

Summary
Here, we determined the relative importance of different transcriptional mechanisms in the genome-reduced bacterium Mycoplasma pneumoniae, by employing an array of experimental techniques under multiple genetic and environmental perturbations. Of the 143 genes tested (21% of the bacterium’s annotated proteins), only 55% showed an altered phenotype, highlighting the robustness of biological systems. We identified nine transcription factors (TFs) and their targets, representing 43% of the genome, and 16 regulators that indirectly affect transcription. Only 20% of transcriptional regulation is mediated by canonical TFs when responding to perturbations. Using a Random Forest, we quantified the non-redundant contribution of different mechanisms such as supercoiling, metabolic control, RNA degradation, and chromosome topology to transcriptional changes. Model-predicted gene changes correlate well with experimental data in 95% of the tested perturbations, explaining up to 70% of the total variance when also considering noise. This analysis highlights the importance of considering non-TF-mediated regulation when engineering bacteria.
Keywords: systems biology, gene regulatory network, transcription regulation, transcription, transcription factors, Mycoplasma pneumoniae
Graphical Abstract
Highlights
-
•
Full comprehensive reconstruction of a bacterial gene regulatory network achieved
-
•
Genome-reduced bacterium Mycoplasma pneumoniae is robust to genetic perturbations
-
•
Large part of transcription regulation in bacteria is transcription-factor independent
-
•
Transcription-factor-independent regulation has a smaller dynamic range
We have achieved a comprehensive reconstruction of a gene regulatory network in a genome-reduced bacterium, Mycoplasma pneumoniae. With this network, we observed that a large part of transcription regulation is determined by non-canonical factors such as DNA supercoiling, riboswitches, and genome organization or RNA-mediated regulation. This analysis highlights the importance of considering non-transcription-factor (TF)-mediated regulation when engineering bacteria.
Introduction
Transcription factors (TFs) are key players in gene regulatory networks. Accordingly, classical studies have generally focused on uncovering their function. Such studies rely on genome annotation and comparative sequence analysis to first identify the TFs (Hecker and Völker, 2001, Mitrophanov and Groisman, 2008, Schmidl et al., 2011), and then, through bottom-up approaches, individually or systematically analyze the TF targets or regulons (Minch et al., 2015), (Lee et al., 2002). These approximations are limited by the fact that many non-canonical regulators and proteins with moonlighting functions (metabolic enzymes (Commichau and Stülke, 2008, Jeffery, 2015) and structural proteins (e.g., nucleoid-associated proteins (NAPs)) (Dillon and Dorman, 2010) are often neglected in such studies.
Transcriptional regulation in bacteria depends on more than just TFs. For example, even in the case of well-studied bacterial models such as Escherichia coli and Bacillus subtilis, less than 40% and 52% of the genes, respectively, seem to be regulated by TFs (not including the targets of the housekeeping sigma-70) (Salgado et al., 2013), (Michna et al., 2016), (Leyn et al., 2013). This is even more remarkable in streamlined genomes such as those of endosymbionts (Brinza et al., 2013). Other factors including structural proteins and NAPs have been shown to provide an additional layer of regulation by affecting DNA topology (Hatfield and Benham, 2002), (Travers and Muskhelishvili, 2005). DNA supercoiling plays an important role in transcriptional regulation. Supercoiling depends on the opposite actions of ATP-independent topoisomerase I and ATP-dependent gyrase. In this way, ATP concentrations regulate supercoiling and gene transcription (Baranello et al., 2012) (Dorman and Dorman, 2016). Local chromosome interacting domains have a role in transcriptional coordination (Dekker et al., 2013, Trussart et al., 2017) and the co-expression of genes in M. pneumoniae (Junier et al., 2016) and other bacteria (Junier and Rivoire, 2016).
Cell signaling in bacteria is often related to small metabolites and second messengers (Shimizu, 2013). Thus, the overall physiology, growth rate, and metabolic activity of a cell are major contributors to transcriptional status (Berthoumieux et al., 2013, Klumpp and Hwa, 2014). For example, in Gram-negative bacteria, the alarmone (p)ppGpp can regulate transcription by interacting with the RNA polymerase core complex (RNAP) (Potrykus and Cashel, 2008). Furthermore, the concentration of certain nucleoside triphosphates (NTPs) regulates transcription in some promoters whose transcript starts with the corresponding NTP (Schneider et al., 2003), (Sojka et al., 2011). Attenuation of transcription by riboswitches and Rho-independent terminators also plays an important role (Barrick and Breaker, 2007). For example, the cold-shock response regulates expression of the infB operon in E. coli through an anti-terminator (Bae et al., 2000). Non-coding RNAs (ncRNAs) are also thought to be involved in transcriptional regulation (Hüttenhofer et al., 2005), (Costa, 2007). Finally, regulation of RNA half-life by RNases plays a role in determining RNA levels. The bacterial RNA degradosome contains the glycolytic enzyme enolase, which depending on the metabolic status of the cell, may change RNA degradation specificity (Cho, 2017). The precise contribution of all these non-TF mechanisms to transcriptional regulation remains unclear and unquantified.
To quantify the contribution of different mechanisms to transcriptional regulation, we selected the genome-reduced bacterium M. pneumoniae, which has retained the basic regulatory machinery for sustaining autonomous life (Lluch-Senar et al., 2015). M. pneumoniae was typically believed to have little regulation (Knudtson and Minion, 1993). However, recent studies have suggested that M. pneumoniae exhibits responses that are known to occur in more complex organisms (Güell et al., 2009). Sequence analysis suggests the existence of 10 putative TFs (HcrA, MPN124; GntR, MPN239; WhiA-like, MPN241; SpxA, MPN266; MraZ, MPN314; Fur, MPN329; YlxM, MPN424, YebC, MPN478; alternative sigma MPN626; and DnaA, MPN686).
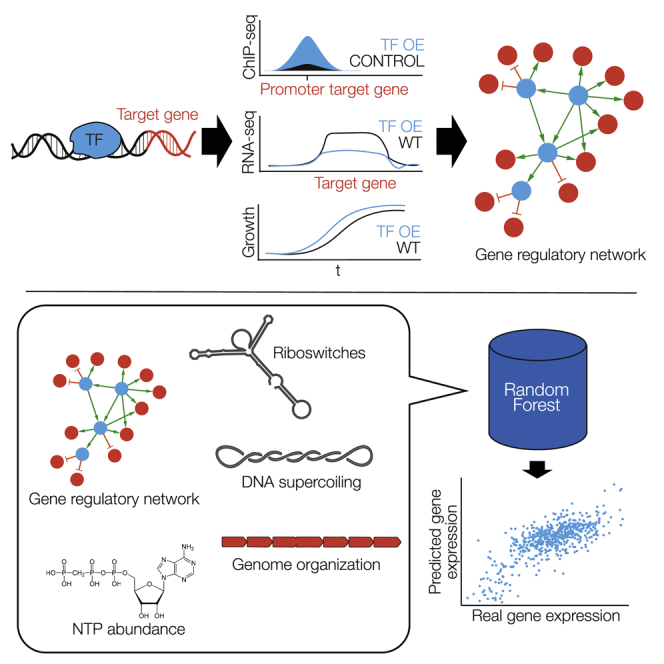
Here, we systematically examined the protein-DNA interactome of M. pneumoniae using classical biochemical techniques. Together with information from the literature, we compiled a list of candidate proteins that could have a role in transcriptional regulation. We identified the DNA-binding sites of each protein by using chromatin immunoprecipitation coupled to ultra-sequencing (ChIP-seq). Then, we determined growth phenotypes of as well as gene expression changes by transcriptomics in strains in which the candidate proteins had been overexpressed or mutated. We confirmed and identified 9 TFs, of which 7 had novel binding motifs. Another 16 proteins not binding DNA produced significant transcriptional changes when overexpressed or mutated. As TFs alone were not able to explain the observed transcriptional landscape, we investigated the contribution of other elements to regulation, including riboswitches, supercoiling, intrinsic terminators, ncRNAs, RNA degradation, and the concentration of the first nucleotide to be incorporated into a transcript (iNTP). We confirmed the existence of some of these ancient, basal mechanisms of regulation and quantified their global contribution to transcriptional regulation in environmental perturbations, showing that these mechanisms play a more important role in regulation than TFs themselves (see Figure 1 for a graphical explanation).
Figure 1.

Schematic Definition of the Workflow to Determine the Mycoplasma pneumoniae Gene Regulatory Network
Results
Identification of the Complete Set of DNA-Binding Proteins
Based on sequence functional annotation, 57 and 106 M. pneumoniae proteins are predicted to interact with DNA and/or RNA, respectively, either directly or as a part of a complex (Table S1). To define the complete set of M. pneumoniae DNA-binding proteins in an unbiased manner, we performed the following experiments: (1) classic DNA affinity chromatography, (2) pull-down experiments with short DNA sequences, and (3) subcellular fractionation to isolate chromatin (Figures 1 and S1A; Table S1; see STAR Methods).
We identified 174 putative DNA and/or RNA-binding proteins that passed the cutoff criteria (see STAR Methods and Figure 1) in at least two different experiments. Out of the 57 proteins predicted to interact with DNA, 11 were not detected by mass spectrometry under any circumstance (see Table S1) (Miravet-Verde et al., 2019). Nine of these eleven are gene duplications of components of the type I restriction enzyme complex, suggesting they are pseudogenes or splinted pieces of an adenine methylase. The other two not found are the alternative sigma factor MPN626 and its target (MPN536, ruvB). These two proteins are not expressed under normal in vitro growth conditions (Burgos and Totten, 2014) (Torres-Puig et al., 2015). 40 passed the selection criteria. Out of the remaining 6 proteins 3 of them (MPN341, pcrA; MPN529, HU; and MPN551, yqaJ) were found to bind to DNA in one experiment. The probable DNA helicase I homolog (MPN340, uvrD1) was not found because it eluted from the RNA and DNA columns at a different salt concentration than the majority of DNA-binding proteins. Only 2 proteins that should interact directly with DNA—the putative TF YebC (MPN478) and the predicted NAP YbaB (MPN275)—were not found to interact with DNA in any of the experiments done.
Additionally, we identified previously described moonlighting proteins with DNA-binding properties, including leucine aminopeptidase (Charlier et al., 2000), Lon protease (Lin et al., 2009), and some metabolic enzymes that can bind DNA or RNA (Commichau and Stülke, 2015) (Table S1). Our approach also identified 73 out of 106 ribosomal and RNA-binding proteins, and is therefore unlikely unable to distinguish effectively between RNA- and DNA-binding proteins. Nonetheless, we cannot rule out the possibility that some ribosomal proteins also bind DNA (Warner and McIntosh, 2009).
Selection of Putative DNA-Binding Proteins for Further Characterization
The preliminary set of 174 putative DNA and/or RNA-binding proteins was reduced to 65 proteins by removing: (1) chaperones known to appear in pull-down approaches (Kühner et al., 2009); (2) surface-exposed proteins that could bind to and/or degrade DNA; and (3) membrane-associated proteins with ATPase activity (we kept some as controls, see Table S1). In addition, we removed housekeeping proteins that are involved in DNA and RNA metabolism. As M. pneumoniae has several duplicated genes, we only kept some representative proteins per duplicated family (see Figure S1B and Table S1). To the remaining 65 proteins, we added the three putative DNA-binding proteins mentioned above (MPN626, YebC, and YbaB).
Finally, we included 79 proteins that could have an indirect effect on transcriptional regulation (signaling proteins and metabolic regulators) or RNA levels (RNases), as well as two negative controls (the surface nuclease MPN133 and the yellow fluorescent protein, YFP). In total, 147 genes were cloned into a minitransposon to overexpress them with a tag in M. pneumoniae (see Table S2). Protein expression was verified by western blot and mass spectrometry (see STAR Methods). As expression was not detected for 3 of these proteins (MPN302, MPN429, and MPN470), 144 proteins were retained for further analysis (21% of the 689 annotated open reading frames) along with YFP (Table S2).
Identification of Protein-DNA-Binding Sites
DNase protection assays reveal all regions of the genome that are covered by protein, ChIP-seq identifies the binding sites of the selected proteins (see STAR Methods and Figures S2A–S2D for reproducibility and validation assays).
We identified 156 unique protection peaks in the DNase protection experiments at exponential and stationary phases (Table S3) and mapped them on transcription start sites (TSSs) (Lloréns-Rico et al., 2015, Yus et al., 2009) (see STAR Methods and Table S4). Of the 156 protected DNA regions, 103 (66%) correspond to promoters (see Figure 2 for examples).
Figure 2.

ChIP-Seq Results for Validating DNA Binding
ChIP-seq selected tracks for different proteins: TFs, RNAP complex, structural, and proteins involved in DNA replication, as well as DNA regions protected from DNase digestion (POD). Vertical lines indicate annotated TSSs and the origin of replication region (OriC). The regions or peaks bound by the studied protein or protected from DNase digestion are shown in the color of the corresponding category. The x axis shows the base position in the genome. The y axis shows the read coverage per base. Binding motifs found for some of the structural proteins are shown in the right.
128 of the 144 proteins were analyzed with ChIP-seq (we excluded membrane proteins and RNases; Table S2). We found specific peaks for 23 proteins (18% of the total tested and 24% of the candidate RNA-/DNA-binding proteins; Table S4). These proteins are either part of the RNAP, RNAP-associated proteins (Kühner et al., 2009), TFs, part of the DNA replication machinery, or structural proteins (Figure 2; Table S3). Analyses with the RNAP core subunits revealed that many of the promoter-proximal DNA sites occupied in the DNase protection assay are bound by RNAP (see Figure 2 and Table S3). In total, 146 of the 156 protected DNA regions were found in at least one of the ChIP-seq experiments, indicating that we have achieved a comprehensive coverage of the DNA-binding sites within the M. pneumoniae chromosome. Out of the ten putative TFs mentioned in the introduction, we found specific peaks at promoters for six of them (SpxA, MraZ, DnaA, Fur, HcrA, and WhiA; see Table S3 and Figure 2; for details see below).
For some moonlighting proteins that bind DNA in other organisms, like Lon (MPN332), we identified several small peaks outside promoter regions without a clear motif. This is probably because they do not have a high DNA specificity (Lin et al., 2009) (Charlier et al., 2000). Other non-RNAP-associated proteins (e.g., MPN555) mimicked the RNAP profile, but with only a few peaks in promoters of highly expressed genes, likely to be artifactual or phantom (Jain et al., 2015). The rest of the tested proteins did not have any significant peaks.
Potential binding sites and the binding motifs of structural proteins are shown in Figure 2 and Table S3. We found DnaA at the oriC as previously suggested (Blötz et al., 2018), as well as at different sites in the first quarter of the chromosome (Figure S2E). We also found specific peaks at the promoters of genes related to nucleotide metabolism and DNA replication (Table S3). The 3 proteins that form the condensin complex (Smc, MPN426; ScpA, MPN300; and ScpB, MPN301) were found to bind to the oriC (Figure 2 and Table S3) as described in B. subtilis (Wilhelm et al., 2015), as well as to a site at the opposite side of the chromosome. This site is close to the attachment organelle (Trussart et al., 2017) and could be the terminus of replication, ter. Thus, it seems that in M. pneumoniae the condensin complex plays the same role as in B. subtilis, aligning the left and right arms of the chromosome. At the oriC, we also found a clear peak for the DnaB1 protein (MPN525) and for the paralog (MPN554; SsbB) of the single-stranded binding protein SsbA (MPN229), suggesting a role for these proteins in chromosome replication. We found specific peaks for the histone-like protein (HU, MPN529) scattered through the chromosome and generally in the middle of genes (HU induces negative supercoiling in circular DNA with the assistance of the topoisomerase). For the ATPase component of the phosphate transporter, PhoU (MPN608), peaks concentrated opposite the oriC, suggesting it could play a role in anchoring the chromosome to the cell membrane (Figure 2).
Characterization of the Transcriptional Changes Induced by Overexpression and/or Mutation of the Selected Proteins
To unveil a regulatory effect of the 143 selected proteins, we analyzed their impact on the global transcriptome using microarrays and/or RNA-seq. We used strains that overexpressed the proteins as well as transposon or dominant-negative point mutant strains (Table S2; Figure S3). On average, overexpression (OE) of the candidate genes resulted in a 4-fold increase in the corresponding protein levels (see STAR Methods, Table S2, and Figure S4A). OE and/or mutation of 24 of the selected DNA-binding proteins led to significant changes in global gene expression (Table S5), but no correlation was found between protein OE levels and the number of transcriptional changes (Figure S4B). 8 of the 10 potential TFs in M. pneumoniae (YebC and SpxA as exceptions) control gene expression when overexpressed as wild-type (WT) or dominant-negative mutants (Figure 3A and Table S5). Regarding the putative TF YebC (Brown et al., 2017), we did not observe any change in the transcriptome upon OE, nor did it bind DNA in the biochemical assays or display specific ChIP-seq peaks, suggesting it is not a TF.
Figure 3.

Classification of the Candidates and DNA Motifs Using the Phenotypic Analyses
(A) The sequence motifs for 9 TFs analyzed in this study (in orange hexagons), as well as their main regulated targets (arrows indicate activation and crosses repression) are shown. We also show the proteins identified as regulators in this study (orange diamonds) and the functional category of the genes they significantly affected (if a regulator affects a single gene in a functional category, we do not show it; see Table S5).
(B) M. pneumoniae phenotypes. Experiments were classified into three groups according to the number of identified transcriptional changes that they displayed. For each group, bar charts show the percentage of genes that display a growth curve phenotype (faster or slower growth and/or medium acidification) with respect to the total.
(C) Inferelator results on the genetic perturbations. The Venn diagram depicts the interactions retrieved manually, those found using Inferelator, and those common to both methods.
(D) Network of the transcriptional phenotypes induced by OE, KO and/or mutants of all genes selected in this study, as well as by environmental perturbations. Lines represent correlations between the experiments above 0.45. Colors represent different clusters of highly interconnected experiments. Clusters enriched in different functional categories are represented in shaded circles. Perturbations are shown in square boxes and overexpressed, mutated or deleted genes in ovals. The sign of fresh medium addition was reversed for visualization in the growth-associated cluster (with which it correlates negatively).
