

Currently, no approved treatments are available to combat infections with nidoviruses, a group of positive-stranded RNA viruses, including important zoonotic and veterinary pathogens. Previously, the cyclophilin inhibitors cyclosporine (CsA) and alisporivir (ALV) were shown to inhibit the replication of diverse nidoviruses (both arteriviruses and coronaviruses), and they may thus represent a class of pan-nidovirus inhibitors. In this study, using the arterivirus prototype equine arteritis virus, we have established that resistance to CsA and ALV treatment is associated with adaptive mutations in two transmembrane subunits of the viral replication machinery, nonstructural proteins 2 and 5. This is the first evidence for the involvement of specific replicase subunits of arteriviruses in the mechanism underlying the inhibition of their replication by cyclophilin inhibitors. Understanding this mechanism of action is of major importance to guide future drug design, both for nidoviruses and for other RNA viruses inhibited by these compounds.
KEYWORDS: alisporivir, arterivirus, cyclosporine, drug resistance, host factors, nidovirus, nonstructural proteins, replication
ABSTRACT
Previously, the cyclophilin inhibitors cyclosporine (CsA) and alisporivir (ALV) were shown to inhibit the replication of diverse RNA viruses, including arteriviruses and coronaviruses, which both belong to the order Nidovirales. In this study, we aimed to identify arterivirus proteins involved in the mode of action of cyclophilin inhibitors and to investigate how these compounds inhibit arterivirus RNA synthesis in the infected cell. Repeated passaging of the arterivirus prototype equine arteritis virus (EAV) in the presence of CsA revealed that reduced drug sensitivity is associated with the emergence of adaptive mutations in nonstructural protein 5 (nsp5), one of the transmembrane subunits of the arterivirus replicase polyprotein. Introduction of singular nsp5 mutations (nsp5 Q21R, Y113H, or A134V) led to an ∼2-fold decrease in sensitivity to CsA treatment, whereas combinations of mutations further increased EAV’s CsA resistance. The detailed experimental characterization of engineered EAV mutants harboring CsA resistance mutations implicated nsp5 in arterivirus RNA synthesis. Particularly, in an in vitro assay, EAV RNA synthesis was far less sensitive to CsA treatment when nsp5 contained the adaptive mutations mentioned above. Interestingly, for increased sensitivity to the closely related drug ALV, CsA-resistant nsp5 mutants required the incorporation of an additional adaptive mutation, which resided in nsp2 (H114R), another transmembrane subunit of the arterivirus replicase. Our study provides the first evidence for the involvement of nsp2 and nsp5 in the mechanism underlying the inhibition of arterivirus replication by cyclophilin inhibitors.
IMPORTANCE Currently, no approved treatments are available to combat infections with nidoviruses, a group of positive-stranded RNA viruses, including important zoonotic and veterinary pathogens. Previously, the cyclophilin inhibitors cyclosporine (CsA) and alisporivir (ALV) were shown to inhibit the replication of diverse nidoviruses (both arteriviruses and coronaviruses), and they may thus represent a class of pan-nidovirus inhibitors. In this study, using the arterivirus prototype equine arteritis virus, we have established that resistance to CsA and ALV treatment is associated with adaptive mutations in two transmembrane subunits of the viral replication machinery, nonstructural proteins 2 and 5. This is the first evidence for the involvement of specific replicase subunits of arteriviruses in the mechanism underlying the inhibition of their replication by cyclophilin inhibitors. Understanding this mechanism of action is of major importance to guide future drug design, both for nidoviruses and for other RNA viruses inhibited by these compounds.
INTRODUCTION
Equine arteritis virus (EAV) is a positive-strand RNA (+RNA) virus that belongs to the order Nidovirales (suborder Arnidovirineae, family Arteriviridae, subfamily Equarterivirinae, genus Alphaarterivirus), a steadily expanding clade of +RNA viruses. The nidovirus order currently comprises 88 formally recognized species that are classified in 9 virus families (1; https://talk.ictvonline.org/taxonomy/) and can infect a striking variety of vertebrate and invertebrate hosts. The order also includes the coronaviruses, like the zoonotic severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome (MERS)-CoV, which can cause lethal respiratory infections in humans (reviewed in references 2 and 3), and several important veterinary pathogens, such as avian, porcine, and bovine coronaviruses and the arterivirus porcine reproductive and respiratory syndrome virus (PRRSV). The arteriviruses EAV and PRRSV continue to cause major economic losses in the swine and equine industries worldwide, respectively (4, 5). Despite extensive efforts during recent years, currently no approved treatments to combat nidovirus infections are available.
EAV is the prototype of the arterivirus family and has a genome size of 12.7 kb (6). About three-quarters of its polycistronic genome is occupied by the viral replicase gene, which is expressed as two polyproteins (pp), pp1a and the C-terminally extended pp1ab, with the synthesis of the latter depending on a programmed ribosomal frameshift that can occur just upstream of the ORF1a stop codon. The posttranslational maturation of pp1a and pp1ab (Fig. 1A) involves extensive proteolytic cleavage (into at least 13 nonstructural proteins [nsps]) by viral proteases residing in nsp1 (PLP1), nsp2 (PLP2), and nsp4 (Mpro). In the cytoplasm of arterivirus-infected cells, most of the viral nsps and viral RNA synthesis are associated with membranous “replication organelles” (ROs) that typically include paired membranes and double-membrane vesicles (DMVs [7, 8; for a review, see reference 9]). Coexpression of the nsp2 and nsp3 transmembrane subunits of the EAV replicase induces the formation of similar double-membrane structures (10). Recently, a role in modulating RO membrane curvature and DMV formation was proposed for the third transmembrane replicase subunit, nsp5 (9). The core of the arteriviral RNA-synthesizing machinery, sometimes also referred to as the replication and transcription complex (RTC), is formed by ORF1b-encoded nsps like nsp9 and nsp10, which include the RNA-dependent RNA-polymerase (RdRp) and helicase (Hel) domains, respectively. In addition, host factors appear to be required to promote efficient viral RNA synthesis (11). The RTC directs viral genome replication as well as the synthesis of a nested set of subgenomic (sg) mRNAs that is used to express the genes encoding the viral structural proteins, which are located in the 3′-proximal quarter of the genome (reviewed in reference 6).
FIG 1.

Positions of CsA resistance-associated mutations in the EAV 5′ UTR and ORF1ab. (A) Map of CsA resistance-associated mutations identified in the consensus sequence of replicates CsA-1, -2, and -3 after seven passages. Depicted are the 5′ UTR and ORF1ab, with replicase polyprotein 1ab amino acid numbers, cleavage sites, and nsp cleavage products indicated. The seven mutations identified in the sequences of the CsA-resistant viruses are indicated with black arrows, and the amino acid position/change in the respective nsp is indicated, as well as the replicate in which the mutation was identified (see also Table 2). nsp5 is highlighted in green. P1 and P2, papain-like proteinases in nsp1 and nsp2, respectively; TM, transmembrane domains; Mpro, main proteinase; NT, nidovirus RdRp-associated nucleotidyltransferase (NiRAN); RdRp, RNA-dependent RNA polymerase; Z, zinc-binding domain; Hel, helicase; U, endoribonuclease; RFS, ribosomal frameshift site. (B) Membrane topology of EAV nsp5 as predicted using the TMHMM topology prediction method within the Geneious software package. The position/change of the four nsp5 mutations identified in replicates CsA-1, -2, and -3 is indicated with a black arrow. (C) Multiple-sequence alignment of nsp5 from selected arteriviruses, performed by Clustal Omega (51). Fully conserved residues are indicated in gray, predicted transmembrane helices (TMHMM topology prediction from Geneious software package) are boxed, and adaptive mutations are marked with an asterisk. EAV, equine arteritis virus (GenBank accession number DQ846750); SHFV, simian hemorrhagic fever virus (AF180391); PRRSV-1, porcine reproductive and respiratory syndrome virus, European genotype (GU737264.2); PRRSV-2, porcine reproductive and respiratory syndrome virus, North American genotype (JX138233); LDV, lactate dehydrogenase-elevating virus (U15146).
Multiple laboratories, including our own, have shown that in cell culture-based infection models, low micromolar concentrations of the FDA-approved cyclophilin inhibitor cyclosporine (CsA) inhibit the replication of a wide variety of nidoviruses, including the arteriviruses EAV and PRRSV (12) as well as SARS-CoV, MERS-CoV, and other CoVs (13–16). CsA inhibits members of the cyclophilin (Cyp) family, which are peptidyl-proline isomerases (PPIases) that act as chaperones and facilitate protein folding and function (reviewed in references 17 and 18). However, CsA also has immunosuppressive properties (19) that would constitute a highly undesirable side effect in the context of antiviral therapy. Therefore, numerous nonimmunosuppressive Cyp inhibitors have been developed. One of these, alisporivir (ALV), has been identified as a potent inhibitor of hepatitis C virus (HCV) and other RNA viruses (reviewed in references 20 and 21). Previously, we showed that also nidovirus replication is sensitive to ALV treatment, using drug concentrations comparable to the half-maximal effective concentration (EC50) previously determined for CsA (22).
The most ubiquitously expressed member of the Cyp family is the cytosolic CypA, which has been proven to be an essential host factor in the replication of, e.g., HCV, human immunodeficiency virus type 1 (HIV-1), and West Nile virus (reviewed in reference 23), as well as the coronaviruses HCoV-NL63 and HCoV-229E (24, 25). However, other coronaviruses seem to be much less affected by depletion of CypA in the infected cell (12, 13, 26). EAV replication, on the other hand, strongly depends on CypA expression, as it was nearly abolished in CypA knockdown or knockout cell cultures (12, 26).
