + H]+ 507.2356, found 507.2355.
(S)-2-Amino-4-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(naphthalen-2-ylmethyl)amino)butanoic Acid (78)
Following the procedure described for compound 57, compound 55 (50 mg, 0.071 mmol) was deprotected to obtain compound 78 as a white powder (28 mg, 65% yield). 1H NMR (400 MHz, D2O) δ 8.03 (bs, 2H), 7.54–6.97 (m, 7H), 5.83 (bs, 1H), 4.55–4.25 (m, 3H), 4.24–4.16 (m, 1H), 4.16–4.09 (m, 1H), 3.65 (s, 2H), 3.49 (s, 1H), 2.46 (br, 2H). 13C NMR (101 MHz, D2O) δ 170.8, 148.3, 146.0, 143.3, 142.2, 132.0, 131.5, 129.7, 127.1, 126.9, 126.6, 126.3, 118.2, 117.6, 114.7, 90.9, 77.5, 73.8, 71.4, 50.7, 24.5. HRMS (ESI): calcd for C25H29N7O5 [M + H]+ 508.2308, found 508.2309.
(2R,3R,4S,5R)-2-(6-Amino-9H-purin-9-yl)-5-((isopropyl(naphthalen-2-ylmethyl)amino)methyl)tetrahydrofuran-3,4-diol (79)
Following the procedure described for compound 57, compound 56 (50 mg, 0.102 mmol) was deprotected to obtain compound 79 as a white powder (36 mg, 65% yield). 1H NMR (400 MHz, D2O) δ 7.86 (s, 1H), 7.64–7.47 (m, 2H), 7.42–7.10 (m, 6H), 5.58 (s, 1H), 4.41–4.35 (m, 1H), 4.29 (br, 1H), 4.16–4.04 (m, 2H), 3.92 (br, 1H), 3.80–3.73(m, 1H), 3.58 (br, 1H), 3.24 (m, 1H), 1.35 (m, 6H). 13C NMR (101 MHz, D2O) δ 148.7, 146.3, 143.6, 143.0, 142.6, 132.2, 131.7, 130.4, 128.3, 127.5, 127.4, 118.0, 90.0, 77.5, 73.9, 71.9, 59.1, 51.7, 49.6, 17.4, 15.9. HRMS (ESI): calcd for C24H28N6O3 [M + H]+ 449.2301, found 449.2299.
tert-Butyl (S)-4-((((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydro-furo-[3,4-d][1,3]dioxol-4-yl)methyl)(naphthalen-2-ylmethyl)amino)-2-bis((tert-butoxycarbonyl)amino)pentanoate (80)
Following the procedure described for compound 33, tert-butyl (R)-2-((tert-butoxycarbonyl)amino)-5-oxopentanoate 28 (312 mg, 0.80 mmol) and compound 32 (300 mg, 0.67 mmol) were coupled to obtain compound 80 as a white powder (319 mg, 58% yield). 1H NMR (400 MHz, CDCl3) δ 8.05 (s, 1H), 7.79 (s, 1H), 7.73 m, (1H), 7.68 (d, J = 8.6 Hz, 2H), 7.61 (s, 1H), 7.45–7.34 (m, 3H), 6.59 (s, 2H), 5.98 (d, J = 2.2 Hz, 1H), 5.27 (dd, J = 6.4, 2.2 Hz, 1H), 4.84 (dd, J = 6.4, 3.1 Hz, 1H), 4.69 (dd, J = 9.6, 5.2 Hz, 1H), 4.37 (m, 1H), 3.80 (br, 1H), 3.57 (br, 1H), 2.76 (m, 1H), 2.64–2.47 (m, 3H), 2.04 (m, 1H), 1.83 (m, 1H), 1.54 (s, 3H), 1.43 (s, 16H), 1.41 (s, 8H), 1.31 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 155.9, 152.9, 152.5, 139.5, 127.7, 127.5, 127.3, 127.2, 125.8, 125.5, 90.8, 85.4, 83.6, 83.41, 82.6, 81.0, 59.0, 55.6, 54.0, 28.0, 27.1, 27.0, 25.3, 23.8. HRMS (ESI): calcd for C43H59N7O9 [M + H]+ 818.4453, found 818.4458.
(S)-2-Amino-5-((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)(naphthalen-2-ylmethyl)amino)pentanoic Acid (81)
Following the procedure described for compound 57, compound 80 (120 mg, 0.15 mmol) was deprotected to obtain compound 81 as a white powder (58 mg, 63% yield). 1H NMR (600 MHz, D2O) δ 8.14 (br, 1H), 7.69–6.93 (m, 8H), 5.93 (br, 1H), 4.59–4.43 (m, 2H), 4.27 (br, 2H), 4.15–3.73 (m, 2H), 3.47 (m, 4H), 2.16–1.90 (m, 4H). 13C NMR (151 MHz, D2O) δ 171.6, 162.9, 162.7, 148.6, 131.8 127.3, 127.1, 126.8, 119.2, 117.3, 115.3, 90.7, 78.0, 74.3, 71.3, 58.8, 52.3, 26.9, 19.4. HRMS (ESI): calcd for C26H31N7O5 [M + H]+ 522.2465, found 522.2468.
