

Abstract
The liver secretes hepcidin (Hepc) into the bloodstream to reduce blood iron levels. Hepc accomplishes this by triggering degradation of the only known cellular iron exporter ferroportin in the gut, macrophages, and liver. We previously demonstrated that systemic Hepc knockout (HepcKO) mice, which have high serum iron, develop retinal iron overload and degeneration. However, it was unclear whether this is caused by high blood iron levels or, alternatively, retinal iron influx that would normally be regulated by retina-produced Hepc. To address this question, retinas of liver-specific and retina-specific HepcKO mice were studied. Liver-specific HepcKO mice had elevated blood and retinal pigment epithelium (RPE) iron levels and increased free (labile) iron levels in the retina, despite an intact blood-retinal barrier. This led to RPE hypertrophy associated with lipofuscin-laden lysosome accumulation. Photoreceptors also degenerated focally. In contrast, there was no change in retinal or RPE iron levels or degeneration in the retina-specific HepcKO mice. These data indicate that high blood iron levels can lead to retinal iron accumulation and degeneration. High blood iron levels can occur in patients with hereditary hemochromatosis or result from use of iron supplements or multiple blood transfusions. Our results suggest that high blood iron levels may cause or exacerbate retinal disease.
Iron is critical for cellular survival, as it is involved in several essential biochemical and metabolic processes. However, iron loading can cause oxidative injury through production of highly reactive hydroxyl free radicals in the Fenton reaction.1, 2 Because of the danger of cellular iron overload, tight iron regulation within all tissues is essential for proper cellular function. The retina, and especially the photoreceptors, is particularly vulnerable to iron-induced oxidative injury because of factors such as the high metabolic rate, high oxygen tension, and the abundance of polyunsaturated fatty acids.3 Retinal iron overload has been implicated in the pathophysiology of retinal degenerative diseases, including age-related macular degeneration, the leading cause of irreversible blindness in individuals >50 years old in developed nations.4 Brain iron overload may also contribute to the development of other neurodegenerative diseases, such as Alzheimer and Parkinson diseases.5, 6, 7 The observation that iron overload occurs in a variety of neurodegenerative diseases suggests that retinal or brain iron dysregulation may play an important role in the development or exacerbation of neurodegeneration and that the processes that promote iron accumulation within neuronal tissue, which are incompletely understood, need to be more thoroughly explored.
Unlike other organs, which are directly exposed to the systemic circulation, the retina should be protected from fluctuations in plasma iron by the presence of the blood-retinal barrier (BRB). Yet, in some human cases and mouse models of hereditary hemochromatosis, a set of genetic diseases associated with high plasma iron levels,8 there are elevated retinal iron levels and retinal degeneration.9, 10 These effects on the retina may result from the high plasma iron, abnormal local iron regulatory mechanisms within the retina, or both. Systemic iron levels are elevated in patients receiving oral or i.v. iron supplementation, common treatments for iron-deficiency anemia. A case study from our laboratory recently demonstrated that the short-term i.v. iron treatment in a 42-year-old patient was associated with the development of early-onset macular degeneration,11 suggesting that high plasma iron levels may be sufficient to cause retinal damage. The goal of this study was to determine whether high plasma iron levels could cause retinal iron overload and degeneration, despite intact local iron regulatory mechanisms within the retina.
Regulation of systemic iron levels is accomplished by several iron-handling proteins that regulate the flux of iron into and out of the systemic iron pool. The iron regulatory hormone, hepcidin (Hepc), is instrumental in regulating these processes. Hepc [alias hepcidin antimicrobial peptide (Hamp)], is a 25–amino acid peptide hormone that is produced primarily by hepatocytes and plays an essential role in regulating systemic iron levels by antagonizing the only known mammalian iron exporter, ferroportin (Fpn). Hepc binds to the extracellular domain of ferroportin on the cell surface, leading to its internalization and degradation, effectively preventing cellular iron export and limiting the amount of iron that gets into the serum or extracellular fluid.12 Hepc serves as the central regulator of systemic iron metabolism by controlling the flux of iron through Fpn from three distinct points, the release of iron from liver stores, the release of iron from macrophages involved in recycling red blood cells, and the transport of iron, absorbed from the diet by enterocytes within the duodenum, into the bloodstream.13 When the systemic iron pool is elevated, Hepc production by hepatocytes increases, preventing additional release of iron into the systemic pool. The Hepc/Fpn axis is responsible for mediating systemic iron levels, and dysregulation of this axis, in diseases such as primary hemochromatosis and anemia of chronic inflammation, leads to disruption of iron homeostasis.