SpxA is an essential protein in M. pneumoniae. In B. subtilis it directs the RNAP to specific promoters upon oxidative stress or redox changes (Nakano et al., 2003). Our ChIP-seq experiment revealed that SpxA is found at promoters (see above and Figure 2), but we were unable to detect any transcriptional changes upon OE. In B. subtilis, drugs that promote disulfide formation (e.g., diamide) were used to identify SpxA targets (Leichert et al., 2003, Nakano et al., 2003). Addition of diamide to M. pneumoniae revealed that SpxA regulates itself and a regulon involved in the oxidative stress response (mpn607, msrA; mpn625, osmC; and mpn662, msrB) (Zhang and Baseman, 2014) as well as other genes (Table S5).
To identify the DNA recognition motifs of the TFs, we combined three sets of data: (1) the ChIP-seq data, (2) upstream sequences of the TSSs of the target genes detected in the transcriptome analysis, and (3) sequence conservation with the closely related species M. genitalium (see STAR Methods and Figure 3A).
We confirmed that HcrA represses the genes involved in heat-shock response (Güell et al., 2009) and that MraZ regulates the cell division operon (Fisunov et al., 2016). Our findings include (1) repression of the DNA polymerase III subunit dnaN and activation of genes involved in nucleotide metabolism, by DnaA. (2) Fur repression of different genes (mpn043, glpF; mpn162; mpn433; mpn561, udk; mpn363, and mpn408). (3) WhiA and YlxM each repress one single ribosomal operon (mpn164-185 and mpn656-660, respectively). (4) GntR regulates metabolic genes involved in arginine metabolism, fermentation, and the pentose phosphate pathway (Fisher’s enrichment test, p = 0.01; 1e−4, and 1e−4, respectively) and (5) the targets of SpxA involved in oxidative stress (see above; Figure 3A).
Regarding proteins that do not bind DNA, 16 of them (hereafter called regulators) produced major transcriptome alterations (≥ 10 genes showing significant changes; see STAR Methods and Figure 3A). GlpQ (MPN420) catalyzes the hydrolysis of glycerophosphocholine (GPC) and leads to the production of glycerol-3-phosphate, a building block for lipids that enters into glycolysis, producing peroxide. The inactivation of glpQ affects the Fur regulon as previously shown (Schmidl et al., 2011), as well as other genes (see Figure 3A and Table S5). Our findings include: (1) the correlation between the changes induced by the inactivation of lactate dehydrogenase (MPN674, Ldh) and of the putative redox chaperone MPN294 (r = 0.72; p < 2.2e−16) (Figure S4C). The majority of changes observed in these strains are related to nucleotide metabolism and overlap with the genes upregulated upon OE of DnaA (Figure 3A and below on Figures 4B and 4C). This suggests that the redox state, which determines the equilibrium between NTP and dNTPs, regulates DnaA. (2) the inactivation of the Mg2+ transporter MPN159 (CorB), the transition metal-binding protein, MPN162, and the recombination protein MPN490 (RecA) correlate significantly (r > 0.67, p < 2.2e−16) (Figure S4D), thus linking metal transport and binding to recombination and expression of surface proteins. The major effect of CorB inactivation is the downregulation of lipoprotein gene expression (p = 1.4e−5; Table S5; Figure 3A).
Figure 4.

Network of Co-regulated Genes in M. pneumoniae
(A) Network of co-expressed genes in M. pneumoniae. Nodes of the network represent genes, and the edges between two nodes indicate co-expression (r > 0.5). Colors of the network nodes represent different clusters of highly interconnected genes (i.e., highly co-expressed). We show the functional categories significantly enriched in each group (Benjamini-Hochberg adjusted p < 0.05). Inset shows the two main expression clusters determined by k-means clustering analysis.
(B) TFs partly explain some of the different clusters of co-regulated genes. Genes targeted by different TFs are marked with different colors.
(C) Transcriptional changes induced by the Ldh and MPN294 (disulfide chaperone) genes are, in the case of upregulation (blue nodes), mainly explained by the DnaA TF and involve genes related to nucleotide metabolism. Red nodes indicate downregulated genes.
(D) Supercoiling driven by heat or cold shock explain the two major clusters of co-regulated genes. Blue nodes: genes upregulated in cold shock and downregulated in heat shock. Red nodes: genes downregulated in cold shock and upregulated in heat shock.
(E) An initial GC dinucleotide at the TSS (blue nodes) explains a large part of the ribosome-associated cluster.
Second Messenger-Mediated Signaling in M. pneumoniae
M. pneumoniae uses three major second messenger nucleotides, (p)ppGpp, c-di-AMP, and AppppA (Ap4A). The stringent response-related alarmone (p)ppGpp is synthesized and degraded by the SpoT enzyme (MPN397). C-di-AMP is synthesized by the essential diadenylate cyclase CdaM (MPN244) and degraded by the phosphodiesterase PdeM (MPN549) (Blötz et al., 2017). The alarmone Ap4A is produced by some tRNA synthases (e.g., those for Ser and Lys) when the corresponding amino acid is missing (Belrhali et al., 1995) and degraded by the Hit1 (MPN273) enzyme. In many bacteria, analogs of serine and valine, serine hydroxamate (SHX) and norvaline (NVAL), respectively, induce the stringent response. This response induces the synthesis of (p)ppGpp, concomitant repression of ribosomal proteins and rRNA operons, and induction of peptide and amino acid transporters (Geiger et al., 2012). In M. pneumoniae, however, the addition of either amino acid analog did not result in a repression of the ribosomal protein operons but rather in the upregulation of the main ribosomal protein operon (mpn164-183) and induction of the oppA gene (mpn456, and its duplicates, mpn457-458) and the oppBCDF operon (mpn215-218), which encodes an ABC peptide transporter. Inactivation of SpoT did not affect the induction of the main ribosomal protein operon or opp genes upon SHX and NVAL addition (Table S6). Thus, (p)ppGpp does not seem to be implicated in the classical amino acid starvation-mediated response. On the other hand, while mild glucose starvation results in the repression of the main ribosomal operons (without affecting the opp operons, [(Güell et al., 2009), see below], this was not the case for the spoT mutant strain (Table S5). This suggests that SpoT regulates expression of ribosomal proteins upon glucose deprivation (as seen in other bacteria (Traxler et al., 2006; Zhang et al., 2016), see below and Figure 3A), agreeing with its role as a hub that integrates various stress signals including fatty acid, iron, and carbon starvation (Hauryliuk et al., 2015).
With respect to the Ap4A alarmone, we overexpressed the Ap4A-degrading enzyme Hit1 and analyzed the transcriptome of the cells in the presence and absence of SHX and NVAL. In contrast to the WT strain, no upregulation of the first genes of the main ribosomal operon or the opp genes was observed (Table S5). This observation suggests that Ap4A and Hit1, rather than (p)ppGpp and SpoT, are involved in the response to amino acid starvation in M. pneumoniae. According to this, Ap4A can be generated by loading defects of some aminoacyl-tRNA synthetases (Belrhali et al., 1995). Indeed, we observed that expression of the hit1 gene and expression of the serS gene, which encodes the Ap4A-producing serine-tRNA synthetase, were significantly anticorrelated (r = −0.36, p = 4.39e−7) (see Figure S4E). This reinforces the functional link between the Ap4A-synthesizing and Ap4A-degrading enzymes.
Phosphorylation-Mediated Signal Transduction in M. pneumoniae
Signal transduction and the resulting transcription regulation may also involve post-translational modifications of proteins, in particular phosphorylation and acetylation. M. pneumoniae has two annotated protein kinases, the HPr kinase HprK (MP223) and the Ser/Thr kinase (PrkC, MPN248) and one protein phosphatase (PrpC, MPN247). Inactivation of either PrkC or PrpC resulted in significant transcriptional changes in some of the previously described targets identified by proteomic analysis (Schmidl et al., 2010) (van Noort et al., 2012). We observed a negative correlation between the transcriptional changes (r = −0.19; p = 6.93e−7) of the prkC and prpC mutants (Figure S4F). Targets included the cell division operon (mpn314–317) and adhesion genes (Table S5; Figure 3A). This indicates that PrkC and PrpC do not only regulate protein levels as previously described (van Noort et al., 2012) but also affect transcriptional regulation in some of their targets. Inactivation or OE of HprK, as well as of the protein acetylase MPN114, resulted in no significant transcriptional effects.
Phenotypic Analysis
To assess the gross phenotypic effects of OE, inactivated genes (KO), and mutant strains, we determined their growth profiles (in total, 169 strains corresponding to the 143 studied M. pneumoniae genes and YFP; Table S2) by measuring cell metabolism (medium acidification) and protein biomass (Table S7). Out of the 143 genes studied, only 42 caused an altered phenotype in at least one mutant strain (in total, 48 strains showed significant changes). The remaining 101 genes did not result in significant growth differences in any condition, suggesting that M. pneumoniae, despite being a genome-reduced bacterium, is a rather robust system. These percentages are similar to those found in the transcriptomics analysis, with those strains showing larger changes in gene expression also exhibiting more severe growth phenotypes (Figure 3B).
Reconstruction of the Gene Regulatory Network
Integration of the different experimental datasets mentioned above revealed a gene regulatory network for M. pneumoniae (hereafter called experimental network), which comprises 1,062 interactions between 25 regulatory proteins (9 TFs and 16 regulators) and the 689 M. pneumoniae genes (Figure 3A). To uncover potential additional levels of regulation, we performed an automated network reconstruction analysis using Inferelator (see STAR Methods) (Bonneau et al., 2006). As input for this analysis, we used the list of 1,062 curated interactions as prior knowledge, as well as the transcriptomics fold changes from all genetic perturbations. The network recovered by Inferelator consisted of 1,036 interactions, 668 (63%) of which were present in the experimentally recovered Gene Regulatory Network (GRN; Figure 3C). 394 interactions from the experimental network were missed by Inferelator, 28% of which involve the TF SpxA. The reason for this is probably that SpxA activity does not rely on its mRNA levels, but rather on redox changes as discussed above. On the other hand, 368 interactions found by Inferelator were not present in our experimental network. These likely correspond to regulatory interactions that are below the threshold we used to specify relevant associations (Figure S4G). Taken together, the application of Inferelator to our dataset expanded our original network to 1,430 regulatory interactions (expanded gene regulatory network) (see STAR Methods; Figure 3C).
The Impact of Environmental Perturbations on Transcription Regulation
To test whether we could explain transcriptional regulation in M. pneumoniae with the expanded gene regulatory network, and to identify environmental conditions that could affect the activity of the TFs and regulators, we exposed cells to 37 environmental perturbations (see Table S6). For 31 cases, we observed major transcriptional changes (≥10 genes showing significant changes in their expression level). For example, heat shock upregulated the HrcA targets but also caused numerous other gene changes that negatively mirror those found under cold-shock conditions (r = −0.3, p = 3.2e−16), indicating a secondary effect not related to HrcA. Medium acidification resembles glucose starvation (Wodke et al., 2013), reducing expression of genes involved in amino acid metabolism and translation (i.e., a growth arrest phenotype) (Yus et al., 2009). Relaxation of DNA supercoiling by novobiocin-induced gyrase inhibition upregulates gyrA (mpn003), gyrB (mpn004), and other genes involved in DNA replication (Figure S5C; Table S6) (El Houdaigui et al., 2019). Antibiotics that target the ribosome and affect protein synthesis, such as macrolides or tetracyclines, caused major changes even at sub-lethal concentrations and short exposure times (less than 15% of generation time). These changes are similar to those caused by thiolutin, a Zn2+ chelator with antibiotic properties (Lauinger et al., 2017) and diamide (Table S6; Figure 3D). This suggests that as reported for other bacteria, antibiotics trigger a redox and/or oxidative stress response (Kohanski et al., 2010). Perturbations affecting the redox state (e.g., diamide, glycerol, hydrogen peroxide, etc.), as well as the inactivation of Ldh and MPN294 (related to redox balance) and GlpQ (related to peroxide production), significantly affect the targets of Fur. The Zn2+ chelator thiolutin (Lauinger et al., 2017) provokes a major upregulation of Fur targets, suggesting that Fur is regulated by Zn2+ (see Table S6). Moreover, addition of the iron chelator bipyridine did not induce the Fur-controlled genes. Taken together, these observations suggest that this TF has been mis-annotated and should therefore be regarded as a member of the Fur-family that uses Zn2+ for signaling (Zur).
To determine whether the phenotypes found in the different perturbations were similar to those caused by OE or mutation of some of the genes studied in this work, we performed a correlation and clustering analysis of all the transcriptomics experiments in this study (see STAR Methods). We found large groups of experiments where we overexpressed or mutated specific genes clustering around specific perturbations (Figure 3D).
Hydrogen peroxide treatment is found together with the experiments where we added GPC or phosphatidylcholine (PC) to the medium. Both compounds are converted to glycerol-3-phosphate, which upon being metabolized produces H2O2 (Hames et al., 2009). In addition, we found the inactivated Ldh or MPN294 mutants, which have a redox phenotype (see above) and the transporter for GPC (glpU; MPN421) (Großhennig et al., 2013) in the same group, representing phenotypes related to oxidative stress. A second major cluster encompasses the heat-shock response and several experiments addressing growth phenotypes (stationary versus exponential phase, or pH 6 that prevents glucose metabolism; addition of fresh medium to the stationary phase provokes exactly the opposite effect). These growth phenotypes could be related to changes in ATP when cells lack a carbon source because of medium acidification. A third major cluster contains the cold-shock response that promotes transcriptional read-through (Junier et al., 2016), which includes OE of one of the gyrase components (GyrA, MPN004). The fourth major cluster includes the mutants of metal transporters, CorB and MPN162, as well as of the RecA gene and it does not have any associated perturbation. The fifth one includes the treatment with puromycin. Then, we have two smaller clusters. One contains the glucose starvation condition that produced a general decrease in the levels of RNA. The second one contains all treatments with ribosome antibiotics as well as the treatment with diamide and the antibiotic thiolutin. Finally, we found some small isolated clusters of experiments such as the one including the novobiocin treatment. At high concentrations, novobiocin releases the RNAP from the chromosome, halting transcription (see Figure S2D).
This analysis showed that although OE or inactivation of the putative regulatory factors provoked only small changes in global gene expression, some tend to mimic the stronger phenotypes produced by environmental perturbations.
The Recovered Gene Regulatory Network Does Not Explain the Changes Found in the Environmental Perturbations
Adaptation to several perturbations was not fully explained by the effects of OE or mutation of the regulatory factors. To address this apparent discrepancy, we tested the ability of the gene regulatory network to explain the changes observed in the environmental perturbations. For this purpose, we used the expanded gene regulatory network encompassing 1,430 regulatory interactions as prior information for Inferelator. For the dataset of environmental perturbations, Inferelator yielded a network with only 230 interactions between any TF or regulator and their respective targets.
We determined the overlap between this network, derived from the environmental perturbations and the expanded gene regulatory network extracted from the OE and mutant experiments. A total of 196 (85%) of the 230 interactions were present in the expanded network. Nevertheless, 1,234 of the original 1,430 interactions from the expanded gene regulatory network were missing (Figure S4H), and this network explains only 21% of the total gene variance in the environmental perturbation experiments. In contrast, the expanded gene regulatory network could explain 53% of the variation in the OE and mutant experiments.
These results suggest that even though the gene regulatory network may be accurate in predicting changes upon gene OE or knock out of TFs and regulators, it cannot be used to predict the transcriptional response to changes in the environment.
A Gene Co-expression Network Highlights the Role of Alternative Mechanisms
To compare the influence of TFs and regulators with that of potential alternative regulatory mechanisms, we performed a global co-expression analysis of all the genes. First, we performed a k-means clustering analysis of all protein-coding genes in this bacterium, using all transcriptomics datasets from this study, including the genetic and environmental perturbations. We first estimated the optimal number of clusters in this dataset, which was estimated to be 2 (see STAR Methods and Figure 4A inset). One of these clusters is enriched in growth-related proteins (p = 6.8e−11), which increase their expression levels in the exponential phase of the growth curve, while the other cluster is enriched in stationary phase proteins that are poorly expressed during exponential growth (p < 2.2e−16).
To find smaller clusters representing more tightly co-regulated genes, we calculated pairwise gene correlation across all conditions tested in this study to assemble a gene correlation network (see STAR Methods). Genes linked together in this network are likely to be co-regulated. We applied community clustering to the network to find groups of highly interconnected genes (see STAR Methods). Some of the resulting groups are enriched in functional categories (adjusted p < 0.05; Figure 4A). The two clusters from the k-means analysis overlap with groups of the smaller clusters from the correlation network (Figure 4A).
Next, we addressed the relationship between the obtained clusters and the expanded gene regulatory network obtained in the genetic perturbation analysis. This analysis revealed that genes regulated by the same TF are generally embedded within the same cluster, with the exception of GntR and SpxA (Figure 4B). For instance, the targets of DnaA are enriched in the cluster related to nucleotide metabolism (p = 4.3e−9). This cluster is specifically affected by regulators that change the redox state (Ldh and MPN294) (Figure 4C; Table S5). However, other clusters are not associated to any TF or regulator, and hence point to alternative regulatory mechanisms.
Relevance of DNA Supercoiling and Genome Organization
DNA supercoiling controls transcription at the global level (Dorman and Dorman, 2016). It has been reported that whereas cold shock promotes DNA supercoiling (Grau et al., 1994) (López-García and Forterre, 1997), heat shock induces DNA relaxation (Krispin and Allmansberger, 1995). Moreover, DNA is known to be negatively supercoiled in exponential phases of growth but more relaxed during the stationary phase (Balke and Gralla, 1987). Thus, we analyzed our data in light of a potential role for DNA supercoiling. We found that heat and cold shock explain the majority of the genes in the two major clusters found by the k-means approach (see inset in Figures 4A and 4D). We observed a negative correlation between cold and heat shock (r = −0.3, p = 3.2e−16; see Figure S5A). In addition, cold shock anti-correlates with gene expression at the stationary growth phase (r = −0.51; p < 2.2e−16) (Figure S5B). These results confirm the association of cold shock and supercoiling through the different growth phases of this bacterium.