Our present study aimed to investigate the molecular details of the inhibition of arterivirus replication by cyclophilin inhibitors. To this end, we first passaged EAV in the presence of inhibitory CsA concentrations to induce resistance to the compound. Subsequently, adaptive mutations that markedly decreased the sensitivity of EAV replication to CsA treatment were found to map to nsp5, one of the transmembrane subunits of the arterivirus replicase. We could show that CsA treatment interferes with EAV RNA synthesis and that the CsA resistance mutations in nsp5 render replication, and in particular RNA synthesis, less sensitive to CsA treatment. Furthermore, we show that resistance to the closely related Cyp inhibitor ALV requires an additional adaptive mutation in nsp2, a second transmembrane subunit of the arterivirus replicase that is essential for both polyprotein processing and RO formation. Our studies implicate both nsp2 and nsp5 in the mechanism underlying the sensitivity of arterivirus replication to cyclophilin inhibitor treatment. Moreover, they suggest that the role of cyclophilins as arterivirus host factors is specifically linked to viral RNA synthesis, possibly through interactions with key players in RO formation.
(This article was submitted to an online preprint archive [27].)
RESULTS
Selection of CsA-resistant EAV mutants.
Previously, we showed that low micromolar CsA concentrations can effectively block EAV and PRRSV replication in cultured cells (12). In this study, we aimed to investigate the mechanism of action by which arterivirus infection is inhibited in the presence of this cyclophilin inhibitor. To this end, wild-type EAV (EAVwt; strain Bucyrus; multiplicity of infection [MOI], 0.005) was serially passaged in Huh7 cells in the presence of increasing concentrations of CsA (4 to 20 μM). Three independent replicates were generated (named EAV CsA-1 to -3), which exhibited clearly decreasing sensitivity to CsA (Fig. 2A). By passage 7 (P7), all three replicates grew in the presence of 20 μM CsA (data not shown), a dose that is approximately 10-fold higher than the EC50 value observed when using EAVwt (Fig. 2B) (12). The CsA sensitivity of the three replicates was measured in an assay that is based on (the inhibition of) EAV-induced cytopathic effect (CPE) in Huh7 cells. Following infection with an MOI of 0.05 in the presence of 0.4 to 50 μM CsA, cell viability was assayed at 3 days postinfection (p.i.). CsA doses above 12.5 μM appeared to be cytotoxic, as the viability of mock-infected CsA-treated cells dropped below 85% that of Huh7 cells not treated with CsA (Fig. 2B, light gray line). The CPE induced by EAVwt infection was inhibited by CsA with an EC50 value of 2.4 μM, whereas treatment with 3.1 μM CsA fully blocked the development of virus-induced CPE. This was in line with our previous results for EAV-infected BHK-21 cells, in which treatment with 4 μM CsA resulted in a 4-log reduction of viral progeny (12). While seven passages in the absence of CsA did not alter the drug sensitivity of the EAVwt control (EC50, 2.1 μM), P7 virus of the CsA-treated EAV replicates CsA-1 to -3 had become 5-fold less sensitive to the compound (Fig. 2B and Table 1). Of note, additional passaging (up to passage 9) did not decrease CsA sensitivity any further.
FIG 2.
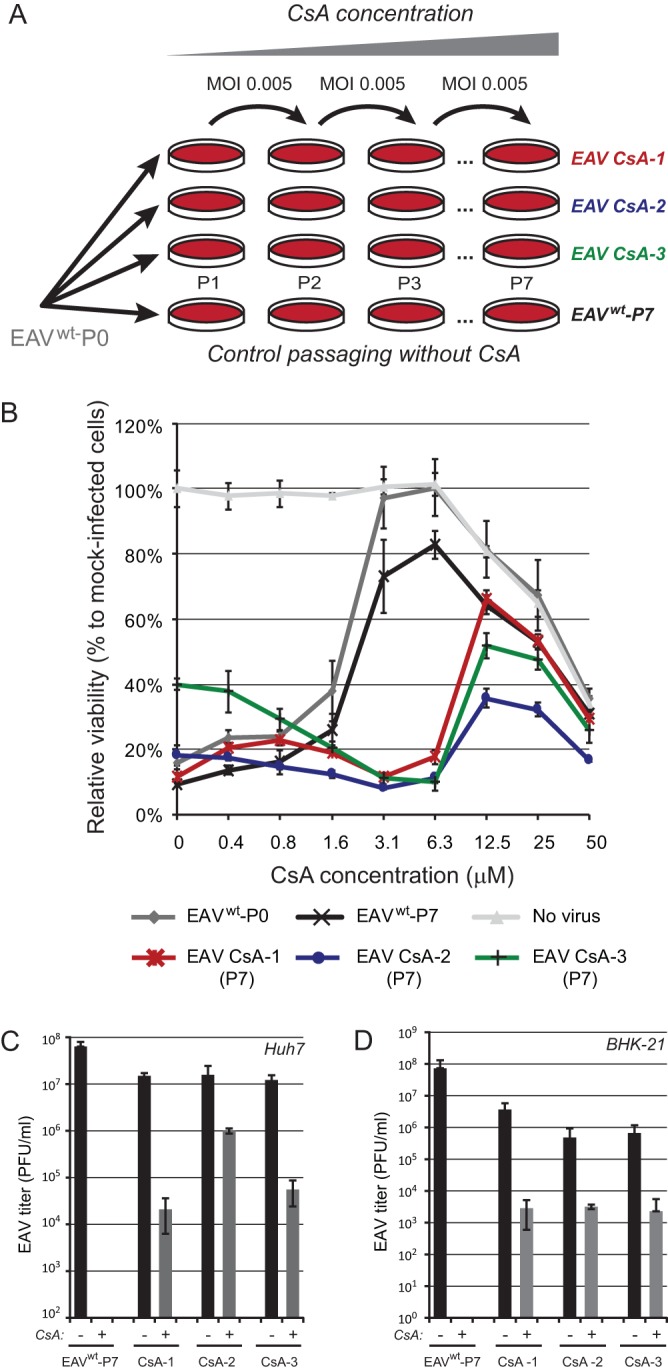
Selection of CsA-resistant EAV mutants. (A) Passaging scheme for wild-type (wt) EAV (strain Bucyrus) in Huh7 cells (MOI, 0.005) to generate three independent CsA-resistant replicates (EAV CsA-1, CsA-2, and CsA-3). Passaging was performed in the presence of increasing CsA concentrations, ranging from 4 μM during passage 1 (P1) to 20 μM during P7. As a control, EAVwt was passaged in the absence of CsA to assess the acquisition of mutations unrelated to CsA resistance. (B) After P7, EAV CsA-1 (red), CsA-2 (blue), and CsA-3 (green) were tested for CsA resistance, while including the original (P0, gray) and passaged (P7, black) EAVwt as controls. Huh7 cells in 96-well plates were infected at an MOI of 0.05 in the presence of 0 to 50 μM CsA. Cells were incubated for 3 days and cell viability was monitored using a commercial assay. Furthermore, the cytotoxicity of CsA treatment only was monitored in parallel in mock-infected Huh7 cells (light gray). Graphs show the results (averages and SD) of a representative experiment performed in quadruplicate. All experiments were repeated at least twice. Huh7 cells (C) or BHK-21 cells (D) were infected with EAV CsA-1, -2, or -3 or with lineage EAVwt P7 (MOI, 0.01). At 1 h p.i., the inoculum was replaced by medium containing either 0.02% DMSO (bars labeled “−”; solvent control) or 4 μM CsA (bars labeled “+”). Medium was collected at 32 h p.i., and EAV progeny titers were determined by plaque assay.
TABLE 1.
CsA sensitivities of serially passaged and engineered CsA-resistant viruses
| Virus | EC50 | Fold resistanceb | Remarks |
|---|---|---|---|
| Wild-type EAV control | |||
| EAVwt | 2.4 ± 0.5 | 1.0 | |
| 2.1 ± 0.2 | 1.0 | ||
| rEAVwt | 1.6 ± 0.1 | 1.0 | |
| EAV CsA-1 | |||
| Passage 7 virus | ∼10.9a | 5.1 | |
| rEAV nsp5Q21R | 4.4 ± 0.2 | 2.9 | |
| rEAV nsp5Y113H | 4.4 ± 0.3 | 2.8 | |
| rEAV nsp5A134V | 4.5 ± 0.3 | 2.9 | |
| rEAV nsp5Y113H+A134V | 9.1 ± 0.6 | 5.9 | Acquisition of second site reversion nsp5 Q21R or Q21H |
| rEAV nsp5Q21R+Y113H+A134V (rEAVQYA) | 7.2 ± 0.7 | 4.7 | |
| EAV CsA-2 | |||
| Passage 7 virus | ∼11.0a | 5.2 | |
| rEAVA43G | 1.8 ± 0.1 | 1.2 | |
| rEAV nsp5Y113H | 4.4 ± 0.3 | 2.8 | |
| rEAV nsp10Y387H | 1.6 ± 0.1 | 1.1 | |
| rEAVA43G + nsp5Y113H + nsp10Y387H | 4.6 ± 0.3 | 3.0 | |
| EAV CsA-3 | |||
| Passage 7 virus | ∼10.2a | 4.8 | |
| rEAV nsp2A199T | 1.7 ± 0.1 | 1.1 | |
| rEAV nsp5L8S | 2.2 ± 0.1 | 1.4 | Reversion to wt nsp5-L8 |
| rEAV nsp5Q21R | 4.4 ± 0.2 | 2.9 | |
| rEAV nsp5L8S+Q21R | 9.1 ± 1.4 | 5.9 | Reversion to wt nsp5-L8 |
| rEAV nsp2A199T + nsp5L8S+Q21R | 8.7 ± 0.5 | 5.6 | Reversion to wt nsp5-L8 |
The EC50 is an estimated value (labeled “ambiguous” during GraphPad data analysis), due to the fact that the EC50 approached the CC50 for CsA in Huh7 cells, leaving only a few data points between the two values.