Although most of Hepc is produced by hepatocytes, small amounts are also produced in organs that do not have any known role in regulating systemic iron levels, including the brain,14 heart,15 kidney,16 pancreas,17 and retina.18 The role of Hepc produced by some of those organs is largely unknown; however, a recent study demonstrated that cardiomyocyte-produced Hepc is necessary for proper cardiac iron regulation and function.15 Our laboratory has previously shown that a systemic Hepc knockout (HepcKO), which leads to high blood iron levels due to unrestricted iron absorption from the diet, results in retinal iron overload and retinal degeneration in mice by the age of 18 months.19 We also demonstrated that in mice with a mutation in Fpn, which causes resistance to Hepc regulation and subsequent serum iron overload, the mutation leads to a similar retinal iron accumulation as observed in the systemic HepcKO mice.20 These transgenic models demonstrate that changes in the Hepc/Fpn axis significantly affect retinal iron levels but fail to determine whether the retinal iron overload observed in these models is the result of long-term exposure to high blood iron levels or, rather, changes in the local retinal iron regulation from loss of retina-produced Hepc.
To test the role of Hepc in the retina, we analyzed the retinal phenotype in two mouse models: the first with a liver-specific deletion of the Hepc (official name, Hamp) gene and the second with a retina-specific deletion of the Hepc gene. In the first model, a liver-specific Hepc knockout mouse strain (LS-HepcKO) has high systemic iron levels, recapitulating the high serum iron conditions of the systemic HepcKO model, while maintaining the ability of the retina to produce its own Hepc. Using this LS-HepcKO model, it was investigated whether the presence of retina-produced Hepc plays a significant role in regulating retinal iron levels and retinal health by comparing these mice with age-matched controls. In the second model, the retina-specific Hepc knockout (RS-HepcKO), it was tested whether loss of retina-produced Hepc alters retinal iron homeostasis in mice with normal serum iron levels.
It is essential to understand how changes in serum iron levels affect retinal health and how the retina can regulate local iron levels when the serum iron levels are normal or elevated. These data will provide a clearer image of how retinal health is affected by fluctuations in serum iron levels and provide insight into the mechanisms that contribute to retinal iron overload in retinal degenerative disease.
Materials and Methods
Generation of Liver-Specific HepcKO Mice
The liver-specific hepcidin knockout mice were generated using a Hepc-floxed (Hepcflox/flox) strain, which was developed as previously described.15 Hepcflox/flox mice were crossed with a strain expressing the Cre-recombinase under the control of the hepatocyte-specific promoter, albumin (Alb-Cre; stock number 003574; Jackson Laboratory, Bar Harbor, ME),21 to generate the hepatocyte-specific HepcKO (Hepcflox/flox;Alb-Cre+; referred to herein as LS-HepcKO). The knockout of Hepc in the liver was verified using real-time quantitative PCR (qPCR) analysis of Hepc mRNA levels (Figure 1). Mice were aged to 15 days, 3 months, 6 months, and 12 months and then euthanized. All mice were fed a standard laboratory diet with 300 ppm iron, given free access to water and food, and maintained in a temperature-controlled room at 21°C to 23°C under dim cyclic light (12:12 hours light-dark cycle). Both LS-HepcKO mice and controls were on a C57BL/6J background, and both males and females were used in this study. All mice were negative for the rd1 and rd8 alleles. Experimental procedures were performed in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmology and Vision Research. All protocols were approved by the animal care review board of the University of Pennsylvania (Philadelphia, PA).
Figure 1.