Inhibition Studies
Expression and purification of full-length wild-type NNMT protein (NNMTwt) were performed as previously described.32 The purity of the enzyme was confirmed using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) with Coomassie blue staining, and NNMT identity was confirmed using SDS-PAGE and Western blotting. Catalytic activity of the recombinant protein was evaluated with 1 unit of enzyme activity representing the formation of 1 nmol of MNA/h of incubation at 37 °C. The specific activity of the batch used in the inhibitory activity assays was 18 665 units/mg of protein at a protein concentration of 0.56 mg/mL. NNMT was used at a final concentration of 100 nM diluted in assay buffer (50 mM Tris buffer (pH 8.4) and 1 mM dithiothreitol). The compounds were dissolved in DMSO and diluted with water to concentrations ranging from 0.1 to 500 μM (DMSO was kept constant at 1.25% final concentration). The compounds were incubated with the enzyme for 10 min at 37 °C before initiating the reaction with a mixture of NA and AdoMet at their KM values of 200 and 8.5 μM, respectively. The formation of MNA was measured after 30 min at 37 °C. The reaction was quenched by addition of 15 μL of the sample to 70 μL of acetonitrile containing 50 nM deutero-methylated nicotinamide as internal standard. The enzymatic activity assays were performed using UHP-HILIC-MS/MS as previously described with minor modifications.24 The UHP-HILIC-MS/MS system consisted of a binary ultra-HPLC system, consisting of two LC-30AD pumps, a SIL30-ACmp auto-sampler, a CTO-20AC column oven, and a DGU-20A5R degasser (all from Shimadzu, ’s-Hertogenbosch, The Netherlands). Isocratic elution was performed after 1 μL injections on a Waters Acquity BEH Amide HILIC column (3.0 × 100 mm, 1.7 μm particle size, Waters, Milford), using water containing 300 μM formic acid and 550 μM NH4OH (pH 9.2) at 40% v/v and acetonitrile at 60% v/v, with a runtime of 3 min. Calibration samples were prepared using 75 μL of internal standard d3-MNA at 50 nM in acetonitrile and 25 μL of an aqueous solution of reference standard MNA with concentrations ranging from 2500 to 1.221 nM. For detection, a Sciex QTRAP 5500 triple quadrupole mass spectrometer, with Analyst 1.6.2 and MultiQuant 3.0.1 software (Sciex, Ontario, Canada), was used. Settings used for the ionization source were as follows: curtain gas, 40 psi; collision gas, “medium”; ionspray voltage, 5000 V; temperature, 600 °C; ion source gas 1, 60 psi; and ion source gas 2, 80 psi. Dwell times were 10 ms, and the entrance potential was set to 10 V; specific parameters of the compounds can be found in Table 2. The whole eluate was transferred to the electrospray probe from 1.0 till 2.8 min using the MS diverter valve. Ratios of the sums of the MNA and d3-MNA transitions were calculated and plotted versus concentration.
Table 2. Tuned MS/MS Parameters for All Quantified Componentsa,b.
| compound | Q1 (m/z) | Q3 (m/z) | DP | CE | CXP |
|---|---|---|---|---|---|
| MNA | 137.101 | 94.0 | 136 | 27 | 12 |
| 92.0 | 136 | 29 | 12 | ||
| 78.0 | 136 | 35 | 10 | ||
| MNA-d3 | 140.128 | 97.1 | 121 | 29 | 12 |
| 95.1 | 121 | 31 | 12 | ||
| 78.0 | 121 | 35 | 10 |
The entrance potential was set at 10 V for all compounds, dwell time was 10 ms.
Q1: quadrupole 1, Q3: quadrupole 3, z: charge, DP: declustering potential, CE: collision energy, CXP: collision cell exit potential.
Isothermal Titration Calorimetry
Expression and purification of full-length wild-type NNMT protein (NNMTwt) were performed as previously described.31 Isothermal titration calorimetry (ITC) measurements were made at 25 °C on a MicroCal ITC200 Instrument (Malvern Instruments) with 2 μL injections. NNMTwt was diluted at 200 μM in ITC buffer [50 mM Tris (pH 8.0), 150 mM NaCl] supplemented with 4% DMSO. Compounds were dissolved in DMSO at 50 mM and diluted to 2 mM in ITC buffer with a final DMSO concentration of 4%. Binding constants were calculated by fitting the data using the ITC data analysis module in Origin 7.0 (OriginLab Corp.).