Fold resistance of the P7 viruses relative to EAVwt; for the engineered rEAV ALV mutants, fold resistance was calculated relative to the rEAVwt value.
Next, we confirmed the reduced CsA sensitivity of CsA-1 to -3 P7 virus by infecting (MOI, 0.01) Huh7 or BHK-21 cells in the presence of 4 μM CsA, a dose that fully inhibits EAVwt replication. Virus production was measured at 48 h p.i., which confirmed that EAVwt P7 infection in the presence of 4 μM CsA indeed did not yield detectable virus progeny (Fig. 2C). At the same CsA concentration, titers of above 104 PFU/ml (EAV CsA-1 and CsA-3) or even around 106 PFU/ml (EAV CsA-2) were reached when infection was done with the P7 viruses from the three CsA-resistant replicates. Similar results were obtained with BHK-21 cells, although that the yields of all three replicates were equally reduced compared to that of EAVwt P7 in this cell line (data not shown).
Most CsA resistance-associated mutations map to EAV nsp5.
In order to identify CsA resistance-associated mutations, the consensus genome sequence of the three P7 CsA-resistant EAV samples was determined following reverse transcription-PCR (RT-PCR) amplification (Table 2 and Fig. 1A). The EAV CsA-1 consensus sequence contained three mutations, all mapping to the nsp5-coding region of ORF1a and resulting in three amino acid substitutions: Q21R (CAA→CGA; nucleotides [nt] 4089 to 4091; mixed with the wt sequence), Y113H (UAC→CAC; nt 4365 to 4367), and A134V (GCU→GUU; nt 4428 to 4430). Remarkably, the Y113H mutation in nsp5 was also encoded by the ORF1a consensus sequence of the EAV CsA-2 population, but now accompanied by a mutation in the 5′ untranslated region (5′ UTR) (A43G) and one in ORF1b, specifying a mutation in the nsp10 helicase domain (Y387H). Replicate CsA-3 contained mutations leading to substitutions in nsp2 (A199T) and again nsp5 (L8S and Q21R). None of the consensus sequences contained any synonymous mutations that had been fixed during passaging. Likewise, no mutations were discovered in the consensus sequence of the EAVwt P7 control, implying that the mutations identified in the CsA-1 to -3 P7 viruses potentially are linked to the established reduced sensitivity to CsA.
TABLE 2.
Mutations identified after serial passaging of EAV in the presence of increasing concentrations of CsA or ALV
| Virus | Mutation in: |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| 5′ UTR | nsp1 | nsp2 | nsp5 |
nsp10 | GP2b | GP4/OR5a protein | |||
| CsA-treated replicates | |||||||||
| EAVwt P7 | |||||||||
| EAV CsA-1 P7 |
Q21R (mixa
) (CAA→CGA) |
Y113H (UAC→CAC) |
A134V (GCU→GUU) |
||||||
| EAV CsA-2 P7 | nt 43 (A→G) |
Y133H (UAC→CAC) |
Y387H (UAC→CAC) |
||||||
| EAV CsA-3 P7 |
A199T (GCU→ACU) |
L8S (UUG→UCG) |
Q21R (CAA→CGA) |
||||||
| ALV-treated replicates | |||||||||
| EAVwt P10 | |||||||||
| EAV ALV-1 P10 | nt 45 (A→G) |
H114R (CAC→CGC) |
Y113H (UAC→CAC) |
A134V (GCU→GUU) |
Y151C/I14V (TATC→TGTC)b |
||||
| EAV ALV-2 P10 | nt 47 (U→C) |
T39N (ACC→AAC) |
H114R (CAC→CGC) |
Q21R (CAA→CGA) |
M62T (AUG→ACG) |
H11L (CAT→CTT) |
|||
Two variants were detected: nsp5 Q21 (wild type) and R21 (mutated sequences).
The open reading frames for EAV GP4 and the ORF5a protein overlap.
To assess the importance of the identified mutations for CsA sensitivity, each mutation was individually reverse engineered into the EAV genome, using full-length cDNA clone pEAN900. Recombinant mutant viruses were launched and their CsA sensitivity was assayed in Huh7 cells using the CPE-based assay outlined above (Fig. 1B). Interestingly, three of the nsp5 mutations (Q21R, Y113H, and A134V) decreased the CsA sensitivity of EAV replication (EC50 values of ∼4.4 μM versus 1.6 μM for the wt control [rEAVwt]). No change in sensitivity was observed for any of the other mutations (Table 1; see also Fig. S1A to H in the supplemental material). Of note, in all mutant viruses carrying the nsp5L8S mutation (from CsA-3), this substitution readily reverted to the wild-type residue within the time frame of the CPE-based assay (data not shown). This most likely explains the marginal decrease in CsA sensitivity observed in this assay for these viruses (Fig. S1C).
The increase in CsA resistance observed for the recombinant mutant viruses with singular mutations appeared to be ∼3-fold smaller than the approximately 5-fold increase observed for the P7 CsA-1 to -3 viruses, which each carried multiple substitutions. Therefore, we also engineered mutant viruses with genome sequences identical to the consensus sequence of these three CsA-resistant virus replicates. For replicate CsA-1, which contained a mix of Q (wt) and R (mutant) at position 21 of nsp5, both variants were engineered and tested. Compared to the viruses carrying singular mutations, mutant rEAV nsp5Y113H+A134V displayed a decreased sensitivity to CsA, which now approached that of the EAV CsA-1 replicate (Fig. 3A and Table 1). When the Q21R mutation was added, the CsA resistance diminished slightly compared to the Y113H A134V double mutant (Fig. 3B and Table 1; EC50 of 9.1 versus 7.2 μM). However, upon passaging and resequencing (data not shown), the double mutant appeared to be genetically instable, which may also explain the mixed sequence found at nsp5 codon 21 in the CsA-1 consensus sequence. For replicate CsA-2, combining the three mutations (in the 5′ UTR, nsp5, and nsp10) into the same genome did not further improve CsA resistance compared to that of rEAV nsp5Y113H (Fig. 3C). This lack of a synergistic effect indicates that the mutations in the 5′ UTR and nsp10 do not contribute substantially to the level of CsA resistance of CsA-2. In the case of EAV CsA-3, combining nsp5L8S, but not nsp2A199T, with nsp5Q21R yielded an ∼2-fold further decrease in CsA sensitivity (EC50 value of 9.1 μM; compare Fig. S1B and E and Fig. 3D and see Table 1), suggesting that the nsp2A199T mutation does not contribute to CsA resistance.
FIG 3.

CsA resistance analysis of engineered rEAV mutants reflecting the consensus sequences of CsA-1, -2, and -3. Huh7 cells in 96-well plates were infected (MOI, 0.05) with rEAVwt (control; black line) or mutants reflecting the consensus sequences of CsA-1, -2, and -3. Cells were incubated for 3 days in the presence of 0 to 50 μM CsA, and cell viability was monitored using a commercial assay. The cytotoxicity of CsA treatment only was monitored in parallel in mock-infected Huh7 cells (light gray line). Above each panel, the EC50 value of CsA inhibition is indicated (see also Table 1). Graphs show the results (average and SD) of a representative experiment that was performed in quadruplicate. All experiments were repeated at least twice.
Overall, the above results clearly implicated various EAV nsp5 mutations in reduced CsA sensitivity. Whereas combining nsp5 mutations increased CsA resistance compared to that of singular nsp5 mutants, the substitutions in the 5′ UTR, nsp2, and nsp10 did not contribute detectably to CsA resistance. CsA-1 mutant rEAV nsp5Q21R+Y113H+A134V was selected for follow-up experiments, as it was genetically stable for at least two additional passages (data not shown) and included the single nsp5 mutation (Y113H) that was key to the CsA resistance of replicate CsA-2. For simplicity, this mutant is referred to as rEAVQYA from this point forward.
rEAVQYA replicates in the presence of CsA.
The CsA resistance of rEAVQYA was analyzed in more detail by infecting Huh7 cells in the presence or absence of 4 μM CsA. Upon high-MOI inoculation (MOI, 3), the replication kinetics of rEAVwt and rEAVQYA in the absence of CsA were very similar (Fig. 4, solid lines). Progeny was released from rEAVwt- and rEAVQYA-infected cells from 12 h p.i. onwards, with rEAVQYA titers being only slightly reduced compared to those for rEAVwt. In the presence of 4 μM CsA, rEAVwt was essentially unable to replicate (Fig. 4, black dashed line) while rEAVQYA was able to produce infectious progeny, although with a delay compared to the CsA-free infection (Fig. 4, red dashed line).
FIG 4.