Validation of LS-HepcKO model. A: Liver Hepc mRNA levels measured by real-time quantitative PCR in LS-HepcKO (Hepcflox/flox;Alb-Cre+) versus controls (Hepc+/+;Alb-Cre+ or Hepcflox/flox;Alb-Cre-) at 3 months. B: Nonheme liver iron quantification (by bathophenanthroline sulfonate assay) in the LS-HepcKO mice compared with controls (Hepc+/+;Alb-Cre+ or Hepcflox/flox;Alb-Cre-) at 3 months. Statistical analysis for liver Hepc mRNA and liver iron quantification was performed using one-way analysis of variance with post hoc pairwise comparisons using the Tukey method. C: Serum iron concentration of LS-HepcKO mice versus controls at 3 and 12 months. D: Serum transferrin (Tf) saturation of LS-HepcKO mice versus controls at 3 and 12 months. For Tf saturation calculation, any value that was calculated to be >100%, because of the presence of nontransferrin bound iron, was recorded as 100% Tf saturation on the graph. E: Body weight of LS-HepcKO versus control mice at 3 months. F: Body weight of LS-HepcKO versus control mice at 12 months. Statistical analysis was performed using two-group, two-sided t-test. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗∗P < 0.0001.
Generation of Retina-Specific HepcKO Mice
To generate the retina-specific HepcKO mice (referred to herein as RS-HepcKO), Hepcflox/flox mice were crossed to a strain expressing mRx-Cre. The mRx-Cre mouse strain was supplied by an author of this work (Z.K.). The mRx promoter is expressed in most cells within the neurosensory retina (NSR) and a subset of retinal pigment epithelium (RPE) cells.22 RS-HepcKO experimental mice had the genotype Hepcflox/flox;mRx-Cre+, and the control group had the genotype Hepc+/+;mRx-Cre+. Mice were aged to 6 and 12 months and euthanized. Both RS-HepcKO mice and controls were on a C57BL/6J background, and both males and females were used in this study. All mice were negative for the rd1 and rd8 alleles.
Dissection of Murine RPE and Retinas for RT-PCR and Western Blot Analysis
Mice were euthanized, and eyes were immediately enucleated. Anterior segments were removed, and retinas were completely dissected away from the underlying RPE. Retinas were then flash frozen and stored at -80°C. RPE cells were isolated from other ocular structures using enzymatic (dispase and hyaluronidase) digestion and mechanical dissection, as previously described.23
qPCR Data
RNA was isolated according to the manufacturer's protocol (RNeasy Kit; Qiagen, Valencia, CA). cDNA was synthesized with TaqMan Reverse Transcription Reagents (Applied Biosystems, Foster City, CA), according to the manufacturer's protocol. Gene expression of Tfrc (transferrin receptor), Dmt1 (divalent metal transporter 1; official name, Slc11a2), and Hepc (hepcidin) was analyzed using qPCR, as previously described.19 Gene expression assays (TaqMan; Applied Biosystems, Foster City, CA) were used for qPCR analysis. Real-time RT-PCR was performed on a commercial sequence detection system (ABI Prism 7500; Applied Biosystems, Darmstadt, Germany). All reactions were performed in technical triplicates (n = 3 to 10 mice per genotype). Probes used were as follows: Tfrc (Mm00441941_m1), Hepc (Mm04231240_s1), and Dmt1 (Mm01308330_s1). Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) served as an internal control (4352932E).
Neurosensory Retinal Protein Extraction and Western Blot Analysis
NSR protein lysates were extracted using Laemmli SDS lysis buffer supplemented with protease/phosphatase inhibitor mixture (Cell Signaling Technology, Danvers, MA). Lysates were treated and run, as described previously.23 Imaging was performed using GE Amersham Imager 600 (GE Healthcare, Chalfont St. Giles, UK). FIJI software (NIH, Bethesda, MD) was used for band densitometry.24 Primary antibodies used were as follows: rat antitransferrin receptor (Serotec, Kidlington, UK), goat antialbumin (Bethyl Laboratories, Montgomery, TX), and rabbit anti-GAPDH (Thermo Fisher Scientific, Philadelphia, PA). Secondary antibodies used were as follows: donkey anti-rabbit (ECL Rabbit IgG, horseradish peroxidase–linked whole antibody) and donkey anti-rat (ECL Rat IgG, horseradish peroxidase–linked whole antibody) (GE Healthcare, Chicago, IL). All primary antibodies were used at a 1:1000 dilution, and all secondary antibodies were used at a 1:5000 dilution. GAPDH served as an internal control.
Fixation of Eyes and Preparation of Eyecups
Eyes were enucleated immediately after sacrifice and fixed for 15 minutes in 4% paraformaldehyde, and eyecups were generated by removing the cornea and lens. Eyecups were dehydrated overnight in 30% sucrose and embedded in Tissue-Tek OCT (Sakura Finetek, Torrance, CA).