Modeling Studies
Docking computations were performed using Autodock 4.2.49 Compounds 1, 2, 78, and 81 were docked into the catalytic pocket of the structure taken from PDB ID: 3ROD.32 Four molecular dynamic simulations were performed with GROMACS 2018.250 using the AMBER03 force field.51 Each structure was immersed in a cubic box using TIP3P water molecules52 and neutralized with counter ions. A production step of 250 ns was carried out using the Parrinello–Rahman algorithm53 for temperature and pressure control, with coupling constants of T = 0.1 ps and P = 2.0 ps, for compounds 1, 2, and 81 and extended to 450 ns for compound 78, to reach equilibrium of the system. Coordinates were saved every 200 ps, and the protein/ligand binding energy was estimated using g_mmpbsa calculations54,55 on the last 50 ns of each trajectory. The conformation of minimal energy in these 50 ns was extracted from the simulations and minimized to represent the interactions between the ligands and NNMT protein.
Enzyme Assay for Selectivity
Methyltransferase inhibition assays were performed as described56 by using commercially available chemiluminescent assay kits for PRMT1 and NSD2 (purchased from BPS Bioscience). The enzymatic reactions were conducted in duplicate at room temperature for 1 h (PRMT1) or 2 h (NSD2) in substrate-coated well plates at a final reaction volume of 50 μL containing the manufacturer’s proprietary assay buffer, AdoMet (at a concentration of 5 times the respective Km value for each enzyme), the methyltransferase enzyme: PRMT1 (100 ng per reaction) and NSD2 (500 ng per reaction), and inhibitor 78. Before addition of AdoMet, the enzyme was first incubated with the inhibitor for 15 min at room temperature. Positive controls were performed in the absence of the inhibitor using water to keep the final volume consistent. Blanks and substrate controls were performed in the absence of the enzyme and AdoMet, respectively. Following the enzymatic reactions, 100 μL of primary antibody (recognizing the respective immobilized methylated product) was added to each well, and the plate was incubated at room temperature for an additional 1 h. Then, 100 μL of secondary horseradish peroxidase (HRP)-conjugated antibody was added to each well, and the plate was incubated at room temperature for additional 30 min. Finally, 100 μL of an HRP substrate mixture was added to the wells, and the luminescence was measured directly by using a standard microplate reader. The luminescence data were normalized with the positive controls defined as 100% activity and blank defined as 0%.
Cell Culture and Treatment with Compounds
The HSC-2 human oral cancer cell line was purchased from the American Type Culture Collection (Rockville, MD) and cultured in Dulbecco’s modified Eagle’s medium/F12 medium, supplemented with 10% fetal bovine serum and 50 μg/mL gentamicin, at 37 °C in a humidified 5% CO2 incubator. Compounds 1, 2, 78, and 81 were tested for their inhibitory effect on cell proliferation of HSC-2 cells. Each compound was dissolved in DMSO at 100 mM concentration. This stock solution was then diluted in culture medium to final concentration values ranging between 1 and 100 μM. For each sample, DMSO was kept constant at 0.1% final concentration.
The day before starting treatment, cells were seeded in 96-well plates, at a density of 1 × 103 cells/well. Cells were allowed to attach overnight and then incubated with compounds at different final concentrations, or with only DMSO, for 24, 48, and 72 h. All experiments were performed in triplicate.
MTT Assay
Cell proliferation was determined using a colorimetric assay with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT). The MTT assay measures the conversion of MTT to insoluble formazan by dehydrogenase enzymes of the intact mitochondria of living cells. HSC-2 cell proliferation was evaluated by measuring the conversion of the tetrazolium salt MTT to formazan crystals upon treatment with compounds or only DMSO for 24, 48, and 72 h. Briefly, cells were incubated for 2 h at 37 °C with 100 μL of fresh culture medium containing 5 μL of the MTT reagent (5 mg/mL in PBS). The medium was removed, and 200 μL of isopropanol were added. The amount of formazan crystals formed correlated directly with the number of viable cells. The reaction product was quantified by measuring the absorbance at 540 nm using an enzyme-linked immunosorbent assay plate reader. Experiments were repeated three times. Results were expressed as percentage of the control (control equals 100% and corresponds to the absorbance value of each sample at time zero) and presented as mean values ± standard deviation of three independent experiments performed in triplicate. Data were analyzed using GraphPad Prism software (GraphPad Software, San Diego, CA). Significant differences between groups were determined using the one-way analysis of variance. A p value <0.05 was considered as statistically significant.