Upon the addition of the gyrase inhibitor novobiocin to M. pneumoniae, we previously observed the disappearance of defined borders between chromosomal interacting domains in the 3D structure of its chromosome (Trussart et al., 2017). This results in a loss of co-regulation and indicates a direct link between supercoiling and chromosome structure.
In five cases, we observed a global anticorrelation between convergent genes considering all conditions tested (r < −0.1; adjusted p < 0.05), indicating transcriptional interference as a result of supercoiling (see an example in Figure S5D).
Regulation by transcriptional read-through, probably due to the anti-terminator function of RNA helicases and cold-shock-regulated proteins (Stülke, 2002) (Bae et al., 2000) has been reported before (Junier et al., 2016). Conversely, we see stronger termination signals and less read-through in heat shock, as well as when using macrolides (see Figure S6 for examples).
RNA-Mediated Regulatory Mechanisms
Several potential regulatory mechanisms rely on nucleotide- and RNA-based signaling. The identity of the initiating nucleotide of a transcript has a significant impact on the expression levels of the corresponding genes (Schneider et al., 2003, Sojka et al., 2011). For the cluster enriched in ribosomal genes (Figure 4A), the TFs WhiA and YlxM (Figure 4B) can only explain part of the regulation, thereby suggesting that additional factors must also play a role. In an analysis of the starting nucleotides (+1 and +2 positions) of all the mRNA transcripts of M. pneumoniae, the operons encoding ribosomal proteins seem to be particularly enriched (12 out of 18 operons) with a GC dinucleotide at positions +1 and +2. Out of these 12 operons, 9 have the position of the GC dinucleotide conserved with M. genitalium. The GC sequence is also found in the rRNA transcript and is conserved in the corresponding B. subtilis operon (Figure S5E). Indeed, ribosomal operons and genes involved in protein translation having the GC dinucleotide correlate significantly, as shown by their proximity in Figure 4E. In B. subtilis, the production of (p)ppGpp results in a decrease in the GTP concentration via inhibition of the guanylate kinase Gmk, and therefore downregulation of rRNAs and mRNAs that start with a G (Kriel et al., 2012). In M. pneumoniae, the stringent response is caused by glucose starvation (see above). The resulting accumulation of (p)ppGpp by SpoT activation likely leads to a decrease in the GTP concentration, and therefore downregulation of the ribosomal operons with a GC. Thus, it seems that the translation cluster is mainly regulated by the concentration of GTP. The importance of the first base of a transcript is illustrated when looking at base conservation of promoters in M. genitalium and M. pneumoniae (Figure S5F).
Furthermore, we identified putative RNA structures that might regulate premature termination of transcription (Dar et al., 2016) (see STAR Methods). The identified structures could regulate the expression of 29 genes under certain conditions (see Table S8 and Figure S5G). Interestingly, the co-regulated oppA and oppBCDF operons share a complex structure at the 5′ UTR. Although we found little sequence conservation when comparing the oppB 5′ UTR region with the equivalent one in M. genitalium, the overall RNA secondary structure of this region is conserved (Figure S5G). It is tempting to speculate that this structure is important for the regulation of the two operons in response to amino acid limitation (see above).
We did not find any significant anticorrelation between antisense RNAs and their overlapping protein-coding gene. This agrees with recent work in which we proposed that the majority of antisense RNAs in M. pneumoniae are the product of transcriptional noise (Lloréns-Rico et al., 2016). In addition, we did not observe any consistent correlation between a particular ncRNA and genes that are not adjacent to it. Thus, we did not find evidence for a transcriptional regulation role for ncRNAs.
Because of the short half-lives of bacterial mRNAs, RNA degradation is an important factor controlling gene expression (Selinger et al., 2003). We observed a mild decrease in RNA abundance, especially of ribosomal genes (p = 1.4e−5), upon OE of RNase III. In contrast, the inactivation of the corresponding gene rnc (mpn545) resulted in a very large increase in the amounts of mRNA (larger for some genes in particular), indicating that this gene plays a major role in the control of RNA half-life (Table S5). We did not detect any transcriptional effect when overexpressing other RNases such as RNase R (MPN243), RNase J1 (MPN280), RNase J2 (MPN621), or RNase Y (MPN269). The last 3 RNases form part of a protein complex, the RNA degradosome, and thus their individual OE may not be sufficient to affect RNA stability (Cho, 2017, Commichau et al., 2009).
Evaluating How Much the Different Regulatory Mechanisms Contribute to Transcriptional Variation
To study the relative effect of TFs, regulators, and alternative regulatory mechanisms in M. pneumoniae, we used a Random Forest regressor (see STAR Methods). To do so, we first associated each gene with its different regulatory mechanisms as described above (Table S8). We included its TFs and regulators as defined by the interactions in the expanded gene regulatory network. In addition, we also included other features that could contribute to transcriptional variation: (2) RNA half-life (Junier et al., 2016); (2) the Pribnow (-10) box (genes with identical Pribnow boxes are more correlated; p value < 0.05, and Figure S5H); and (3) AT content of the 5′ UTR and the sequences around the Pribnow box (Yus et al., 2017) (Table S8). With the set of features assigned to each gene, we fitted a Random Forest to each of the perturbations to predict gene changes in each experiment (Table 1; see STAR Methods).
Table 1.
Results of the Random Forest Model for Each Experimental Condition Studied
| Experiment | Adjusted R2 (Variance Explained) | Spearman Correlation (Predicted versus Real) |
|---|---|---|
| Thioguanine | 0.533 | 0.775 |
| Sanguinarine | 0.454 | 0.719 |
| Fresh media | 0.500 | 0.717 |
| Thiolutin | 0.418 | 0.680 |
| Chloramphenicol | 0.498 | 0.678 |
| Osmostress | 0.507 | 0.679 |
| Mitomycin C | 0.489 | 0.687 |
| Norfloxacin | 0.550 | 0.673 |
| Macrolides | 0.510 | 0.683 |
| Glycerol | 0.455 | 0.656 |
| Growth | 0.546 | 0.660 |
| Tetracyclines | 0.467 | 0.645 |
| Cytochalasin B | 0.478 | 0.594 |
| Spectinomycin | 0.425 | 0.601 |
| Puromycin | 0.505 | 0.665 |
| Gencitabine | 0.338 | 0.556 |
| Peroxide | 0.365 | 0.591 |
| Shx | 0.423 | 0.563 |
| CCCP | 0.352 | 0.565 |
| Norvalin | 0.520 | 0.562 |
| Triton X | 0.405 | 0.571 |
| pH 6 | 0.281 | 0.536 |
| Bipyridine | 0.253 | 0.422 |
To test this approach, for each perturbation included in our study, we calculated the Spearman correlation coefficient between the actual fold changes and the ones predicted by the Random Forest (see STAR Methods and Figures S7A and S7B for additional validation). The average correlation between the Random Forest predicted and the observed fold changes across all experiments was r = 0.62 (sd = 0.07). Moreover, 22 out of 23 analyzed experiments (see STAR Methods) show a correlation greater than 0.5 (and p < 2e−16). This means that we can determine the transcriptional variation in these conditions by using a set of descriptors for each gene (three examples are shown in Figure 5A). In our dataset, we can explain 45% of gene expression variability using the Random Forest classifier, and in some individual experiments this value increases up to 55% (Figure 5B).
Figure 5.

Variance Explained in Experimental Perturbations
(A) The upper panel shows correlation plots between the predicted and the experimental fold changes for three different perturbations: entry into stationary phase, glycerol, and thioguanine addition. The lower panel shows the variance explained by each canonical or alternative mechanism for these experiments.
(B) The violin plots show, for each experiment, how much variance (in %) can be explained by each regulatory mechanism. The total variance explained in the entire dataset (not accounting for noise) is 45%.
Some of the studied features, such as chromosome organization, seem to have a relatively important and constant contribution across the majority of conditions tested (Figure 5B). Genetic perturbations caused by TFs and regulators, can have a different impact according to the environmental conditions. Of the total variance across the 24 perturbations tested, only around 9% can be attributed to TFs and regulators (see Figure 5B). However, in some individual experiments the TF response plays a more important role, explaining up to 18% of the variance (see Figure 5A).
We validated the results of the Random Forest by exploring the predictions on the clustered experiments from Figure 3D, to determine if different clusters of correlated experiments (formed by both genetic and environmental perturbations) were regulated by the same mechanisms. First, we observed that the contributions of the different regulatory mechanisms were more similar in experiments from the same cluster than from different clusters (p value = 8.9e−12). Then, we observed the specific differences between clusters. As in the analysis of the individual perturbations, we also observed that some factors such as the CIDs have a constant and important effect in all the clusters. Others, such as the TFs and regulators, are more variable and depend on the cluster (Figure S8).
To determine to which extent the different cellular processes are regulated by different mechanisms, we classified the genes into various functional classes (Figure S7C; Table S1). We observed that different cellular processes have quite distinct modes of regulation. Some processes such as arginine metabolism, metabolic homeostasis, adhesins and attachment organelle, protein secretion, oxidative homeostasis, and TFs have a large proportion of non-regulated genes. In contrast, other processes such as DNA recombination, protein homeostasis, the pentose phosphate pathway, and fermentation are largely regulated by TFs. Interestingly, non-canonical regulation is not distributed equally among the different processes. Genes related to protein synthesis (i.e., ribosomes, rRNA, and ribosomal protein modifications, tRNA modifications, and protein synthesis factors) share an initiating GC dinucleotide at the transcriptional start site. Many categories related to metabolism and transport have a number of genes controlled by RNA structure (see the regulation of the opp operons above; Table S8). This shows that different cellular processes have preferentially adopted specific regulatory mechanisms to tightly control transcription of their genes (see Figure S7C).
The Impact of Noise on Transcriptional Variation
An intrinsic feature of each complex system is the existence of noise. This has also been observed with transcription in M. pneumoniae (Güell et al., 2009) (Miravet-Verde et al., 2017). To estimate the proportion of variance in the environmental perturbations that is explained by noise, we performed 2 analyses. First, we looked at the RNA-seq control experiments in which gene expression of the WT strain with an empty transposon insertion was measured. Any variance not explained by the model can be attributed to noise, whether it be biological or technical. On average, the non-explained variance for all genes in our RNA-seq control experiments (i.e., with no gene expression changes) was 27.9% (see STAR Methods).
Measured noise can vary across experiments and can be relatively higher in conditions that have low or no changes compared with conditions with many significant changes. As such, we further explored the noise observed in two specific perturbations (addition of glycerol and addition of diamide), which had five biological replicates each and large significant transcriptional changes. For this, we fitted the Random Forest to each of the five replicates and calculated the variance explained in the same replicate as well as in the remaining replicates. The decrease in the variance explained in the different replicates, as compared to the same replicate can be associated to noise. In the glycerol experiments, this decrease accounts for 10% of the variance, while in the diamide experiments, it accounts for 14% of the variance (Figure S7A).
This means that even in well correlated replicates of experiments, there is a significant contribution of noise to transcriptional variation (10%–14%). The relative contribution of noise can be even higher (up to 27.9%) in perturbations with low changes, as shown by the analysis of RNA-seq controls.
Discussion
Here, we comprehensively analyzed the mechanisms involved in transcription regulation in the genome-reduced bacterium M. pneumoniae (Figure 6). We first performed an unbiased screen for all possible DNA-binding proteins. We then characterized nine TFs (Fur, MraZ, DnaA, HrcA, YlxM, WhiA, GntR, SpxA, and MPN626) along with their binding motifs, nine structural DNA-binding proteins (Smc, ScpA and ScpB, PhoU, HU, SsbB, MraZ, and DnaB1 and DnaA; some of them also TFs), and 16 regulators. We observed that the majority of our M. pneumoniae strains did not show any phenotypic or transcriptional changes upon OE or inactivation of different genes, suggesting great robustness of its gene expression machinery. Similar robustness has also been observed with other bacteria (Isalan et al., 2008, Reuß et al., 2017). We found that 296 protein-coding genes (43% of the genome) were directly regulated by one of the nine TFs in at least one condition studied. This is similar to E. coli and B. subtilis, where 40% and 52% of the genes, respectively, are regulated by TFs (Gama-Castro et al., 2016, Leyn et al., 2013, Michna et al., 2016, Salgado et al., 2013).
Figure 6.

Cellular Processes and Their Respective Regulatory Mechanisms
(A) A simplified version of the gene regulatory network of M. pneumoniae, incorporating both canonical and alternative regulatory mechanisms. Different cellular processes preferentially use one regulatory mechanism over another to control the expression of genes.
(B) Simplified schema of the structural proteins and their binding regions in the chromosome of M. pneumoniae.
Regarding structural proteins, we found the Smc-ScpA-ScpB complex (condensin) located at the oriC. In B. subtilis Smc–ScpAB is recruited to the oriC by the ParB (Spo0J) protein bound to parS sites (Wilhelm et al., 2015). M. pneumoniae lacks ParB and Spo0J genes, suggesting that in this bacterium other components could recruit the Smc–ScpAB complex, such as the DnaB1 protein (MPN525), found in the same region. Binding to regions in the proximity of the oriC may act as sinks for DnaA when replication starts (Ishikawa et al., 2007). The single-stranded protein SsbB is found enriched at the opposite site of the OriC as well as co-localizing with the Smc-ScpA/B complex, suggesting a possible role in DNA replication. In Figure 6B, we show a scheme with the distribution of these proteins.
We observed that a large part of transcription regulation is determined by non-canonical factors such as DNA supercoiling and genome organization or RNA-mediated regulation. Previous comprehensive studies have aimed at quantifying the contribution of individual TFs to the expression changes of genes (Bonneau et al., 2007). Our study revealed that in the majority of cases, no correlation exists between the expression of a TF or regulator and the resulting regulation of the target genes. The reason is that RNA levels of a TF may not change under different environmental conditions, but instead the protein is (in)activated by adopting a different conformation (such as HrcA (Susin et al., 2004)), via effector binding (such as Fur with Zn2+), or by post-translational modification (such as SpxA with disulfide bond formation). We found that the actions of TFs and regulators could explain on average only as little as 9% of the variance of any of the experiment, while 36% of the experimental variance was explained by other regulatory mechanisms. In total, we have assigned 45% of the variance to the regulatory mechanisms studied here and estimated that up to 28% of the unexplained variance can be associated with the noise of gene expression. We cannot discard the possibility that we have missed some TFs that are only relevant under very special conditions. However, a systematic sequence comparison of all M. pneumoniae promoters both with themselves and with those of the closely related M. genitalium did not reveal additional clear sequence motifs. Thus, although we could have missed a TF that controls only one operon, our conclusion that non-TF transcriptional regulation dominates in M. pneumoniae is still valid.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Flag Monoclonal Antibody | Sigma | Cat#F3165; RRID: AB_262044 |
| Bacterial and Virus Strains | ||
| Mycoplasma pneumoniae M129 | Colleague | Richard Herrmann |
| Mycoplasma pneumoniae strains | this paper | Mendeley: [doi:10.17632/xf6y59gz6c.1] |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 6-Thioguanine | Sigma | Cat#A4882 |
| Azithromycin | Sigma | Cat#75199 |
| CCCP | Sigma | Cat#C2759 |
| Ciprofloxacin | Sigma | Cat#17850 |
| Clarythromycin | Sigma | Cat#C9742 |
| Cytochalasin B | Sigma | Cat#C8273 |
| DCCD | Sigma | Cat#D80002 |
| Diamide | Sigma | Cat#D3648 |
| Doxycycline | Sigma | Cat#D9891 |
| Erythromycin | Sigma | Cat#E5389 |
| Gemcitabine | Sigma | Cat#G6423 |
| Levofloxacin | Sigma | Cat#28266 |
| Minocycline | Sigma | Cat#M9511 |
| Mitomycin C | Sigma | Cat#M4287 |
| Norfloxacin | Sigma | Cat#N9890 |
| Plasmocin | Invivogen | Cat#ant-mpp |
| Pyocyanin | Sigma | Cat#P0046 |
| Sanguinarine | Sigma | Cat#S5890 |
| Spectinomycin | Sigma | Cat#S4014 |
| Spiramycin | Sigma | Cat#S9132 |
| Streptomycin | Sigma | Cat#S6501 |
| T-butyl hydroperoxide | Sigma | Cat#458139 |
| Tetracycline | Sigma | Cat#T3258 |
| Thiolutin | Fermentek | Cat#87-11-6 |
| Valinomycin | Sigma | Cat#V0627 |
| Critical Commercial Assays | ||
| NebNExt Ultra kit | New England Biolabs | Cat#E7370L |
| RNA Isolation Kit: RNeasy Mini Kit | Qiagen | Cat#74004 |
| RNA Isolation Kit: miRNeasy mini kit | ||
| TruSeq smallSmall RNA Sample Prep Kit | Illumina | Cat#RS-200-0012 |
| Phosphopeptide Enrichment kit | Thermo Scientific | Cat#88301 |
| Deposited Data | ||
| RNA-Seq: Phenome analysis of Mycoplasma pneumoniae | this paper | ArrayExpress E-MTAB-3771 |
| RNA-seq: Transcriptome analysis of Mycoplasma pneumoniae I | this paper | ArrayExpress E-MTAB-3772 |
| RNA-seq: Transcriptome analysis of Mycoplasma pneumoniae II | ||
| RNA-seq: Transcriptome analysis of Mycoplasma pneumoniae III | this paper | ArrayExpress E-MTAB-4642 |
| DNase proteccion: Protein occupancy of Mycoplasma pneumoniae chromosome | this paper | ArrayExpress E-MTAB-3783 |
| RNA-seq: Transcriptome analysis of Mycoplasma pneumoniae IV | this paper | ArrayExpress E-MTAB-4784 |
| ChIP-seq of Mycoplasma pneumoniae putative Transcription factors | this paper | ArrayExpress E-MTAB-5944 |
| RNA-seq: 5'′-end mapping of totalTotal Mycoplasma pneumoniae RNA | this paper | ArrayExpress E-MTAB-6124 |
| RNA-seq: Transcriptome analysis of Mycoplasma pneumoniae V | this paper | ArrayExpress E-MTAB-6229 |
| RNA-seq: Transcriptome analysis of Mycoplasma pneumoniae VI | this paper | ArrayExpress E-MTAB-7153 |
| Proteomics: Mycoplasma pneumoniae Chromatin isolation | this paper | ProteomeXchange XD007672 |
| Proteomics: DNA Affinity chromatography on Mycoplasma pneumoniae extracts I (RNA elution) | this paper | ProteomeXchange PXD007674 |
| Proteomics: DNA Affinity chromatography on Mycoplasma pneumoniae extracts II (cellulose column) | this paper | ProteomeXchange PXD007676 |
| Proteomics: DNA Affinity chromatography on Mycoplasma pneumoniae extracts III (DNA column) | this paper | ProteomeXchange PXD007677 |
| Proteomics: TF overexpression and mutant data | this paper | ProteomeXchange PXD007551, PXD007537, PXD007545, PXD007557, PXD007558, PXD007560, PXD007561, PXD007565, PXD007566, PXD007658, PXD007658. |
| Oligonucleotides | ||
| Oligonucleotides | this paper | Mendeley: [doi:10.17632/xf6y59gz6c.1] |
| Recombinant DNA | ||
| Plasmids | this paper | Mendeley: [doi:10.17632/xf6y59gz6c.1] |
| Software and Algorithms | ||
| Xcalibur software v3.0.63 | Thermo Fisher Scientific | https://www.thermofisher.com/order/catalog/product/OPTON-30487 |
| Proteome Discoverer software suite v2.0 | Thermo Fisher Scientific | https://www.thermofisher.com/order/catalog/product/OPTON-30795 |
| MAQ software | Li et al., 2008 | N/A |
| Inferelator software | Bonneau et al., 2006 | https://github.com/simonsfoundation/inferelator_ng |
| R version 3.5.1 | R Core Team, 2018 | |
| tidyverse (R packages ; version 1.2.1) | Wickham et al., 2017 | R package |
| ggpubr (R package ; version 0.2) | Kassambara, 2018 | R package |
| Gdata (R package ; version 2.18.0) | Warnes et al., 2017 | R package |
| DESeq2 (R package ; version 1.22.2) | Love et al., 2014 | R package |
| NbClust (R package ; version 3.0) | Charrad et al., 2014 | R package |
| randomForest (R package ; version 4.6-14) | Liaw and Wiener, 2002 | R package |
| Cytoscape (Version 3.6.0) | Shannon et al., 2003 | https://cytoscape.org/ |
| clusterMaker2 (Cytoscape plugin, version 1.2.1) | Morris et al., 2011 | Cytoscape plugin |
Lead Contact and Materials Availability
This study generated the constructs and Mycoplasma cell lines listed in Table S2. These are available upon request with no restrictions. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Luis Serrano (luis.serrano@crg.eu).