Immunofluorescence
Immunohistochemistry was performed on cryosections (10 μm thick), as described previously.19 Antibody used goat antialbumin (dilution 1:200; Bethyl Laboratories) and rabbit antilight ferritin (E17; dilution 1:2500; a gift from Paolo Arosio, Ph.D., University of Brescia, Brescia, Italy). Control sections were treated identically but with omission of primary antibody. Sections were analyzed by fluorescence microscopy using identical exposure parameters across genotype using a Nikon Eclipse 80i microscope (Nikon Instruments, Melville, NY), and images were acquired using NIS-BR Elements software version 4.1 (Nikon Instruments). Pixel density analysis of the l-ferritin (Ft-L) stain was completed using an open-source image-processing package (FIJI software).24
Quantitative Liver Iron Detection
Livers from experimental and age-matched control mice were frozen on dry ice. Dried tissue was digested overnight at 65°C in acid digest solution (0.1% trichloroacetic acid and 0.03 mol/L HCl). After digestion, samples were centrifuged, and supernatant (20 μL) was added to 1 mL of chromogen reagent [2.25 mol/L sodium acetate pretreated with Chelex 100 (Bio-Rad, Hercules, CA), 0.01% bathophenanthroline sulfonate, and 0.1% thioglycolic acid]. The absorbances were read at 535 nm. Iron levels were calculated by comparing absorbances of tissue-chromogen samples with serial dilutions of iron standard (Sigma-Aldrich, St. Louis, MO).
iCP-MS data
NSR and RPE samples were analyzed for iron and zinc using an inductively coupled mass spectrometer (iCP-MS; Nexion 300D; Perkin Elmer, Shelton, CT) at the Pennsylvania Animal Diagnostic Laboratory System (PADLS) New Bolton Center Toxicology Laboratory, University of Pennsylvania, School of Veterinary Medicine (Kennett Square, PA), as described previously.25 NSR and RPE tissues for iCP-MS analysis were collected from mice that had been perfused with saline.
Morphologic Analysis
Eyes were enucleated, fixed in 2% paraformaldehyde–2% glutaraldehyde overnight, and embedded in JB-4 plastic (Polysciences, Warrington, PA). Sections (3 μm thick) were cut through the sagittal plane and stained with toluidine blue, as previously described.19 Stained sections were observed and imaged using bright-field illumination (model TE300; Nikon, Inc., Tokyo, Japan). To measure outer nuclear layer (ONL) thickness, a retinal section adjacent to the optic nerve head was analyzed (n = 3 per genotype). ONL thickness was determined by counting nuclei per row at 200-μm intervals to 2200 μm superior and inferior to the optic nerve head. To determine the percentage of the retina that contained hypertrophic RPE cells, one slide per retina (n = 3 per genotype) that contained the optic nerve head was chosen. The total length of the RPE in one retinal section was determined. The total length of the RPE that contained hypertrophic RPE (defined as an RPE cell that is >15 μm in height) was determined, and the percentage of the total retinal length that contained hypertrophic RPE cells was calculated. All morphologic analysis was performed by a masked observer (B.H.B. or W.S.).
Electron Microscopy
Electron microscopy on retinal samples was performed as described previously.26 Briefly, after enucleation, eyes were fixed in 2% paraformaldehyde/2% glutaraldehyde overnight at 4°C. The anterior segment was removed, and the posterior portion of each eye was cut into small wedge-shaped pieces and post-fixed in 1% osmium tetroxide/0.1 mol/L sodium cacodylate buffer, dehydrated, and embedded in EMbed-812 (Electron Microscopy Sciences, Hatfield, PA). Ultrathin sections (60 to 80 nm thick) were stained and examined with a JEOL1010 transmission electron microscope (JEOL Ltd, Tokyo, Japan). Images were acquired with Advanced Microscopy Techniques Image Capture software version 602 (Advanced Microscopy Techniques Corp., Woburn, MA) and were rotated and cropped with Adobe Photoshop CS5 (Adobe Systems Inc., San Jose, CA).
