

In this study, Steinfeld et al. investigated why the vast majority of eukaryotes have two Rad51/RecA family DNA recombinases: Rad51, which is ubiquitously expressed, and Dmc1, which is meiosis-specific. They show that this difference can be attributed to three amino acids conserved only within the Dmc1 lineage of the Rad51/RecA family and propose that Dmc1 lineage-specific amino acids involved in the stabilization of heteroduplex DNA joints with mismatch-containing base triplets may contribute to normal meiotic recombination.
Keywords: DNA repair, Dmc1, homologous recombination, meiosis, Rad51
Abstract
The vast majority of eukaryotes possess two DNA recombinases: Rad51, which is ubiquitously expressed, and Dmc1, which is meiosis-specific. The evolutionary origins of this two-recombinase system remain poorly understood. Interestingly, Dmc1 can stabilize mismatch-containing base triplets, whereas Rad51 cannot. Here, we demonstrate that this difference can be attributed to three amino acids conserved only within the Dmc1 lineage of the Rad51/RecA family. Chimeric Rad51 mutants harboring Dmc1-specific amino acids gain the ability to stabilize heteroduplex DNA joints with mismatch-containing base triplets, whereas Dmc1 mutants with Rad51-specific amino acids lose this ability. Remarkably, RAD-51 from Caenorhabditis elegans, an organism without Dmc1, has acquired “Dmc1-like” amino acids. Chimeric C. elegans RAD-51 harboring “canonical” Rad51 amino acids gives rise to toxic recombination intermediates, which must be actively dismantled to permit normal meiotic progression. We propose that Dmc1 lineage-specific amino acids involved in the stabilization of heteroduplex DNA joints with mismatch-containing base triplets may contribute to normal meiotic recombination.
Homologous recombination (HR) enables the exchange of genetic information between DNA molecules and is a major driving force in evolution. HR plays essential roles in double-strand DNA break (DSB) repair (Symington et al. 2014), the rescue of stalled or collapsed replication forks (Cox et al. 2000; Symington et al. 2014), and meiosis (Neale and Keeney 2006; Brown and Bishop 2014). During HR, a presynaptic ssDNA is paired with the complementary strand of a homologous dsDNA, resulting in displacement of the noncomplementary strand (Kowalczykowski 2015; Morrical 2015), and the resulting D-loop intermediates can then be channeled through several mechanistically distinct pathways to complete repair (Paques and Haber 1999; Symington et al. 2014). The DNA pairing reactions that take place during HR are promoted by the Rad51/RecA family of DNA recombinases, which are ATP-dependent proteins that form extended helical filaments on DNA, referred to as presynaptic complexes (Kowalczykowski 2015; Morrical 2015; Prentiss et al. 2015). Crystal structures of RecA-ssDNA presynaptic and RecA-dsDNA postsynaptic complexes reveal that the DNA is organized into near B-form base triplets separated by ∼8 Å between adjacent triplets (Chen et al. 2008; Prentiss et al. 2015). This structural organization likely underpins homology recognition mechanisms and the ability of the Rad51/RecA family of recombinases to promote DNA strand invasion in 3-nucleotide (nt) steps (Ragunathan et al. 2011; Lee et al. 2015, 2017; Prentiss et al. 2015; Qi et al. 2015).
In mitotic cells, HR is used primarily for the repair of spontaneous DNA breaks, such as those associated with DNA replication errors (Paques and Haber 1999; Cox et al. 2000; Symington et al. 2014). Meiotic HR is used to repair programmed DSBs generated by the Spo11 complex and is biased toward interhomolog recombination to allow for the formation of crossovers necessary for accurate chromosome segregation in the first meiotic division (Neale and Keeney 2006; Brown and Bishop 2014; Thacker et al. 2014; Zhu and Keeney 2014; Hunter 2015; Marsolier-Kergoat et al. 2018).
Rad51 is the only recombinase in mitotic cells, whereas both Rad51 and Dmc1 are expressed during meiosis in the vast majority of eukaryotes (Neale and Keeney 2006; Brown and Bishop 2014; Hunter 2015). In the budding yeast Saccharomyces cerevisiae, Dmc1 is responsible for catalyzing interhomolog recombination during meiosis, while Rad51 promotes Dmc1 presynaptic filament assembly and participates in intersister repair that gives rise to noncrossover outcomes (Neale and Keeney 2006; Lao et al. 2013; Brown and Bishop 2014). Dmc1 mediates the intersister recombination that occurs contemporaneously with interhomolog recombination (Cloud et al. 2012). Rad51 is inactive during this period and becomes active only after induction of Ndt80 and destruction of the synaptonemal complex (Argunhan et al. 2017; Prugar et al. 2017). If activated prematurely, Rad51 can mediate interhomolog recombination, but not as efficiently as Dmc1 (Lao et al. 2013; Callender et al. 2016). It is clear that Rad51 and Dmc1 interact with different subsets of accessory factors (Brown and Bishop 2014); some examples include the protein complexes Mei5/Sae3 and Hop2/Mnd1, which interact with Dmc1 (Tsubouchi and Roeder 2002; Chen et al. 2004; Hayase et al. 2004; Tsubouchi and Roeder 2004; Petukhova et al. 2005; Ferrari et al. 2009); Rad54 and Rdh54, which show distinct genetic and biochemical properties in combination with Rad51 and Dmc1 (Klein 1997; Nimonkar et al. 2012); and Hed1, which is a Rad51-specific binding protein that blocks Rad54 interactions with Rad51 (Tsubouchi and Roeder 2006; Busygina et al. 2008). In the fission yeast Schizosaccharomyces pombe, the protein Rad22 (an ortholog of S. cerevisiae Rad52) activates Rad51 (also called Rhp51) but inhibits Dmc1, highlighting another example of a recombinase-specific accessory factor (Murayama et al. 2013). Interestingly, S. pombe Rad51 and Dmc1 promote four-strand exchange reactions, mimicking the formation and branch migration of Holliday junctions, but Rad51 promotes exchange in the 3′-to-5′ direction whereas Dmc1 promotes exchange with the opposite polarity, which could have implications for crossover production in meiosis (Murayama et al. 2008, 2011). However, there are few other biochemical or biophysical differences between Rad51 and Dmc1 that might help to explain the possible origins or potential evolutionary advantages of using different recombinases during mitosis and meiosis (Neale and Keeney 2006; Sheridan et al. 2008; Brown and Bishop 2014).
Interestingly, biophysical studies have shown that Rad51-ssDNA filaments can bind to dsDNA fragments containing short tracts of sequence microhomology to yield heteroduplex DNA joints, the lifetime of which scales in 3-nt increments consistent with a mechanism involving the stabling pairing of base triplet interactions (Lee et al. 2015, 2016, 2017; Qi et al. 2015). However, the introduction of a single nucleotide mismatch within one of the base triplets causes a reduction in binding lifetime of the heteroduplex DNA joint commensurate with the loss of one base triplet pairing interaction (Lee et al. 2015, 2017). In contrast, Dmc1-ssDNA can tolerate base triplets bearing single, double, or triple mismatches and even abasic sites with no change in the binding lifetimes of the resulting heteroduplex DNA intermediates relative to reactions with fully paired heteroduplex intermediates (Lee et al. 2015, 2017). These findings suggest that Dmc1 can stabilize mismatched base triplets within heteroduplex DNA joints, whereas Rad51 cannot (Lee et al. 2015, 2017; Borgogno et al. 2016). Similarly, genetic studies also support the notion that Dmc1 can stabilize mismatch-containing recombination intermediates, whereas Rad51 cannot (Callender et al. 2016). We and others have hypothesized that the ability of Dmc1 to stabilize imperfectly paired recombination intermediates might reflect an intrinsic difference between the two eukaryotic recombinases (Lee et al. 2015, 2017; Callender et al. 2016). However, the molecular basis for these differences and their biological implications remained unexplored.
Here, we use structural analysis and bioinformatics to identify Rad51 and Dmc1 lineage-specific amino acid residues that contribute to their unique responses to mismatched base triplets. Based on these analyses, we swapped Rad51 lineage-specific amino acid residues present at the putative DNA-binding interfaces with their lineage-specific counterparts from Dmc1 and vice versa. Single-molecule biophysical analysis of these chimeric recombinases reveals that the differential responses of S. cerevisiae and human Rad51 and Dmc1 to mismatches can be attributed to three lineage-specific amino acid residues within DNA-binding loop L1. Mating type (MAT) switching analysis provides genetic evidence that these L1 residues affect recombination between divergent sequences in vivo. Remarkably, Caenorhabditis elegans RAD-51 L1 amino acid residues more closely resemble Dmc1. Accordingly, wild-type (wt) C. elegans RAD-51 stabilizes mismatch-containing substrates, as is observed for yeast and human Dmc1, whereas mutation of C. elegans L1 residues to their “canonical” Rad51 counterparts abolishes mismatch stabilization. Moreover, worms expressing this chimeric RAD-51 recombinase are proficient for mitotic DNA repair but can accumulate aberrant postsynaptic intermediates during meiosis that must be dismantled by RTEL-1 or HELQ-1 to allow for normal progression through meiosis. Together, our results show that highly conserved lineage-specific amino acid residues in the L1 DNA-binding domain are in part responsible for the differential response of Rad51 and Dmc1 to mismatched sequences and suggest that these amino acid residues may play important roles in mitotic and meiotic recombination.
Results
Identification of Rad51 and Dmc1 lineage-specific amino acid residues
The Rad51 and Dmc1 lineages within the Rad51/RecA family of recombinases arose early in the evolutionary history of eukaryotes (Story et al. 1993; Lin et al. 2006; Chintapalli et al. 2013; Brown and Bishop 2014). These proteins remain closely related; for instance, S. cerevisiae Rad51 (ScRad51) and Dmc1 (ScDmc1) share 56% sequence similarity and 45% sequence identity (Neale and Keeney 2006; Brown and Bishop 2014). For brevity, we use the nomenclature Rad51 and Dmc1 as general designations and ScRad51, ScDmc1, hRAD51 (human RAD51), and hDMC1 (human DMC1) when referring to specific recombinases. Rad51 and Dmc1 form similar filaments on ssDNA, and both promote DNA strand invasion (Neale and Keeney 2006; Brown and Bishop 2014). However, Dmc1 can stabilize imperfectly paired base triplets, whereas Rad51 cannot (Lee et al. 2015, 2017). We speculated that Dmc1-specific amino acid residues might be responsible for this differential response to mismatches. Furthermore, we presumed that residues responsible for this effect might fulfill three criteria: (1) They should be conserved within the Dmc1 lineage of the recombinase family. (2) They should be absent from the Rad51 lineage. (3) They would likely be within one of the two known DNA-binding motifs, DNA-binding loop 1 (L1) or DNA-binding loop 2 (L2), which are present in all recombinase family members (Story et al. 1993; Chen et al. 2008). Given these criteria, we sought to determine whether Rad51 and Dmc1 harbor lineage-specific residues within the L1 and L2 DNA-binding loops.