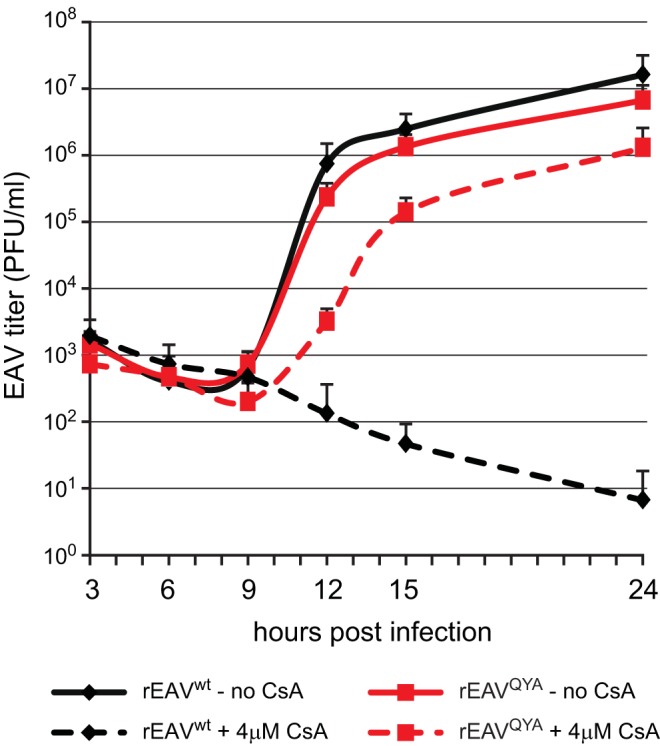
Virus yields of rEAVwt and rEAVQYA infection in the absence or presence of CsA. Shown are growth kinetics of rEAVwt and rEAVQYA in Huh7 cells (MOI, 3). Infections were performed in the absence or presence of 4 μM CsA. EAV yields at the indicated time points were determined by plaque assay (averages and SD are given [n = 3]).
Neither CsA treatment nor nsp5 mutations affect the morphology of EAV-induced replication organelles.
It was previously reported that Cyp inhibitor treatment affects the morphology of the “membranous web,” the RO that is formed upon HCV infection and with which viral RNA synthesis is associated (28, 29). It was striking that the EAV mutations that promote CsA resistance all map to the nsp5 transmembrane protein, which has been suggested to play a regulatory role in the formation of arterivirus ROs (9), of which the peculiar DMVs are a key feature (7, 30). We therefore used electron microscopy (EM) to investigate whether CsA treatment or the mutations in nsp5 affect EAV-induced DMV formation. Analysis of Huh7 cells (11 h p.i.) infected with either rEAVwt (Fig. 5B) or rEAVQYA (Fig. 5C) did not reveal obvious differences in DMV amounts, size, or ultrastructure. As replication of rEAVQYA in the presence of CsA was delayed compared to the replication in untreated cells, we analyzed cells infected with this mutant at 14 h p.i. (Fig. 4). In the presence of CsA, no DMVs were observed for rEAVwt (Fig. 5E), while the morphology and size of the DMVs in rEAVQYA-infected cells were similar to the structures observed upon infection of untreated cells infected with rEAVwt or rEAVQYA (Fig. 5F). Notably, severe compound-induced dilation of endoplasmic reticulum (ER) membranes was observed, a phenomenon that was independent of EAV infection, as it was also observed in CsA-treated mock-infected Huh7 cells (compare Fig. 5A to C and D to F). Taken together, these observations strongly suggest that CsA treatment interferes with a very early stage of arterivirus infection, i.e., prior to or during RO formation. Apparently, adaptive mutations in nsp5 can compensate for the detrimental effects of CsA treatment, which may either target one of the viral players directly or affect a host factor involved in arterivirus replication, like cyclophilin A (see above).
FIG 5.

Adaptive mutations in EAV nsp5 do not affect the morphology of virus-induced double membrane vesicles. Huh7 cells were infected with rEAVwt (B and E) or rEAVQYA (C and F) at an MOI of 5 or were mock infected (A and D). After an incubation of 11 h (untreated [A to C]) or 14 h (CsA treated [D to F]), cells were fixed and processed for transmission electron microscopy. DMV formation is very similar upon infection with rEAVwt and rEAVQYA, while CsA treatment induces dilated ER both in mock-infected and in infected cells. Scale bars: 2 μm (left images) and 500 nm (middle and right images).
rEAVQYA replication continues to depend on CypA.
We previously showed that EAV replication was reduced in cells depleted of CypA by small interfering RNA (siRNA) treatment (12, 26) and strongly affected in a CypA knockout cell line (Huh7-CypAKO) produced using CRISPR-Cas9 technology (26). The latter cells were used to investigate whether the replication of rEAVQYA still depended on CypA expression. We infected both the Huh7-CypAKO cells and the parental Huh7 cells with wt and mutant virus (MOI, 0.01) and compared progeny titers at 32 h p.i. At this time point, rEAVwt yields were almost 4-log reduced in the knockout cells (Fig. 6). Replication of rEAVQYA was inhibited to the same extent, indicating that despite its reduced CsA sensitivity, replication of this mutant continued to depend on the presence of CypA (Fig. 6).
FIG 6.
rEAVQYA does not replicate in the absence of CypA. (A) Virus yields of rEAVwt and rEAVQYA (MOI, 0.01) at 32 h after infection of parental Huh7 cells and Huh7 CypAKO cells. EAV yields were determined by plaque assay (averages and SD [n = 3]).
nsp5 mutations reduce the inhibitory effect of CsA on EAV RNA synthesis.
Previously, we showed that EAV RNA synthesis is effectively inhibited by CsA (12). This led us to analyze whether the nsp5QYA mutations reduce the inhibitory effect of CsA on EAV RNA synthesis. For this purpose, BHK-21 cells were infected with rEAVwt or rEAVQYA and viral RNA synthesis was assayed by metabolic labeling with [3H]uridine from 6.5 to 7.5 h p.i. (Fig. 7A and B). Cells were also treated with dactinomycin to block cellular transcription. [3H]uridine incorporation into viral RNA was quantified by isolation of intracellular RNA and liquid scintillation counting (Fig. 7A). This revealed that RNA synthesis by the mutant virus was approximately 40% of wt virus activity, which is in line with the observed delayed growth kinetics and reduced titers of the mutant. However, in the presence of 8 μM CsA (which was added 1 h prior to the pulse-labeling), rEAVwt RNA synthesis was completely blocked, while rEAVQYA RNA synthesis was hardly affected (Fig. 7A).
FIG 7.

rEAVQYA RNA synthesis is impaired but less sensitive to CsA. (A and B) Viral RNA synthesis in BHK-21 cells infected with rEAVwt or rEAVQYA was metabolically labeled between 6.5 and 7.5 h p.i. using [3H]uridine. This was done in the presence or absence of 8 μM CsA and dactinomycin. Total intracellular RNA was isolated at 7.5 h p.i. (A) The total incorporation of 3H label was quantified by liquid scintillation counting. (B) ROs were semipurified from lysates of rEAVwt- or rEAVQYA-infected BHK-21 cells (MOI, 5) at 7.5 h p.i. and were used in an in vitro RNA synthesis assay (IVRA) in which [32P]CTP was incorporated into viral RNA products. Reactions were performed in the presence of increasing concentrations of CsA (indicated above the lanes) and were terminated after 100 min. Labeled RNA products were isolated, separated in a denaturing formaldehyde agarose gel, and visualized by phosphorimaging. The positions of the genomic RNA (RNA1) and subgenomic RNAs (positions 2 to 7) are indicated on the left side of the gel. (C) Hybridization analysis of RNA synthesis in rEAVwt- and rEAVQYA-infected cells. Intracellular RNA was isolated at 7.5 h p.i. from rEAVwt- and rEAVQYA-infected BHK-21 cells and analyzed in a denaturing formaldehyde agarose gel. The EAV RNA was visualized by hybridization to a 32P-labeled oligonucleotide probe (see Materials and Methods) complementary to the 3′ end of EAV genome and sg mRNAs. The positions of the genomic RNA (RNA1) and subgenomic mRNAs 2 to 7 are indicated on the left side of the gel. Subgenomic RNA abundance was measured by phosphorimaging-based quantification of RNA bands and is given relative to the abundance of RNA1, which was placed at 100%.
Next, we assayed the RNA-synthesizing activity of semipurified ROs from rEAVwt- and rEAVQYA-infected cells using a previously developed in vitro RNA synthesis assay (IVRA) (11). The incorporation of [32P]CTP into viral RNA was analyzed in the presence of various CsA concentrations. In the absence of the compound, in vitro synthesis of rEAVwt genomic and sg RNAs was observed (Fig. 7B, lane 1), which was clearly reduced when the assay was performed in the presence of ≥8 μM CsA (lanes 3 to 6). In line with the [3H]uridine metabolic labeling experiment, the RNA-synthesizing complexes from rEAVQYA-infected cells were insensitive to treatment with up to 16 μM CsA (Fig. 7B, lanes 7 to 12), thus directly linking the effect of the adaptive nsp5 mutations to the overall activity of the arterivirus RTC.
To analyze the RNAs produced by wt and mutant viruses, intracellular RNA from rEAVwt- and rEAVQYA-infected BHK-21 cells (isolated at 7.5 h p.i.) was subjected to a hybridization analysis using a 32P-labeled probe that recognizes all EAV mRNAs (Fig. 7C). An overall decrease in the amount of mutant viral RNAs was visible, but the relative abundances of individual sg mRNAs and genomic RNA were similar, indicating that the resistance-associated mutations resulted in a general RNA synthesis defect.
Resistance to the nonimmunosuppressive CsA analog alisporivir requires a combination of mutations in EAV nsp5 and nsp2.
Previously, we established that EAV replication can also be inhibited by the nonimmunosuppressive CsA analog Debio-064 (12). More recently, we reported the inhibition of coronavirus replication in cell culture by the related CsA analog alisporivir (ALV) (22), a drug that was explored as a host-directed antiviral treatment option for chronic HCV infection (21). ALV lacks the immunosuppressive properties of CsA, while retaining a high affinity for cyclophilins. We established that ALV is able to block also the replication of EAVwt with an EC50 value of 4.5 ± 0.2 μM, an efficiency that is comparable to that observed for CsA (Table 3).
TABLE 3.