Perls Staining for Iron
Perls stain for iron was performed on plastic sections (3 μm thick) or cryosections (10 μm thick), as previously described.27 For cryosections, slides were incubated in 5% potassium ferrocyanide in 5% aqueous hydrochloric acid for 45 minutes at 65°C and then bleached using potassium permanganate and oxalic acid. For plastic sections, slides were bleached using potassium permanganate and oxalic acid and then incubated in 5% potassium ferric ferrocyanide and 5% hydrochloric acid solution for 30 minutes at room temperature. Sensitivity for iron detection was enhanced by subsequent incubation of tissue in purple peroxidase substrate for 25 minutes at room temperature (VIP; Vector Laboratories, Inc., Burlingame, CA). Slides were then washed in 1× phosphate-buffered saline. Slides were examined on a Nikon Eclipse 80i microscope, and images were acquired using NIS-BR Elements software.
Serum Iron Concentration and Tf Saturation
Blood was collected from anesthetized animals by retro-orbital bleeding, placed into BD microtainer blood collection tubes (BD Biosciences, San Jose, CA), and spun down for 30 minutes at 1000 × g. Serum was collected and stored at −20°C. Serum Fe status was analyzed by quantifying total serum iron and transferrin saturation using an Iron/TIBC testing kit (Pointe Scientific, Inc., Canton, MI). For some of the mice with serum iron overload, the Tf saturation calculation was >100% because of the presence of non-transferrin bound iron. If Tf saturation was >100%, it was recorded as 100% Tf saturation.
Fundus Imaging
Mice were anesthetized with an i.p. injection of (in mg/kg body weight): 80 ketamine (Par Pharmaceutical, Spring Valley, NY), 10 xylazine (Lloyd Inc., Shenandoah, IA), and 2 acepromazine (Boehringer Ingelheim Vetmedica, Inc., St. Joseph, MO). Pupils were then dilated with 1% tropicamide (Akorn, Inc., Lake Forest, IL). Once anesthetized adequately, mice were placed on a metal stage. Color and autofluorescence images were acquired using a fundus camera (Micron III; Phoenix Research Laboratories, Inc., Pleasanton, CA).
Electroretinography
Electroretinography recordings followed procedures described previously.28, 29 In brief, mice were dark adapted overnight and then anesthetized with an i.p. injection of (in mg/kg body weight): 80 ketamine, 10 xylazine, and 2 acepromazine. Pupils were dilated with 1% tropicamide saline solution (Akorn, Inc., Lake Forest, IL). Two electrodes made of UV transparent plastic with embedded platinum wires were placed in electrical contact with the corneas. A platinum wire loop placed in the mouth served as the reference and ground electrode. The electroretinograms were then recorded (Espion Electrophysiology System; Diagnosys LLC, Lowell, MA). The apparatus was modified by the manufacturer for experiments with mice by substituting light-emitting diodes with emission maximum at 365 nm for standard blue ones. The stage was positioned in such a way that the mouse's head was located inside the stimulator (ColorDome; Diagnosys LLC), thus ensuring uniform full-field illumination. The flash intensities for recordings of rod a- and b-waves were 500 and 0.01 scotopic candelas m−2second, delivered by the white xenon flash and green (510-nm maximum) LED, respectively. The white flash intensity of the cone b-wave is 500 scotopic candelas m−2second, with a rod-suppressing steady green background of 30 scot m−2second.
Statistical Analysis
Means ± SEM were calculated for each group. Two-group, two-tailed t-test was used for statistical analysis between two groups. For multiple comparisons, one-way analysis of variance, with post hoc pairwise comparisons using the Tukey method, was used. Comparison of photoreceptor nuclei counts was performed with two-way analysis of variance with post hoc pairwise comparisons using Bonferroni's adjustment. Comparisons of Tfrc and Dmt1 mRNA levels in the NSR and RPE over time between the LS-HepcKO and control mice were performed using two-way analysis of variance with post hoc pairwise comparisons using Tukey adjustment. All statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA).
Results
Validation of the LS-HepcKO Model
Elevated blood and liver iron levels observed in the LS-HepcKO (Hepcflox/flox;Alb-Cre+) mice used in this study recapitulate the phenotype that has been previously described.30 For validation of the LS-HepcKO model, two control groups were compared with LS-HepcKO experimental mice. Control groups were Hepcflox/flox;Alb-Cre- and Hepc+/+;Alb-Cre+. Compared with either control group, LS-HepcKO mice had significantly reduced liver Hepc mRNA levels (Figure 1A) and elevated liver iron levels (Figure 1B), consistent with previous findings.30 Because the two control groups, Hepc+/+;Alb-Cre+ and Hepcflox/flox;Alb-Cre-, did not differ from each other for either of these measures, only the Hepc+/+;Alb-Cre+ control mice were generated for subsequent experiments and compared with experimental Hepcflox/flox;Alb-Cre+ mice, derived from the same crosses.