L1 and L2 were originally identified from examination of the Escherichia coli RecA structure (Story et al. 1993). Therefore, we usd E. coli RecA as a model to verify the boundaries of the L1 and L2 motifs (Supplemental Fig. S1; Chen et al. 2008). E. coli RecA shares 26.5% and 25.6% sequence identity with ScRad51 and ScDmc1, respectively, and the core domain of E. coli RecA coaligns with the core domain of ScRad51 with root mean square deviation of ∼1.5 Å (Conway et al. 2004). We then mapped these regions onto a primary structure alignment of Rad51 and Dmc1 from S. cerevisiae, Homo sapiens, Pneumocystis carinii, Oryza sativa, Mus musculus, Entamoeba histolytica, Toxoplasma gondii, and Sus scrofa (Fig. 1A). From this initial comparison, we identified four amino acid residues within L1 and five amino acid residues within L2 that are conserved within either the Rad51 lineage or the Dmc1 lineage but are divergent between the two recombinases (Fig. 1A). For ScRad51, these lineage-specific amino acid residues correspond to L1 residues T288, A298, M301, and H302 and L2 residues V328, Q330, V331, D332, and N348. The ScDmc1 lineage-specific amino acids include L1 residues V224, E234, Q237, and K238 and L2 residues Q264, D266, P267, G268, and H285. We validated this initial assignment by analysis of 600 Rad51 protein sequences and 270 Dmc1 sequences (Fig. 1B,C; Supplemental Table S1).
Figure 1.

Identification of Rad51 and Dmc1 L1 and L2 lineage-specific amino acids. (A) Location and sequences of the L1 and L2 DNA-binding loops from RecA, Rad51, and Dmc1. Amino acids conserved in all three lineages are highlighted in red, Rad51 lineage-specific amino acids are highlighted in blue, and Dmc1 lineage-specific amino acids are highlighted in green. Included in the alignments are recombinases from S. cerevisiae, H. sapiens, P. carinii, O. sativa, M. musculus, E. histolytica, T. gondii, and S. scrofa. Asterisks denote L1 and L2 lineage-specific amino acids. (B) Conservation and identity of L1 amino acids based on analysis of 600 Rad51 and 270 Dmc1 sequences. The analyzed positions correspond to ScRad51 amino acids T288, A298, M301, and H302 and ScDmc1 amino acids V224, E234, Q237, and K238. Color-coding indicates Rad51-like, Dmc1-like, and other amino acids (see Supplemental Table S1 for a complete list of all Rad51 and Dmc1 lineage-specific L1 and L2 amino acid residues). (C) Conservation and identity of L2 amino acids.
Biochemical characterization of Rad51 and Dmc1 chimeras
If Dmc1 lineage-specific residues present within L1, L2, or both are responsible for mismatch tolerance, then mutation of these residues to those present in Rad51 might abolish this property. Conversely, mutation of the Rad51 lineage-specific residues to their Dmc1 counterparts might enable Rad51 to stabilize mismatched recombination intermediates. To test these hypotheses, we designed chimeric recombinases by swapping the entire L1 and L2 motifs (Supplemental Table S2). For brevity, we assigned names to the mutants based on the identity of the altered residues (e.g., ScDmc1-RL1 refers to S. cerevisiae Dmc1 harboring the ScRad51 L1, hRAD51-DL12 refers to human RAD51 with the amino acid residues from hDMC1 L1 and L2, etc.) (Supplemental Table S2). All mutants behaved like their wt counterparts during expression and purification, with the exception of the hDMC1 chimeras, which were significantly less soluble than the wt protein (data not shown); therefore, we were unable to analyze these hDMC1 mutants.
Each chimeric protein was tested for ATP hydrolysis and DNA strand exchange activity (Supplemental Fig. S2A). Most of the chimeras retained DNA-dependent ATPase activity, albeit typically at a lower level relative to the wt recombinases, and the single loop swaps exhibited greater ATP hydrolysis activity than the double loop swaps (Supplemental Fig. S2A). DNA strand exchange assays revealed that the mutant proteins with a single chimeric loop swap exhibited activity comparable with that of their wt counterparts (Supplemental Fig. S2B,C). However, chimeric recombinases in which both loops were swapped were deficient in strand exchange activity. In particular, ScDmc1-RL12 was significantly compromised for both strand exchange and ATP hydrolysis (Supplemental Fig. S2). Interestingly, ScDmc1-RL1 exhibited approximately threefold more strand exchange activity compared with wt Dmc1 (Supplemental Fig. S2B,C). These findings indicate that the identity of the lineage-specific amino acid residues contributes to recombinase interactions with DNA substrates and also show that it is technically possible to swap the Rad51 and Dmc1 L1 or L2 regions without completely abolishing basic protein activities in vitro, although, as specified above, the resulting mutant proteins do show some differences in ATPase and strand exchange activity levels (Supplemental Fig. S2).
Assembly of presynaptic filaments with chimeric recombinases
The chimeric recombinases were tested for the ability to assemble into stable presynaptic complexes using ssDNA curtain assays (Supplemental Fig. S3). As reported, the addition of wt (unlabeled) Rad51 or Dmc1 results in ATP-dependent displacement of RPA-GFP from the ssDNA, reflecting the assembly of the presynaptic complexes (Supplemental Fig. S3; Ma et al. 2017). The RPA-GFP reappears when ATP (or both ATP and Ca2+ in the case of ScDmc1 and hRAD51) is flushed from the sample chamber, reflecting presynaptic complex disassembly (Supplemental Fig. S3; Ma et al. 2017). Most of the chimeras assembled into presynaptic filaments, which remained stable for ≥30 min so long as ATP (and Ca2+ when appropriate) was in the reaction buffer (Supplemental Fig. S3; Supplemental Table S3). One exception was ScDmc1-RL12, which failed to assemble into stable filaments (data not shown) and was not characterized further.
Chimeric recombinases exhibit base triplet stepping
We developed a ssDNA curtain assay for visualizing DNA strand exchange intermediates at the single-molecule level (Lee et al. 2015, 2016, 2017; Qi et al. 2015; Qi and Greene 2016; Ma et al. 2017). In brief, a series of Atto565-labeled dsDNA substrates (70 bp) harboring 8- to 15-nt tracts of microhomology targeted toward unique sequences in the M13 ssDNA are incubated with the presynaptic complexes, and unbound dsDNA is flushed away (Fig. 2A,B). The resulting intermediates are visualized by total internal reflection fluorescence microscopy (TIRFM), and dsDNA dissociation rates are obtained from the survival probabilities of the bound dsDNA fragments (Lee et al. 2015, 2016, 2017; Qi et al. 2015; Qi and Greene 2016; Ma et al. 2017). Using this assay, we showed that RecA, Rad51, and Dmc1 stabilize paired heteroduplex intermediates in 3-nt increments; each base triplet “step” coincides with an energetic signature (ΔΔG‡) of 0.3 kBT, corresponding to an ∼30% change in the dissociation rates (Lee et al. 2015, 2017; Qi et al. 2015). Importantly, presynaptic complexes prepared with the chimeric recombinases could bind the Atto565-labeled dsDNA, the resulting dsDNA dissociation rates were comparable with those measured for the wt proteins, and the dissociation rates also varied in 3-nt increments (Fig. 2C–J; Supplemental Fig. S4, S5). We conclude that the chimeric Rad51 and Dmc1 recombinases possess dsDNA-binding and base triplet stepping attributes similar to those determined for their wt counterparts.
Figure 2.
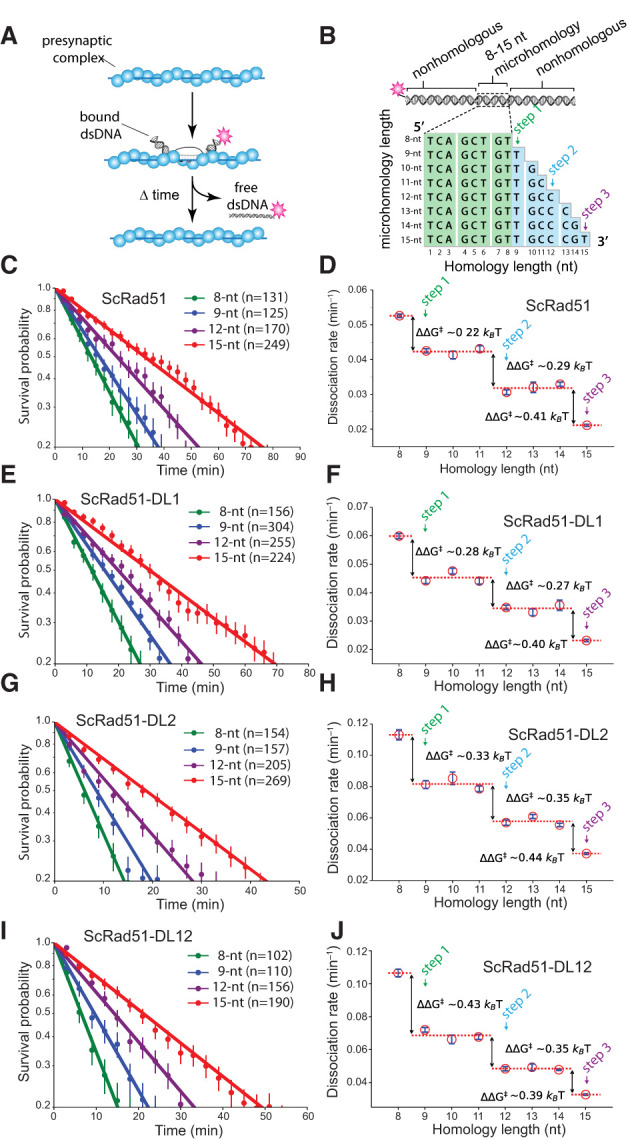
Chimeric recombinases undergo base triplet stepping. (A) Schematic of dsDNA capture assay. (B) Illustration of dsDNA substrates used for base triplet stepping assays. The 8-nt tract of microhomology highlighted in green is required for efficient binding, and the nucleotides highlighted in blue represent incremental increases in the microhomology length. Color-coded designations here and in all subsequent figure panels indicate the length of homology at which steps 1–3 are detected (Lee et al. 2015, 2016, 2017). (C,D) Survival probabilities (C) and dissociation rate data (D) obtained from the survival probability plots for wt ScRad51. (E,F) Survival probabilities (E) and dissociation rate data (F) for ScRad51-DL1. (G,H) Survival probabilities (G) and dissociation rate data (H) for ScRad51-DL2. (I,J) Survival probabilities (I) and dissociation rate data (J) for ScRad51-DL12. For all dissociation rate graphs, arrows indicate stepwise reductions in dissociation rates coincident with recognition of the third base of each triplet, dashed lines report the mean rate for each step, and the free energy changes (ΔΔG‡) associated with each triplet step are indicated. Here and throughout, error bars for survival probability plots represent 70% confidence intervals (CIs) obtained through bootstrap analysis, error bars for the dissociation rate data represent mean ± standard deviation (SD), the number of events used to calculate these values are shown in the survival probability panels, and the color-coded designations indicating steps 1–3 are indicated.
Dmc1 L1 lineage-specific amino acid residues regulate mismatch stabilization
We next asked how the chimeric recombinases responded to DNA mismatches. These assays are conducted by calculating the ΔΔG‡ values for a given recombinase in reactions with fully paired dsDNA oligonucleotide substrates (as described above) compared with reactions with the same recombinase using dsDNA oligonucleotide substrates that have single nucleotide mismatches introduced at defined locations (Fig. 3A; Lee et al. 2015, 2017). Rad51, RecA, and Dmc1 require perfect Watson-Crick pairing interactions to stabilize base triplets located at the terminal positions of tracts of microhomology tracts embedded within the dsDNA; in the absence of perfect pairing, they fail to take a step (Fig. 3A, panel i; Lee et al. 2015, 2017). Rad51 and RecA also require perfect Watson-Crick base-pairing interactions to stabilize base triplets located at internal positions, and the presence of a mismatch at these internal positions is revealed as the loss of a base triplet step (Fig. 3A, panel ii; Lee et al. 2015, 2017). In contrast, Dmc1 can stabilize mismatches at internal positions (Fig. 3A, panel iii; Lee et al. 2015, 2017). Indeed, Dmc1 can stabilize internal base triplets containing single, double, and triple mismatches and even abasic sites so long as these imperfect triplets are flanked by homologous sequences (Lee et al. 2017).