Experimental Model and Subject Details
M. pneumoniae strain M129 (passage 33–34) was grown in modified Hayflick medium and transformed by electroporation with the pMT85 transposon as previously described (Yus et al., 2009). Briefly, cells were split 1:10, washed twice with 10 ml, and collected in 300 μl Electroporation buffer (8 mM Hepes·HCl, 272 mM sucrose, pH 7.4) after 72 hours. Cells (50 μl) were electroporated with 5 μg plasmid in 1-mm gapped cuvettes at 1.25 kV, 100 Ω, and 25 μF (Gene Pulser Xcel Electroporator, Bio-Rad). Cells were recovered in Hayflick for 2 h at 37 °C, diluted 1:5 in Hayflick with 200 μg ml-1 gentamycin, selected for three days and then maintained with 80 μg ml-1 gentamycin. The cell lines used are detailed in Table S2. Identity of the inserted material was validated by RNA-seq. All plasmids used in this work are in Online Table 1.
Method Details
Duplicated Proteins and Promoter Alignment with Mycoplasma genitalium
To identify duplicated proteins in Mycoplasma pneumoniae we run Blast for each protein against the rest of the protein sequences annotated at NCBI. In Figure S1B we show sequence alignment for some of the duplicated genes having the DUF16 domain.
We aligned the Mycoplasma pneumoniae and Mycoplasma genitalium by identifying the ortologous genes and then doing a DNA blast alignment using the M. pneumoniae promoter sequences corrected manually when we could identify the Pribnow boxes (TATAAT, TAGAAT, TAAAAT, TACAAT) (see Online Figure 1).
DNA Affinity Column
M. pneumoniae cells were diluted 1:10 in Hayflick and grown for 3 days at 37°C in a 300-cm2 flask. Cells were washed twice with ice-cold phosphate buffer saline (PBS: 150 mM NaCl, 10 mM Potassium Phosphate buffer pH 7.4), and collected in 5 ml of lysis buffer (50 mM Tris·HCl, 1 M NaCl, 1 mM CaCl2, 1 mM EDTA, 0.1% Triton X-100, 1 mM DTT, pH 8) supplemented with a protease inhibitor cocktail (Roche). The buffer contained 1 M NaCl in order to release the proteins from the DNA. Cell extracts were centrifuged for 30 min at 100,000 × g and 4°C (Beckman ultracentrifuge), and the soluble fraction was diluted 10 times with 50 mM Tris·HCl, 1 mM CaCl2, 1 mM EDTA, pH 8 (to dilute out salt and detergent) before filtering the lysate through a 22-μm filter. A DNA-cellulose column was compacted and assembled (2 g, Sigma, D8515, in 8 ml of TE, 10 mM Tris·HCl, 1mM EDTA, pH 7.9) and run on an Äkta Xpress (GE Healthcare) in equilibration buffer (50 mM Tris·HCl, 0.1 M NaCl, 1 mM CaCl2, 1 mM EDTA, pH 8), before binding of the cleared cell lysate. Once the protein signal was stabilized at zero, the column was washed with a moderate ionic strength buffer (equilibration buffer plus 200 mM NaCl), and then nucleic acid-binding proteins were eluted with 1 M NaCl in equilibration buffer (for elution of DNA-binding proteins), or 5 mg ml-1 yeast ribonucleic acid (RNA, Sigma, R8501) in 0.1 NaCl buffer (for elution of proteins with affinity for RNA). Protein elution was monitored at 280 nm and 0.5-ml fractions were collected. Fractions from the elution profile were run on a 4–12% SDS-PAGE gel. Fractions with greater amounts of protein were concentrated by TCA precipitation before submission for MS identification (see below). A cellulose resin was used as a negative control for unspecific binding. “Peak” and “E1” are the two main 1 M NaCl elution fractions from the DNA column; “A3” and “A6” are the main elution peaks after RNA addition to the DNA column (Online Table 2).
Chromatin Isolation
DNA binding properties were assessed by ultracentrifugation, employing a sucrose cushion according to a previously described method (Prasad and Dritschilo, 1992) with only minor modifications. Briefly, a 300-cm2 flask was grow for 3 days as above, washed with PBS and lysis was performed using 2 ml of lysis buffer (10 mM Tris·HCl, 1 mM EDTA, 1% Nonidet P-40, pH 8 plus a protease inhibitor cocktail from Roche). To follow the chromatin, 0.2 μl of Sybersafe (Invitrogen) was added and 1 ml of lysate was loaded on top of a 20%–40% sucrose cushion (in TE: 10 mM Tris·HCl, 1 mM EDTA). Chromatin was fractionated by ultracentrifugation in a Ti45 rotor (Beckman) at 30,000 rpm and 4°C for 18 h and collected from the interphase with the assistance of a UV light. After pelleting by centrifugation at 100,000 × g for 1 h, the supernatant was discarded and the pellet was resuspended in digestion buffer (50 mM Tris·HCl, 0.3 M NaCl, 1 mM MgCl2, pH 7.5) plus 8 U rDNase I (Ambion), for 1 h at room temperature (RT) to release the DNA-binding proteins. After spinning down for 30 min at 14,000 rpm and 4°C in a tabletop centrifuge, both the supernatant and pellet were analyzed by SDS-electrophoresis. Samples were concentrated by TCA precipitation before submission for MS identification (see below). “Supernatant” is the supernatant after DNase treatment, “pellet” is that pellet after washing it with PBS and resuspending it in sample buffer (Online Table 2).
DNA Pull-Down
This protocol was adapted from (Masternak et al., 2000). The oligos can be found here (Online Table 3). First, from a 3-day, 300-cm2 culture flask, cells were washed with ice-cold PBS, scrapped in PBS plus 0.1% glucose at 4°C, and centrifuged for 10 min in a tabletop centrifuge. The pellet was resuspended in 2 ml lysis buffer (1 M NaCl, 50 mM Hepes·NaOH, 0.1% NP 40, 6 mM MgCl2, 1 mM EGTA, pH 7.5) plus a protease inhibitor cocktail (EDTA-free, Roche), and passed throw a G25 needle 10 times prior to clearance by spinning 30 min in a tabletop centrifuge at maximum speed and 4°C. The supernatant was diluted 1:10 in dilution buffer (50 mM Hepes·NaOH, 1 mM EGTA, 6 mM MgCl2 pH 7.5) and 4.4 ml were used per assay. Sepharose-streptavidin beads (M-280, Invitrogen) were bound to biotinylated oligos as follows. First, forward and reverse oligos at 50 μM were annealed in Annealing buffer (10 mM Tris·HCl, 50 mM NaCl, 1 mM EDTA, pH 8.0) in a PCR machine: 95°C 2 min, 52°C 10 min, 4°C. Then 20 μl of annealed oligos were mixed and incubated with equilibrated (TE) beads for 30 min at 4°C in a roller. Beads were washed with binding buffer (50 mM Hepes·NaOH, 1 mM EGTA, 0.1 M NaCl, pH 7.5) and incubated with lysate for 1 h or O/N at 4°C. Formaldehyde was added to 1% and proteins and DNA were fixed for 10 min at RT. Crosslinking was stopped with glycine (100 mM final) for 5 min at RT. Beads were washed once with 1 ml binding buffer, 3X with 1 ml wash buffer 1 (50 mM Hepes·NaOH, 1 mM EGTA, 0.2 M NaCl, 6 M Urea, 0.2% SDS, pH 7.5) and 3X with 1 ml wash buffer 2 (50 mM Hepes·NaOH, 1mM EGTA, 0.2 M NaCl, pH 7.5). Material was eluted/un-crosslinked with 50 μl of elution buffer (1% SDS, 10 mM Tris·HCl, 1 mM EDTA pH 8.0) at 65°C for 15 min and 95°C for 5 min more. Finally, the eluent was visualized on an SDS electrophoresis gel after staining with Instant Blue Commassie (Expedeon). Optimal pull-downs were submitted to proteomics (see below) without the need for additional concentration.
Proteomics
Total protein lysates from M. pneumoniae were obtained by breaking the cells with 200 μl of lysis buffer (4% SDS, 0.1 M DTT and 0.1 M Hepes·NaOH). Total protein extracts (two biological replicates) were digested with trypsin and subsequently analyzed by MS. Briefly, samples were dissolved in 6 M urea, reduced with DTT (10 mM at 37°C, 60 min), and alkylated with iodoacetamide (20 mM at 25°C, 30 min). Samples were diluted 10-fold with 0.2 M NH4HCO3 before being digested at 37°C overnight with trypsin (in a protein:enzyme ratio of 10:1). Peptides generated in the digestion were desalted, evaporated to dryness, and dissolved in 300 μl of 0.1% formic acid.
An aliquot of 2.5 μl of each fraction (amounts ranging from 0.17 to 4 μg) was run on an LTQ-Orbitrap Velos (ThermoFisher) fitted with a nanospray source (Thermofisher) after a nanoLC separation in an EasyLC system (Proxeon). Peptides were separated in a reverse phase column, 75 μm x 150 mm (Nikkyo Technos Co., Ltd.) with a gradient of 5% to 35% acetonitrile in 0.1% formic acid for 60 min at a flow rate of 0.3 ml min-1. The Orbitrap Velos was operated in positive ion mode with the nanospray voltage set at 2.2 kV and the source temperature at 325°C. The instrument was externally calibrated using Ultramark 1621 for the FT mass analyzer and the background polysiloxane ion signal at m/z 445.120025 was used as lock mass. The instrument was operated in data-dependent acquisition (DDA) mode and full-MS scans were acquired in all experiments over a mass range of m/z 350–2,000, with detection in the Orbitrap mass analyzer set at a resolution setting of 60,000. Fragment ion spectra produced via collision-induced dissociation (CID) were acquired in the ion trap mass analyzer. In each cycle of data-dependent analysis, the top 20 most intense ions with multiple charges above a threshold ion count of 5,000 were selected for fragmentation at a normalized collision energy of 35% following each survey scan. All data were acquired with the Xcalibur 2.1 software. Total extract (20 μg) was also digested and desalted, and 1 μg of the resulting peptides were analyzed in an Orbitrap Velos Pro in the same conditions as the fractions but using a longer gradient (120 min).
The data were searched using an internal version of the search algorithm Mascot against a database that contained all the putative M. pneumoniae open reading frames (ORFs) larger than 19 amino acids (MPNHomoContTrans19). The data has been filtered using a 5% false discovery rate (FDR). Protein grouping was not applied in the results and we quantified the proteins using the following parameters: i) only unique peptides without miss cleavage; ii) only peptides with “Protein Group=1”. Protein top 3 area was calculated using the average of the 3 most abundant unique peptides per protein group. Only unique peptides corresponding to ORFs for which we could identify an RNA transcript were considered. In Online Table 4 we show the fold changes and p-val per gene, for all proteomics experiments done in this study.
ROC Curves
Gold sets of DNA- and RNA-binding proteins were constructed using information from the literature. For instance, even for bona fide DNA-binding protein complexes, only the protein that was binding directly to the nucleic acid was considered as a true DNA-binding protein.
For each of the experiments that were performed to identify DNA/RNA-binding proteins, we calculated whether there was any enrichment in DNA/RNA-binding proteins in the fractions obtained. To do so, first we averaged the areas obtained in MS among biological and technical replicates.
In the DNA affinity column, we determined the ratio between each of the fractions and the extract for each protein, using the aforementioned averages. In the case of chromatin isolation, we calculated the ratio between the supernatant fractions S2 (only RNase treatment) and S3 (RNase and DNase treatment), and the pellet for each of the candidates. For the oligonucleotide pull-down experiments, we calculated the ratio between each of the pull-downs and the extract, for each protein, and then we took the median value.
Once all the ratios were calculated, we used the gold set to calculate ROC curves for each of the ratios. This enabled us to determine how well they discriminate DNA- and RNA-binding proteins. While a good separation between DNA- and RNA-binding proteins could not be obtained, four of the fractions displayed area under the curve (AUC) values higher than 0.75: fractions E1 and A6 of the DNA columns, the supernatant/pellet ratio in the chromatin isolation, and the DNA motif pull-downs (Figure S1A). Thus, these experiments are able to discriminate between the DNA/RNA-binding proteins and the negative data in our gold set. For each of these experiments, we calculated the threshold in which this separation is maximized as the threshold maximizing the difference between the true positive rate and the false positive rate. The cutoff values for the different experiments are as follows: E1 fraction = 0.56; A6 fraction = 0.69; Supernatant/pellet = 0.19; DNA motif pull-downs = 1.7.
With these cutoff values, we determined which DNA/RNA-binding proteins met the criteria for each of the experiments selected. To obtain a consensus, we chose as preliminary candidates all those proteins that were regarded as DNA/RNA-binding in at least two out of the four experiments (probability ≥0.5).
Protein Expression
In general, proteins were FLAG-tagged (DYKDDDKG) on their N- or C-terminus, and expression was confirmed by Western blot using M2 monoclonal anti-FLAG antibodies (Sigma). In some cases, when the tag was foreseen to interfere with the protein function, proteins were expressed without a tag. In general, the MPN665|tuf promoter was used for overexpression unless otherwise indicated (for cases in which the protein was toxic, the endogenous promoter was used instead). In a few cases, TAP (Tandem affinity purification)-tagged clones from Anne-Claude Gavin’s collection (EMBL) were used (Kühner et al., 2009) (see Table S2).
Dominant-Negative Mutants or Deletions
In some cases, point mutants were obtained to generate dominant-negative effects on the endogenous genes. In some cases, such as for SpxA (Nakano et al., 2010) or SpoT (Hogg et al., 2004), these mutations were taken from the literature. For the rest, if the protein of interest itself dimerizes and the dimeric status is essential for its activity, mutations were introduced into the DNA-binding site. In this way, the mutant copy could sequester the wildtype copy. To select which residues to mutate, we inspected the 3D structure of the target protein (or a close orthologue) with DNA and mutated one of the residues that strongly interacts with the DNA molecule (in general, interacting Lys or Arg residues were mutated to Asp or Glu). Finally, some phosphomimetic mutants were generated by changing the phosphorylated Ser or Thr to an aspartic or glutamic acid, respectively. A non-phosphorylable mutant was created in parallel, replacing the same residue for Ala (see the alignments of the conserved and mutated positions in Figure S3).
Transposon Insertion Mutants Obtained by Haystack Mutagenesis
For the isolation of M. pneumoniae mutants, we used a collection of strains carrying insertions of transposon Tn4001 (Halbedel et al., 2006). The presence of the desired mutant was assayed by PCR using one primer that hybridizes to the transposon (directed outwards), and a second primer specific for the gene of interest (see the oligos used for the screenings in Online Table 3, the RNA-seq profile plots in Online Figure 2 and the description of the mutants in Table S2). In Online Table 5 we show the insertion site for all transposon experiments as well as the changes in gene expression of the adjacent genes.
Environmental Perturbations
A minimum of two replicates were used for each condition.
Novobiocin Treatment (RNA Half-Life)
We pulsed M. pneumoniae cells with 100 μg ml-1 novobiocin for 30 min at 37°C. We then removed the drug by changing to medium alone, and took samples at the indicated time points after transcription arrest.
Novobiocin Titration (Supercoiling Analysis)
We treated M. pneumoniae cells with increasing concentrations of novobiocin (0, 1, 5, 10, 50 and 100 μg ml-1) for 30 min. After the treatment, total RNA was extracted, and we performed RNA sequencing as detailed above.