The LS-HepcKO mice also had elevated serum iron levels (Figure 1C) and elevated serum transferrin saturation (Figure 1D) compared with controls at both the 3- and 12-month time points. There was no observed weight difference between the LS-HepcKO mice and controls at 3 months (Figure 1E). However, at 12 months, the LS-HepcKO mice of both sexes had reduced weights compared with controls (Figure 1F), most likely resulting from iron-induced organ damage.
LS-HepcKO Mice Develop RPE Iron Overload after Long-Term Exposure to Elevated Serum Iron Levels
To determine how the loss of liver-produced Hepc affects RPE iron levels, LS-HepcKO mice and controls were assessed at four ages: 15 days, 3 months, 6 months, and 12 months, using both direct and indirect methods. Changes in levels of the iron importer transferrin receptor at the mRNA (Tfrc) or protein (Tfr) levels are an indirect, but highly validated, measure of cellular iron levels. Tfrc is regulated by the iron regulatory protein/iron responsive element system so that when labile (free) iron levels are high, Tfrc and Tfr are reduced.31, 32
At the ages of 3, 6, and 12 months, there was a significant decrease in Tfrc in LS-HepcKO mice compared with age-matched controls, indicating elevated labile iron levels (Figure 2A). There was no difference in the Tfrc mRNA levels in the RPE between the LS-HepcKO mice and controls at the earliest, 15-day age. In addition, because iron accumulation is a normal age-related change,33 the control mice had diminished Tfrc with increasing age. There was no difference in RPE Tfrc levels in LS-HepcKO mice among the 3-, 6-, and 12-month time points, suggesting that either most of the RPE iron accumulation occurred by 3 months or Tfrc mRNA levels were already maximally diminished at the age of 3 months and, therefore, relative Tfrc mRNA levels were no longer useful as an indicator of further iron accumulation.
Figure 2.

LS-HepcKO mice have retinal iron accumulation in the retinal pigment epithelium (RPE). A: Graphs of relative transferrin receptor (Tfrc) mRNA levels, determined by real-time quantitative PCR, in RPE of LS-HepcKO mice versus controls (Hepc+/+;Alb-Cre+) at the indicated ages. Groups marked with a different symbol are significantly different from each other (P < 0.05), as determined by two-way analysis of variance with post hoc Tukey adjustment. B: Relative Dmt1 mRNA levels in RPE of LS-HepcKO mice versus controls (Hepc+/+;Alb-Cre+ or Hepcflox/+;Alb-Cre+). C–H: Perls histochemical iron staining of cryosections at 3, 6, and 12 months. Control mice do not show any labeling in the RPE at any of the time points (C–E). LS-HepcKO mice show progressively stronger labeling in the RPE with age (F–H, black arrowheads). Inductively coupled mass spectrometry of RPE from LS-HepcKO and control mice at 6 months (I and J). I: There is significantly more iron within the RPE of the LS-HepcKO mice when compared with controls. J: There is also an elevated iron/zinc ratio in the RPE of LS-HepcKO mice compared with controls. Data are expressed as means ± SEM (A, B, I, and J). n = 3 per genotype (C–H). ∗P < 0.05, ∗∗P < 0.01 versus control (I and J). Scale bars = 50 μm (C–H).
Dmt1 is another cellular iron transporter that is regulated in the same way as Tfrc through the iron regulatory protein/iron responsive element system.32 Dmt1, an iron/proton symporter, is localized to both the endosome and plasma membranes and is involved in the release of iron from acidified endosomes after holo-Tf (diferric transferrin) is internalized through receptor-mediated endocytosis. Dmt1 mRNA levels are normally decreased in response to high intracellular iron levels. In addition to the decrease in Tfrc mRNA levels, there was also a decrease in Dmt1 mRNA levels in the 3-, 6-, and 12-month LS-HepcKO RPE compared with age-matched controls (Figure 2B).