Figure 3.

The Dmc1 L1 DNA-binding loop allows for mismatch stabilization. (A) Schematic illustration of a dsDNA substrate (shown in blue; the noncomplementary strand is omitted for clarity) bound to a presynaptic ssDNA (in red). A single mismatch is indicated with a star (★) and is positioned in either the terminal base triplet (panel i), an internal base triplet that is not stabilized (as with Rad51; panel ii), or an internal triplet that is stabilized (as with Dmc1; panel iii) (Lee et al. 2015). The binding steps relative to triplet length are highlighted, where step 1 corresponds to the initial binding interaction, whereas steps 2 and 3 reflect the changes in dissociation rates that occur for each 3-nt increase in length. (B) Schematic of dsDNA substrates with mismatches positioned within the terminal base triplet. The locations and identities of the mismatches are highlighted as an underlined magenta “X.” (C) Schematic of dsDNA substrates with mismatches positioned within the internal triplet. Assays with terminal mismatch substrates for wt and chimeric versions of ScRad51 (D), hRAD51 (E), and ScDmc1 (F). Assays with internal mismatch substrates for wt and chimeric versions of ScRad51 (G), hRAD51 (H), and ScDmc1 (I). In D–I, the location and identity of the mismatched nucleotide are highlighted in magenta and underlined. Error bars represent mean ± SD. All ΔΔG‡ values for the mismatched substrates for each protein were calculated to relative reactions performed with the same protein and the fully paired substrates. In A–C, color-coded designations indicate the length of homology at which steps 1–3 are detected for fully homologous substrates (see Fig. 2B,D), and in D–H, the corresponding free energy changes associated with each step (for fully homologous substrates) are indicated with color-coded dashed lines.
Consistent with previous results, none of the recombinases was capable of stabilizing a base triplet located at the terminal position of a 12-nt tract of microhomology (Fig. 3B), and the resulting substrates exhibited dissociation rates similar to those measured for a substrate with only 9 nt of microhomology (Fig. 3D–F). We next tested the chimeric recombinases with mismatch-containing base triplets that were juxtaposed to a single perfectly paired triplet with a 15-nt tract of microhomology (Fig. 3C). As shown previously, ScRad51 and hRAD51 could step over internal mismatches but could not stabilize the internal mismatched triplet, instead yielding dissociation rates comparable with a substrate bearing only 12 nt of microhomology (Fig. 3G,H; Lee et al. 2015, 2017). In contrast, ScDmc1 and hDMC1 yielded dissociation rates comparable with the corresponding substrate bearing 15 nt of perfect microhomology (Fig. 3I; Lee et al. 2015). Remarkably, ScRad51-DL1, ScRad51-DL12, hRAD51-DL1, and hRAD51-DL12 could stabilize the mismatched substrates similar to wt Dmc1 (Fig. 3G,H). In contrast, ScRad51-DL2 and hRAD51-DL2 were unable to stabilize internal mismatches and instead exhibited behaviors more comparable with wt Rad51 (Fig. 3G,H). Moreover, ScDmc1-RL1 was unable to stabilize the mismatched substrates and instead exhibited behavior similar to ScRad51 (Fig. 3I). Finally, ScRad51-DL2, ScDmc1-DL2, and hRAD51-DL2 all exhibited responses to the mismatch-containing triplets comparable with their wt counterparts (e.g., ScRad51 and ScRad51-DL2 behaved similarly and ScDmc1 and ScDmc1-RL2 behaved similarly, as did hRAD51 and hRAD51-DL2) (Fig. 3G–I). These findings demonstrate that Rad51 chimeras harboring L1 amino acid residues from Dmc1 attain the ability to stabilize mismatched base triplets, whereas Dmc1 chimeras harboring L1 residues from Rad51 lose the ability to stabilize mismatch-containing base triplets.
Three Dmc1 L1 amino acid residues contribute to DNA mismatch stabilization
ScRad51-DL1 has a total of six amino acid residues from ScDmc1 (Supplemental Tables S1, S2), and the complete ScRad51-DL1 swap mutant has compromised DNA-binding and ATPase activity relative to ScRad51 (Supplemental Fig. S2A, S3; Supplemental Table S1). Therefore, in an effort to identify a mutant protein that might more closely resemble the general biochemical characteristics of wt ScRad51, we sought to determine whether a smaller subset of these L1 residues could confer the ability to stabilize mismatches. We focused on the ScRad51 chimeric point mutants T288V, A298E, M301Q, and H302K because these residues were most conserved across the Rad51 or Dmc1 lineages (Fig. 1A,B). The resulting mutants displayed filament assembly and disassembly kinetics that closely resembled the wt protein (Supplemental Table S3). Each mutant exhibited dissociation rates for the Atto565-dsDNA fragments harboring the 9-, 12-, and 15-nt tracts of microhomology, which were essentially indistinguishable from wt ScRad51 (Supplemental Table S4). As with all recombinases, the point mutants were unable to stabilize mismatches present at the terminal position of an embedded tract of microhomology (Fig. 4A,C). ScRad51-T288V behaved like wt ScRad51, as it was able to step over the internal mismatches but did not stabilize the mismatch (Fig. 4B,D). However, ScRad51-M301Q stabilized mismatches regardless of the relative position of the mismatch within the base triplet, exhibiting behavior comparable with wt ScDmc1 (Fig. 4D). Interestingly, both ScRad51-A298E and ScRad51-H302K mutants were able to stabilize an internal mismatch located at the edge of a base triplet (nucleotide position 12 in Fig. 4B), but neither mutant could stabilize an internal mismatch located at the center of the triplet (nucleotide position 11 in Fig. 4B,D); these properties were confirmed using an alternative set of dsDNA substrates targeted to a different region of the presynaptic ssDNA (data not shown; Lee et al. 2015). However, a ScRad51-A298E, H302K double mutant (Supplemental Table S3) could stabilize internal mismatched base triplets regardless of whether the mismatch was located at the center or edge of the triplet (Fig. 4D). These results suggest that ScRad51 L1 amino acids A298, M301, and H302, when mutated to the corresponding Dmc1 L1 residues, can all contribute to mismatch stabilization.
Figure 4.
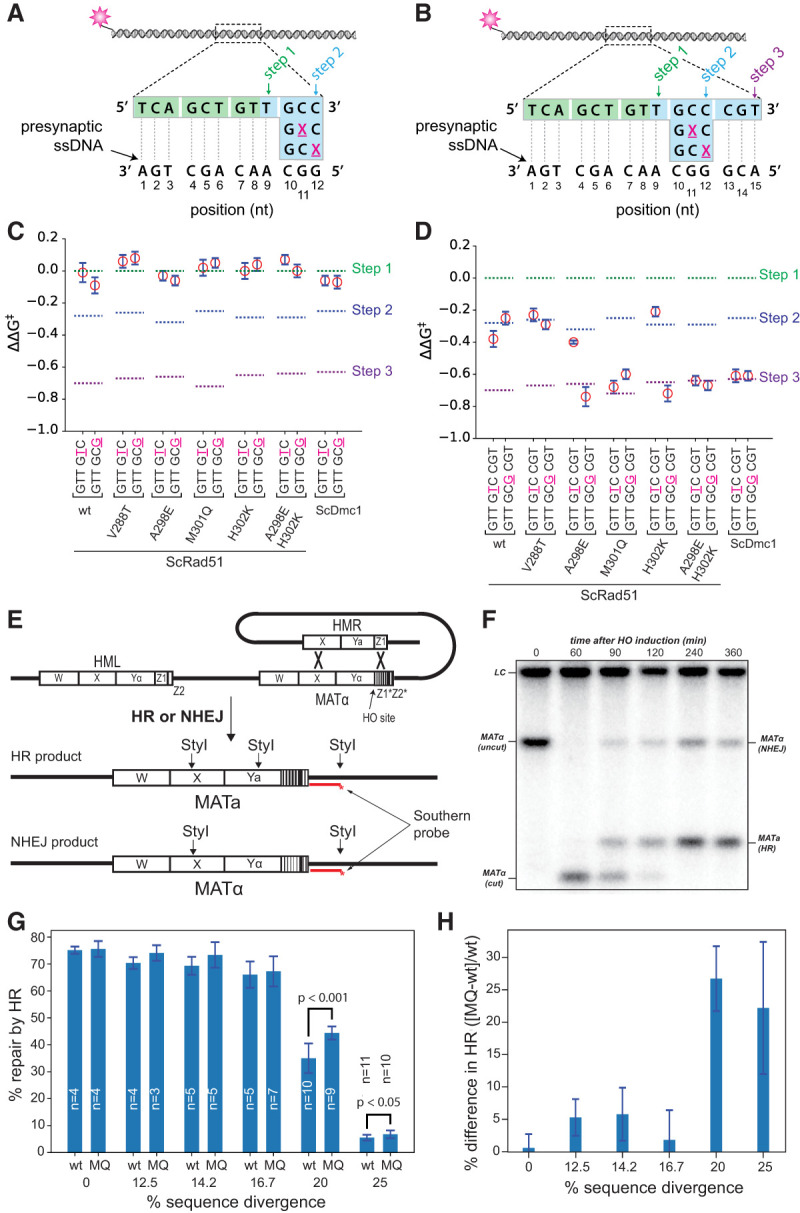
L1 amino acid identity influences recombination between divergent DNA sequences. Schematic illustrations of dsDNA sequences used to analyze the effects of terminal (A) and internal (B) mismatched triplets. (C) Terminal mismatch assays for the ScRad51 point mutants. (D) Internal mismatch assays for the ScRad51 point mutants. In C and D, data for wt ScRad51 are reproduced from Figure 3, D and G, and data for ScDmc1 are reproduced from Figure 3, F and I, for comparison. (E) Schematic illustration of the MAT switching assay highlighting the products that arise from nonhomologous end joining (NHEJ) and HR. (F) Example of a genomic blot analysis of StyI-digested DNA after galactose induction of HO endonuclease. (G) Bar graph showing the fraction of repair products attributed to HR for strains expressing either wt ScRad51 or ScRad51-M301Q for templates with varying levels of sequence divergence. Each bar represents the mean ± SD from n independent experiments, as indicated. The statistical significance for the observed differences between wt and mutant for the 20% and 25% sequence divergence data sets are indicated. P-values from an unpaired two-tailed Student's t-test are indicated. (H) Bar graph illustrating the percent difference for HR-mediated MAT switching between ScRad51-M301Q and wt Rad51. Each bar represents the mean ± SEM calculated from data in F. In A and B, color-coded designations indicate the length of homology at which steps 1–3 are detected for fully homologous substrates, and in C and D, the corresponding free energy changes associated with each step (for fully homologous substrates) are indicated with color-coded dashed lines.
Genetic characteristics of ScRad51 chimeras
A major challenge in understanding why eukaryotes have two recombinases is that Rad51 and Dmc1 cannot simply be replaced for one another because of the many mitotic- and meiotic-specific cofactors necessary for each of their in vivo functions, respectively (Neale and Keeney 2006; Brown and Bishop 2014). However, the chimeric proteins described here offer the opportunity to examine potential benefits of the dual recombinase system within the context of mutants that have a well-defined biochemical characteristic; namely, the ability or inability to stabilize mismatched recombination intermediates.