ALV sensitivities of serially passaged and engineered ALV-resistant viruses
| Virus | EC50 | Fold resistanceb |
|---|---|---|
| Wild-type EAV control | ||
| EAVwt | 4.5 ± 0.2 | 1.0 |
| rEAVwt | 2.3 ± 0.3 | 1.0 |
| EAV ALV-1 | ||
| Passage 10 virus | 33.8 ± 5.8 | 7.4 |
| rEAV nsp2H114R | 4.6 ± 0.1 | 2.0 |
| rEAV nsp5A134V | 4.3 ± 0.1 | 1.9 |
| rEAV nsp2H114R + nsp5A134V | 8.9 ± 0.3 | 3.9 |
| rEAV nsp2H114R + nsp5A134V + GP4Y151C | 9.7 ± 0.5 | 4.2 |
| EAV ALV-2 | ||
| Passage 10 virus | ∼12.7a | 2.8 |
| rEAV nsp2H114R | 4.6 ± 0.1 | 2.0 |
| rEAV nsp5Q21R+M62T | 3.5 ± 0.2 | 1.9 |
| rEAV nsp2H114R + nsp5Q21R+M62T | 14.3 ± 1.0 | 6.2 |
| rEAV nsp2H114R + nsp5Q21R+M62T + GP2H11L | 13.2 ± 5.5 | 5.8 |
| rEAVQYA + nsp2H114R | ∼20.0a | 8.7 |
The EC50 is an estimated value (labeled “ambiguous” during GraphPad data analysis), due to the fact that the EC50 approached the CC50 for ALV in Huh7 cells, leaving only a few data points between the two values.
Fold resistance of both P10 viruses relative to EAVwt; for the engineered rEAV ALV mutants fold resistance was calculated relative to the rEAVwt value.
To explore the development of ALV resistance in EAV, two independent passaging experiments in Huh7 cells were performed (EAV ALV-1 and ALV-2) in the presence of increasing ALV concentrations (from 4 μM in passage 1 to 75 μM in passage 10), similar to the approach used to generate CsA-resistant viruses (Fig. 2). By passage 10, a CPE-based assay revealed that compared to that of the EAVwt P10 virus, the sensitivity to ALV was reduced 7.4- and 2.8-fold for EAV replicates ALV-1 and ALV-2, respectively (Fig. 8A and Table 3). Interestingly, the P10 samples of EAV replicates ALV-1 and -2 were ∼4-fold less sensitive to CsA as well (Fig. 8B). Analysis of the ALV-1 and -2 P10 consensus sequences revealed that each population contained multiple mutations (Table 2). These included singular mutations in the 5′ UTR, in ORF1a (nsp1 region), and in the structural protein-coding region. Strikingly, the P10 viruses from both replicates also carried two substitutions in nsp5. The two nsp5 mutations in ALV-1 (Y113H and A134V) had previously been identified in replicate CsA-1 (see above), whereas ALV-2 also contained a mutation present in CsA-1, Q21R. Strikingly, both ALV-resistant replicates contained the same mutation (H114R) within the papainlike protease (PLP2) domain of nsp2. We reverse engineered the ALV-1 and -2 mutations into the viral genome and assayed ALV sensitivity of the mutant viruses. Singular nsp2 and nsp5 mutants displayed a reduced ALV sensitivity, but only the nsp5 mutants were also less sensitive to CsA (Fig. 9 and Table 3). Subsequently, combining nsp5 and nsp2 mutations had a synergistic effect and confirmed the importance of the common nsp2H114R substitution for ALV resistance. Mutations in the 5′ UTR and nsp1 did not further reduce the sensitivity to ALV (data not shown), nor did the mutations in the structural protein-coding region contribute to ALV resistance (Fig. 9 and Table 3).
FIG 8.

Passaging in the presence of ALV reduces EAV sensitivity toward both ALV and CsA. Virus was passaged at increasing ALV concentrations, ranging from 4 μM during passage 1 (P1) to 75 μM during P10. P10 harvests of EAV replicates ALV-1 and ALV-2 were tested for resistance to ALV (A) and CsA (B) treatment. Huh7 cells in 96-well plates were infected at an MOI of 0.05 in the presence of 0 to 50 μM ALV or CsA. Cells were incubated for 3 days, and cell viability was monitored using a commercial assay. In addition, the cytotoxicity of CsA treatment was monitored in parallel in mock-infected Huh7 cells. Graphs show the results (average and SD) of a representative experiment that was performed in quadruplicate. All experiments were repeated at least twice.
FIG 9.

Analysis of ALV and CsA sensitivities of engineered rEAV ALV-resistant mutants. Huh7 cells in 96-well plates were infected with rEAVwt (control) and rEAV ALV-resistant mutants (MOI, 0.05), grouped per ALV replicate, in the presence of 0 to 50 μM ALV (A and B) or CsA (C and D). Cells were incubated for 3 days and cell viability was monitored using a commercial assay. The cytotoxicity of ALV or CsA treatment only was monitored in parallel in mock-infected Huh7 cells (light gray line). Graphs show the results (average and SD) of a representative experiment that was performed in quadruplicate. All experiments were repeated at least twice.
Finally, to further corroborate the importance of nsp2H114R for ALV resistance, this mutation was transferred to the rEAVQYA triple mutant already carrying the nsp5 mutations identified in our studies into CsA resistance (see above). As anticipated, while the triple mutant was marginally resistant to ALV (most likely due to the presence of the nsp5A134V mutation; see Fig. 10A), the addition of nsp2H114R rendered the rEAVQYA virus nearly insensitive to ALV treatment, while also retaining its reduced CsA sensitivity (Fig. 10). These results clearly confirmed that the highest level of ALV resistance is achieved by introducing a combination of adaptive mutations in EAV nsp2 and nsp5.
FIG 10.
EAV resistance to ALV requires an additional mutation in nsp2. (A and B) Huh7 cells in 96-well plates were infected with rEAVwt (control) and the rEAV mutants rEAV-nsp2H114R, rEAVQYA, and rEAVQYA-nsp2H114R in the presence of 0 to 50 μM ALV (A) or CsA (B). Cells were incubated for 3 days and cell viability was monitored using a commercial assay. In addition, the cytotoxicity of ALV or CsA treatment only was monitored in parallel in mock-infected Huh7 cells (light gray line). Graphs show the results (average and SD) of a representative experiment that was performed in quadruplicate. All experiments were repeated at least twice.
DISCUSSION
This study demonstrates that adaptive mutations in the transmembrane replicase subunit nsp5 can reduce the sensitivity of EAV replication to CsA treatment. Independently, experiments using the CsA analog ALV pointed in the same direction, in addition to implicating nsp2 in ALV sensitivity and resistance. Substitutions in nsp5 and nsp2 enable EAV to tolerate CsA or ALV concentrations that would normally block virus replication (Fig. 2, 3, 9, and 10). The largest EC50 increase, about 5-fold, was achieved when multiple substitutions in nsp5 (and nsp2, in the case of ALV) were combined (Tables 1 and 3). We previously showed that CsA inhibits EAV RNA synthesis and hypothesized that this may be the major determinant for its inhibitory effect (12). Indeed, a combination of three of the acquired CsA resistance mutations in nsp5 (Q21R, Y113H, and A134V) facilitated a certain level of EAV RNA synthesis in the presence of a CsA dose that blocks wt virus activity. This was observed in both virus-infected cells and an in vitro assay for EAV RNA synthesis (Fig. 7).
Unfortunately, arterivirus nsp5 has not been studied in great detail, and little is known about its functions and interactions during virus replication. The protein is one of three ORF1a-encoded transmembrane subunits that are presumed to drive the formation of the membranous ROs supporting viral RTC activity in the infected cell (7, 9, 30). In expression systems, nsp2 and nsp3 interact and their combined expression suffices to induce the conversion of ER membranes into double-membrane vesicles that strikingly resemble those observed in arterivirus-infected cells. Although nsp5 appears to be dispensable for the basic interactions leading to these membrane transformations (10), the protein was recently postulated to play a regulatory role by modulating membrane curvature and DMV formation (9).
The 162-amino acid nsp5 of EAV is largely hydrophobic and contains four predicted membrane-spanning domains and a cytosolic C-terminal domain (Fig. 1) (9). In infected cells, nsp5 is mainly present within a number of long-lived processing intermediates of the pp1a and pp1ab replicase polyproteins (31, 32), in particular nsp5-7 and nsp3-8 but also larger intermediates. The latter extend into the ORF1b-encoded part of the replicase polyprotein, which includes key enzymes for arterivirus RNA synthesis (31, 32). Consequently, nsp5 may have an important role as an “interaction platform” that could target and/or anchor other RTC subunits to membranes. Being part of several larger processing intermediates, nsp5 may potentially perform such a role in cis, prior to the proteolytic release of those other subunits from nsp5-containing precursors. Obviously, also host cell proteins involved in arterivirus RNA synthesis are likely to become associated with the membrane-bound viral RTC and may thus interact with nsp5. Such factors may include CypA and other members of the Cyp family that bind CsA and ALV, although these compounds may also directly target EAV replicase subunits. In any case, our EM analysis of CsA-treated, EAV-infected cells, which were devoid of DMVs (Fig. 5), confirmed that the drug treatment blocks an early and basic step in the viral replicative cycle and that this block can apparently be circumvented by the acquisition of a few adaptive mutations in nps5.