Other Treatments
Diamide catalyzes the formation of disulfide bridges between proximal Cys residues, thereby favoring SpxA conformational change and revealing its targets (Rojas-Tapias and Helmann, 2018). However, it also affects genes regulated by DnaA and Fur upon redox stress (these genes were removed as targets of SpxA).
Glucose starvation: cell culture medium was removed completely, and new Hayflick medium without glucose was added. Cells were incubated for 3–5 h at 37°C to exhaust all the remaining glucose (Hayflick contains approximately 5 mM glucose).
Amino acid starvation: half of the medium was taken, and 200 mg of DL-serine hydroxamate (to 10 mg ml-1) was added to the medium to dissolve it before adding it back to the cells. Cells were then incubated cells for 15–30 min at 37°C. Alternatively, norvaline was added at 0.5 mg ml-1, or minimal medium without amino acids and peptides was used (Yus et al., 2009).
Temperature: cells were changed to a water bath at 15oC (cold-shock) for 15 min or at 43°C (heat-shock) for 20 min.
Fe2+ depletion: The iron chelator 2,2′-bipyridyl was added directly to the flask at 3 mM, and cells were incubated for 30–60 min at 37°C.
Glycerol: glycerol was added directly to the flask at 0.1% or 1%, and incubated for 30 min at 37°C. In some cases, sucrose was used as a control.
Low PH: medium was changed to Hayflick at pH 6 and incubated for 1 h.
Osmotic shock: NaCl was added to 300 mM and incubated for 1 h.
Oxidative stress: H2O2 was added directly to the flask to 0.5%, and incubated for 15 min at 37°C.
Antibiotics and other drugs:
| Treatment | Perturbant | Concentration | Duration (min) |
|---|---|---|---|
| G metabolism | 6-Thioguanine 200 μg ml-l | 30 | |
| Protein synthesis inhibition | Azithromycin 0.0078 μg ml-1 | 60 | |
| PMF uncoupler | CCCP | 2 mM | 30 |
| Protein synthesis inhibition | Chloramphenicol 20 μg ml-1 | 60 | |
| DNA damage | Ciprofloxacin 1 μg ml-1 | 60 | |
| Protein synthesis inhibition | Clarythromycin | 0.015 μg ml-1 | 60 |
| Cell cycle | Cytochalasin B | 75 μg ml-1 | 60 |
| PMF∗ uncoupler | DCCD | 0.4 mM | 60 |
| Redox balance | Diamide 1 mM | 30 | |
| Protein synthesis inhibition | Doxycycline 0.3 μg ml-1 | 60 | |
| Protein synthesis inhibition | Erythromycin 0.0156μg ml-1 | 60 | |
| G metabolism | Gemcitabine 50 μg ml-1 | 30 | |
| Oxidative stress | Hydrogen peroxide | 0.1% | 20 |
| Oxidative stress | Hydrogen peroxide | 0.5% | 20 |
| DNA damage | Levofloxacin 0.75 μg ml-1 | 60 | |
| Protein synthesis inhibition | Minocycline 0.3 μg ml-1 | 60 | |
| DNA damage | Mitomycin C 5 μg ml-1 | 60 | |
| DNA damage | Mitomycin C 0.5 μg ml-1 | 60 | |
| DNA damage | Norfloxacin 10 μg ml-1 | 60 | |
| DNA gyrase | Plasmocin | 1 μg ml-1 | 60 |
| Oxidative stress | Pyocyanin | 3 μg ml-1 | 60 |
| FtsZ inhibitor | Sanguinarine | 28 μM | 60 |
| Protein synthesis inhibition | Spectinomycin 5 μg ml-1 | 60 | |
| Protein synthesis inhibition | Spiramycin 0.5 μg ml-1 | 60 | |
| Protein synthesis inhibition | Streptomycin 5 μg ml-1 | 60 | |
| Oxidative stress | T-butyl hydroperoxide 0.1 mM | 60 | |
| Protein synthesis inhibition | Tetracycline 0.3 μg ml-1 | 60 | |
| RNAP inhibitor/Zn2+ chelator | Thiolutin | 2.5 μg ml-1 | 60 |
| Membrane integrity | Triton X-100 | 0.01% | 60 |
| Potassium ionophore | Valinomycin | 0.1 mM | 60 |
PMF: Proton motive force
Chromatin Immunoprecipitation
We adapted the protocol from Buratowski’s lab (Keogh and Buratowski, 2004). From a pre-culture, M. pneumoniae cells were split 1:10 in a 300-cm2 flask and grown for 4 days at 37°C. When indicated, cells were collected at this point (stationary phase), or they were scrapped and seeded in 40 ml of fresh Hayflick in a 150-cm2 flask and incubated 6 h longer at 37°C (exponential phase). In some cases, a perturbant was introduced prior to fixation (see above). Formaldehyde was added at a final concentration of 1% (16% stock, Pierce), incubated for 10 min at room temperature and quenched by adding glycine to 100 mM, for 5 min at RT. Cells were washed twice with ice-cold PBS, scraped in 5 ml PBS and spun for 5 min at 4°C and 8,000g. The pellet was lysed by adding 1 ml of ysis buffer (50 mM Hepes·KOH, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, pH 7.5) plus a protease inhibitor cocktail (Pierce) at 4 °C for 5 min. Chromatin was sheared by ultrasonication using Covaris with the following settings: Duty Cycle, 20%; Intensity, 5; Cycles/Burst, 200; Time, 15 min; Water level, 15. Approximately 200-bp fragments were generated and cell debris were removed by centrifugation at 16,000 × g and 4°C for 10 min. The NaCl concentration of the supernatant was adjusted to 275 mM. The beads (50 μl) were pre-blocked with 0.5% bovine serum albumin (BSA) in PBS for 15 min at RT. Sepharose-protein-G was bound to either 10 μl of 1 mg ml-1 mouse IgG (control, Sigma), or 10 μl anti-FLAG (M2 monoclonal) for FLAG-tagged proteins. In the case of TAP-tagged proteins, 50 μl sepharose-IgG was used, and no control was included. About 0.5–1 mg chromatin per reaction was added and incubated over night at 4°C. Then, the following washes were done: 1x FA wash buffer 1 (FA lysis buffer with 275 mM NaCl), 1x FA wash buffer 2 (FA lysis buffer with 500 mM NaCl), 1x FA wash buffer 3 (10 mM Tris·HCl, 250 mM LiCl, 1 mM EDTA, 0.5% Nonidet P-40, 0,5% sodium deoxycholate, pH 8), and finally TE. Then, the immunoprecipitated material was extracted with 250 μl of FA elution buffer (50 mM Tris·HCl, 1% SDS, 10 mM EDTA, pH 7.5) and incubated for 10 min at 65°C. The beads were added to a micro-spin column (Bio-Rad) in order to collect the beads' death volume by centrifugation. Then, 5 μl of 20 mg ml-1 proteinase K was added, and tubes were incubated for 15 min at 55°C and 10 min at 95°C before letting them cool at RT, to elute the samples. To purify and extract the DNA, we used a phenol/chloroform extraction protocol and ethanol precipitation. Precipitated DNA was resuspended in 10 mM Tris·HCl, pH 8.0 and measured with PicoGreen in a Qubit fluorometer (DNA High sensitivity kit, Invitrogen). At least 8 ng of material was submitted for DNA ultra-sequencing. Briefly, DNA was ligated to DNA paired-end adapters with the NebNExt Ultra kit (by the CRG Genomics Core facility). Samples were also sequenced at the facility (see below).
All proteins were tested at early exponential phase of growth (6 hours). Reproducibility of ChIP-seq profiles was assessed by comparing biological replicas performed at different times (6 h and 96h) of the sigma factor SigA (MPN352) and the TF SpxA (MPN266) (see Methods). In general, all proteins were FLAG tagged at either the N- or C-terminal ends, except some components of the RNAP that were TAP tagged (see Table S2). The putative effect of tags was assessed by fusing the two tags to the same protein. Independently of the tag used, peaks associated to proteins from the core RNAP complex (RpoB MPN516 and RpoA MPN191 TAP-tagged; and SigA MPN352 FLAG-tagged) as well as the associated to the component of the RNA polymerase, SpxA (FLAG-tagged), were found at promoter sites. Both tags yielded similar results, with 199 common peaks between the SigA and at least one of the two RNAP subunits (Table S3). Similarly, we found 167 common peaks between the SpxA and the RNAP subunits. In both cases, more than 91% of these common peaks corresponded to annotated promoters (Table S3). When comparing the ChIP-seq profiles to gene expression, we found a significant correlation between the height of the promoter-associated peaks of the RNAP subunits and the expression level of those genes (Figure S2B). Some of the promoter-associated RNAP peaks changed their relative height between 6 h and 96 h, coinciding with changes in gene expression (Figure S2C). Additionally, we observed that all RpoA promoter-associated peaks disappeared when incubating the cells with a gyrase inhibitor (novobiocin) that reduces DNA negative supercoiling (Figure S2D). These results validated the RNAP associated peaks.
Transcriptomics
M. pneumoniae cells at various stages of growth, overexpressing different regulators or being exposed to various perturbations were washed with PBS and collected immediately in lysis buffer. Antibiotics were omitted before the last inoculation to avoid unwanted phenotypes. In all cases, we have at least two biological replicates per experiment.
As for microarray analysis, cells were collected in RTL buffer plus 143 mM beta-mercaptoethanol (RNeasy kit, Qiagen) and RNA extraction, cDNA synthesis and labeling were performed as described (Güell et al., 2009). In the case of RNA-seq, Qiazol was used to lyse the cells. RNA isolation was performed following the manufacturer’s instructions (miRNeasy kit from Qiagen), and an in-column DNase I treatment was included. RNA was measured using a Nanodrop (Thermo) and integrity was confirmed on a Bioanalyzer (Agilent). A paired-end directional strand-specific RNA-seq protocol (Illumina) was applied for sequencing library preparation at the CRG ultrasequencing facility. Briefly, 1 μg of total RNA was fragmented to ~100–150 nt using the NEB Next Magnesium RNA Fragmentation Module (EB). Treatments with Antarctic phosphatase and PNK (both from NEB) were performed in order to make the 5’ and 3’ ends of the RNA available for adapter ligation. Samples were further processed using the TruSeq small RNA Sample Prep Kit (ref. RS-200-0012, Illumina) for stranded RNA-seq, according to the manufacturer's protocol. In summary, 3’ adapters and subsequently 5’ adapters were ligated to the RNA. cDNA was synthesized using reverse transcriptase (SuperScript II, Invitrogen) and a specific primer (RNA RT Primer) complementary to the 3’ RNA adapter. cDNA was further amplified by PCR using indexed adapters supplied in the kit. Finally, size selection of the libraries was performed using 6% Novex TBE Gels (Life Technologies). Fragments with insert sizes of 100–130 bp were cut from the gel, and cDNA was precipitated and eluted in 10 μl of elution buffer. dsDNA samples were cluster amplified and sequenced in the HiSeq 2000 platform (Illumina). In Online Table 6 we show the fold changes and p-val per gene, for all genetic experiments.
Transcriptional analysis was performed at the exponential phase, 32 candidates were also tested at other timepoints (late exponential: 24 hours, or stationary: 48 and 96 hours)(Table S2).
mRNA Quantification by RT-qPCR
To monitor promoter regulation, reporter chimeras with YFP-Venus were built. As YFP seems to be a very stable protein in M. pneumoniae, gene expression was followed by retro-transcription (RT) followed by real time or quantitative PCR (qPCR). Briefly, RNA was purified as above, and 1 μg was hybridized to 2 μg random hexamers (Invtrogen) by heating to 65°C for 5 min and quick chilling on ice in a 11 μl total volume. Retrotranscription was performed by adding 4 μl 5X first-strand buffer, 2 μl 0.1 M DTT, 1 μl RNase OUT (40 units/μl, Promega), 1 μl 10 mM dNTP mix, and 1 μl SuperScript II RT (200 units, Invitrogen), and incubating for 50 min at 42°C before inactivation at 70°C for 15 min. A 2x GoTaq qPCR mastermix was used (Promega) with 0.5 ng cDNA per 10 μl reaction and 0.5 μM oligos, and run on a Lightcycler 480 (Roche). Oligonucleotides can be found in Online Table 3. Ribosomal RNA (16S) was used as a reference.
Growth Curves
To obtain equal amounts of each sample, initial inocula for the growth curves were quantified. Briefly, cells were grown for 3 days in 25-cm2 flasks, collected in 1 ml medium and 100 μl used for quantification using the BCA (bicinchoninic acid) protein assay kit (Pierce, see below). Same amounts of total protein (1 μg) were aliquoted per well in a 96-multiwell plate in duplicates. Two hundred μl of Hayflick medium was added per well and the cells were incubated in a Tecan Infinite plate reader at 37°C. The “growth index” (absorbance 430/560 nm, settle time at 300 msec and number of flashes equal to 25) was obtained every hour for 5 days as published (Yus et al., 2009). To quantify growth, we determined two slopes of the growth curve. The first one is based on the time interval from 10 to 30 h (“early slope”) and the second one on the whole growth curve (“late”). The early slope was determined by considering the maximum median of the slope between two time points (Equation 1) separated by three time measurements over successive periods of 30 time points. The late slope was determined by considering the maximum median value of the slope between two time points separated by four time measurements (Equation 2) over successive periods of 30 time points.
| Early Slope = (value (time [i]) – value (time[i+3)]) / (time[i]-time[i+3] | (Equation 1) |
| Late Slope = (value (time [i]) – value (time[i+4)]) / (time[i]-time[i+4] | (Equation 2) |
The early slope is more representative of growth, while the late slope reflects the metabolic activity.
On the other hand, biomass was quantified at 48 h (early stationary phase) by inoculating twin 96-well plates, in the same conditions as above. After incubation for two days at 37°C, medium was aspirated, and cells were carefully washed twice with 200 μl PBS and lysed with 100 μl lysis buffer (10 mM Tris·HCl, 6 mM MgCl2, 1 mM EDTA, 100 mM NaCl, 0.1% Tx-100, pH 8, and 1× Protease Inhibitor Cocktail, Roche) at 4°C. In the same first 96-well plate, cell lysates were kept on ice and extracted protein was quantified using the BCA Protein Assay Kit (Pierce, see below).
The protein concentrations at 48 h and early slope are more representative of growth, while the late slope and the A430/560 value at midpoint reflect the metabolic activity. These four parameters of growth and metabolism were analyzed for each batch of experiments. Outliers larger than quartile 3 (Q3) by at least 1.5 times the interquartile range (IQR), or smaller than Q1 by at least 1.5 times the IQR were removed to calculate the mean and the standard deviation of each of the parameters for each batch. Values larger or smaller than the mean by at least 2 times the standard deviation of each parameter were considered to determine fast- and slow-growing/metabolizing clones, respectively.
The data can be found in Online Table 7.
5′-RNA End Mapping
To obtain transcriptome-wide mRNA 5’ ends (TSSs), we amplified cDNA using a random primer by RT-PCR. Specifically, RT_read2 at 2.5 μM was hybridized to 5 μg of total RNA and the RT reaction was carried out as above (see “mRNA quantification by RT-qPCR”). Once the reaction was finished, RNA was removed by first adding 1 μl of 4 N NaOH during 5 min at 95°C, then neutralizing it with 2 μl of 1 M Tris·HCl (pH 8.0). Then, we added 1 μl of RNase cocktail (Ambion) and incubated the sample at 37°C for 30 min. After this, the reaction was brought to 50 μl with H2O and the ssDNA was cleaned with 1 volume of AMPure beads as above. Finally, we eluted with 25 μl of EB and proceeded to the ssDNA ligation. We prepared a 50-μl reaction with 5 μl of 10x buffer, 1 μl of ATP, 2.5 μl of MnCl2, 2 μl of CircLigase (Epicenter), 0.84 μl of 100 μM linker (L_read1_N6) and 15 μl ssDNA (from previously step). This mix was incubated at 65°C for 120 min, and then the ligase inactivated at 85°C for 15 min. The reaction was cleaned with 1 volume of AMPure beads as above and eluted with 25 μl of EB. The final step involved a PCR reaction to introduce Illumina sequences and thus prepare the libraries for sequencing. This reaction was performed with 4 μl of 5x Buffer, 0.4 μl of dNTPs, 0.4 μl of Phusion HF polymerase (NEB), 0.5 μM of primers (F_PEu and indexed R_PE) and 5 μl of ssDNA in a 20-μl final reaction volume. We PCR-amplified this during 20 cycles (elongation at 60°C). Each PCR product was checked on a 2% agarose/TAE gel, and once the correct band was obtained (approximately 250 bp), we repeated the same reaction but in a 50-μl final volume. The PCR was cleaned and selected by size using AMPure beads (as above), and libraries were quantified using the Illumina KAPA quantification kit according to the manufacturer’s instructions (Kapa Biosystems).
Ultrasequencing
A sample of all indexed libraries was prepared at 4 or 10 nM in 10 μl of H2O. All libraries were subjected to quality control using a Bioanalyzer High Sensitivity DNA Assay chip (Agilent). Twelve ChIP-seq samples, six RNA-seq samples and two 5’-end specific RNA-seq samples were multiplexed. dsDNA samples was cluster-amplified and sequenced in the HiSeq 2500 platform (Illumina) at the CRG Genomics Core facility.
Phosphoproteomics
Protein Sample Preparation
260 μg of protein from each sample (in 8 M urea, 50 mM NH4HCO3) were reduced with dithiothreitol (780 nmol, 1 h, 37°C) and alkylated in the dark with iodoacetamide (1,560 nmol, 30 min, 25 °C). First, the resulting protein extract was diluted with 200 mM NH4HCO3 to 2 M urea and digested with 26 μg LysC (Wako, cat # 129-02541) overnight at 37°C, and then to 1 M urea and digested with 26 μg of trypsin (Promega, cat # V5113) for 8 h at 37°C. The peptide mixture was acidified with formic acid and desalted with a MacroSpin C18 column (The Nest Group, Inc) prior to LC-MS/MS analysis. Samples were enriched for phosphopeptides with the Pierce TiO2 Phosphopeptide Enrichment kit (Thermo Scientific, cat # 88301). Finally, the peptide mixture was acidified with formic acid and desalted with a MicroSpin C18 column (The Nest Group, Inc) prior to LC-MS/MS (liquid chromatography–mass spectrometry) analysis.