For genetic testing of the chimeric ScRad51 proteins, we constructed S. cerevisiae strains in which the chromosomal RAD51 gene was replaced with rad51 mutants containing Dmc1 lineage-specific amino acids. Western blot analysis confirmed that all proteins were expressed (Supplemental Fig. S6). In addition, wt ScRad51, ScRad51-DL1, ScRad51-A298E, and ScRad51-M301Q all supported some level of YFP-Rad54 DNA repair focus formation upon exposure to ionizing radiation (Supplemental Fig. S7A,B). Since the assembly of DNA repair-specific Rad54 foci is known to be dependent on Rad51-ssDNA filaments (Lisby et al. 2004), these results provide evidence that the mutant Rad51 proteins could form presynaptic filaments in vivo, although the A298E mutant was compromised in focus formation compared with wt ScRad51. In contrast, ScRad51-H302K and the ScRad51-A298E, H302K double mutant were unable to support YFP-Rad54 focus formation, suggesting that these mutants were defective in presynaptic filament assembly in cells (Supplemental Fig. S7B). We next asked whether ScRad51 chimeras could support cell growth on media containing the DNA-damaging agents methyl methanesulfonate (MMS) or zeocin (Supplemental Fig. S7C). Importantly, strains expressing either ScRad51-A298E or ScRad51-M301Q exhibited near wt levels of cell growth in the presence of MMS or zeocin (Supplemental Fig. S7B), demonstrating that these point mutants retain function in vivo. As expected, ScRad51-H302K and ScRad51-A298E, H302K were compromised for growth on plates with MMS or zeocin (Supplemental Fig. S7C). Interestingly, ScRad51-DL1 was also compromised for growth on MMS or zeocin plates even though this chimera was functional in vitro and supported Rad54 focus formation (Supplemental Fig. S2, S3, S7B,C). We speculate that swapping the entire Rad51 L1 motif may hinder some downstream step in the HR pathway.
Lineage-specific amino acid residues contribute to recombination in yeast
We used a modified version of the mating type switching assay to determine whether lineage-specific L1 residues might contribute to recombination fidelity. In this assay, the MAT locus is cleaved by the HO endonuclease, and Rad51-mediated recombination takes place between the cleaved MAT locus and either the HMRa or HMRα donor locus (Haber 2012; Mehta et al. 2017). Strand invasion initiates from the Z-box within the MAT locus, which is homologous to sequences in the donor loci (Fig. 4E; Haber 2012; Mehta et al. 2017). The cleaved intermediates can be repaired by HR or nonhomologous end joining (NHEJ), and the identity of the resulting products can be defined by genomic blot analysis (Fig. 4F). To examine the effects of mismatches on recombination, we introduced point mutations at every eighth, seventh, sixth, fifth, or fourth position within the Z-box, corresponding to 12.5%, 14.2%, 16.7%, 20%, or 25% sequence divergence between donor and acceptor loci, respectively (Fig. 4E; Supplemental Table S5).
For the strain expressing wt ScRad51, MAT switching was remarkably tolerant of mismatches (Fig. 4G). For instance, with the wt templates (0% divergence) 75.1% ± 1.33% of the repair products could be attributed to HR-mediated repair, while 66.1% ± 4.75% of the repair products could be attributed to HR for templates with 16.7% sequence divergence (Fig. 4G). However, HR efficiency drops markedly for templates with mismatches at every fifth (20% divergence) and fourth (25% divergence) position, yielding values of 35.0% ± 4.75% and 5.52% ± 1.46%, respectively (Fig. 4G). These observations are in good agreement with previous reports indicating that wt ScRad51 supports efficient break-induced replication (BIR) for templates with similar levels of sequence divergence (Anand et al. 2017).
Consistent with the MMS and zeocin resistance assays, ScRad51-DL1, ScRad51-H302K, and the ScRad51-A298E, H302K double mutant were all compromised for MAT switching even at 0% divergence, and these defects were exacerbated at higher levels of sequence divergence (Supplemental Fig. S7D,F,G). In contrast, the ScRad51-M301Q point mutant supported levels of HR-mediated repair comparable with wt ScRad51 for the substrates with up to 16.7% divergence. Remarkably, at 20% sequence divergence, representing the midpoint of the curve for wt ScRad51 (Fig. 4G), there was a 26.7% ± 5.00% (Student's t-test, P < 0.001) increase in HR-mediated repair by ScRad51-M301Q relative to wt ScRad51, and there was a 22.2% ± 10.2% (Student's t-test, P < 0.05) increase in HR for the template with 25% sequence divergence (Fig. 4G,H). These findings, together with our biophysical data, demonstrate that ScRad51-M301Q, which can stabilize mismatched HR intermediates in vitro, also supports a higher recombination frequency for mismatched substrates in vivo. Interestingly, although ScRad51-A298E was functional for MAT switching, this mutant showed no differences in HR efficiency compared with wt ScRad51 for templates with increasing sequence divergence (Supplemental Fig. S7E). Given this outcome, it is notable that ScRad51-A298E could only stabilize mismatches located at the edge of a base triplet (Fig. 4B,D), and the divergent Z-box sequences will always have mismatches at both the center and edge positions of the mismatched base triplets (irrespective of the frame of reference) (Supplemental Table S5). The inability of ScRad51-A298E to stabilize mismatches located in the center of a base triplet could explain why this particular mutant does not behave like ScRad51-M301Q in the MAT switching assays.
C. elegans RAD-51 behaves like Dmc1 in vitro
Some eukaryotes, such as Caenorhabditis sp., have lost the DMC1 gene, although the reasons for this loss remain uncertain (Brown and Bishop 2014). Surprisingly, inspection of RAD-51 from Caenorhabditis sp. revealed that the lineage-specific residues present in L1 were not the same as “canonical” Rad51 (we use the term “canonical” to identify Rad51 from species that have both recombinases) but instead more closely resembled Dmc1 (Fig. 5A). If our hypothesis regarding the role of L1 in mismatch stabilization is correct, then C. elegans RAD-51 (CeRAD-51) may stabilize mismatches, whereas a RAD-51 mutant in which the “Dmc1-like” amino acids were converted to the Rad51 lineage-specific residues might lose the ability to stabilize mismatches.
Figure 5.

C. elegans RAD-51 has Dmc1-like amino acids and dsDNA-binding properties. (A) Comparison of Caenorhabditis sp. RAD-51 sequences (C. elegans, Caenorhabditis remanei, and Caenorhabditis brenneri) with Rad51 and Dmc1 sequences from organisms with both recombinases (S. cerevisiae, H. sapiens, and O. sativa). Color-coding is the same as shown in Figure 1A. Base triplet stepping data for wt CeRAD-51 (B) and the CeRAD-51 triple mutant (C). (D) Terminal mismatch triplet assays for CeRAD-51 and CeRAD-51-TM (CeRAD-51 N246S, E256A, K260H triple mutant protein). Data for wt ScRad51 and ScDmc1 are shown for comparison and are reproduced from Figure 3, D and F. (E) Internal mismatch triplet assays for CeRAD-51 and CeRAD-51-TM. Data for wt ScRad51 and ScDmc1 are shown for comparison and are reproduced from Figure 3, D, F, G, and I. In D and E, the free energy changes associated with each step (for fully homologous substrates) are indicated with color-coded dashed lines.
To test this hypothesis, we made a CeRAD-51 N246S, E256A, K260H triple mutant protein (corresponding to ScRad51 amino acids T288, A298, and H302, respectively) (Fig. 5A), which we refer to as CeRAD-51-TM for brevity. Both CeRAD-51 (Taylor et al. 2016) and CeRAD-51-TM retain similar biochemical activity, although the ssDNA- and dsDNA-binding activity of CeRAD-51-TM is modestly reduced (Supplemental Fig. S8A,B). CeRAD-51 and CeRAD-51-TM also form presynaptic complexes in the ssDNA curtains with similar assembly and disassembly kinetics, although CeRAD-51-TM binds ∼21% more slowly and dissociates ∼26% more quickly than CeRAD-51 (Supplemental Fig. S8C,D; Supplemental Table S3). Assays with Atto565-dsDNA fragments confirmed that both CeRAD-51 proteins exhibited base triplet stepping (Fig. 5B,C; Supplemental Fig. S8E,F), and, as with the other recombinases, wt and triple mutant RAD-51 were unable to stabilize mismatches present at the end of an embedded tract of microhomology (Fig. 5D). However, in contrast to ScRad51 and hRAD51, wt CeRAD-51 could stabilize mismatches located at an internal position within the embedded tract of microhomology (Fig. 5E). This finding demonstrates that wt CeRAD-51 does not behave like “canonical” Rad51 when presented with a mismatched substrate; rather, it responds similarly to Dmc1. However, the ability to stabilize mismatched base triplets was abolished for the CeRAD-51-TM (Fig. 5E). Remarkably, CeRAD-51 could also promote D-loop formation with mismatched substrates (32% sequence divergence), albeit at low efficiency, whereas CeRAD-51-TM lacks this activity (Supplemental Fig. S8G,H). Together, these findings provide additional support for the premise that lineage-specific L1 residues in Dmc1 confer the ability to stabilize mismatched base triplets within the context of heteroduplex strand exchange intermediates.
Chimeric CeRAD-51 gives rise to toxic recombination intermediates
To investigate the importance of the L1 lineage-specific amino acids in vivo, we performed genome editing by CRISPR–Cas9 to introduce three substitution mutations (N246S, E256A, and K260H) into the C. elegans rad-51 gene. The resulting rad-51(knu529) strain (Supplemental Fig. S9A,B) was assessed for evidence of meiotic dysfunction, genome instability, and loss of fecundity. Deletion of rad-51 results in defective meiotic DSB repair and embryonic lethality in worms (Alpi et al. 2003; Martin et al. 2005). In contrast, the rad-51(knu529) strain exhibited brood sizes, embryonic viability, and meiotic chromosome nondisjunction rates (as assessed by the frequency of males) comparable with N2(Wt) control strains (Fig. 6A,B). The lack of an apparent phenotype associated with the rad-51(knu529) allele indicates that CeRAD-51-TM is a functional recombinase in vivo.
Figure 6.

C. elegans RAD-51 requires Dmc1-like amino acids to avoid formation of toxic HR intermediates during meiosis. (A) Increased recombination between heterologous sequences induced by rtel-1 or brc-1 depletion is suppressed in the rad-51(knu529) background, indicating loss of tolerance for DNA sequence heterology during RAD-51-TM-mediated recombination in vivo. The “-RNAi” label corresponds to conditions in which the C. elegans strain was treated with control bacteria lacking an expression plasmid for RNAi. P-values by χ2. (n.s.) P > 0.05; (*) P ≤ 0.05; (**) P ≤ 0.01; (***) P ≤ 0.001. (B) Brood size in strains of the indicated genotype. Progeny of five to 12 worms were evaluated. P-values by Mann-Whitney test. (n.s.) P > 0.05; (*) P ≤ 0.05; (**) P ≤ 0.01. (C) Percentage of hatched eggs after 24 h in strains of the indicated genotype. Progeny of five to 12 worms were evaluated. P-values by Mann-Whitney test. (n.s.) P > 0.05; (****) P ≤ 0.0001. (D) C. elegans germline with marked zones used to score meiotic RAD-51 focus formation. Quantification of meiotic RAD-51 focus formation in the different zones of the worm germline in strains of the indicated genotype. helq-1; rad-51(knu529) and rtel-1; rad-51(knu529) display persistent RAD-51 foci in late stages of meiosis. Between 67 and 548 cells were quantified for each zone in two independent experiments for each genotype. P-values by Mann-Whitney test. (n.s.) P > 0.05; (*) P ≤ 0.05; (****) P ≤ 0.0001. (E) Representative images of different compartments of the C. elegans germline. (Blue) DAPI staining; (green) RAD-51 staining. Scale bar, 5 µm.