Four substitutions in nsp5 were implicated in CsA resistance (L8S, Q21R, Y113H, and A134V), of which only the L8 residue is conserved among arteriviruses (Fig. 1C). Interestingly, this residue is also part of a potential CypA binding motif (GPxL, as identified in reference 33), constituting a potential interaction site between the two proteins. However, during infection, this L8S mutation quickly reverted, and therefore the contribution of the L8S substitution to CsA resistance remains unclear. All other three nsp5 mutations (Q21R, Y113H, and A134V) are present in predicted transmembrane domains (Fig. 1B) and therefore may not be directly involved in interactions of nsp5 with Cyps or other proteins. There are no experimental data available on the membrane topology of nsp5, but upon in silico modeling these nsp5 mutations did not appreciably alter the predicted conformation of the various transmembrane domains (data not shown). In addition, our EM analysis of DMV formation upon rEAV nsp5QYA infection did not reveal any ultrastructural differences in either the absence or presence of CsA (Fig. 5). Clearly, more detailed information on the three-dimensional (3D) structure of nsp5 is needed to assess the impact of the mutations identified in this study on the protein’s conformation(s) and interactions at the molecular level.
Efficient EAV replication is dependent on the presence of CypA, as it is severely reduced in cells depleted of this host factor (Fig. 6) (12, 26). We previously postulated a role for CypA in EAV RTC formation or function, upon demonstrating the cosedimentation of CypA with EAV RTCs in density gradients. Interestingly, no CypA cosedimentation with RTCs was detected in the presence of CsA (12). Unfortunately, coimmunoprecipitation experiments using lysates from either ectopic expression of the EAV nsp2-7 polyprotein in 293T cells or EAV-infected cells could not confirm an interaction between nsp5 and CypA, even when using mild conditions for cell lysis and immunoprecipitation (data not shown). This could obviously be due to technical reasons, e.g., the interaction being very weak or short-lived, but an alternative explanation may be that the interaction of CypA with the EAV RTC is not directly via nsp5, or that it depends on factors that were lacking in our assays.
The fact that all relevant adaptive mutations in EAV nsp5 map inside the first or last of its four (predicted) transmembrane domains provides a link to studies with HCV and Cyp inhibitors. For this virus, it was postulated that the interaction between CypA and NS5A is involved in the formation and stabilization of the RO, the so-called membranous web with which viral RNA synthesis is associated (28, 29). A direct interaction has been demonstrated between CypA and the proline-rich domain 2 of NS5A (34, 35). Presumably, CypA catalyzes the proper folding of NS5A, which, in turn, enhances the direct binding of RNA to NS5A, or recruitment of the NS5B-RdRp to the membrane structures, ultimately leading to efficient RNA synthesis (36). HCV mutations associated with resistance to Cyp inhibitors map to NS5A, close to the cleavage site between NS5A and NS5B, and either slow down processing of the NS5A-5B junction or render replication less dependent on CypA (37). Interestingly, NS5A mutations that confer resistance to Cyp inhibitors do not reduce CypA-NS5A binding (38).
It has been shown that CsA can be inserted into and destabilize membrane structures, with a preference for sphingomyelin-rich membranes (39, 40). In addition, several reports have been published that show that CsA treatment induces the unfolded protein response (UPR) and endoplasmic reticulum (ER) stress (41, 42). Indeed, severe dilation of ER membranes was observed in CsA-treated Huh7 cells (compare, for instance, mock-infected cells in Fig. 5A and D). CsA may thus also affect the stability of virus-induced membrane structures and by doing so inhibit viral RNA synthesis, for example, by disrupting the integrity of the viral RTC or its association with the ROs. Integration of mutant nsp5 in EAV RTCs may stabilize the RTC complex and allow it to tolerate higher CsA concentrations before it disassembles and loses its RNA synthesis activity.
Interestingly, despite the fact that CsA and ALV both are cyclophilin inhibitors, EAV resistance to ALV requires an additional mutation in the nsp2 protease domain PLP2: H114R (Fig. 10). This suggests that compared to CsA, ALV has an additional target that is relevant for EAV replication, either directly or indirectly. Residue H114 is positioned in helix α4 in domain I of the PLP2 structure, which is distant from both the protease’s active site and the surface involved in ubiquitin binding (43), a feature linked to PLP2’s secondary function as a deubiquitinase involved in arteriviral innate immune evasion. Unfortunately, only the structure of the PLP2 domain of nsp2 has been resolved, and the overall fold and membrane topology of the subunit of EAV remain unclear. Our study implicates nsp5 in viral RNA synthesis, either directly or indirectly, and provides evidence that mutations in nsp5 are involved in reduced sensitivity to CsA, while the same mutations in combination with the nsp2 H114R substitution contribute to ALV resistance. Future studies should aim to elucidate the difference between the modes of action of ALV and CsA and the exact role of nsp5 and nsp2 in arterivirus sensitivity and resistance to Cyp inhibitors.
MATERIALS AND METHODS
Cell culture, EAV infection, and virus titration.
BHK-21 cells (44), Huh7 cells (14), and 293T cells (45) were cultured as described previously. A cell culture-adapted derivative of the EAV Bucyrus isolate (46) was used to infect BHK-21 and Huh7 cell monolayers at 37°C as described previously (9, 44, 47). EAV titers in cell culture supernatants were determined by plaque assay on BHK-21 cells (44). Huh7 cells lacking CypA expression (Huh7-CypAKO cells), generated by transfection with a pLentiCRISPR v2 plasmid and a CypA-specific guide RNA, were described previously (26).
Compounds.
The cyclophilin inhibitors CsA (Sigma) and alisporivir (Novartis and Debiopharm, Switzerland) were dissolved in dimethyl sulfoxide (DMSO) or ethanol, respectively. Both drugs were stored at −20°C as a 20 mM stock in aliquots for single use.
Isolation of drug-resistant EAV mutants.
To obtain CsA- or ALV-resistant mutants, EAV was passaged (MOI, 0.005) in Huh7 cells in the presence of increasing compound concentrations (4 to 20 μM for CsA and 4 to 75 μM for ALV). For CsA, passaging was repeated independently three times, resulting in EAV replicates CsA-1, CsA-2, and CsA-3. For ALV, two independent ALV-resistant replicates (EAV ALV-1 and ALV-2) were obtained. As a control, in parallel, virus was also passaged in the absence of CsA or ALV (EAVwt) to screen for adaptive mutations unrelated to drug treatment, which might arise during cell culture passaging of EAV. CsA or ALV concentrations were increased in the following passage at the moment that in drug-treated, EAV-infected cells, virus-induced CPE appeared at the same moment as in the EAVwt control, indicating that virus replication was not affected by that particular drug concentration. Drug-specific toxicity was monitored in cells that were left uninfected. Resistance to CsA or ALV was monitored using the cell-based antiviral screening assay described below.
Cell culture-based assay for inhibition of EAV infection.
Huh7 cells were seeded in transparent 96-well plates at a density of 104 per well. After overnight incubation at 37°C, 100 μl of 1.5× concentrated compound dilution and 50 μl of EAV inoculum in Dulbecco modified Eagle medium (DMEM)–2% fetal calf serum (FCS) were added to each well (MOI, 0.05). The final compound concentrations in the medium that were tested ranged from 0.4 to 50 μM. DMSO (0.25%, CsA) and ethanol (0.25%, ALV) were used as solvent controls. At 3 days postinfection (p.i.), differences in cell viability due to virus-induced cytopathic effect (CPE) or compound-specific side effects were analyzed using the CellTiter 96 AQueous nonradioactive cell proliferation assay (Promega), as described previously (14). The cytotoxic effects of treatment with compound only were monitored in parallel in plates containing mock-infected cells. The half-maximal effective concentration (EC50) and the compound-specific toxicity (50% cytotoxic concentration [CC50]) were calculated with GraphPad Prism 6 software, using the nonlinear regression model.
Identification of adaptive mutations in the EAV genome.
To isolate viral RNA, Huh7 cells (10-cm2 dish) were infected with wild-type (wt) EAV, EAVwt P7, EAV CsA replicates, or EAV ALV replicates (MOI of 0.1). Cells were incubated for 16 h at 37°C and lysed in 1 ml of TriPure (Sigma-Aldrich), and intracellular RNA was isolated according to the manufacturer’s instructions. Reverse transcription (RT) was performed with the ThermoScript RT-PCR system (Thermo Fisher Scientific) according to the manufacturer’s instructions, using random hexamers (Promega) and 10 μM primer 5′-CGCCGTTTTTTTTTTTTTTTTTTTTTTTTT-3′. Subsequently, the complete EAV genome was amplified as seven PCR amplicons by using MyTaq DNA polymerase (Bioline) and sequenced (primer sequences used in PCR are available upon request).
EAV reverse genetics.
Resistance-associated adaptive mutations (as listed in Table 2) were introduced into shuttle plasmids by standard site-directed PCR mutagenesis. After sequence verification, restriction fragments containing the desired mutations were transferred to full-length EAV cDNA clone pEAN900, a derivative of pEAV030 (48) containing a few translationally silent mutations to engineer restriction sites. Virus derived from clone pEAN900 has a wt phenotype, as confirmed by a side-by-side comparison with pEAV030-derived virus in growth curve experiments and plaque assays (data not shown). In vitro-transcribed RNA derived from wt or mutant pEAN900 was electroporated into BHK-21 cells using Amaxa nucleofector technology (Lonza), as described previously (49). Recombinant virus was harvested upon complete CPE, typically between 48 and 72 h posttransfection (p.t.). The presence of the engineered mutations was confirmed by performing one additional virus passage and sequencing RT-PCR products obtained after isolation and RT-PCR amplification of intracellular viral RNA.
Electron microscopy.