Quantitative Measurements of MNA Levels in Cultured Cells
The analysis was performed as previously described57 with minor modifications. Cellular MNA levels were determined using the same UHP-HILIC-MS/MS employed for the inhibition studies, as described above. To determine the effect of compound 78 on NNMT activity in the HSC-2 oral cancer cell line, used cells were treated with 78 at 100 μM (final DMSO content 0.1%) and incubated for 24, 48, or 72 h. The day prior to starting treatment, cells were seeded in 6-well plates, at a density of 3 × 104 cells/well. Cells were allowed to attach overnight and were then incubated with compound 78. All experiments were performed in duplicate. Following treatment, medium was removed, and adherent cells were trypsinized and harvested by centrifugation at 1000g for 3 min at 4 °C. Supernatant was then discarded and cell pellets were stored at −80 °C until further use. The extraction of MNA from the cell pellets was performed as previously described.58 Briefly, 100 μL of acetonitrile containing 50 nM d3-MNA (as internal control) was added to the cell pellets, and the cells were lyzed for 20 min at room temperature with mild shaking. Then, 50 μL of purified water was added, followed by mixing, and the resulting cell debris was centrifuged for 10 min at 5000 rpm. Then, 100 μL of the resulting supernatant was transferred to a 96-well plate and analyzed for MNA content.
Glossary
Abbreviations
- DCE
1,2-dichloroethane
- DIBAL-H
diisobutylaluminum hydride
- DMAP
4-dimethylaminopyridine
- Kd
dissociation constant
- IC50
half-maximal inhibitory concentration
- ITC
isothermal titration calorimetry
- MNA
1-methyl-nicotinamide
- NA
nicotinamide
- NNMT
nicotinamide N-methyltransferase
- NNMTwt
wild-type NNMT
- PDC
pyridinium dichromate
- SAH
S-adenosyl-l-homocysteine
- SAM
S-adenosyl-l-methionine
- Trt
triphenylmethyl (trityl)
- UHP-HILIC
ultra-high-performance hydrophilic liquid interaction chromatography
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.9b00413.
Analytical data for all new compounds including 1H and 13C NMR spectra; supporting figures for enzyme inhibition curves, molecular modeling results, and cellular MNA determination; supporting tables for selectivity assays and estimated binding energies (PDF)
Molecular formula strings for all new compounds (CSV)
Author Contributions
∇ Y.G. and M.J.v.H. contributed equally to this work.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Yongzhi Gao gratefully acknowledges the financial support provided by a scholarship from the Chinese Scholarship Council (CSC, file No. 201506270162). Jian Jin acknowledges support by grants R01CA218600, R01CA230854, R01GM122749 and R01HD088626 from the U.S. National Institutes of Health.
The authors declare no competing financial interest.
Supplementary Material
References
- Alston T. A.; Abeles R. H. Substrate Specificity of Nicotinamide Methyltransferase Isolated from Porcine Liver. Arch. Biochem. Biophys. 1988, 260, 601–608. 10.1016/0003-9861(88)90487-0. [DOI] [PubMed] [Google Scholar]
- van Haren M. J.; Toraño J. S.; Sartini D.; Emanuelli M.; Parsons R. B.; Martin N. I. A Rapid and Efficient Assay for the Characterization of Substrates and Inhibitors of Nicotinamide N -Methyltransferase. Biochemistry 2016, 55, 5307–5315. 10.1021/acs.biochem.6b00733. [DOI] [PubMed] [Google Scholar]
- Thomas M. G.; Sartini D.; Emanuelli M.; van Haren M. J.; Martin N. I.; Mountford D. M.; Barlow D. J.; Klamt F.; Ramsden D. B.; Reza M.; Parsons R. B. Nicotinamide N-Methyltransferase Catalyses the N-Methylation of the Endogenous-Carboline Norharman: Evidence for a Novel Detoxification Pathway. Biochem. J. 2016, 473, 3253–3267. 10.1042/BCJ20160219. [DOI] [PubMed] [Google Scholar]