Next, we investigated whether the rad-51(knu529) strain displays intolerance to mismatches in the context of chromosome pairing during meiotic HR. Since C. elegans is a highly inbred organism, the lack of an apparent phenotype of the rad-51(knu529) allele may be partially explained by low DNA sequence divergence in the C. elegans N2 strain. To circumvent this potential issue, we took advantage of the recently described heterozygous mln-1 inversion system (León-Ortiz et al. 2018) on chromosome II. The inverted mIn1 region is flanked by gfp and rol-1 markers, while the noninverted chromosome carries the dpy-25 allele. Rare heterologous recombination (het-rec) events occurring between this synapsed 8-Mb inverted sequence in meiosis can be observed by scoring recombination between the visible markers (Fig. 6A). In wt and rad-51(knu529) strains, het-rec events are extremely rare. However, it has been shown that depletion of RTEL-1 or BRC-1 in the N2(wt) strain leads to a significant increase in the frequency of het-rec events, which are mismatch-dependent (León-Ortiz et al. 2018). Given the latter, this system can be used to assess the mismatch tolerance of C. elegans RAD-51. When compared with N2(Wt), depletion of RTEL-1 or BRC-1 yielded a significantly reduced frequency of het-rec events in the rad-51(knu529) strain. Given that RAD-51-TM is intolerant to mismatches, these results suggest that the reduction in het-rec events seen in the rad-51(knu529) strain likely reflects an inability to promote pairing reactions between mismatch-containing sequences within the mIn-1 inversion. Importantly, scoring the visible markers dpy-17/unc-36, carried on a fully homologous genetic interval on chromosome III, did not reveal any significant difference in recombination frequencies between N2(Wt) and rad-51(knu529) strains (Supplemental Fig. S9C). These observations suggest that the reduced het-rec frequencies in rad-51(knu529) background arise due to lower mismatch tolerance of CeRAD-51-TM rather than an overall impairment of its recombinase activity. Furthermore, since deletion of the mismatch repair gene msh-2 suppresses het-rec events occurring in the rtel-1 background (León-Ortiz et al. 2018), our data further support the importance of mismatch tolerance and recognition in this process.
Given the suppression of the rtel-1 phenotype in the het-rec assay, we next assessed whether the rad-51(knu529) allele could also suppress other rtel-1-associated phenotypes, including decreased brood size, reduced fecundity, and DNA damage sensitivity (Barber et al. 2008). Contrary to expectation, we instead observed a synthetic phenotype when rtel-1 and rad-51(knu529) were combined, including a further decrease in brood size (Fig. 6B), elevated levels of embryonic lethality (Fig. 6C), and increased sensitivity to genotoxins (Supplemental Fig. S9D). We also observed a modest increase in chromosomal aberrations in diakinesis (Supplemental Fig. S9E,F). RECQL5 was implicated previously in suppressing HR by dismantling RAD51 filaments (Hu et al. 2007), while RTEL1 has been shown to disrupt D-loop HR intermediates to alter the outcome of meiotic DSB repair (Barber et al. 2008; Youds et al. 2010). C. elegans RECQL5 does not seem to play a role in suppressing additional meiotic crossovers or het-rec events (Barber et al. 2008; Youds et al. 2010). Importantly, no discernable genetic interaction was observed when rad-51(knu529) was combined with a deletion in rcq-5 (Fig. 6B,C).
Next, we analyzed the rtel-1; rad-51(knu529) strain for alterations in RAD-51 focus formation and/or resolution in the C. elegans germline. Premeiotic S phase at the distal end of the germline is followed by progressive stages of meiosis I, which are defined by specific chromosome alterations associated with homologous chromosome alignment, pairing, synapsis, and chiasmata formation (Fig. 6D). Recombination intermediates formed by RAD-51 at SPO-11-induced meiotic DSBs are repaired to produce crossovers or noncrossovers and can be monitored by the appearance and timely resolution of RAD-51 foci in late pachytene/early diplotene. Quantification of RAD-51 foci in each of these zones revealed that both the timing and levels of RAD-51 focal accumulation at meiotic DSBs were comparable between N2(Wt) and the rtel-1; rad-51(knu529) strain, further supporting the conclusion that RAD-51-TM is a functional recombinase in vivo. However, in contrast to the N2(Wt) and rad-51(knu529) strain alone, RAD-51 foci persisted into late stages of meiotic prophase in the rtel-1; rad-51(knu529) strain (Fig. 6D,E). This phenotype is reminiscent of the phenotype observed in helq-1; rtel1-1 double mutants that also present with reduced brood sizes, embryonic lethality, and persistent RAD-51 foci (Ward et al. 2010). Prompted by this similarity, we crossed rad-51(knu529) into the helq-1 background. Intriguingly, the helq-1; rad-51(knu529) strain also displayed persistence of RAD-51 foci in late pachytene and diplotene phases of meiosis (Fig. 6D,E). The helq-1; rad-51(knu529) strain also showed a significant brood size reduction (Fig. 6B), increased numbers of unhatched eggs (Fig. 6C), and a modest effect on chromosomal aberrations in diakinesis (Supplemental Fig. S9E,F).
Taken together, our data reveal that CeRAD-51-TM is a functional recombinase proficient for meiotic HR in vivo but is unable to tolerate mismatch-containing heterologous DNA sequences during strand invasion. Since RTEL-1 and HELQ-1 are believed to act to dismantle postsynaptic recombination intermediates, the synthetic genetic interactions with rtel-1 and helq-1 also reveal a dependence on postsynaptic recombination regulators for the survival and effective repair of meiotic DSBs in the rad-51(knu529) strain. This is suggestive of the presence of toxic recombination intermediates arising in the rad-51(knu529) strain, which must be effectively removed by RTEL-1 or HELQ-1 to allow meiotic progression.
Discussion
Here we explored the structural elements that contribute to the differential response of Rad51 and Dmc1 to mismatch-containing base triplets. We propose that these structural and functional differences represent a fundamental distinction between the Rad51 and Dmc1 lineages of the Rad51/RecA family of DNA recombinases. Here, we discuss possible implications of these findings with respect to recombinase structures, recombination mechanisms, and the evolution of Rad51/RecA family members.
Lineage-specific amino acid residues help determine recombinase behaviors
L1 and L2 DNA-binding loops of the Rad51/RecA family of DNA recombinases contain amino acids that are specifically conserved within either the Rad51 lineage or the Dmc1 lineage but not both. Our data demonstrate that three amino acid residues within the L1 DNA-binding loop regulate the response of Rad51 and Dmc1 to mismatched base triplets. Mutations that swap the identities of these residues alter the response of the respective recombinase to match that of its paralog, and this behavior appears to be conserved between recombinases from both S. cerevisiae and humans. We speculate that within the Rad51 lineage, these residues may help enhance recombination fidelity, whereas the identity of these residues within the Dmc1 lineage may contribute to meiosis, perhaps by allowing for more favorable recombination between polymorphic parental alleles. Indeed, a single point mutation in Rad51 (M301Q) is sufficient to recapitulate Dmc1-like mismatch stabilization in vitro and also enhances recombination between divergent sequences in vivo. However, we also note that the precise spatial geometry of the L1 DNA-binding loop is likely to be influenced by its native context, in particular its connectivity to the rest of the recombinase core domain. Thus, it is possible that the mismatch tolerance observed for Rad51-M301Q may not exactly match that of Dmc1. Indeed, Dmc1 itself may prove to be even more mismatch-tolerant in vivo than the Rad51 chimeras harboring Dmc1 residues. Although the natural prevalence of sequence polymorphisms between parental alleles would not approach the high levels of sequence divergence in our MAT switching assays, we anticipate that more subtle differences in recombination efficiency involving templates with lower divergence may be important when considered over long evolutionary time scales. Moreover, MAT switching is a highly efficient HR-dependent process that is greatly facilitated by local chromosomal architecture (Haber 2012), which may make MAT switching inherently more tolerant to mismatches. It is possible that less efficient HR-mediated repair events could exhibit even more pronounced effects at lower levels of sequence divergence.
Interestingly, ScRad51-DL2 retains basic biochemical functions but does not support Rad54 focus formation or allow for growth on MMS or zeocin plates (data not shown). The disparity between the in vivo and in vitro activities of this chimera suggest that lineage-specific amino acids in L2 may be important for assembly of the native presynaptic complex. Finally, in addition to the L1 and L2 amino acids described in this study, we also identified ∼19 lineage-specific amino acid residues present in other regions of the two recombinases (data not shown). We anticipate that many of these residues may mediate Rad51- and Dmc1-specific protein–protein contacts. Exploring the roles of these lineage-specific amino acids may yield further insights into the differential properties of the two eukaryotic recombinases.
Recombination between divergent sequences
Rad51/RecA family recombinases require at least consecutive 8 nt of microhomology for efficient recognition of short dsDNA substrates in vitro (Hsieh et al. 1992; Ragunathan et al. 2011; Danilowicz et al. 2015; Qi et al. 2015). However, the efficiency of BIR (Anand et al. 2017) and MAT switching (this study) indicate that the in vivo requirements for donor DNA recognition and strand invasion are significantly less stringent. Two factors may account for these differences. First, in vivo recombination requires many other proteins, a number of which may influence the activities of Rad51 (Symington et al. 2014; Kowalczykowski 2015). Second, long substrates likely allow for multiple simultaneous interactions, the cumulative effects of which may circumvent the kinetic benefits observed in vitro for shorter substrates bearing a single contiguous tract of homology. For example, the Z-box with mismatches at every eighth position has 40 adjacent 7-nt tracts of microhomology separated from one another by single mismatches, and the Z-box with mismatches at every sixth position has 52 adjacent 5-nt tracts of microhomology (Supplemental Table S5). At present, the lengths and complex sequence compositions of these substrates preclude detailed biophysical analysis in ssDNA curtain assays, and we cannot yet recapitulate the protein composition of a native presynaptic complex in vitro. Future work will be necessary to understand how these parameters influence the efficiency and kinetics of HR reaction mechanisms.
Our data reveal that the chimeric ScRad51-M301Q mutant, which stabilizes mismatches in biophysical assays, can also support more efficient HR-mediated repair during MAT switching for a Z-box bearing ≥20% sequence divergence. It is interesting to note that for wt ScRad51, we observed the largest decline in HR efficiency only for templates with ≥20% divergence (Fig. 4G,H). Inspection of these DNA sequences reveals an interesting feature that coincides with this dramatic change in HR efficiency; namely, for templates ranging up to 16.7% sequence divergence, the mismatched base triplets are never adjacent to one another, but this is not true for the templates with ≥20% sequence divergence, which instead are comprised of repeating patterns in which there are always two or three mismatched triplets adjacent to one another (Supplemental Table S5). One possible explanation for the marked decline in HR efficiency at ≥20% sequence divergence is that Rad51 may have particular difficulties in promoting recombination when two or more adjacent base triplets contain mismatches, whereas Dmc1 (or ScRad51 harboring Dmc1 L1 amino acids) may be more tolerant of these substrates.
What are the mechanistic impacts of mismatches on recombination?