Huh7 cells were infected with rEAVwt or rEAVQYA, or were mock infected, and either treated with 8 μM CsA at 1 h p.i. or left untreated. Cells were fixed at 11 h p.i. (no CsA) or 14 h p.i. (8 μM CsA) in 1.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) for 1 h. Subsequently, samples were stained during 1-h incubation steps, first with 1% OsO4 in 0.1 M cacodylate buffer and then with 1% uranyl acetate. Samples were then dehydrated in a graded ethanol series (70% to 100% ethanol) and embedded in Epon LX-112 resin (Ladd Research) that was polymerized at 60°C. Sections with a thickness of 100 nm were collected on carbon-coated Pioloform EM grids and poststained with lead citrate and a saturated aqueous solution of uranyl acetate. Images were collected on a Tecnai12 TWIN electron microscope (Thermo Fisher Scientific [formerly FEI]) operating at 120 kV using a OneView 4k high-frame-rate camera (Gatan).
Isolation of EAV RTC-containing replication organelles and in vitro RNA synthesis assays.
Membrane-associated EAV RTCs were isolated from BHK-21 cells, and in vitro RNA synthesis assays (IVRAs) were performed essentially as described previously (11). In short, approximately 5 × 107 EAV-infected BHK-21 cells (MOI, 5) were harvested at 7.5 h p.i. and lysed using a ball-bearing homogenizer (Isobiotek) with a 16-μm clearance. Lysates were centrifuged for 10 min at 1,000 × g to obtain a postnuclear supernatant (PNS). A standard IVRA mixture contained PNS (the equivalent of 6 × 104 infected cells) and a 5× concentrated CsA stock (final concentration, 4 to 32 μM) or RTC dilution buffer (control), and IVRA was performed in the presence of [α-32P]CTP that was incorporated in the newly synthesized EAV RNA. Assay mixtures were incubated at 30°C for 100 min and terminated by the addition of 60 μl of 5% LiDS-LET-ProtK. After a 15-min incubation at 42°C, the 32P-labeled reaction products were isolated and separated in a 1.5% denaturing agarose gel. Agarose gels were dried and reaction products were visualized by phophorimaging using a Typhoon 9410 imager (GE Healthcare). Incorporation of label was quantified using ImageQuant TL software.
Analysis of viral RNA synthesis.
Metabolic labeling of viral RNA with [3H]uridine was performed essentially as described previously (50). Briefly, 4 × 105 BHK-21 cells in 4-cm2 wells were infected with rEAVwt or rEAVQYA (MOI, 5). At 5.5 h p.i., DMEM containing 8% FCS, 5 μg/ml of dactinomycin (Sigma-Aldrich), and 8 μM CsA or 0.02% DMSO (solvent control) was given. At 6.5 h p.i., 100 μCi of [3H]uridine was added to 300 μl of culture medium. Cells were lysed at 7.5 h p.i. and intracellular 3H-labeled RNAs were isolated (Tripure), separated by denaturing agarose gel electrophoresis, and visualized by fluorography. The total incorporation of 3H label was determined using liquid scintillation counting. For hybridization analysis of intracellular RNA from EAV-infected cells, RNA was first separated by denaturing RNA agarose gel electrophoresis. Subsequently, the gel was dried and hybridization was performed using a 32P-labeled oligonucleotide probe (5′-TTGGTTCCTGGGTGGCTAATAACTACTT-3′) recognizing the 3′ end of the EAV genome, as described previously (11).
Supplementary Material
ACKNOWLEDGMENTS
We thank Ettore Ullo, Jayron Habibe, Jessika Zevenhoven-Dobbe, and Diede Oudshoorn for technical assistance and helpful discussions, Grégoire Vuagniaux for critically reading the manuscript, and Novartis (Basel, Switzerland) and DebioPharm (Lausanne, Switzerland) for providing alisporivir.
This research was supported in part by the EU-FP7-Health project SILVER (grant 260644).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00490-19.
REFERENCES
- 1.Siddell SG, Walker PJ, Lefkowitz EJ, Mushegian AR, Adams MJ, Dutilh BE, Gorbalenya AE, Harrach B, Harrison RL, Junglen S, Knowles NJ, Kropinski AM, Krupovic M, Kuhn JH, Nibert M, Rubino L, Sabanadzovic S, Sanfaçon H, Simmonds P, Varsani A, Zerbini FM, Davison A. 2019. Additional changes to taxonomy ratified in a special vote by the International Committee on Taxonomy of Viruses (October 2018). Arch Virol 164:943–946. doi: 10.1007/s00705-018-04136-2. [DOI] [PubMed] [Google Scholar]
- 2.de Wit E, van Doremalen N, Falzarano D, Munster VJ. 2016. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol 14:523–534. doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cui J, Li F, Shi ZL. 2019. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol 17:181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holtkamp DJ, Kliebenstein JB, Neumann EJ. 2013. Assessment of the economic impact of porcine reproductive and respiratory syndrome virus on United States pork producers. J Swine Health Prod 21:72–84. [Google Scholar]
- 5.Balasuriya UB, Carossino M. 2017. Reproductive effects of arteriviruses: equine arteritis virus and porcine reproductive and respiratory syndrome virus infections. Curr Opin Virol 27:57–70. doi: 10.1016/j.coviro.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 6.Snijder EJ, Kikkert M, Fang Y. 2013. Arterivirus molecular biology and pathogenesis. J Gen Virol 94:2141–2163. doi: 10.1099/vir.0.056341-0. [DOI] [PubMed] [Google Scholar]
- 7.Knoops K, Barcena M, Limpens RW, Koster AJ, Mommaas AM, Snijder EJ. 2012. Ultrastructural characterization of arterivirus replication structures: reshaping the endoplasmic reticulum to accommodate viral RNA synthesis. J Virol 86:2474–2487. doi: 10.1128/JVI.06677-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang W, Chen K, Zhang X, Guo C, Chen Y, Liu X. 2018. An integrated analysis of membrane remodeling during porcine reproductive and respiratory syndrome virus replication and assembly. PLoS One 13:e0200919. doi: 10.1371/journal.pone.0200919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van der Hoeven B, Oudshoorn D, Koster AJ, Snijder EJ, Kikkert M, Bárcena M. 2016. Biogenesis and architecture of arterivirus replication organelles. Virus Res 220:70–90. doi: 10.1016/j.virusres.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Snijder EJ, van Tol H, Roos N, Pedersen KW. 2001. Non-structural proteins 2 and 3 interact to modify host cell membranes during the formation of the arterivirus replication complex. J Gen Virol 82:985–994. doi: 10.1099/0022-1317-82-5-985. [DOI] [PubMed] [Google Scholar]
- 11.van Hemert MJ, de Wilde AH, Gorbalenya AE, Snijder EJ. 2008. The in vitro RNA synthesizing activity of the isolated arterivirus replication/transcription complex is dependent on a host factor. J Biol Chem 283:16525–16536. doi: 10.1074/jbc.M708136200. [DOI] [PubMed] [Google Scholar]
- 12.de Wilde AH, Li Y, van der Meer Y, Vuagniaux G, Lysek R, Fang Y, Snijder EJ, van Hemert MJ. 2013. Cyclophilin inhibitors block arterivirus replication by interfering with viral RNA synthesis. J Virol 87:1454–1464. doi: 10.1128/JVI.02078-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Wilde AH, Zevenhoven-Dobbe JC, van der Meer Y, Thiel V, Narayanan K, Makino S, Snijder EJ, van Hemert MJ. 2011. Cyclosporin A inhibits the replication of diverse coronaviruses. J Gen Virol 92:2542–2548. doi: 10.1099/vir.0.034983-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Wilde AH, Raj VS, Oudshoorn D, Bestebroer TM, van Nieuwkoop S, Limpens RWAL, Posthuma CC, van der Meer Y, Bárcena M, Haagmans BL, Snijder EJ, van den Hoogen BG. 2013. MERS-coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon-alpha treatment. J Gen Virol 94:1749–1760. doi: 10.1099/vir.0.052910-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pfefferle S, Schöpf J, Kögl M, Friedel CC, Müller MA, Carbajo-Lozoya J, Stellberger T, von Dall’Armi E, Herzog P, Kallies S, Niemeyer D, Ditt V, Kuri T, Züst R, Pumpor K, Hilgenfeld R, Schwarz F, Zimmer R, Steffen I, Weber F, Thiel V, Herrler G, Thiel H-J, Schwegmann-Weßels C, Pöhlmann S, Haas J, Drosten C, von Brunn A. 2011. The SARS-coronavirus-host interactome: identification of cyclophilins as target for pan-coronavirus inhibitors. PLoS Pathog 7:e1002331. doi: 10.1371/journal.ppat.1002331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanaka Y, Sato Y, Sasaki T. 2013. Suppression of coronavirus replication by cyclophilin inhibitors. Viruses 5:1250–1260. doi: 10.3390/v5051250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang P, Heitman J. 2005. The cyclophilins. Genome Biol 6:226. doi: 10.1186/gb-2005-6-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dunyak BM, Gestwicki JE. 2016. Peptidyl-proline isomerases (PPIases): targets for natural products and natural product-inspired compounds. J Med Chem 59:9622–9644. doi: 10.1021/acs.jmedchem.6b00411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schreiber SL, Crabtree GR. 1992. The mechanism of action of cyclosporin A and FK506. Immunol Today 13:136–142. doi: 10.1016/0167-5699(92)90111-J. [DOI] [PubMed] [Google Scholar]