- Pissios P. Nicotinamide N -Methyltransferase: More Than a Vitamin B3 Clearance Enzyme. Trends Endocrinol. Metab. 2017, 28, 340–353. 10.1016/j.tem.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J.; Kim L. J. Y.; Wang X.; Wu Q.; Sanvoranart T.; Hubert C. G.; Prager B. C.; Wallace L. C.; Jin X.; Mack S. C.; Rich J. N. Nicotinamide Metabolism Regulates Glioblastoma Stem Cell Maintenance. JCI Insight 2017, 2, 1–23. 10.1172/jci.insight.90019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn B. B.; Alhonen L.; Pulinilkunnil T. C.; Cen Y.; Puigserver P.; Wang Y.; Monia B. P.; Asara J. M.; Zhang L.; Kong D.; Rodgers J. T.; Yang Q.; Gong F.; Pirinen E.; Sauve A. A.; Bhanot S.; Banks A. S.; Kraus D.; Peroni O. D. Nicotinamide N-Methyltransferase Knockdown Protects against Diet-Induced Obesity. Nature 2014, 508, 258–262. 10.1038/nature13198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Klooster J. P.; Sotiriou A.; Boeren S.; Vaessen S.; Vervoort J.; Pieters R. Type 2 Diabetes-Related Proteins Derived from an in Vitro Model of Inflamed Fat Tissue. Arch. Biochem. Biophys. 2018, 644, 81–92. 10.1016/j.abb.2018.03.003. [DOI] [PubMed] [Google Scholar]
- Khalil E. M.; Mackie B. D.; Mao Y. Methyltransferases: Key Regulators in Cardiovascular Development and Disease. Ann. Vasc. Med. Res. 2016, 3, 1032–1039. [Google Scholar]
- Fedorowicz A.; Mateuszuk Ł.; Kopec G.; Skórka T.; Kutryb-Zając B.; Zakrzewska A.; Walczak M.; Jakubowski A.; Łomnicka M.; Słomińska E.; Chlopicki S. Activation of the Nicotinamide N-Methyltransferase (NNMT)-1-Methylnicotinamide (MNA) Pathway in Pulmonary Hypertension. Respir. Res. 2016, 17, 108 10.1186/s12931-016-0423-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulanovskaya O. A.; Zuhl A. M.; Cravatt B. F. NNMT Promotes Epigenetic Remodeling in Cancer by Creating a Metabolic Methylation Sink. Nat. Chem. Biol. 2013, 9, 300–306. 10.1038/nchembio.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palanichamy K.; Kanji S.; Gordon N.; Thirumoorthy K.; Jacob J. R.; Litzenberg K. T.; Patel D.; Chakravarti A. NNMT Silencing Activates Tumor Suppressor PP2A, Inactivates Oncogenic STKs, and Inhibits Tumor Forming Ability. Clin. Cancer Res. 2017, 23, 2325–2334. 10.1158/1078-0432.CCR-16-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Wang Y.; Li G.; Yu H.; Xie X. Down-Regulation of Nicotinamide N-Methyltransferase Induces Apoptosis in Human Breast Cancer Cells via the Mitochondria-Mediated Pathway. PLoS One 2014, 9, e89202 10.1371/journal.pone.0089202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartini D.; Muzzonigro G.; Milanese G.; Pierella F.; Rossi V.; Emanuelli M. Identification of Nicotinamide N-Methyltransferase as a Novel Tumor Marker for Renal Clear Cell Carcinoma. J. Urol. 2006, 176, 2248–2254. 10.1016/j.juro.2006.07.046. [DOI] [PubMed] [Google Scholar]
- Sartini D.; Santarelli A.; et al. Nicotinamide N-Methyltransferase Upregulation Inversely Correlates with Lymph Node Metastasis in Oral Squamous Cell Carcinoma. Mol. Med. 2007, 13, 415–421. 10.2119/2007-00035.Sartini. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons R. B.; Smith S. W.; Waring R. H.; Williams A. C.; Ramsden D. B. High Expression of Nicotinamide N-Methyltransferase in Patients with Idiopathic Parkinson’s Disease. Neurosci. Lett. 2003, 342, 13–16. 10.1016/S0304-3940(03)00218-0. [DOI] [PubMed] [Google Scholar]
- Parsons R. B.; Williams A. C.; Waring R. H.; Ramsden D. B.; Smith M.-L. Expression of Nicotinamide N-Methyltransferase (E.C. 2.1.1.1) in the Parkinsonian Brain. J. Neuropathol. Exp. Neurol. 2002, 61, 111–124. 10.1093/jnen/61.2.111. [DOI] [PubMed] [Google Scholar]
- Thomas M. G.; Saldanha M.; Mistry R. J.; Dexter D. T.; Ramsden D. B.; Parsons R. B. Nicotinamide N-Methyltransferase Expression in SH-SY5Y Neuroblastoma and N27 Mesencephalic Neurones Induces Changes in Cell Morphology via Ephrin-B2 and Akt Signalling. Cell Death Dis. 2013, 4, e669 10.1038/cddis.2013.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Haren M. J.; Thomas M. G.; Sartini D.; Barlow D. J.; Ramsden D. B.; Emanuelli M.; Klamt F.; Martin N. I.; Parsons R. B. The Kinetic Analysis of the N-Methylation of 4-Phenylpyridine by Nicotinamide N-Methyltransferase: Evidence for a Novel Mechanism of Substrate Inhibition. Int. J. Biochem. Cell Biol. 2018, 98, 127–136. 10.1016/j.biocel.2018.03.010. [DOI] [PubMed] [Google Scholar]