Chromatography and Mass Spectrometry Analysis
The peptide mixtures were analyzed using an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific) coupled to an EasyLC (Thermo Scientific, Proxeon). Peptides were loaded directly onto the analytical column at a flow rate of 1.5–2 μl min-1 using a wash volume of four times the injection volume, and were separated by reversed-phase chromatography using a 50-cm column with an inner diameter of 75 μm that was packed with 2-μm C18 particles (Thermo Scientific). Chromatographic gradients started at 95% buffer A (0.1% formic acid in H2O) and 5% buffer B (0.1% formic acid in acetonitrile) with a flow rate of 300 nl min-1 and gradually increased to 22% buffer B in 120 min and then to 35% buffer B in 11 min. After each analysis, the column was washed for 10 min with 5% buffer A and 95% buffer B.
The mass spectrometer was operated in DDA mode, and full MS scans with 1 microscan at a resolution of 120,000 were used over a mass range of m/z 350–1,500 with detection in the Orbitrap. Auto gain control (AGC) was set to 2e5, and dynamic exclusion to 60 s. In each cycle of DDA analysis, ions charged 2 to 7 above a threshold ion count of 1e4 were selected for fragmentation at normalized collision energy of 28%. Fragment ion spectra produced via high-energy collision dissociation (HCD) were acquired in the Ion Trap with AGC set to 3e4, an isolation window of 1.6 m/z, and a maximum injection time of 40 ms. All data were acquired using Xcalibur software v3.0.63.
Data Analysis
Proteome Discoverer software suite (v2.0, Thermo Fisher Scientific) and the Mascot search engine (v2.5, Matrix Science) (Perkins et al., 1999) were used for peptide identification and quantification. Samples were searched against a M. pneumoniae database with a list of common contaminants and all the corresponding decoy entries (87,059 entries). Trypsin was chosen as the enzyme and a maximum of three miscleavages were allowed. Carbamidomethylation (cysteine) was set as a fixed modification, whereas oxidation (methionine), acetylation (N-terminal), and phosphorylation (serine, threonine and tyrosine) were used as variable modifications. Searches were performed using a peptide tolerance of 7 ppm, and a product ion tolerance of 0.5 Da. Resulting data files were filtered by an FDR < 5%.
Total Protein Determination
Fresh seed cultures were inoculated using a 1:100 dilution in 25-cm2 tissue culture flasks containing 5 ml Hayflick medium without antibiotics. All measurements were performed in duplicates at the indicated time points. To extract total cellular protein at the selected time points, medium was removed and cells were washed twice with PBS. Subsequently, cells were scraped and pelleted by centrifugation at 14,100 × g for 10 min. PBS was removed and pelleted cells were stored at -70°C for further processing. After collecting samples twice per day for seven days, frozen pelleted cells were suspended in 60 μl of lysis buffer (4% SDS, 0.1 M Hepes·NaOH). Cell lysates were kept on ice and disrupted using a Bioruptor sonication system (Diagenode, B01010004) with an On/Off interval time of 30/30 sec at high frequency for 10 min. Finally, cell lysates were spun, pipetted up and down to complete lysis, and extracted protein was quantified using the Pierce BCA Protein Assay Kit. Cell lysates taken from those time points after 48 h of growth were diluted five-fold to be within the working range of the assay. The standards were prepared with Pierce bovine serum albumin (BSA) standard (Thermo Scientific, Cat # 23209) diluted at different concentrations with lysis buffer, following the manufacturer’s instructions. For each sample and BSA standard, 25 μl was added in duplicate to a 96-well plate. Then, 200 μl of BCA working solution was added to each well, and mixed for 30 sec inside an Infinite M200 Tecan plate reader. Samples were incubated at 37°C for 30 min and, after cooling down to RT, absorbance at 562 nm was measured using the Infinite M200 Tecan plate reader. The known concentrations of the BSA standards were used to make the standard curve and interpolate the protein concentration for each sample.
Mapping Ultrasequencing Data
Resulting raw reads from RNA sequencing were mapped to the M. pneumoniae reference genome (NC_000912, NCBI) with Bowtie2 (default parameters, and 1 mismatch allowed) (Langmead and Salzberg, 2012). Only reads mapping to a unique position of the genome were considered. Counts per gene were extracted from the mapping .sam files using HTSeq-count (Anders et al., 2015) and our custom genome annotation (Lluch-Senar et al., 2015).
In the case of ChIP-seq, the libraries were single end. The plus and minus strand data were mapped using MAQ software (default parameters, and 1 mismatch allowed) (Li et al., 2008), and piled separately in order to reconstruct the original peaks (see below).
TSS mapping reads (see “TSS mapping”) were mapped using MAQ software (as above) but only the first position of the read (5′ of RNA) was taken into account for further analysis.
Quantification and Statistical Analysis
ChIP-seq Analysis
Two curves were obtained for each ChIP-seq experiment, corresponding to the reads mapped to the plus and minus strand of the M. pneumoniae chromosome. Additionally, each immunoprecipitation experiment is accompanied by a control experiment (IgG), in which only the secondary antibody was used for the immunoprecipitation. In the case the IgG was not available, we used the average of the remaining IgGs sequenced in the same batch. For each of the experimental curves, we normalized the profile of the IgG using the signal from the corresponding IP to equate their baseline levels. After normalization, the control signal was subtracted from the experiment profile. With the resulting profile, noise was modeled to fit a Gaussian distribution centered on zero and with a standard deviation varying across experiments. According to this distribution, all values with probability larger than 1e-3 of being noise were set to zero, to remove the noise.
Peak calling was performed separately in each of the profiles for the plus and the minus strand, and was performed with the Matlab findpeaks function. A custom R implementation of this function was used for our analyses. The parameters used in the peak calling were manually adjusted, with the following values: slope threshold = 4e-8 (minimum peak slope); amplitude threshold = 10 (minimum peak width); smoothing width = 15 (number of points to consider when smoothing the curve); and peak group=15 (number of points used to fit the peak). Additional filters were used to discard false positive peaks. We removed those peaks that were detected in the IP-IgG curve, but not found in the IP curve. Furthermore, to find those peaks that significantly differed from the control ones, we discarded those peaks in which the ratio between the IP-IgG peak and the IgG peak was smaller than two. We also removed those peaks that were most likely phantom peaks (Jain et al., 2015), identifying bins of 100 bps for which a peak was identified in at least 75% of experiments.
After peak calling in each of the strands, the data from both of them was merged. In ChIP-seq, the same peaks are expected to be found in both strands. However, as the DNA fragments whose ends are sequenced are usually larger than the sequencing read length, their positions tend to be displaced from one another and usually do not overlap. Thus, we associated each peak from the plus strand to a peak in the minus strand, provided that the distance between them was smaller than 300 bps. The actual peak position was attributed to the mid-point between the two partner peaks. The average distance between these partner peaks was calculated for each experiment. Single peaks with no partners in the opposite strand were discarded. Finally, a score was assigned to each of the peaks, describing how well the pair matches this average distance.
The same procedure was repeated using the IgG as the sample and the IP as a control. The ratio between the number of peaks found in this analysis and the number of peaks found in the previous one was used as an empirical FDR value. In this way, we could distinguish between the experiments that yield reliable peaks and those that yield mostly noise.
In all experiments, we found a set of common peaks (“phantom-peaks” (Jain et al., 2015)) that, in general, correspond to very high peaks found in the RNAP ChIP-seq experiments. These peaks were not considered in the analysis of the different ChIP-seq experiments. The genome positions in the bins (100 bases each) in which we identified phantom peaks are the following: 85101, 150651, 172301, 195501, 217501, 261601, 265701, 364101, 385701, 420201, 427501, 470401, 483301, 541701, 543101, 546301, 636401, 636501, 648901, 690401, 715901, 799801 and 799901.
Gene Expression Analysis
Differential expression analysis in RNA-seq experiments was performed using DESeq2, grouping all biological replicates for each experiment and including the effect of the conditions, time of the experiment (6 h or 96 h) and batch in the experimental design. For experiments in which RNA decay was observed, we applied a size factor correction based on their rRNA levels, as rRNA was assumed to be stable. In the remaining experiments, default size factors were determined by DESeq2 (Love et al., 2014).
Significant changes were considered when the absolute fold change was above 0.6 and the multiple-test corrected p-value was below 0.01. After this first filtering, we used a more relaxed filter to also consider significant those genes that did not pass our first criteria but were part of an operon in which at least one gene passed the filters, and the changes of all genes in the operon were in the same direction (either all upregulated or all downregulated). Finally, we manually rescued some changes that, despite not meeting our stringent criteria, were supported by proteomics data.
Operons were defined by correlation analysis of consecutive genes in the genome, using all the RNA-seq experiments in this study to define these correlations. We defined operons as groups of consecutive genes fulfilling two conditions. First, all genes in an operon had to be correlated over a certain value; and second, pairs of consecutive genes in these groups had to be correlated over a certain threshold, larger than the previous one. Both values were determined empirically to maximize the number of operons having only one annotated TSS (threshold for consecutive genes = 0.74; threshold for non-consecutive genes = 0.5). Transcription units are listed in Online Table 8.
Differential expression in microarrays was calculated via a two-sided t-test assuming non-equal variance for each gene. Criteria for assessing biological significance were the same as above.
For experiments with replicates in RNA-seq and microarrays, a consensus was determined for each gene only if in both cases the direction of the change is the same and in at least one of them the change is statistically significant. The list of datasets used for each condition can be found in Online Table 9.
To identify regulators, we established a threshold, setting a minimal number of targets in transcriptomics. To establish this threshold, we calculated the distribution of the number of transcriptomics changes per experiment. This distribution has a large number of zeros and one large outlier (MPN545-KO, which affects RNA degradation and thus targets the majority of genes in M. pneumoniae). After removing these extreme values, we observed a bimodal distribution. We fitted a mixture model using the mixtools package in R (Tatiana Benaglia et al., 2009), which fitted two normal distributions intersecting in N=9 changes. Thus, we considered all strains with 10 or more changes in transcriptomics as putative regulators (except for the TFs, for which we found a common motif).
Gene Regulatory Network Reconstruction and Validation
From the ChIP-seq and genetic perturbation transcriptomics data we manually curated a set of 1,062 transcription factor/regulator target interactions, which we refer to as “Manual Gene Regulatory Network”. To verify how well this network recapitulates the changes observed in these experiments, we used the Inferelator software to reconstruct a gene regulatory network from our data. As inputs, we used the Manual Gene Regulatory Network as prior information, together with the list of identified transcription factors and regulators and the set of genetic perturbation transcriptomics experiments. We ran Inferelator with 20 bootstraps, and filtered the results from the resulting network by the parameters beta.non.zero>10 and ombined_confidences>0.5 to ensure the filtered interactions appeared in at least half of the bootstrap iterations. As a gold standard to validate the performance of the network, we used the same manual network from the prior information. The resulting network (“Inferelator Network”) contained 1,036 interactions, 668 of which were common to the Manual Gene Regulatory Network.
The Manual Gene Regulatory Network and the Inferelator Network were merged (by pulling the interactions listed in each of them) to obtain an expanded network with 1,430 interactions. To study how well this expanded network explained the changes observed in the perturbation experiments, we ran Inferelator again using the expanded network and the list of transcription factors and regulators as prior knowledge together with the set of environmental perturbation experiments. From these environmental perturbation experiments, those with no major changes in transcription (<10 genes significantly changing) were excluded. As a gold standard, we included the same expanded network from the prior knowledge. We ran Inferelator with the same parameters as above, and filtered the results using the same criteria. The resulting perturbations network contained 230 interactions. The variance recapitulated by the Inferelator networks is calculated as the sum of the variance explained in each gene by each interaction (the sum of the column var.exp.median in the Inferelator output) divided by the total number of genes in M. pneumoniae (688). In Online Tables 10 and 11 we show the Inferelator output for the genetic perturbations and the environmental perturbations, respectively.
Gene Correlation Network Reconstruction
To identify sets of genes that are co-regulated under a broad set of conditions, we performed correlation analyses of the fold changes observed in the set of genetic or environmental perturbations. To do so, we used a set of 190 environmental and genetic perturbations. This set comprises all OE/KO/mutant strains together with the perturbation experiments, after removal of those whose mean change in gene expression is not zero (i.e., novobiocin, as it causes RNA degradation).
First, we performed k-means clustering of the fold-change matrix. We used the NbClust R function first to determine the optimal number of clusters (resulting to be k=2) and applied k-means clustering to the dataset to map all the genes to 2 major clusters of co-expression. To explore the sub-structure of our data, we constructed a network of highly co-regulated genes by establishing an edge between any two genes showing a correlation higher than 0.5 across our experiment set. Using this approach, we obtained a network comprising 576 nodes and 1,839 edges. To facilitate visualization and interpretation of this network, we clustered it to find groups of highly interconnected nodes. To do so, we used the Girvan-Newman algorithm, as implemented in the ClusterMaker plugin for cytoscape (GLay clustering) (Newman and Girvan, 2004, Smoot et al., 2011, Su et al., 2010). This algorithm finds communities of nodes that are highly interconnected, and removes the edges between different communities. This is done by computing the betweenness centrality of all the edges in the network, and removing the edges with the largest values. We identified 28 clusters, ranging from 2 to 140 genes each. We performed Fisher’s exact tests to determine significant enrichments in any functional category for each cluster. In Online Table 12 we show the overall expression correlation for all gene pairs from experiments done in this work.
RNA Degradation
RNA half-lives were determined from a time-course novobiocin treatment, as previously described (Junier et al., 2016).
Supercoiling Analysis
By inhibiting the gyrase complex, novobiocin is capable of inducing changes in DNA supercoiling. We used the transcriptional changes that occurred under different concentrations of novobiocin to identify genes that behaved in the same manner, but differently from a standard RNA degradation-induced decay. After read mapping and gene expression calculation, we normalized the data considering that the rRNA does not change its expression in these experiments due to its high stability. We scaled the expression values of each gene by subtracting the mean value of the five experiments and dividing by their standard deviation. With the scaled values, we computed the correlation matrix for all the genes of M. pneumoniae. We performed k-means clustering in our data with five centers to find the patterns corresponding to the different groups of genes (Figure S5C shows four of the groups, whilst the fifth one shows no changes at all; Table S8 contains information regarding the group assignation).
Riboswitch Scan
To find potential riboswitches in the genome of M. pneumoniae, we analyzed those 5’-UTRs in the genome whose length is between 60 and 500 bases. We analyzed 284 RNA-seq experiments of genetic and environmental perturbations, with two replicates each, and did not discard any experiment for this analysis. For each experiment, we identified the annotated TSSs and defined three different regions around each of them: i) the previous region covering 100 bps upstream of the TSS; ii) the riboswitch region spanning the 5′-UTR; and iii) the gene region including 150 bps starting at the end of the riboswitch region. If the transcript terminates before the end of the gene region, this was shortened to match the length of the transcript. We calculated the expression values for each of these regions in the 284 experiments and in their corresponding control samples.
To annotate a putative riboswitch that regulates premature termination of transcription, we expected to find differential expression when comparing the riboswitch and the gene regions. Therefore, we calculated the expression ratio between the riboswitch and the gene for all the selected 5′-UTRs in all conditions. We removed any data points in which the expression of the riboswitch or the gene was smaller than 5 log2 CPKM, as in these points, small fluctuations due to experimental noise leads to large changes in the ratios.
As putative riboswitches, we selected cases in which the riboswitch-to-gene ratio was larger than two standard deviations of the whole distribution, considering all the experiments and trimming 1% of the observations at both ends to remove outliers. We applied two further filters to increase the specificity of our search. First, the TSS must be active in the condition where the riboswitch is found. Therefore, we compared the riboswitch region with the previous region, and only kept those cases in which the riboswitch expression was significantly larger than the one of the previous regions (using a t-test and filtering by fold-change and p-value). Second, the riboswitch must behave differently between the condition in which it is identified and its corresponding control experiment. We applied a t-test to compare the riboswitch-to-gene ratio of the sample and the control, and only kept those cases in which the difference was significant.
After this filtering, we identified a set of 29 riboswitches regulated in specific conditions. We then plotted the RNA-seq profiles of each of these putative riboswitches in all the conditions tested, to manually curate and validate our results, and to identify further conditions that did not pass our initial filtering criteria (Online Table 13 and Online Figure 3).
Variance Explained Using Random Forests
To determine the percentage of the variance explained by transcription factors, regulators, and other regulatory mechanisms, we first compiled all the regulatory information that was relevant for each gene in M. pneumoniae. All features compiled for each gene can be found in Table S8. Genes were classified as heat- or cold-shock-activated if they were upregulated in the given condition, while downregulated in the other. Genes changing in the same direction in both conditions remained unclassified. We fitted the Random Forest to each experiment instead of to each gene because we did not measure the effectors of some of the studied alternative mechanisms (for instance the GTP concentration, or the small variations in temperature) and as a consequence, we were unable to include these effectors as “regulatory mechanisms” in Inferelator or similar workflows. Even though we did not measure the values of these effectors in each single experiment, we have evidence (shown above) of the genes affected by them. Thus, fitting the Random Forest enabled us to estimate the relative contribution of each effector to transcriptional variation in our experiments.
For this purpose, we used the environmental perturbation experiments. Of the 37 perturbation experiments, we discarded those in which the total RNA amount varied (such as novobiocin addition) and those used to define some of the regulatory features. For this reason, we discarded cold-shock and heat-shock perturbations, as well as diamide, which was used to define the targets of the transcription factor SpxA. We also discarded those perturbation experiments with no transcriptomics phenotype (<10 significant changes). After removing these experiments, we were left with a list of 23 experiments that we used for the analysis of the variance. For each of these experiments, we performed Random Forest regression to fit the observed fold changes to the features extracted for each gene. This was done using the R package Random Forest (Liaw and Wiener, 2002). Random Forests were chosen because they are not prone to overfitting. The parameters of the Random Forests were the following: number of trees=500; and number of variables randomly sampled at each split of a tree=14 (a third of the total number of variables in the model). From the Random Forest output, we scaled the values of variable importance to the pseudo R-squared obtained in the fitting. This pseudo R-squared is calculated as 1 minus the mean square error (MSE) of the out-of-bag (OOB) samples divided by the variance of the response variable Y (1-MSE/Var(Y)). The scaled values of variable importance can thus be interpreted as the percentage of variance explained by each feature. We also calculated the correlation between the actual fold changes and the ones predicted by the model (out-of-bag prediction, using the samples that were discarded in the model training for each point).