- 20.de Wilde AH, Pham U, Posthuma CC, Snijder EJ. 2018. Cyclophilins and cyclophilin inhibitors in nidovirus replication. Virology 522:46–55. doi: 10.1016/j.virol.2018.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naoumov NV. 2014. Cyclophilin inhibition as potential therapy for liver diseases. J Hepatol 61:1166–1174. doi: 10.1016/j.jhep.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 22.de Wilde AH, Falzarano D, Zevenhoven-Dobbe JC, Beugeling C, Fett C, Martellaro C, Posthuma CC, Feldmann H, Perlman S, Snijder EJ. 2017. Alisporivir inhibits MERS- and SARS-coronavirus replication in cell culture, but not SARS-coronavirus infection in a mouse model. Virus Res 228:7–13. doi: 10.1016/j.virusres.2016.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frausto SD, Lee E, Tang H. 2013. Cyclophilins as modulators of viral replication. Viruses 5:1684–1701. doi: 10.3390/v5071684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carbajo-Lozoya J, Ma-Lauer Y, Malesevic M, Theuerkorn M, Kahlert V, Prell E, von Brunn B, Muth D, Baumert TF, Drosten C, Fischer G, von Brunn A. 2014. Human coronavirus NL63 replication is cyclophilin A-dependent and inhibited by non-immunosuppressive cyclosporine A-derivatives including Alisporivir. Virus Res 184C:44–53. doi: 10.1016/j.virusres.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.von Brunn A, Ciesek S, von Brunn B, Carbajo-Lozoya J. 2015. Genetic deficiency and polymorphisms of cyclophilin A reveal its essential role for human coronavirus 229E replication. Curr Opin Virol 14:56–61. doi: 10.1016/j.coviro.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Wilde AH, Zevenhoven-Dobbe JC, Beugeling C, Chatterji U, de Jong D, Gallay P, Szuhai K, Posthuma CC, Snijder EJ. 2018. Coronaviruses and arteriviruses display striking differences in their cyclophilin A-dependence during replication in cell culture. Virology 517:148–156. doi: 10.1016/j.virol.2017.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Wilde AH, Boomaars-van der Zanden AL, de Jong AWM, Barcéna M, Snijder EJ, Posthuma CC. 2019. Adaptive mutations in replicase transmembrane subunits can counteract inhibition of equine arteritis virus RNA synthesis by cyclophilin inhibitors. bioRxiv 10.1101/587261. [DOI] [PMC free article] [PubMed]
- 28.Madan V, Paul D, Lohmann V, Bartenschlager R. 2014. Inhibition of HCV replication by cyclophilin antagonists is linked to replication fitness and occurs by inhibition of membranous web formation. Gastroenterology 146:1361–1372.e9. doi: 10.1053/j.gastro.2014.01.055. [DOI] [PubMed] [Google Scholar]
- 29.Chatterji U, Bobardt M, Tai A, Wood M, Gallay PA. 2015. Cyclophilin and NS5A inhibitors, but not other anti-hepatitis C virus (HCV) agents, preclude HCV-mediated formation of double-membrane-vesicle viral factories. Antimicrob Agents Chemother 59:2496–2507. doi: 10.1128/AAC.04958-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pedersen KW, van der MY, Roos N, Snijder EJ. 1999. Open reading frame 1a-encoded subunits of the arterivirus replicase induce endoplasmic reticulum-derived double-membrane vesicles which carry the viral replication complex. J Virol 73:2016–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Snijder EJ, Wassenaar AL, Spaan WJ. 1994. Proteolytic processing of the replicase ORF1a protein of equine arteritis virus. J Virol 68:5755–5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wassenaar AL, Spaan WJ, Gorbalenya AE, Snijder EJ. 1997. Alternative proteolytic processing of the arterivirus replicase ORF1a polyprotein: evidence that NSP2 acts as a cofactor for the NSP4 serine protease. J Virol 71:9313–9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Piotukh K, Gu W, Kofler M, Labudde D, Helms V, Freund C. 2005. Cyclophilin A binds to linear peptide motifs containing a consensus that is present in many human proteins. J Biol Chem 280:23668–23674. doi: 10.1074/jbc.M503405200. [DOI] [PubMed] [Google Scholar]
- 34.Foster TL, Gallay P, Stonehouse NJ, Harris M. 2011. Cyclophilin A interacts with domain II of hepatitis C virus NS5A and stimulates RNA binding in an isomerase-dependent manner. J Virol 85:7460–7464. doi: 10.1128/JVI.00393-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ngure M, Issur M, Shkriabai N, Liu HW, Cosa G, Kvaratskhelia M, Gotte M. 2016. Interactions of the disordered domain II of hepatitis C virus NS5A with cyclophilin A, NS5B, and viral RNA show extensive overlap. ACS Infect Dis 2:839–851. doi: 10.1021/acsinfecdis.6b00143. [DOI] [PubMed] [Google Scholar]
- 36.Coelmont L, Hanoulle X, Chatterji U, Berger C, Snoeck J, Bobardt M, Lim P, Vliegen I, Paeshuyse J, Vuagniaux G, Vandamme AM, Bartenschlager R, Gallay P, Lippens G, Neyts J. 2010. DEB025 (Alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A. PLoS One 5:e13687. doi: 10.1371/journal.pone.0013687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaul A, Stauffer S, Berger C, Pertel T, Schmitt J, Kallis S, Zayas M, Lopez MZ, Lohmann V, Luban J, Bartenschlager R. 2009. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog 5:e1000546. doi: 10.1371/journal.ppat.1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Badillo A, Receveur-Brechot V, Sarrazin S, Cantrelle FX, Delolme F, Fogeron ML, Molle J, Montserret R, Bockmann A, Bartenschlager R, Lohmann V, Lippens G, Ricard-Blum S, Hanoulle X, Penin F. 2017. Overall structural model of NS5A protein from hepatitis C virus and modulation by mutations confering resistance of virus replication to cyclosporin A. Biochemistry 56:3029–3048. doi: 10.1021/acs.biochem.7b00212. [DOI] [PubMed] [Google Scholar]
- 39.Azouzi S, El Kirat K, Morandat S. 2010. The potent antimalarial drug cyclosporin A preferentially destabilizes sphingomyelin-rich membranes. Langmuir 26:1960–1965. doi: 10.1021/la902580w. [DOI] [PubMed] [Google Scholar]
- 40.Dynarowicz-Latka P, Wnetrzak A, Makyla-Juzak K. 2015. Cyclosporin A in membrane lipids environment: implications for antimalarial activity of the drug—the Langmuir monolayer studies. J Membr Biol 248:1021–1032. doi: 10.1007/s00232-015-9814-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bouvier N, Flinois JP, Gilleron J, Sauvage FL, Legendre C, Beaune P, Thervet E, Anglicheau D, Pallet N. 2009. Cyclosporine triggers endoplasmic reticulum stress in endothelial cells: a role for endothelial phenotypic changes and death. Am J Physiol Renal Physiol 296:F160–F169. doi: 10.1152/ajprenal.90567.2008. [DOI] [PubMed] [Google Scholar]
- 42.Ram BM, Ramakrishna G. 2014. Endoplasmic reticulum vacuolation and unfolded protein response leading to paraptosis like cell death in cyclosporine A treated cancer cervix cells is mediated by cyclophilin B inhibition. Biochim Biophys Acta 1843:2497–2512. doi: 10.1016/j.bbamcr.2014.06.020. [DOI] [PubMed] [Google Scholar]
- 43.van Kasteren PB, Bailey-Elkin BA, James TW, Ninaber DK, Beugeling C, Khajehpour M, Snijder EJ, Mark BL, Kikkert M. 2013. Deubiquitinase function of arterivirus papain-like protease 2 suppresses the innate immune response in infected host cells. Proc Natl Acad Sci U S A 110:E838–E847. doi: 10.1073/pnas.1218464110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nedialkova DD, Gorbalenya AE, Snijder EJ. 2010. Arterivirus Nsp1 modulates the accumulation of minus-strand templates to control the relative abundance of viral mRNAs. PLoS Pathog 6:e1000772. doi: 10.1371/journal.ppat.1000772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Kasteren PB, Beugeling C, Ninaber DK, Frias-Staheli N, van Boheemen S, García-Sastre A, Snijder EJ, Kikkert M. 2012. Arterivirus and nairovirus ovarian tumor domain-containing deubiquitinases target activated RIG-I to control innate immune signaling. J Virol 86:773–785. doi: 10.1128/JVI.06277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bryans JT, Crowe ME, Doll ER, McCollum WH. 1957. Isolation of a filterable agent causing arteritis of horses and abortion by mares; its differentiation from the equine abortion (influenza) virus. Cornell Vet 47:3–41. [PubMed] [Google Scholar]
- 47.de Vries AA, Chirnside ED, Horzinek MC, Rottier PJ. 1992. Structural proteins of equine arteritis virus. J Virol 66:6294–6303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Dinten LC, den Boon JA, Wassenaar AL, Spaan WJ, Snijder EJ. 1997. An infectious arterivirus cDNA clone: identification of a replicase point mutation that abolishes discontinuous mRNA transcription. Proc Natl Acad Sci U S A 94:991–996. doi: 10.1073/pnas.94.3.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beerens N, Snijder EJ. 2007. An RNA pseudoknot in the 3′ end of the arterivirus genome has a critical role in regulating viral RNA synthesis. J Virol 81:9426–9436. doi: 10.1128/JVI.00747-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Knoops K, Swett-Tapia C, van den Worm SH, Te Velthuis AJ, Koster AJ, Mommaas AM, Snijder EJ, Kikkert M. 2010. Integrity of the early secretory pathway promotes, but is not required for, severe acute respiratory syndrome coronavirus RNA synthesis and virus-induced remodeling of endoplasmic reticulum membranes. J Virol 84:833–846. doi: 10.1128/JVI.01826-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chojnacki S, Cowley A, Lee J, Foix A, Lopez R. 2017. Programmatic access to bioinformatics tools from EMBL-EBI update: 2017. Nucleic Acids Res 45:W550–W553. doi: 10.1093/nar/gkx273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.