- Aksoy S.; Szumlanski C. L.; Weinshilboum R. M. Human Liver Nicotinamide N-Methyltransferase. CDNA Cloning, Expression, and Biochemical Characterization. J. Biol. Chem. 1994, 269, 14835–14840. [PubMed] [Google Scholar]
- Horning B. D.; Suciu R. M.; Ghadiri D. A.; Ulanovskaya O. A.; Matthews M. L.; Lum K. M.; Backus K. M.; Brown S. J.; Rosen H.; Cravatt B. F. Chemical Proteomic Profiling of Human Methyltransferases. J. Am. Chem. Soc. 2016, 138, 13335–13343. 10.1021/jacs.6b07830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruf S.; Hallur M. S.; Anchan N. K.; Swamy I. N.; Murugesan K. R.; Sarkar S.; Narasimhulu L. K.; Putta V. P. R. K.; Shaik S.; Chandrasekar D. V.; Mane V. S.; Kadnur S. V.; Suresh J.; Bhamidipati R. K.; Singh M.; Burri R. R.; Kristam R.; Schreuder H.; Czech J.; Rudolph C.; Marker A.; Langer T.; Mullangi R.; Yura T.; Gosu R.; Kannt A.; Dhakshinamoorthy S.; Rajagopal S. Novel Nicotinamide Analog as Inhibitor of Nicotinamide N-Methyltransferase. Bioorg. Med. Chem. Lett. 2018, 28, 922–925. 10.1016/j.bmcl.2018.01.058. [DOI] [PubMed] [Google Scholar]
- Kannt A.; Rajagopal S.; Kadnur S. V.; Suresh J.; Bhamidipati R. K.; Swaminathan S.; Hallur M. S.; Kristam R.; Elvert R.; Czech J.; Pfenninger A.; Rudolph C.; Schreuder H.; Chandrasekar D. V.; Mane V. S.; Birudukota S.; Shaik S.; Zope B. R.; Burri R. R.; Anand N. N.; Thakur M. K.; Singh M.; Parveen R.; Kandan S.; Mullangi R.; Yura T.; Gosu R.; Ruf S.; Dhakshinamoorthy S. A Small Molecule Inhibitor of Nicotinamide N-Methyltransferase for the Treatment of Metabolic Disorders. Sci. Rep. 2018, 8, 3660 10.1038/s41598-018-22081-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelakantan H.; Wang H. Y.; Vance V.; Hommel J. D.; McHardy S. F.; Watowich S. J. Structure-Activity Relationship for Small Molecule Inhibitors of Nicotinamide N-Methyltransferase. J. Med. Chem. 2017, 60, 5015–5028. 10.1021/acs.jmedchem.7b00389. [DOI] [PubMed] [Google Scholar]
- van Haren M. J.; Taig R.; Kuppens J.; Sastre Toraño J.; Moret E. E.; Parsons R. B.; Sartini D.; Emanuelli M.; Martin N. I. Inhibitors of Nicotinamide N-Methyltransferase Designed to Mimic the Methylation Reaction Transition State. Org. Biomol. Chem. 2017, 15, 6656–6667. 10.1039/C7OB01357D. [DOI] [PubMed] [Google Scholar]
- Lerner C.; Masjost B.; Ruf A.; Gramlich V.; Jakob-Roetne R.; Zürcher G.; Borroni E.; Diederich F. Bisubstrate Inhibitors for the Enzyme Catechol-O-Methyltransferase (COMT): Influence of Inhibitor Preorganisation and Linker Length between the Two Substrate Moieties on Binding Affinity. Org. Biomol. Chem. 2003, 1, 42–49. 10.1039/B208690P. [DOI] [PubMed] [Google Scholar]
- Paulini R.; Trindler C.; Lerner C.; Brändli L.; Schweizer W. B.; Jakob-Roetne R.; Zürcher G.; Borroni E.; Diederich F. Bisubstrate Inhibitors of Catechol O-Methyltransferase (COMT): The Crucial Role of the Ribose Structural Unit for Inhibitor Binding Affinity. ChemMedChem 2006, 1, 340–357. 10.1002/cmdc.200500065. [DOI] [PubMed] [Google Scholar]
- Mori S.; Iwase K.; Iwanami N.; Tanaka Y.; Kagechika H.; Hirano T. Development of Novel Bisubstrate-Type Inhibitors of Histone Methyltransferase SET7/9. Bioorg. Med. Chem. 2010, 18, 8158–8166. 10.1016/j.bmc.2010.10.022. [DOI] [PubMed] [Google Scholar]
- Dowden J.; Hong W.; Parry R. V.; Pike R. A.; Ward S. G. Toward the Development of Potent and Selective Bisubstrate Inhibitors of Protein Arginine Methyltransferases. Bioorg. Med. Chem. Lett. 2010, 20, 2103–2105. 10.1016/j.bmcl.2010.02.069. [DOI] [PubMed] [Google Scholar]
- Van Haren M.; Van Ufford L. Q.; Moret E. E.; Martin N. I. Synthesis and Evaluation of Protein Arginine N-Methyltransferase Inhibitors Designed to Simultaneously Occupy Both Substrate Binding Sites. Org. Biomol. Chem. 2015, 13, 549–560. 10.1039/C4OB01734J. [DOI] [PubMed] [Google Scholar]
