
Fig. 3.
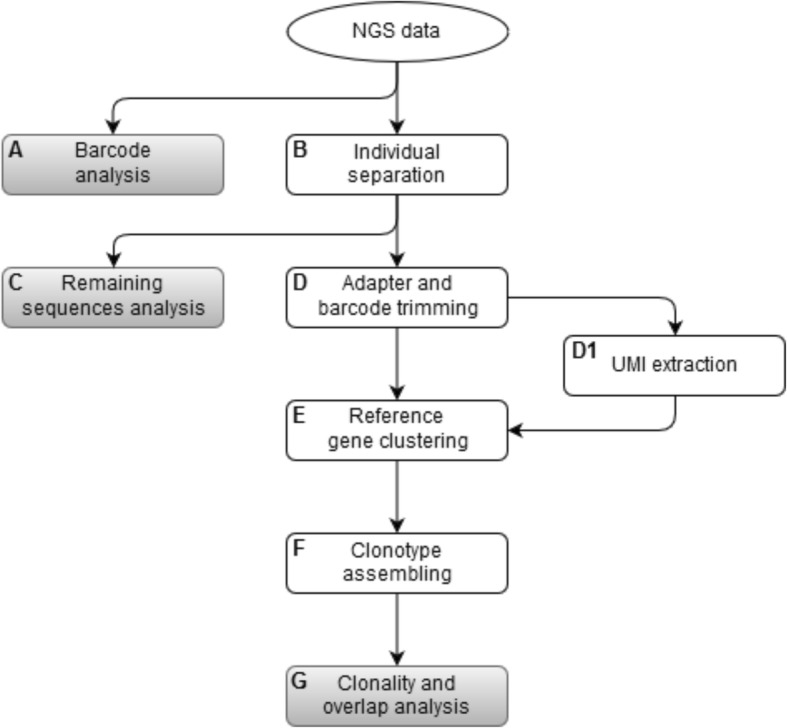
Bioinformatic Flowchart. A pipeline for the analysis of T cell repertoires. A: First, barcode analysis is done as well as separating the sequences belonging to the different individuals (B). C: Reads that cannot be assigned to a specific individual are stored in a separate FASTQ file for later investigation regarding the origin of these sequences. D: Adapters and barcodes from sequences are trimmed. During this step the UMIs are determined and stored in a separate file (D1). E: Afterwards the sequences are clustered with respect to their reference genes and UMIs. F/G: Clonotypes are assembled and clonality, diversity and repertoire overlap analysis is performed