We can envision at least two general mechanisms by which DNA mismatches could affect recombination: (1) by altering the intrinsic stability of the heteroduplex DNA intermediates or (2) by altering the structure of the resulting nucleoprotein complex, perhaps making these intermediates more susceptible to disruption by regulatory enzymes. We do not favor the hypothesis that a small number of mismatches greatly alters the intrinsic stability of the heteroduplex intermediates. Although a single mismatch impacts the binding lifetimes of dsDNA fragments in our biophysical assays, on the whole, these are all still relatively long-lived intermediates (e.g., for ScRad51, a 70-bp dsDNA fragment with 12 nt of microhomolgy, has a lifetime of ∼33 min; increasing the microhomology length to 15 nt yields a lifetime of ∼47 min, and introduction of a single mismatch within the 15-nt tract of microhomology reduces the lifetime to ∼35 min). This conclusion is also consistent with studies demonstrating that bacterial RecA is surprisingly tolerant of mismatches in vitro (Danilowicz et al. 2015). We cannot yet predict how our in vitro observations will scale for the longer in vivo substrates or currently measure the equivalent biophysical parameters in vitro with longer substrates. Nevertheless, the available biophysical data imply that mismatched HR intermediates are not intrinsically unstable. Thus, we favor the hypothesis that mismatches may somehow alter the structure of the nucleoprotein complexes, thus rendering them more susceptible to disruption by regulatory enzymes. Several proteins are known to dissociate HR intermediates, including the S. cerevisiae helicases Srs2 and Sgs1 (BLM in humans), and the postreplicative mismatch repair machinery also plays a role in minimizing HR between divergent sequences (Spell and Jinks-Robertson 2004; Sugawara et al. 2004; Spies and Fishel 2015; Branzei and Szakal 2017; Lorenz 2017). One possibility is that these or other enzymes may recognize some distinct mismatch-dependent structural feature that enables them to more readily act on Rad51-bound intermediates, whereas Dmc1 may shield mismatched intermediates from these enzymes (Fig. 7A). This hypothesis is consistent with the observed synthetic lethality between the CeRAD-51-TM mutant and RTEL-1 and HELQ-1 (see below).
Figure 7.

Potential mechanism of mismatch stabilization and L1 conservation among different Rad51/RecA family members. (A) Model for differences between Rad51 and Dmc1 interactions involving imperfectly paired HR intermediates. (B,C) Snapshots taken from MD simulations of hRAD51 (B) and hRAD51 harboring three Dmc1 lineage-specific amino acids substitutions (A240E M243Q H244K) (C), suggesting that the Dmc1 amino acids are better positioned to contact the incoming complementary DNA strand. Insets highlight potential protein contacts (red dashed lines) with the ribose ring of the phosphate backbone. (D) Comparison of L1 and L2 sequences from E. coli RecA; “canonical” Rad51 and Dmc1 from organisms harboring both recombinases; Rad51 from four Drosophilia sp. (Drosophilia melanogaster, Drosophilia virilis, Drosophilia mojavenis, and Drosophilia simulans); Rad51 sequences designated as “other” from Ustilago maydis, Sodaria macrospora, and Neurospora crassa; and RadA from Haloferax volcanii, Pyrococcus furiousus, Methanococcus maripaludis, Methanococcus voltae, and Sulfolobus solfataricus. Asterisks denote the amino acids that contribute to mismatch stabilization for Dmc1.
Potential mechanisms of DNA mismatch stabilization
Three lineage-specific residues clustered together within L1 DNA-binding loop contribute mismatch stabilization by Dmc1. We proposed that these amino acids may allow Dmc1 to make compensatory DNA contacts that are independent of Watson-Crick pairing interactions. Consistent with this hypothesis, molecular dynamics (MD) simulations of wt hRAD51 and an hRAD51 chimeric mutant bearing three hDMC1 residues in the L1 domain—corresponding to the mutations A241E, M244K and H245K (numbering based on hRAD51) (Fig. 1A)—reveal that the introduction of these Dmc1-specific residues results in more intimate contacts between L1 and the phosphate backbone of the complementary strand of the postsynaptic complex (Fig. 7B,C). The notion that Dmc1 residues contact the phosphate backbone of the nascent DNA joint is also consistent with the observation that Dmc1 can stabilize base triplets bearing abasic sites (Lee et al. 2017). At present, these simulations must be interpreted with caution, given that they are based on structural data that lack sufficient information to fully describe the relevant protein–nucleic acid interfaces. In the case of hDMC1, the crystal structures lack DNA (Kinebuchi et al. 2004). For hRAD51, existing cryo-electron microscopy structures lack sufficient resolution to accurately define the L1 protein–DNA interface (Short et al. 2016; Xu et al. 2017). Similarly, although there is a crystal structure of ScRad51 bound to DNA, the L1 and L2 contacts are not visible (Conway et al. 2004). Finally, there is as yet no structural information regarding how recombinases interact with a DNA joint that harbors mismatches, and our simulations do not account for the potential influence of mismatches on recombinase-DNA interactions. Regardless, the MD simulations, together the observation that Dmc1 can stabilize abasic sites (Lee et al. 2017), are consistent with the general notion that the lineage-specific amino acid residues responsible for the differential responses of Rad51 and Dmc1 to mismatches are positioned to interact with the phosphate backbone of the complementary DNA strand within the nascent DNA joint.
C. elegans RAD-51 requires Dmc1-like amino acids
Ecdysozoans, such as Caenorhabditis sp. and Drosophila sp., possess RAD51 but have lost DMC1 as well as genes encoding core meiotic proteins necessary for Dmc1 activity (e.g., Hop2/Mnd1) (Villeneuve and Hillers 2001; Ramesh et al. 2005; Lin et al. 2006; Chintapalli et al. 2013; Hunter 2015). It remains unknown why these organisms have lost DMC1, and the evolutionary implications of this loss remain unexplored. However, Caenorhabditis sp. and Drosophila sp. have among the highest rates of evolution for the RAD51 genes (Lin et al. 2006). Interestingly, we show that the lineage-specific amino acid residues within the L1 DNA-binding loop of RAD-51 from Caenorhabditis sp. have adapted to more closely match Dmc1 and that CeRAD-51 can stabilize mismatched base triplets in vitro. Thus, the loss of DMC1 may have caused, or allowed for, adaptation of CeRAD-51 to become more “Dmc1-like” with respect to the L1 DNA-binding loop. In accordance with the importance of these “Dmc1-like” residues, we found that the chimeric CeRad51-TM mutant, which bears canonical Rad51-like amino acids in the L1 loop, is a functional recombinase in vivo but exhibits an inability to promote recombination through highly divergent sequences in vivo and a synthetic phenotype with antirecombinases (RTEL1 and HELQ-1). These data indicate that the identity of the native “Dmc1-like” amino acids present in the L1 loops of wt CeRAD-51 cannot be converted to amino acids found in the canonical Rad51 lineage without affecting the fidelity of meiotic HR and increasing the frequency of toxic recombination intermediates that places a dependence on motor proteins such as RTEL-1 and HELQ-1. We speculate that mismatches that would normally be masked by CeRAD-51 are not properly masked by the CeRAD-51-TM, yielding a dead-end D-loop intermediate that must be dismantled by RTEL-1 or HELQ-1. Thus, the most important effect of the canonical Dmc1 amino acid residues may be with respect to differences in their ability to mask mismatched heteroduplexes from aberrant processing (Fig. 7A). Interestingly, Drosophila sp. Rad51 L1 amino acids closely match those found in canonical Rad51 (Fig. 7D). However, N301 (numbering based on S. cerevisiae Rad51 for comparison) in Drosophila Rad51 does not match the methionine found in canonical Rad51 but is instead more similar to the glutamine from Dmc1 (Fig. 7D). Thus, Drosophila Rad51 may also have a similar requirement for “Dmc1-like” amino acids in the L1 DNA-binding loop.
Among organisms that have Dmc1, chromosome pairing is initiated through recombination-dependent mechanisms (Neale and Keeney 2006; Brown and Bishop 2014; Hunter 2015). In contrast, both C. elegans and D. melanogaster have evolved alternative, recombination-independent mechanisms for initiating chromosome pairing during meiosis (Villeneuve and Hillers 2001; Gerton and Hawley 2005). However, use of alternative pairing mechanisms is not necessarily the defining attribute of organisms lacking Dmc1. For instance, Ustilago maydis, Sodaria macrospora, and Neurospora crassa have all lost the DMC1 gene but require recombination to initiate chromosome pairing during meiosis (Storlazzi et al. 2003). Interestingly, U. maydis, S. macrospora, and N. crassa all possess canonical Rad51 lineage-specific amino acids in the L1 DNA-binding loop (Fig. 7D). Finally, it should be noted that the L1 (and L2) amino acids from bacterial RecA are highly divergent from the eukaryotic recombinases (Figs. 1A, 7D). Therefore, the mechanisms by which RecA interacts with the DNA intermediates may differ from those that define the action of the eukaryotic recombinases.
Potential origins of the dual-recombinase paradigm
Rad51 and Dmc1 arose early in eukaryotic evolution from a gene duplication event involving an ancestral archaeal RadA recombinase, and this gene duplication event may have coincided with or allowed for the emergence of meiosis and sexual reproduction (Ramesh et al. 2005; Lin et al. 2006; Chintapalli et al. 2013). Some extant archaea, such as Haloferax volcanii, undergo a conjugation process involving the exchange of highly divergent genetic information via HR, and this conjugation process bears some resemblance to the eukaryotic meiotic program (Mevarech and Werczberger 1985; Rosenshine et al. 1989; Papke et al. 2004; Cohan and Aracena 2012; Naor et al. 2012; Naor and Gophna 2013). Most intriguingly, the RadA L1 DNA-binding loop amino acid residues located at the positions involved in mismatch stabilization are all identical to those found in the Dmc1 lineage of the Rad51/RecA family (Fig. 7D). One speculative possibility is that a “lower-fidelity” recombinase (i.e., more “Dmc1-like”) may have predated the “higher-fidelity” Rad51 present in modern eukaryotic lineages, and the emergence of Rad51 may have allowed eukaryotes to take better advantage of HR as a high-fidelity DNA repair pathway.
Materials and methods
Sequence and crystal structure alignments
Crystal structures of E. coli RecA (1CMX) (Chen et al. 2008), ScRad51 (1SZP) (Conway et al. 2004), hDMC1 (1V5W) (Kinebuchi et al. 2004), and hRAD51 (5H1C) (Xu et al. 2017) were obtained from the RCSB Protein Data Bank and aligned in MacPyMOL (PyMOL molecular graphics system, version 1.8). All protein sequences were obtained from the NCBI database, and sequence alignments were performed using National Institutes of Health COBALT (Papadopoulos and Agarwala 2007). Most of the sequenced RAD51 and DMC1 genes have not been experimentally validated; therefore, to help ensure the alignments reflected in information from bona fide Rad51 and Dmc1 proteins, we restricted our analysis to proteins that were ≥200 amino acids in length and also contained the highly conserved L1 motif GRGEL (or GRGDL; corresponding to amino acids 294–298 in ScRad51). Sequences that did not fulfill these two criteria were excluded from the analysis. Aligned sequences were further analyzed for common features and annotated using ESPript 3.0 (Robert and Gouet 2014).