After calculating the variance explained by each feature in each of the 23 experiments, we computed the variability explained in the entire dataset. We summed the variance explained by each factor across the 23 experiments, and normalized it by the percentage of variance (over the total dataset) represented by each of the experiments. In this way, we could calculate the total variance explained by these features, and how much variability could be explained by any of them.
To validate the Random Forest, we defined all changes in the perturbation experiments whose absolute log2 fold change is above 0.6 as “true regulatory events”, in an attempt to be stringent and consider only truly regulated changes (even if missing some, we wanted to make sure that smaller changes due to noise were not included). For the “true regulatory events”, the Random Forest predicts the direction of the change correctly in 88% of the total cases; whilst for changes below 0.6 log2, the accuracy of the Random Forest in predicting the direction of the change decreased to 68%, suggesting that the Random Forest is indeed capable of predicting correctly the gene regulation.
For two of the features analyzed, Pribnow motif and chromosomal interacting domains, we performed a randomization of their values across all the genes to determine whether the effect found in this analysis was significant or simply due to the large number of categories within each of these features. In each randomization, we kept the operon organization, meaning that all the genes of the same operon kept the same Pribnow motif or CID. For the Pribnow motif, we also maintained the same Pribnow motif in clusters of duplicated genes. After resampling the Pribnow motif and CID values, we fitted a Random Forest regression to each of the 23 experiments in the analysis, using the randomized Pribnow/CIDs. A total of 100 randomizations were performed for each case. We then compared the results of the Random Forest using the randomized values to the results of the Random Forest using the original values. In this way we were able to determine whether the variance explained by these features was or not an artifact resulting from their large number of categories (Figure S7B).
Variance Explained by Noise
To determine the contribution of intrinsic/experimental noise to the variance, we took the RNA-seq control experiments used in this study (i.e., gene expression of a WT strain with an empty transposon insertion). A total of 42 different experiments from 23 different batches were used. In these experiments, RNA-seq counts per gene should be the same, and their differences should only be due to a batch effect and the different sequencing depths, that can be addressed normalizing by a library size factor. Thus, for each gene, we fitted a linear model including these two parameters, with the R lm function as lm (counts ~ batch + sizeFactor). The variance not explained by the model (calculated as 1 – adjusted R-squared) can be attributed to noise, either biological or experimental.
The variance explained by noise in the above setting represents the observable upper limit in experiments that do not show any significant change in their expression profile. Since this measurement is relative, in experiments with minor or no changes, the relative effect of noise will be higher than in experiments with a large number of significant changes. To explore the latter situation, we used two perturbation experiments with a large number of changes and highly correlated biological replicates: i) the addition of glycerol and ii) the addition of diamide (5 biological replicates each). In each experiment, and for each of the replicates, we performed the Random Forest prediction as stated above, and calculated the variance explained in the same as well as in the remaining replicates not used for the fitting. The decrease in the variance explained in the different replicates as compared to the same replicate can be attributed to noise.
To test the robustness of the method to the noise this, we repeated the Random Forest analysis on the 23 environmental perturbations, but discarding the gene changes with absolute log2 fold smaller than 0.4 (which could considered noise). The variance explained when ignoring these small changes decreases only slightly (3%), suggesting that this method is quite robust to the levels of noise observed in M. pneumoniae.
Statistics
Unless otherwise stated, the following statistics were used. To determine correlations, a Spearman rank order correlation coefficient was used. Statistical significance was assessed using Benjamini-Hochberg corrected p-values lower than 0.05. Statistical details specific for each of the different analyses can be found in the corresponding Methods section.
Data and Code Availability
Sequencing data have been deposited in the NCBI Short Read Archive (http://www.ebi.ac.uk/arrayexpress) as datasets E-MTAB-3771, E-MTAB-3772, E-MTAB-3773, E-MTAB-4642, E-MTAB-4784 and E-MTAB-7153 (RNA-seq), E-MTAB-6124 (5′-end RNA mapping), and E-MTAB-5944 and E-MTAB-3783 (ChIP-seq and POD, respectively).
Proteomics data have been submitted to ProteomeXchange via the PRIDE database (http://www.ebi.ac.uk/pride) and assigned the identifiers: XD007672, Mycoplasma pneumoniae Chromatin isolation; PXD007674, DNA Affinity chromatography on Mycoplasma pneumoniae extracts I (RNA elution); PXD007676, DNA Affinity chromatography on Mycoplasma pneumoniae extracts II (cellulose column); PXD007677, DNA Affinity chromatography on Mycoplasma pneumoniae extracts III (DNA column). TF overexpression and mutant data: PXD007551, PXD007537, PXD007545, PXD007557, PXD007558, PXD007560, PXD007561, PXD007565, PXD007566, PXD007658, and PXD007658.
Additional online figures and tables can be accessed through Mendeley (https://data.mendeley.com/datasets/xf6y59gz6c/draft?a=17c16d3d-9698-43c8-b791-a036b7c5f9c3).
Acknowledgments
We acknowledge the staff of the Genomics Core facility for their assistance. Also, we thank the CRG/UPF Proteomics Unit, which is part of the Plataforma de Recursos Biomoleculares y Bioinformáticos (Instituto de Salud Carlos III), supported by grant PT13/0001.
The project was supported by funds from the Fundación Marcelino Botín and the Spanish Ministerio de Economía y Competitividad (BIO2007-61762). This project was financed by Instituto de Salud Carlos III and co-financed by Federación Española de Enfermedades Raras under grant agreement PI10/01702 and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program, under grant agreements no 634942 (MycoSynVac) and 670216 (MYCOCHASSIS). We acknowledge support from the Spanish Ministry of Economy and Competitiveness to the EMBL partnership, Centro de Excelencia Severo Ochoa.
We also acknowledge the support of the CERCA Program from the Generalitat de Catalunya, Spain.
The authors thank Tony Ferrar for help with the growth curve screening and manuscript editing, Robert J. Nichols for help determining drug conditions, Alejandro Nadra for initial experiments with heparin columns that inspired this work, Julia Busse for the help with mutant isolation, and Dr. Ben Lehner for critical reading of the manuscript. We appreciate the feedback from the Serrano lab members.
Author Contributions
E.Y. performed the overexpression mutants, the ChIP-seq, and the POD assays. S.M. was involved in the DNA chromatography experiments. C.G. performed the majority of the growth curve phenotypic assays. C.B. and J.S. isolated the transposon knock-out mutants, and H.E. performed the glucose starvation analysis of the SpoT KO mutant. M.L.-S. helped with the analysis of the ChIP-seq data. V.L.-R. was responsible for the computational analysis. L.S. supervised the entire work. M.L.-S and J.S. proofread the manuscript. E.Y., V.L.-R., and L.S. wrote the manuscript and designed the figures.
Declaration of Interests
The authors declare no competing interests.
Published: August 21, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cels.2019.07.001.
Contributor Information
Eva Yus, Email: eva.yus@crg.eu.
Verónica Lloréns-Rico, Email: veronica.llorens@crg.eu.
Luis Serrano, Email: luis.serrano@crg.eu.
Supplemental Information
References
- Anders S., Pyl P.T., Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae W., Xia B., Inouye M., Severinov K. Escherichia coli CspA-family RNA chaperones are transcription antiterminators. Proc. Natl. Acad. Sci. USA. 2000;97:7784–7789. doi: 10.1073/pnas.97.14.7784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balke V.L., Gralla J.D. Changes in the linking number of supercoiled DNA accompany growth transitions in Escherichia coli. J. Bacteriol. 1987;169:4499–4506. doi: 10.1128/jb.169.10.4499-4506.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranello L., Levens D., Gupta A., Kouzine F. The importance of being supercoiled: how DNA mechanics regulate dynamic processes. Biochim. Biophys. Acta. 2012;1819:632–638. doi: 10.1016/j.bbagrm.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick J.E., Breaker R.R. The distributions, mechanisms, and structures of metabolite-binding riboswitches. Genome Biol. 2007;8:R239. doi: 10.1186/gb-2007-8-11-r239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belrhali H., Yaremchuk A., Tukalo M., Berthet-Colominas C., Rasmussen B., Bösecke P., Diat O., Cusack S. The structural basis for seryl-adenylate and Ap4A synthesis by seryl-tRNA synthetase. Structure. 1995;3:341–352. doi: 10.1016/s0969-2126(01)00166-6. [DOI] [PubMed] [Google Scholar]
- Berthoumieux S., de Jong H., Baptist G., Pinel C., Ranquet C., Ropers D., Geiselmann J. Shared control of gene expression in bacteria by transcription factors and global physiology of the cell. Mol. Syst. Biol. 2013;9:634. doi: 10.1038/msb.2012.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blötz C., Lartigue C., Valverde Timana Y., Ruiz E., Paetzold B., Busse J., Stülke J. Development of a replicating plasmid based on the native oriC in Mycoplasma pneumoniae. Microbiology. 2018;164:1372–1382. doi: 10.1099/mic.0.000711. [DOI] [PubMed] [Google Scholar]
- Blötz C., Treffon K., Kaever V., Schwede F., Hammer E., Stülke J. Identification of the components involved in cyclic Di-AMP signaling in Mycoplasma pneumoniae. Front. Microbiol. 2017;8:1328. doi: 10.3389/fmicb.2017.01328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonneau R., Facciotti M.T., Reiss D.J., Schmid A.K., Pan M., Kaur A., Thorsson V., Shannon P., Johnson M.H., Bare J.C. A predictive model for transcriptional control of physiology in a free living cell. Cell. 2007;131:1354–1365. doi: 10.1016/j.cell.2007.10.053. [DOI] [PubMed] [Google Scholar]
- Bonneau R., Reiss D.J., Shannon P., Facciotti M., Hood L., Baliga N.S., Thorsson V. The Inferelator: an algorithm for learning parsimonious regulatory networks from systems-biology data sets de novo. Genome Biol. 2006;7:R36. doi: 10.1186/gb-2006-7-5-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinza L., Calevro F., Charles H. Genomic analysis of the regulatory elements and links with intrinsic DNA structural properties in the shrunken genome of Buchnera. BMC Genomics. 2013;14:73. doi: 10.1186/1471-2164-14-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown L., Villegas J.M., Elean M., Fadda S., Mozzi F., Saavedra L., Hebert E.M. YebC, a putative transcriptional factor involved in the regulation of the proteolytic system of Lactobacillus. Sci. Rep. 2017;7:8579. doi: 10.1038/s41598-017-09124-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos R., Totten P.A. MG428 is a novel positive regulator of recombination that triggers mgpB and mgpC gene variation in Mycoplasma genitalium. Mol. Microbiol. 2014;94:290–306. doi: 10.1111/mmi.12760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlier D., Kholti A., Huysveld N., Gigot D., Maes D., Thia-Toong T.L., Glansdorff N. Mutational analysis of Escherichia coli PepA, a multifunctional DNA-binding aminopeptidase. J. Mol. Biol. 2000;302:411–426. doi: 10.1006/jmbi.2000.4067. [DOI] [PubMed] [Google Scholar]
- Charrad M., Ghazzali N., Boiteau V., Niknafs A. NbClust: An R Package for Determining the Relevant Number of Clusters in a Data Set. Journal of Statistical Software. 2014;61(6):1–36. [Google Scholar]
- Cho K.H. The structure and function of the Gram-positive bacterial RNA degradosome. Front. Microbiol. 2017;8:154. doi: 10.3389/fmicb.2017.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commichau F.M., Rothe F.M., Herzberg C., Wagner E., Hellwig D., Lehnik-Habrink M., Hammer E., Völker U., Stülke J. Novel activities of glycolytic enzymes in Bacillus subtilis: interactions with essential proteins involved in mRNA processing. Mol. Cell. Proteomics. 2009;8:1350–1360. doi: 10.1074/mcp.M800546-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commichau F.M., Stülke J. Trigger enzymes: bifunctional proteins active in metabolism and in controlling gene expression. Mol. Microbiol. 2008;67:692–702. doi: 10.1111/j.1365-2958.2007.06071.x. [DOI] [PubMed] [Google Scholar]
- Commichau F.M., Stülke J. Trigger enzymes: coordination of metabolism and virulence gene expression. Microbiol. Spectrosc. 2015;3 doi: 10.1128/microbiolspec.MBP-0010-2014. [DOI] [PubMed] [Google Scholar]
- Costa F.F. Non-coding RNAs: lost in translation? Gene. 2007;386:1–10. doi: 10.1016/j.gene.2006.09.028. [DOI] [PubMed] [Google Scholar]
- Dar D., Shamir M., Mellin J.R., Koutero M., Stern-Ginossar N., Cossart P., Sorek R. Term-seq reveals abundant ribo-regulation of antibiotics resistance in bacteria. Science. 2016;352:aad9822. doi: 10.1126/science.aad9822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J., Marti-Renom M.A., Mirny L.A. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat. Rev. Genet. 2013;14:390–403. doi: 10.1038/nrg3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon S.C., Dorman C.J. Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nat. Rev. Microbiol. 2010;8:185–195. doi: 10.1038/nrmicro2261. [DOI] [PubMed] [Google Scholar]
- Dorman C.J., Dorman M.J. DNA supercoiling is a fundamental regulatory principle in the control of bacterial gene expression. Biophys. Rev. 2016;8:209–220. doi: 10.1007/s12551-016-0205-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Houdaigui B., Forquet R., Hindré T., Schneider D., Nasser W., Reverchon S., Meyer S. Bacterial genome architecture shapes global transcriptional regulation by DNA supercoiling. Nucleic Acids Res. 2019;47:5648–5657. doi: 10.1093/nar/gkz300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisunov G.Y., Evsyutina D.V., Semashko T.A., Arzamasov A.A., Manuvera V.A., Letarov A.V., Govorun V.M. Binding Site of MraZ transcription factor in Mollicutes. Biochimie. 2016;125:59–65. doi: 10.1016/j.biochi.2016.02.016. [DOI] [PubMed] [Google Scholar]
- Gama-Castro S., Salgado H., Santos-Zavaleta A., Ledezma-Tejeida D., Muñiz-Rascado L., García-Sotelo J.S., Alquicira-Hernández K., Martínez-Flores I., Pannier L., Castro-Mondragón J.A. RegulonDB version 9.0: high-level integration of gene regulation, coexpression, motif clustering and beyond. Nucleic Acids Res. 2016;44:D133–D143. doi: 10.1093/nar/gkv1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger T., Francois P., Liebeke M., Fraunholz M., Goerke C., Krismer B., Schrenzel J., Lalk M., Wolz C. The stringent response of Staphylococcus aureus and its impact on survival after phagocytosis through the induction of intracellular PSMs expression. PLoS Pathog. 2012;8:e1003016. doi: 10.1371/journal.ppat.1003016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau R., Gardiol D., Glikin G.C., de Mendoza D. DNA supercoiling and thermal regulation of unsaturated fatty acid synthesis in Bacillus subtilis. Mol. Microbiol. 1994;11:933–941. doi: 10.1111/j.1365-2958.1994.tb00372.x. [DOI] [PubMed] [Google Scholar]
- Großhennig S., Schmidl S.R., Schmeisky G., Busse J., Stülke J. Implication of glycerol and phospholipid transporters in Mycoplasma pneumoniae growth and virulence. Infect. Immun. 2013;81:896–904. doi: 10.1128/IAI.01212-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güell M., van Noort V., Yus E., Chen W.H., Leigh-Bell J., Michalodimitrakis K., Yamada T., Arumugam M., Doerks T., Kühner S. Transcriptome complexity in a genome-reduced bacterium. Science. 2009;326:1268–1271. doi: 10.1126/science.1176951. [DOI] [PubMed] [Google Scholar]
- Halbedel S., Busse J., Schmidl S.R., Stülke J. Regulatory protein phosphorylation in Mycoplasma pneumoniae. A PP2C-type phosphatase serves to dephosphorylate HPr(Ser-P) J. Biol. Chem. 2006;281:26253–26259. doi: 10.1074/jbc.M605010200. [DOI] [PubMed] [Google Scholar]
- Hames C., Halbedel S., Hoppert M., Frey J., Stülke J. Glycerol metabolism is important for cytotoxicity of Mycoplasma pneumoniae. J. Bacteriol. 2009;191:747–753. doi: 10.1128/JB.01103-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfield G.W., Benham C.J. DNA topology-mediated control of global gene expression in Escherichia coli. Annu. Rev. Genet. 2002;36:175–203. doi: 10.1146/annurev.genet.36.032902.111815. [DOI] [PubMed] [Google Scholar]
- Hauryliuk V., Atkinson G.C., Murakami K.S., Tenson T., Gerdes K. Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nat. Rev. Microbiol. 2015;13:298–309. doi: 10.1038/nrmicro3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecker M., Völker U. General stress response of Bacillus subtilis and other bacteria. Adv. Microb. Physiol. 2001;44:35–91. doi: 10.1016/s0065-2911(01)44011-2. [DOI] [PubMed] [Google Scholar]
- Hogg T., Mechold U., Malke H., Cashel M., Hilgenfeld R. Conformational antagonism between opposing active sites in a bifunctional RelA/SpoT homolog modulates (p)ppGpp metabolism during the stringent response [corrected] Cell. 2004;117:57–68. doi: 10.1016/s0092-8674(04)00260-0. [DOI] [PubMed] [Google Scholar]
- Hüttenhofer A., Schattner P., Polacek N. Non-coding RNAs: hope or hype? Trends Genet. 2005;21:289–297. doi: 10.1016/j.tig.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Isalan M., Lemerle C., Michalodimitrakis K., Horn C., Beltrao P., Raineri E., Garriga-Canut M., Serrano L. Evolvability and hierarchy in rewired bacterial gene networks. Nature. 2008;452:840–845. doi: 10.1038/nature06847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa S., Ogura Y., Yoshimura M., Okumura H., Cho E., Kawai Y., Kurokawa K., Oshima T., Ogasawara N. Distribution of stable DnaA-binding sites on the Bacillus subtilis genome detected using a modified ChIP-chip method. DNA Res. 2007;14:155–168. doi: 10.1093/dnares/dsm017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain D., Baldi S., Zabel A., Straub T., Becker P.B. Active promoters give rise to false positive 'Phantom Peaks'. Nucleic Acids Res. 2015;43:6959–6968. doi: 10.1093/nar/gkv637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery C.J. Why study moonlighting proteins? Front. Genet. 2015;6:211. doi: 10.3389/fgene.2015.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junier I., Rivoire O. Conserved units of co-expression in bacterial genomes: an evolutionary insight into transcriptional regulation. PLoS One. 2016;11:e0155740. doi: 10.1371/journal.pone.0155740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junier I., Unal E.B., Yus E., Lloréns-Rico V., Serrano L. Insights into the mechanisms of basal coordination of transcription using a genome-reduced bacterium. Cell Syst. 2016;2:391–401. doi: 10.1016/j.cels.2016.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassambara, A. (2018). ggpubr: ‘ggplot2’ Based Publication Ready Plots. R package version 0.2. https://CRAN.R-project.org/package=ggpubr.