- van Haren M. J.; Sbardella G.; Martin N. I.; Troffer-Charlier N.; Marechal N.; Cavarelli J.; Cianciulli A. Transition State Mimics Are Valuable Mechanistic Probes for Structural Studies with the Arginine Methyltransferase CARM1. Proc. Natl. Acad. Sci. 2017, 114, 3625–3630. 10.1073/pnas.1618401114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babault N.; Allali-Hassani A.; Li F.; Fan J.; Yue A.; Ju K.; Liu F.; Vedadi M.; Liu J.; Jin J. Discovery of Bisubstrate Inhibitors of Nicotinamide N-Methyltransferase (NNMT). J. Med. Chem. 2018, 61, 1541–1551. 10.1021/acs.jmedchem.7b01422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y.; Sartini D.; Pozzi V.; Wilk D.; Emanuelli M.; Yee V. C. Structural Basis of Substrate Recognition in Human Nicotinamide N-Methyltransferase. Biochemistry 2011, 50, 7800–7808. 10.1021/bi2007614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters N. J. Preclinical Pharmacokinetics and Pharmacodynamics of Pinometostat (EPZ-5676), a First-in-Class, Small Molecule S-Adenosyl Methionine Competitive Inhibitor of DOT1L. Eur. J. Drug Metab. Pharmacokinet. 2017, 42, 891–901. 10.1007/s13318-017-0404-3. [DOI] [PubMed] [Google Scholar]
- af Gennäs G. B.; Talman V.; Aitio O.; Ekokoski E.; Finel M.; Tuominen R. K.; Yli-Kauhaluoma J. Design, Synthesis, and Biological Activity of Isophthalic Acid Derivatives Targeted to the C1 Domain of Protein Kinase C. J. Med. Chem. 2009, 52, 3969–3981. 10.1021/jm900229p. [DOI] [PubMed] [Google Scholar]
- Cho S. D.; Park Y. D.; Kim J. J.; Falck J. R.; Yoon Y. J. Facile Reduction of Carboxylic Acids, Esters, Acid Chlorides, Amides and Nitriles to Alcohols or Amines Using NaBH4/BF3·Et2O. Bull. Korean Chem. Soc. 2004, 25, 407–409. 10.5012/bkcs.2004.25.3.407. [DOI] [Google Scholar]
- Maruoka H.; Muto T.; Tanaka T.; Imajo S.; Tomimori Y.; Fukuda Y.; Nakatsuka T. Development of 6-Benzyl Substituted 4-Aminocarbonyl-1,4-Diazepane-2,5-Diones as Orally Active Human Chymase Inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 3435–3439. 10.1016/j.bmcl.2007.03.085. [DOI] [PubMed] [Google Scholar]
- Pola Chemical Industries Inc. ; Yokoyama K.; Kimura M.; Tamai M.; Saitoh Y.; Kato T.; Ikeda Y.. Melanin Production Inhibitor. U.S. Patent 8,846,012, Sept 30, 2014.
- Schreiber K. C.; Fernandez V. P. The Lithium Aluminum Hydride Reduction of Some N-Substituted Succinimides. J. Org. Chem. 1961, 26, 1744–1747. 10.1021/jo01065a012. [DOI] [Google Scholar]
- Colombo R.; Mingozzi M.; Belvisi L.; Arosio D.; Piarulli U.; Carenini N.; Perego P.; Zaffaroni N.; De Cesare M.; Castiglioni V.; Scanziani E.; Gennari C. Synthesis and Biological Evaluation (in Vitro and in Vivo) of Cyclic Arginine–Glycine–Aspartate (RGD) Peptidomimetic–Paclitaxel Conjugates Targeting Integrin AVβ3. J. Med. Chem. 2012, 55, 10460–10474. 10.1021/jm301058f. [DOI] [PubMed] [Google Scholar]
- Chi Y.; English E. P.; Pomerantz W. C.; Horne W. S.; Joyce L. A.; Alexander L. R.; Fleming W. S.; Hopkins E. A.; Gellman S. H. Practical Synthesis of Enantiomerically Pure β 2-Amino Acids via Proline-Catalyzed Diastereoselective Aminomethylation of Aldehydes. J. Am. Chem. Soc. 2007, 129, 6050–6055. 10.1021/ja070063i. [DOI] [PubMed] [Google Scholar]
- Wernic D.; DiMaio J.; Adams J. Enantiospecific Synthesis of L-.Alpha.-Aminosuberic Acid. Synthetic Applications in Preparation of Atrial Natriuretic Factor Analogs. J. Org. Chem. 1989, 54, 4224–4228. 10.1021/jo00278a046. [DOI] [Google Scholar]
- Seta R.; Mascitti M.; Campagna R.; Sartini D.; Fumarola S.; Santarelli A.; Giuliani M.; Cecati M.; Lo Muzio L.; Emanuelli M. Overexpression of Nicotinamide N-Methyltransferase in HSC-2 OSCC Cell Line: Effect on Apoptosis and Cell Proliferation. Clin. Oral Investig. 2019, 23, 829–838. 10.1007/s00784-018-2497-8. [DOI] [PubMed] [Google Scholar]
- Kim M.-H.; Chun K.; Choi J.-W.; Joe B.-Y.; Park S.-W.; Kim K. H.; Oh B.-K.; Choi J.-H.. Tricyclic Derivatives or Pharmaceutically Acceptable Salts Thereof, Their Preparations and Pharmaceutical Compositions Containing Them. U.S. Patent 2007/0179143A1, Aug 2, 2007.