Single-molecule dsDNA-binding assays
All single-molecule dsDNA-binding experiments were performed as described previously (Lee et al. 2015, 2017; Qi et al. 2015). In brief, presynaptic filaments were assembled, as described above, by injecting the indicated recombinase at a concentration of 2 µM in the presence of 2 mM ATP followed by a 20-min incubation at 30°C. Free protein was then flushed from the sample chamber, followed by an injection of HR buffer (as indicated above for each recombinase), and the reactions were incubated for an additional 10 min. Unbound dsDNA was then quickly flushed from the sample chamber using a 30-sec wash at 1 mL/min; flow was then reduced to 0.2 mL/min, and images (90-msec integration) were collected at 30- to 60-sec intervals for 1–2 h, as described previously (Lee et al. 2015, 2017; Qi et al. 2015). The data collection intervals were optimized relative to the overall lifetime of each dsDNA substrate, and the laser was shuttered between acquired images to minimize photobleaching. Kymographs were then generated from the resulting movies using Fiji. Survival probabilities were determined from analysis of the resulting kymographs by measuring the time (dwell time) that each molecule of Atto565-dsDNA remained bound to the presynaptic complexes after flushing the unbound DNA from the sample chamber. Error bars for the survival probability measurements and binding distributions represent 70% confidence intervals obtained through bootstrap analysis, providing a close approximation of expectations for one standard deviation from the mean (Lee et al. 2015; Qi et al. 2015). All reported ΔΔG‡ values were calculated from the dissociation rate data for the Atto565-dsDNA substrates, as described (Lee et al. 2015; Qi et al. 2015).
Free energy calculations
All free energy calculations were performed using to the following equation, as described previously (Lee et al. 2015, 2016, 2017; Qi et al. 2015; Qi and Greene 2016; Ma et al. 2017):
where kb is the Boltzmann constant, T is temperature, and ΔΔG‡ is the free energy difference between two escape processes described by the dissociation rates and . The experimentally measured data used to calculate ΔΔG‡ were the dissociation rate data for the different oligonucleotide substrates (see Supplemental Table S4), which were obtained from the survival probability analysis for each different recombinase with the different dsDNA oligonucleotide substrates. The reported ΔΔG‡ values for analysis of base triplet stepping are indicated by double-ended arrows, and the step 1, step 2, and step 3 designations are also highlighted by color-coded schematics and arrowheads. For analysis of mismatch-bearing oligonucleotides, the ΔΔG‡ assigned for the first step was set to zero (“step 1” in figure diagrams in Figs. 2B, 3A–B), and the step 1–3 designations reflect the values obtained for fully paired substrates for each specific protein (as indicated by dashed color-coded lines).
Additional Materials and Methods are in the Supplemental Material.
Acknowledgments
We are grateful to members of the Greene, Boulton, Rothstein, Symington, and Sung laboratories for helpful conversations. We thank Daniel Duzdevich for discussion and experimental assistance, and Johannes Stigler for assistance with data analysis. This work was supported in part by National Institutes of Health (NIH) training grants F31CA210663 (to J.B.S.) and T32GM007088 (to M.J.S.); NIH grants R35GM118026 (to E.C.G.), R35GM126997 (to L.S.S.), R35GM118180 (to R.R.), R01ES007061 (to P.S.), and P01CA92584 (to P.S. and E.C.G); and National Science Foundation grant MCB-1817315 (to E.C.G.). J.B.C. is the Mark Foundation for Cancer Research Fellow for the Damon-Runyon Cancer Research Foundation (DRG 2310-17). The Boulton laboratory is supported by the Francis Crick Institute, which receives its core funding from Cancer Research UK (FC0010048), the UK Medical Research Council (FC0010048), and the Wellcome Trust (FC0010048). S.J.B. is also the recipient of a European Research Council (ERC) Advanced Investigator Grant (TelMetab) and Wellcome Trust Senior Investigator and Collaborative Grant (to S.J.B and E.C.G.).
Author contributions: J.B.S, O.B., S.J.B., and E.C.G. designed the study. J.B.S. conducted all single-molecule experiments with yeast and human recombinases and the mutant C. elegans RAD-51 and conducted the MAT switching assays. O.B. expressed and purified wt and mutant C. elegans RAD-51 and conducted bulk biochemical assays and all in vivo C. elegans studies. Z.Q. conducted single-molecule experiments with wt C. elegans RAD-51. Y.K. conducted all bulk biochemical ATPase and strand exchange assays. J.B.S. cloned all of the yeast and human protein expression plasmids. J.B.S., Y.K., and W.Z. purified all yeast and human recombinases. T.T. performed molecular dynamics simulations. J.B.C. conducted Western blot analysis for protein expression levels. A.A.-Z. and M.J.S. conducted in vivo assays for ScRad51 drug resistance and Rad54 focus formation, respectively. J.B.S., O.B., S.J.B., and E.C.G. cowrote the manuscript with input from all coauthors.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.328062.119.
References
- Alpi A, Pasierbek P, Gartner A, Loidl J. 2003. Genetic and cytological characterization of the recombination protein RAD-51 in Caenorhabditis elegans. Chromosoma 112: 6–16. 10.1007/s00412-003-0237-5 [DOI] [PubMed] [Google Scholar]
- Anand R, Beach A, Li K, Haber J. 2017. Rad51-mediated double-strand break repair and mismatch correction of divergent substrates. Nature 544: 377–380. 10.1038/nature22046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argunhan B, Leung WK, Afshar N, Terentyev Y, Subramanian VV, Murayama Y, Hochwagen A, Iwasaki H, Tsubouchi T, Tsubouchi H. 2017. Fundamental cell cycle kinases collaborate to ensure timely destruction of the synaptonemal complex during meiosis. EMBO J 36: 2488–2509. 10.15252/embj.201695895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber LJ, Youds JL, Ward JD, McIlwraith MJ, O'Neil NJ, Petalcorin MI, Martin JS, Collis SJ, Cantor SB, Auclair M, et al. 2008. RTEL1 maintains genomic stability by suppressing homologous recombination. Cell 135: 261–271. 10.1016/j.cell.2008.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgogno MV, Monti MR, Zhao W, Sung P, Argaraña CE, Pezza RJ. 2016. Tolerance of DNA mismatches in Dmc1 recombinase-mediated DNA strand exchange. J Biol Chem 291: 4928–4938. 10.1074/jbc.M115.704718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, Szakal B. 2017. Building up and breaking down: mechanisms controlling recombination during replication. Crit Rev Biochem Mol Biol 52: 381–394. 10.1080/10409238.2017.1304355 [DOI] [PubMed] [Google Scholar]
- Brown MS, Bishop DK. 2014. DNA strand exchange and RecA homologs in meiosis. Cold Spring Harb Perspect Biol 7: a016659. 10.1101/cshperspect.a016659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busygina V, Sehorn MG, Shi IY, Tsubouchi H, Roeder GS, Sung P. 2008. Hed1 regulates Rad51-mediated recombination via a novel mechanism. Genes Dev 22: 786–795. 10.1101/gad.1638708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callender TL, Laureau R, Wan L, Chen X, Sandhu R, Laljee S, Zhou S, Suhandynata RT, Prugar E, Gaines WA, et al. 2016. Mek1 down regulates Rad51 activity during yeast meiosis by phosphorylation of Hed1. PLoS Genet 12: e1006226. 10.1371/journal.pgen.1006226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YK, Leng CH, Olivares H, Lee MH, Chang YC, Kung WM, Ti SC, Lo YH, Wang AH, Chang CS, et al. 2004. Heterodimeric complexes of Hop2 and Mnd1 function with Dmc1 to promote meiotic homolog juxtaposition and strand assimilation. Proc Natl Acad Sci 101: 10572–10577. 10.1073/pnas.0404195101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Yang H, Pavletich NP. 2008. Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature 453: 489–494. 10.1038/nature06971 [DOI] [PubMed] [Google Scholar]
- Chintapalli SV, Bhardwaj G, Babu J, Hadjiyianni L, Hong Y, Todd GK, Boosalis CA, Zhang Z, Zhou X, Ma H, et al. 2013. Reevaluation of the evolutionary events within recA/RAD51 phylogeny. BMC Genomics 14: 240. 10.1186/1471-2164-14-240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloud V, Chan YL, Grubb J, Budke B, Bishop DK. 2012. Rad51 is an accessory factor for Dmc1-mediated joint molecule formation during meiosis. Science 337: 1222–1225. 10.1126/science.1219379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohan FM, Aracena S. 2012. Prokaryotic sex: eukaryote-like qualities of recombination in an Archaean lineage. Curr Biol 22: R601–R602. 10.1016/j.cub.2012.06.052 [DOI] [PubMed] [Google Scholar]
- Conway AB, Lynch TW, Zhang Y, Fortin GS, Fung CW, Symington LS, Rice PA. 2004. Crystal structure of a Rad51 filament. Nat Struct Mol Biol 11: 791–796. 10.1038/nsmb795 [DOI] [PubMed] [Google Scholar]
- Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ. 2000. The importance of repairing stalled replication forks. Nature 404: 37–41. 10.1038/35003501 [DOI] [PubMed] [Google Scholar]
- Danilowicz C, Yang D, Kelley C, Prevost C, Prentiss M. 2015. The poor homology stringency in the heteroduplex allows strand exchange to incorporate desirable mismatches without sacrificing recognition in vivo. Nucleic Acids Res 43: 6473–6485. 10.1093/nar/gkv610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari SR, Grubb J, Bishop DK. 2009. The Mei5-Sae3 protein complex mediates Dmc1 activity in Saccharomyces cerevisiae. J Biol Chem 284: 11766–11770. 10.1074/jbc.C900023200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerton JL, Hawley RS. 2005. Homologous chromosome interactions in meiosis: diversity amidst conservation. Nat Rev Genet 6: 477–487. 10.1038/nrg1614 [DOI] [PubMed] [Google Scholar]
- Haber JE. 2012. Mating-type genes and MAT switching in Saccharomyces cerevisiae. Genetics 191: 33–64. 10.1534/genetics.111.134577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayase A, Takagi M, Miyazaki T, Oshiumi H, Shinohara M, Shinohara A. 2004. A protein complex containing Mei5 and Sae3 promotes the assembly of the meiosis-specific RecA homolog Dmc1. Cell 119: 927–940. 10.1016/j.cell.2004.10.031 [DOI] [PubMed] [Google Scholar]
- Hsieh P, Camerini-Otero CS, Camerini-Otero RD. 1992. The synapsis event in the homologous pairing of DNAs: RecA recognizes and pairs less than one helical repeat of DNA. Proc Natl Acad Sci 89: 6492–6496. 10.1073/pnas.89.14.6492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Raynard S, Sehorn MG, Lu X, Bussen W, Zheng L, Stark JM, Barnes EL, Chi P, Janscak P, et al. 2007. RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev 21: 3073–3084. 10.1101/gad.1609107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter N. 2015. Meiotic recombination: the essence of heredity. Cold Spring Harb Perspect Biol 7: a016618. 10.1101/cshperspect.a016618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinebuchi T, Kagawa W, Enomoto R, Tanaka K, Miyagawa K, Shibata T, Kurumizaka H, Yokoyama S. 2004. Structural basis for octameric ring formation and DNA interaction of the human homologous-pairing protein Dmc1. Mol Cell 14: 363–374. 10.1016/S1097-2765(04)00218-7 [DOI] [PubMed] [Google Scholar]