- Keogh M.C., Buratowski S. Using chromatin immunoprecipitation to map cotranscriptional mRNA processing in Saccharomyces cerevisiae. Methods Mol. Biol. 2004;257:1–16. doi: 10.1385/1-59259-750-5:001. [DOI] [PubMed] [Google Scholar]
- Klumpp S., Hwa T. Bacterial growth: global effects on gene expression, growth feedback and proteome partition. Curr. Opin. Biotechnol. 2014;28:96–102. doi: 10.1016/j.copbio.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudtson K.L., Minion F.C. Construction of Tn4001lac derivatives to be used as promoter probe vectors in mycoplasmas. Gene. 1993;137:217–222. doi: 10.1016/0378-1119(93)90009-r. [DOI] [PubMed] [Google Scholar]
- Kohanski M.A., Dwyer D.J., Collins J.J. How antibiotics kill bacteria: from targets to networks. Nat. Rev. Microbiol. 2010;8:423–435. doi: 10.1038/nrmicro2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriel A., Bittner A.N., Kim S.H., Liu K., Tehranchi A.K., Zou W.Y., Rendon S., Chen R., Tu B.P., Wang J.D. Direct regulation of GTP homeostasis by (p)ppGpp: a critical component of viability and stress resistance. Mol. Cell. 2012;48:231–241. doi: 10.1016/j.molcel.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krispin O., Allmansberger R. Changes in DNA supertwist as a response of Bacillus subtilis towards different kinds of stress. FEMS Microbiol. Lett. 1995;134:129–135. doi: 10.1111/j.1574-6968.1995.tb07926.x. [DOI] [PubMed] [Google Scholar]
- Kühner S., van Noort V., Betts M.J., Leo-Macias A., Batisse C., Rode M., Yamada T., Maier T., Bader S., Beltran-Alvarez P. Proteome organization in a genome-reduced bacterium. Science. 2009;326:1235–1240. doi: 10.1126/science.1176343. [DOI] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauinger L., Li J., Shostak A., Cemel I.A., Ha N., Zhang Y., Merkl P.E., Obermeyer S., Stankovic-Valentin N., Schafmeier T. Thiolutin is a zinc chelator that inhibits the Rpn11 and other JAMM metalloproteases. Nat. Chem. Biol. 2017;13:709–714. doi: 10.1038/nchembio.2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T.I., Rinaldi N.J., Robert F., Odom D.T., Bar-Joseph Z., Gerber G.K., Hannett N.M., Harbison C.T., Thompson C.M., Simon I. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- Leichert L.I., Scharf C., Hecker M. Global characterization of disulfide stress in Bacillus subtilis. J. Bacteriol. 2003;185:1967–1975. doi: 10.1128/JB.185.6.1967-1975.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyn S.A., Kazanov M.D., Sernova N.V., Ermakova E.O., Novichkov P.S., Rodionov D.A. Genomic reconstruction of the transcriptional regulatory network in Bacillus subtilis. J. Bacteriol. 2013;195:2463–2473. doi: 10.1128/JB.00140-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Ruan J., Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw A., Wiener M. Classification and regression by randomForest. Res. News. 2002;2:5. [Google Scholar]
- Lin Y.C., Lee H.C., Wang I., Hsu C.H., Liao J.H., Lee A.Y., Chen C., Wu S.H. DNA-binding specificity of the Lon protease alpha-domain from Brevibacillus thermoruber WR-249. Biochem. Biophys. Res. Commun. 2009;388:62–66. doi: 10.1016/j.bbrc.2009.07.118. [DOI] [PubMed] [Google Scholar]
- Lloréns-Rico V., Cano J., Kamminga T., Gil R., Latorre A., Chen W.H., Bork P., Glass J.I., Serrano L., Lluch-Senar M. Bacterial antisense RNAs are mainly the product of transcriptional noise. Sci. Adv. 2016;2:e1501363. doi: 10.1126/sciadv.1501363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloréns-Rico V., Lluch-Senar M., Serrano L. Distinguishing between productive and abortive promoters using a random forest classifier in Mycoplasma pneumoniae. Nucleic Acids Res. 2015;43:3442–3453. doi: 10.1093/nar/gkv170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lluch-Senar M., Delgado J., Chen W.-H., Lloréns-Rico V., O'Reilly F.J., Wodke J.A., Unal E.B., Yus E., Martínez S., Nichols R.J. Defining a minimal cell: essentiality of small ORFs and ncRNAs in a genome-reduced bacterium. Mol. Syst. Biol. 2015;11:780. doi: 10.15252/msb.20145558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-García P., Forterre P. DNA topology in hyperthermophilic archaea: reference states and their variation with growth phase, growth temperature, and temperature stresses. Mol. Microbiol. 1997;23:1267–1279. doi: 10.1046/j.1365-2958.1997.3051668.x. [DOI] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masternak K., Muhlethaler-Mottet A., Villard J., Zufferey M., Steimle V., Reith W. CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Genes Dev. 2000;14:1156–1166. [PMC free article] [PubMed] [Google Scholar]
- Michna R.H., Zhu B., Mäder U., Stülke J. SubtiWiki 2.0–an integrated database for the model organism Bacillus subtilis. Nucleic Acids Res. 2016;44:D654–D662. doi: 10.1093/nar/gkv1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minch K.J., Rustad T.R., Peterson E.J., Winkler J., Reiss D.J., Ma S., Hickey M., Brabant W., Morrison B., Turkarslan S. The DNA-binding network of Mycobacterium tuberculosis. Nat. Commun. 2015;6:5829. doi: 10.1038/ncomms6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miravet-Verde S., Ferrar T., Espadas-García G., Mazzolini R., Gharrab A., Sabido E., Serrano L., Lluch-Senar M. Unraveling the hidden universe of small proteins in bacterial genomes. Mol. Syst. Biol. 2019;15:e8290. doi: 10.15252/msb.20188290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miravet-Verde S., Lloréns-Rico V., Serrano L. Alternative transcriptional regulation in genome-reduced bacteria. Curr. Opin. Microbiol. 2017;39:89–95. doi: 10.1016/j.mib.2017.10.022. [DOI] [PubMed] [Google Scholar]
- Mitrophanov A.Y., Groisman E.A. Signal integration in bacterial two-component regulatory systems. Genes Dev. 2008;22:2601–2611. doi: 10.1101/gad.1700308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris J.H., Apeltsin L., Newman A.M., Baumbach J., Wittkop T., Su G., Bader G.D., Ferrin T.E. clusterMaker: a multi-algorithm clustering plugin for Cytoscape. BMC bioinformatics. 2011;12(1):436. doi: 10.1186/1471-2105-12-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano M.M., Lin A., Zuber C.S., Newberry K.J., Brennan R.G., Zuber P. Promoter recognition by a complex of Spx and the C-terminal domain of the RNA polymerase alpha subunit. PLoS One. 2010;5:e8664. doi: 10.1371/journal.pone.0008664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano S., Küster-Schöck E., Grossman A.D., Zuber P. Spx-dependent global transcriptional control is induced by thiol-specific oxidative stress in Bacillus subtilis. Proc. Natl. Acad. Sci. USA. 2003;100:13603–13608. doi: 10.1073/pnas.2235180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman M.E., Girvan M. Finding and evaluating community structure in networks. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2004;69:026113. doi: 10.1103/PhysRevE.69.026113. [DOI] [PubMed] [Google Scholar]
- Perkins D.N., Pappin D.J., Creasy D.M., Cottrell J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Potrykus K., Cashel M. (p)ppGpp: still magical? Annu. Rev. Microbiol. 2008;62:35–51. doi: 10.1146/annurev.micro.62.081307.162903. [DOI] [PubMed] [Google Scholar]
- Prasad S.C., Dritschilo A. High-resolution two-dimensional electrophoresis of nuclear proteins: a comparison of HeLa nuclei prepared by three different methods. Anal. Biochem. 1992;207:121–128. doi: 10.1016/0003-2697(92)90512-6. [DOI] [PubMed] [Google Scholar]
- Reuß D.R., Altenbuchner J., Mäder U., Rath H., Ischebeck T., Sappa P.K., Thürmer A., Guérin C., Nicolas P., Steil L. Large-scale reduction of the Bacillus subtilis genome: consequences for the transcriptional network, resource allocation, and metabolism. Genome Res. 2017;27:289–299. doi: 10.1101/gr.215293.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas-Tapias D.F., Helmann J.D. Induction of the Spx regulon by cell wall stress reveals novel regulatory mechanisms in Bacillus subtilis. Mol. Microbiol. 2018;107:659–674. doi: 10.1111/mmi.13906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado H., Peralta-Gil M., Gama-Castro S., Santos-Zavaleta A., Muñiz-Rascado L., García-Sotelo J.S., Weiss V., Solano-Lira H., Martínez-Flores I., Medina-Rivera A. RegulonDB v8.0: omics data sets, evolutionary conservation, regulatory phrases, cross-validated gold standards and more. Nucleic Acids Res. 2013;41:D203–D213. doi: 10.1093/nar/gks1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidl S.R., Gronau K., Pietack N., Hecker M., Becher D., Stülke J. The phosphoproteome of the minimal bacterium Mycoplasma pneumoniae: analysis of the complete known Ser/Thr kinome suggests the existence of novel kinases. Mol. Cell. Proteomics. 2010;9:1228–1242. doi: 10.1074/mcp.M900267-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidl S.R., Otto A., Lluch-Senar M., Piñol J., Busse J., Becher D., Stülke J. A trigger enzyme in Mycoplasma pneumoniae: impact of the glycerophosphodiesterase GlpQ on virulence and gene expression. PLoS Pathog. 2011;7:e1002263. doi: 10.1371/journal.ppat.1002263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider D.A., Ross W., Gourse R.L. Control of rRNA expression in Escherichia coli. Curr. Opin. Microbiol. 2003;6:151–156. doi: 10.1016/s1369-5274(03)00038-9. [DOI] [PubMed] [Google Scholar]
- Selinger D.W., Saxena R.M., Cheung K.J., Church G.M., Rosenow C. Global RNA half-life analysis in Escherichia coli reveals positional patterns of transcript degradation. Genome Res. 2003;13:216–223. doi: 10.1101/gr.912603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu K. Metabolic regulation of a bacterial cell system with emphasis on Escherichia coli metabolism. ISRN Biochem. 2013;2013:645983. doi: 10.1155/2013/645983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoot M.E., Ono K., Ruscheinski J., Wang P.L., Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sojka L., Kouba T., Barvík I., Sanderová H., Maderová Z., Jonák J., Krásny L. Rapid changes in gene expression: DNA determinants of promoter regulation by the concentration of the transcription initiating NTP in Bacillus subtilis. Nucleic Acids Res. 2011;39:4598–4611. doi: 10.1093/nar/gkr032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stülke J. Control of transcription termination in bacteria by RNA-binding proteins that modulate RNA structures. Arch. Microbiol. 2002;177:433–440. doi: 10.1007/s00203-002-0407-5. [DOI] [PubMed] [Google Scholar]
- Su G., Kuchinsky A., Morris J.H., States D.J., Meng F. GLay: community structure analysis of biological networks. Bioinformatics. 2010;26:3135–3137. doi: 10.1093/bioinformatics/btq596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin M.F., Perez H.R., Baldini R.L., Gomes S.L. Functional and structural analysis of HrcA repressor protein from Caulobacter crescentus. J. Bacteriol. 2004;186:6759–6767. doi: 10.1128/JB.186.20.6759-6767.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatiana Benaglia D.C., Hunter D.R., Young D.S. Mixtools: an R package for analyzing mixture models. J. Stat. Softw. 2009;32:29. [Google Scholar]
- Torres-Puig S., Broto A., Querol E., Piñol J., Pich O.Q. A novel sigma factor reveals a unique regulon controlling cell-specific recombination in Mycoplasma genitalium. Nucleic Acids Res. 2015;43:4923–4936. doi: 10.1093/nar/gkv422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers A., Muskhelishvili G. Bacterial chromatin. Curr. Opin. Genet. Dev. 2005;15:507–514. doi: 10.1016/j.gde.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Traxler M.F., Chang D.E., Conway T. Guanosine 3',5'-bispyrophosphate coordinates global gene expression during glucose-lactose diauxie in Escherichia coli. Proc Natl Acad Sci U S A. 2006;103(7):2374–2379. doi: 10.1073/pnas.0510995103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trussart M., Yus E., Martinez S., Baù D., Tahara Y.O., Pengo T., Widjaja M., Kretschmer S., Swoger J., Djordjevic S. Defined chromosome structure in the genome-reduced bacterium Mycoplasma pneumoniae. Nat. Commun. 2017;8:14665. doi: 10.1038/ncomms14665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Noort V., Seebacher J., Bader S., Mohammed S., Vonkova I., Betts M.J., Kühner S., Kumar R., Maier T., O'Flaherty M., Rybin V., Schmeisky A., Yus E., Stülke J., Serrano L., Russell R.B., Heck A.J., Bork P., Gavin A.C. Cross-talk between phosphorylation and lysine acetylation in a genome-reduced bacterium. Mol Syst Biol. 2012;8:571. doi: 10.1038/msb.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner J.R., McIntosh K.B. How common are extraribosomal functions of ribosomal proteins? Mol. Cell. 2009;34:3–11. doi: 10.1016/j.molcel.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnes, G.R., Bolker, B., Gorjanc, G., Grothendieck, G., Korosec, A., Lumley, T., MacQueen, D., Magnusson, D., Rogers, J., and others (2017). gdata: Various R Programming Tools for Data Manipulation. R package version 2.18.0. https://CRAN.R-project.org/package=gdata.
- Wickham, H. (2017). tidyverse: Easily Install and Load the ‘Tidyverse’. R package version 1.2.1. https://CRAN.R-project.org/package=tidyverse.
- Wilhelm L., Bürmann F., Minnen A., Shin H.C., Toseland C.P., Oh B.H., Gruber S. SMC condensin entraps chromosomal DNA by an ATP hydrolysis dependent loading mechanism in Bacillus subtilis. Elife. 2015;4 doi: 10.7554/eLife.06659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wodke J.A.H., Puchałka J., Lluch-Senar M., Marcos J., Yus E., Godinho M., Gutiérrez-Gallego R., dos Santos V.A., Serrano L., Klipp E. Dissecting the energy metabolism in Mycoplasma pneumoniae through genome-scale metabolic modeling. Mol. Syst. Biol. 2013;9:653. doi: 10.1038/msb.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yus E., Maier T., Michalodimitrakis K., van Noort V., Yamada T., Chen W.H., Wodke J.A., Güell M., Martínez S., Bourgeois R. Impact of genome reduction on bacterial metabolism and its regulation. Science. 2009;326:1263–1268. doi: 10.1126/science.1177263. [DOI] [PubMed] [Google Scholar]
- Yus E., Yang J.S., Sogues A., Serrano L. A reporter system coupled with high-throughput sequencing unveils key bacterial transcription and translation determinants. Nat. Commun. 2017;8:368. doi: 10.1038/s41467-017-00239-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Baseman J.B. Functional characterization of osmotically inducible protein C (MG_427) from Mycoplasma genitalium. J. Bacteriol. 2014;196:1012–1019. doi: 10.1128/JB.00954-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T., Zhu J., Wei S., Luo Q., Li L., Li S., Tucker A., Shao H., Zhou R. The roles of RelA/(p)ppGpp in glucose-starvation induced adaptive response in the zoonotic Streptococcus suis. Sci Rep. 2016;6:27169. doi: 10.1038/srep27169. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data have been deposited in the NCBI Short Read Archive (http://www.ebi.ac.uk/arrayexpress) as datasets E-MTAB-3771, E-MTAB-3772, E-MTAB-3773, E-MTAB-4642, E-MTAB-4784 and E-MTAB-7153 (RNA-seq), E-MTAB-6124 (5′-end RNA mapping), and E-MTAB-5944 and E-MTAB-3783 (ChIP-seq and POD, respectively).
Proteomics data have been submitted to ProteomeXchange via the PRIDE database (http://www.ebi.ac.uk/pride) and assigned the identifiers: XD007672, Mycoplasma pneumoniae Chromatin isolation; PXD007674, DNA Affinity chromatography on Mycoplasma pneumoniae extracts I (RNA elution); PXD007676, DNA Affinity chromatography on Mycoplasma pneumoniae extracts II (cellulose column); PXD007677, DNA Affinity chromatography on Mycoplasma pneumoniae extracts III (DNA column). TF overexpression and mutant data: PXD007551, PXD007537, PXD007545, PXD007557, PXD007558, PXD007560, PXD007561, PXD007565, PXD007566, PXD007658, and PXD007658.
Additional online figures and tables can be accessed through Mendeley (https://data.mendeley.com/datasets/xf6y59gz6c/draft?a=17c16d3d-9698-43c8-b791-a036b7c5f9c3).