- Ewa B.; MacIej W.; Marcin S.; Grzegorz D.; Michał Z.; Jan P.; Józef O. The Development of First Staphylococcus Aureus SplB Protease Inhibitors: Phosphonic Analogues of Glutamine. Bioorg. Med. Chem. Lett. 2012, 22, 5574–5578. 10.1016/j.bmcl.2012.07.011. [DOI] [PubMed] [Google Scholar]
- Chen S.; Zhao X.; Chen J.; Chen J.; Kuznetsova L.; Wong S. S.; Ojima I. Mechanism-Based Tumor-Targeting Drug Delivery System. Validation of Efficient Vitamin Receptor-Mediated Endocytosis and Drug Release. Bioconjugate Chem. 2010, 21, 979–987. 10.1021/bc9005656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berk S. C.; Kreutzer K. A.; Buchwald S. L. A Catalytic Method for the Reduction of Esters to Alcohols. J. Am. Chem. Soc. 1991, 113, 5093–5095. 10.1021/ja00013a074. [DOI] [Google Scholar]
- Liu F.; Zha H. Y.; Yao Z. J. Synthesis of a New Conformation-Constrained L-Tyrosine Analogue as a Potential Scaffold for SH2 Domain Ligands. J. Org. Chem. 2003, 68, 6679–6684. 10.1021/jo0340152. [DOI] [PubMed] [Google Scholar]
- Floyd N.; Vijayakrishnan B.; Koeppe J. R.; Davis B. G. Thiyl Glycosylate of Olefinic Proteins: S-Linked Glycoconjugate Synthesis. Angew. Chem., Int. Ed. 2009, 48, 7798–7802. 10.1002/anie.200903135. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Ponder J. W.; Case D. A. Force Fields for Protein Simulations. Adv. Protein Chem. 2003, 66, 27–85. 10.1016/S0065-3233(03)66002-X. [DOI] [PubMed] [Google Scholar]
- Mahoney M. W.; Jorgensen W. L. A Five-Site Model for Liquid Water and the Reproduction of the Density Anomaly by Rigid, Nonpolarizable Potential Functions. J. Chem. Phys. 2000, 112, 8910–8922. 10.1063/1.481505. [DOI] [Google Scholar]
- Parrinello M.; Rahman A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. 10.1063/1.328693. [DOI] [Google Scholar]
- Kumari R.; Kumar R.; Lynn A. G_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- Baker N. A.; Sept D.; Joseph S.; Holst M. J.; McCammon J. A. Electrostatics of Nanosystems: Application to Microtubules and the Ribosome. Proc. Natl. Acad. Sci. 2001, 98, 10037–10041. 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedadi M.; Barsyte-lovejoy D.; Liu F.; Rival-gervier S.; Allali-hassani A.; Labrie V.; Wigle T. J.; DiMaggio P. A.; Wasney G. A.; Siarheyeva A.; Dong A.; Tempel W.; Wang S.-C.; Chen X.; Chau I.; Mangano T. J.; Huang X.-p.; Simpson C. D.; Pattenden S. G.; Norris J. L.; Kireev D. B.; Tripathy A.; Roth B. L.; Janzen W. P.; Garcia B. A.; Petronis A.; Ellis J.; Brown P. J.; Frye S. V.; Arrowsmith C. H.; Jin J. A Chemical Probe Selectively Inhibits G9a and GLP Methyltransferase and Genanylgeranyltransferase I with Antitumor Activity against Brest Cancer in Vivo. Nat. Chem. Biol. 2011, 7, 566–574. 10.1038/nchembio.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelakantan H.; Vance V.; Wetzel M. D.; Wang H.-Y. L.; McHardy S. F.; Finnerty C. C.; Hommel J. D.; Watowich S. J. Selective and Membrane-Permeable Small Molecule Inhibitors of Nicotinamide N-Methyltransferase Reverse High Fat Diet-Induced Obesity in Mice. Biochem. Pharmacol. 2018, 147, 141–152. 10.1016/j.bcp.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Policarpo R.; Decultot L.; May E.; Kuzmic P.; Carlson S.; Huang D.; Chu V.; Wright B.; Dhakshinamoorthy S.; Kannt A.; Rani S.; Dittakavi S.; Gaudet R.; Shair M.. High-Affinity Alkynyl Bisubstrate Inhibitors of Nicotinamide N-Methyltransferase (NNMT). 2019, chemrxiv.org e-Print archive. https://chemrxiv.org/articles/High-Affinity_Alkynyl_Bisubstrate_Inhibitors_of_Nicotinamide_N-Methyltransferase_NNMT_/8010302/1 10.26434/chemrxiv.8010302.v1 (accessed May 28, 2019). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.