- Klein HL. 1997. RDH54, a RAD54 homologue in Saccharomyces cerevisiae, is required for mitotic diploid-specific recombination and repair and for meiosis. Genetics 147: 1533–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczykowski SC. 2015. An overview of the molecular mechanisms of recombinational DNA repair. Cold Spring Harb Perspect Biol 7: a016410. 10.1101/cshperspect.a016410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lao JP, Cloud V, Huang CC, Grubb J, Thacker D, Lee CY, Dresser ME, Hunter N, Bishop DK. 2013. Meiotic crossover control by concerted action of Rad51–Dmc1 in homolog template bias and robust homeostatic regulation. PLoS Genet 9: e1003978. 10.1371/journal.pgen.1003978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Terakawa T, Qi Z, Steinfeld JB, Redding S, Kwon Y, Gaines WA, Zhao W, Sung P, Greene EC. 2015. DNA RECOMBINATION. Base triplet stepping by the Rad51/RecA family of recombinases. Science 349: 977–981. 10.1126/science.aab2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Qi Z, Greene EC. 2016. ATP hydrolysis promotes duplex DNA release by the RecA presynaptic complex. J Biol Chem 291: 22218–22230. 10.1074/jbc.M116.740563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Steinfeld JB, Qi Z, Kwon Y, Sung P, Greene EC. 2017. Sequence imperfections and base triplet recognition by the Rad51/RecA family of recombinases. J Biol Chem 292: 11125–11135. 10.1074/jbc.M117.787614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- León-Ortiz AM, Panier S, Sarek G, Vannier JB, Patel H, Campbell PJ, Boulton SJ. 2018. A Distinct class of genome rearrangements driven by heterologous recombination. Mol Cell 69: 292–305.e6. 10.1016/j.molcel.2017.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Kong H, Nei M, Ma H. 2006. Origins and evolution of the recA/RAD51 gene family: evidence for ancient gene duplication and endosymbiotic gene transfer. Proc Natl Acad Sci 103: 10328–10333. 10.1073/pnas.0604232103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Barlow JH, Burgess RC, Rothstein R. 2004. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 118: 699–713. 10.1016/j.cell.2004.08.015 [DOI] [PubMed] [Google Scholar]
- Lorenz A. 2017. Modulation of meiotic homologous recombination by DNA helicases. Yeast 34: 195–203. 10.1002/yea.3227 [DOI] [PubMed] [Google Scholar]
- Ma CJ, Steinfeld JB, Greene EC. 2017. Single-stranded DNA curtains for studying homologous recombination. Methods Enzymol 582: 193–219. 10.1016/bs.mie.2016.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsolier-Kergoat MC, Khan MM, Schott J, Zhu X, Llorente B. 2018. Mechanistic view and genetic control of DNA recombination during meiosis. Mol Cell 70: 9–20.e6. 10.1016/j.molcel.2018.02.032 [DOI] [PubMed] [Google Scholar]
- Martin JS, Winkelmann N, Petalcorin MI, McIlwraith MJ, Boulton SJ. 2005. RAD-51-dependent and -independent roles of a Caenorhabditis elegans BRCA2-related protein during DNA double-strand break repair. Mol Cell Biol 25: 3127–3139. 10.1128/MCB.25.8.3127-3139.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Beach A, Haber JE. 2017. Homology requirements and competition between Gene conversion and break-induced replication during double-strand break repair. Mol Cell 65: 515–526.e3. 10.1016/j.molcel.2016.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mevarech M, Werczberger R. 1985. Genetic transfer in Halobacterium volcanii. J Bacteriol 162: 461–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrical SW. 2015. DNA-pairing and annealing processes in homologous recombination and homology-directed repair. Cold Spring Harb Perspect Biol 7: a016444. 10.1101/cshperspect.a016444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama Y, Kurokawa Y, Mayanagi K, Iwasaki H. 2008. Formation and branch migration of Holliday junctions mediated by eukaryotic recombinases. Nature 451: 1018–1021. 10.1038/nature06609 [DOI] [PubMed] [Google Scholar]
- Murayama Y, Tsutsui Y, Iwasaki H. 2011. The fission yeast meiosis-specific Dmc1 recombinase mediates formation and branch migration of Holliday junctions by preferentially promoting strand exchange in a direction opposite to that of Rad51. Genes Dev 25: 516–527. 10.1101/gad.1997511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama Y, Kurokawa Y, Tsutsui Y, Iwasaki H. 2013. Dual regulation of Dmc1-driven DNA strand exchange by Swi5-Sfr1 activation and Rad22 inhibition. Genes Dev 27: 2299–2304. 10.1101/gad.218693.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naor A, Gophna U. 2013. Cell fusion and hybrids in archaea: prospects for genome shuffling and accelerated strain development for biotechnology. Bioengineered 4: 126–129. 10.4161/bioe.22649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naor A, Lapierre P, Mevarech M, Papke RT, Gophna U. 2012. Low species barriers in halophilic archaea and the formation of recombinant hybrids. Curr Biol 22: 1444–1448. 10.1016/j.cub.2012.05.056 [DOI] [PubMed] [Google Scholar]
- Neale MJ, Keeney S. 2006. Clarifying the mechanics of DNA strand exchange in meiotic recombination. Nature 442: 153–158. 10.1038/nature04885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimonkar AV, Dombrowski CC, Siino JS, Stasiak AZ, Stasiak A, Kowalczykowski SC. 2012. Saccharomyces cerevisiae Dmc1 and Rad51 proteins preferentially function with Tid1 and Rad54 proteins, respectively, to promote DNA strand invasion during genetic recombination. J Biol Chem 287: 28727–28737. 10.1074/jbc.M112.373290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos JS, Agarwala R. 2007. COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics 23: 1073–1079. 10.1093/bioinformatics/btm076 [DOI] [PubMed] [Google Scholar]
- Papke RT, Koenig JE, Rodriguez-Valera F, Doolittle WF. 2004. Frequent recombination in a saltern population of Halorubrum. Science 306: 1928–1929. [DOI] [PubMed] [Google Scholar]
- Paques F, Haber JE. 1999. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 63: 349–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petukhova GV, Pezza RJ, Vanevski F, Ploquin M, Masson JY, Camerini-Otero RD. 2005. The Hop2 and Mnd1 proteins act in concert with Rad51 and Dmc1 in meiotic recombination. Nat Struct Mol Biol 12: 449–453. 10.1038/nsmb923 [DOI] [PubMed] [Google Scholar]
- Prentiss M, Prévost C, Danilowicz C. 2015. Structure/function relationships in RecA protein-mediated homology recognition and strand exchange. Crit Rev Biochem Mol Biol 50: 453–476. 10.3109/10409238.2015.1092943 [DOI] [PubMed] [Google Scholar]
- Prugar E, Burnett C, Chen X, Hollingsworth NM. 2017. Coordination of double strand break repair and meiotic progression in yeast by a Mek1–Ndt80 negative feedback loop. Genetics 206: 497–512. 10.1534/genetics.117.199703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Z, Greene EC. 2016. Visualizing recombination intermediates with single-stranded DNA curtains. Methods 105: 62–74. 10.1016/j.ymeth.2016.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Z, Redding S, Lee JY, Gibb B, Kwon Y, Niu H, Gaines WA, Sung P, Greene EC. 2015. DNA sequence alignment by microhomology sampling during homologous recombination. Cell 160: 856–869. 10.1016/j.cell.2015.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragunathan K, Joo C, Ha T. 2011. Real-time observation of strand exchange reaction with high spatiotemporal resolution. Structure 19: 1064–1073. 10.1016/j.str.2011.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh MA, Malik SB, Logsdon JM Jr. 2005. A phylogenomic inventory of meiotic genes; evidence for sex in Giardia and an early eukaryotic origin of meiosis. Curr Biol 15: 185–191. [DOI] [PubMed] [Google Scholar]
- Robert X, Gouet P. 2014. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42: W320–W324. 10.1093/nar/gku316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenshine I, Tchelet R, Mevarech M. 1989. The mechanism of DNA transfer in the mating system of an archaebacterium. Science 245: 1387–1389. 10.1126/science.2818746 [DOI] [PubMed] [Google Scholar]
- Sheridan SD, Yu X, Roth R, Heuser JE, Sehorn MG, Sung P, Egelman EH, Bishop DK. 2008. A comparative analysis of Dmc1 and Rad51 nucleoprotein filaments. Nucleic Acids Res 36: 4057–4066. 10.1093/nar/gkn352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short JM, Liu Y, Chen S, Soni N, Madhusudhan MS, Shivji MK, Venkitaraman AR. 2016. High-resolution structure of the presynaptic RAD51 filament on single-stranded DNA by electron cryo-microscopy. Nucleic Acids Res 44: 9017–9030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spell RM, Jinks-Robertson S. 2004. Examination of the roles of Sgs1 and Srs2 helicases in the enforcement of recombination fidelity in Saccharomyces cerevisiae. Genetics 168: 1855–1865. 10.1534/genetics.104.032771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spies M, Fishel R. 2015. Mismatch repair during homologous and homeologous recombination. Cold Spring Harb Perspect Biol 7: a022657. 10.1101/cshperspect.a022657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storlazzi A, Tesse S, Gargano S, James F, Kleckner N, Zickler D. 2003. Meiotic double-strand breaks at the interface of chromosome movement, chromosome remodeling, and reductional division. Genes Dev 17: 2675–2687. 10.1101/gad.275203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Story RM, Bishop DK, Kleckner N, Steitz TA. 1993. Structural relationship of bacterial RecA proteins to recombination proteins from bacteriophage T4 and yeast. Science 259: 1892–1896. 10.1126/science.8456313 [DOI] [PubMed] [Google Scholar]
- Sugawara N, Goldfarb T, Studamire B, Alani E, Haber JE. 2004. Heteroduplex rejection during single-strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. Proc Natl Acad Sci 101: 9315–9320. 10.1073/pnas.0305749101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS, Rothstein R, Lisby M. 2014. Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics 198: 795–835. 10.1534/genetics.114.166140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MRG, Špírek M, Jian Ma C, Carzaniga R, Takaki T, Collinson LM, Greene EC, Krejci L, Boulton SJ. 2016. A polar and nucleotide-dependent mechanism of action for RAD51 paralogs in RAD51 filament remodeling. Mol Cell 64: 926–939. 10.1016/j.molcel.2016.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker D, Mohibullah N, Zhu X, Keeney S. 2014. Homologue engagement controls meiotic DNA break number and distribution. Nature 510: 241–246. 10.1038/nature13120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubouchi H, Roeder GS. 2002. The Mnd1 protein forms a complex with hop2 to promote homologous chromosome pairing and meiotic double-strand break repair. Mol Cell Biol 22: 3078–3088. 10.1128/MCB.22.9.3078-3088.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubouchi H, Roeder GS. 2004. The budding yeast mei5 and sae3 proteins act together with dmc1 during meiotic recombination. Genetics 168: 1219–1230. 10.1534/genetics.103.025700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubouchi H, Roeder GS. 2006. Budding yeast Hed1 down-regulates the mitotic recombination machinery when meiotic recombination is impaired. Genes Dev 20: 1766–1775. 10.1101/gad.1422506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve AM, Hillers KJ. 2001. Whence meiosis? Cell 106: 647–650. 10.1016/S0092-8674(01)00500-1 [DOI] [PubMed] [Google Scholar]
- Ward JD, Muzzini DM, Petalcorin MI, Martinez-Perez E, Martin JS, Plevani P, Cassata G, Marini F, Boulton SJ. 2010. Overlapping mechanisms promote postsynaptic RAD-51 filament disassembly during meiotic double-strand break repair. Mol Cell 37: 259–272. 10.1016/j.molcel.2009.12.026 [DOI] [PubMed] [Google Scholar]
- Xu J, Zhao L, Xu Y, Zhao W, Sung P, Wang HW. 2017. Cryo-EM structures of human RAD51 recombinase filaments during catalysis of DNA-strand exchange. Nat Struct Mol Biol 24: 40–46. 10.1038/nsmb.3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youds JL, Mets DG, McIlwraith MJ, Martin JS, Ward JD NJON, Rose AM, West SC, Meyer BJ, Boulton SJ. 2010. RTEL-1 enforces meiotic crossover interference and homeostasis. Science 327: 1254–1258. 10.1126/science.1183112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Keeney S. 2014. Zip it up to shut it down. Cell Cycle 13: 2157–2158. 10.4161/cc.29747 [DOI] [PMC free article] [PubMed] [Google Scholar]