Perls Prussian blue staining was used to assess ferric iron stores in the LS-HepcKO and control mice at the 3-, 6-, and 12-month ages. Although the Perls histochemical stain was not sensitive enough to detect iron in the NSR, in the LS-HepcKO mice, it detected elevated iron levels in the RPE and choroid. There was no RPE Perls staining at any of the time points in the control mice (Figure 2, C–E). In contrast, there was Perls staining in the RPE of the LS-HepcKO mice at all three ages, which indicates that ferric iron levels are higher within the RPE of the LS-HepcKO mice (Figure 2, F–H). The intensity of the Perls staining in the RPE increased over time in the LS-HepcKO mice, with the 3-month time point having the lightest staining (Figure 2F) and the 12-month RPE having the darkest staining (Figure 2H), indicating a progressive RPE iron loading with age.
Another method to measure tissue iron levels directly is iCP-MS. To use this method, mice were perfused with saline to remove the potentially confounding influence of iron in the blood. RPE and NSR were isolated, and iCP-MS for iron and zinc was performed. There were significantly higher levels of iron within the RPE of the LS-HepcKO mice compared with controls (Figure 2I), and this increase was sustained when iron levels were normalized to zinc levels (Figure 2J). The decrease in RPE Tfrc and Dmt1 mRNA levels in the LS-HepcKO mice indicates that there is an increase in iron in the labile iron pool, whereas the increase in RPE iron, as measured by iCP-MS, indicates an increase in total intracellular iron levels.
LS-HepcKO Mice Have Elevated Labile Iron in the NSR after Long-Term Exposure to Elevated Serum Iron Levels
Similar methods were used to assess the iron levels within the NSR of the LS-HepcKO mice. There was a decrease in NSR Tfrc mRNA levels in the LS-HepcKO mice compared with controls at the 3-, 6-, and 12-month ages; and like the RPE, there was no change in Tfrc mRNA levels between the LS-HepcKO and control NSRs at the age of 15 days (Figure 3A). Tfr protein levels were also compared between the LS-HepcKO and control mice using Western blot analysis at 12 months. Tfr protein levels were decreased in the LS-HepcKO NSR compared with controls (Figure 3B). There was no decrease in Dmt1 mRNA levels in the 3-, 6-, and 12-month LS-HepcKO NSR compared with age-matched controls (Figure 3C), probably because Dmt1 is a less sensitive measure of elevated labile iron levels than Tfrc.
Figure 3.

LS-HepcKO mice have labile iron accumulation in the neurosensory retina (NSR). A: Graphs of relative transferrin receptor (Tfrc) mRNA levels, determined by real-time quantitative PCR (qPCR), in NSR of LS-HepcKO mice versus controls (Hepc+/+;Alb-Cre+) at the indicated ages. Groups marked with a different symbol are significantly different from each other (P < 0.05), as determined by two-way analysis of variance with post hoc Tukey adjustment. B: Western blot analysis showing Tfr protein levels in the NSR in the LS-HepcKO mice versus controls at 12 months and relative pixel density of Western blot analysis Tfr bands corrected for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). C: Relative Dmt1 mRNA levels in NSR of LS-HepcKO mice versus controls (Hepc+/+;Alb-Cre+ or Hepcflox/+;Alb-Cre+). D–O: Ferritin-L (Ft-L) immunolabeling of retinal sections. D, F, H, J, L, and N: At all three ages (3, 6, and 12 months), there is increased Ft-L immunolabeling in the NSR of LS-HepcKO mice (F, J, and N) compared with age-matched controls (D, H, and L). P: Pixel density of Ft-L immunolabeling in the NSR was quantified. Q: Relative levels of Hepc mRNA in LS-HepcKO NSR versus controls, determined by qPCR at 6 months. R and S: Inductively coupled mass spectrometry of NSR from LS-HepcKO and control mice at 6 months. R: There is no difference in iron (ppm) within the NSR of the LS-HepcKO mice when compared with controls. S: There is no change in the iron/zinc ratio in the NSR of LS-HepcKO mice compared with controls. Data are expressed as means ± SEM (A–C and P–S). ∗P < 0.05, ∗∗P < 0.01 (B and Q). Scale bars = 25 μm (D–O). GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer; RPE, retinal pigment epithelium.
To further assess changes in the labile iron pool within the NSR, immunolabeling of the iron storage protein, Ft-L, in the retina was assessed at the 3-, 6-, and 12-month ages. Ft-L, like Tfr and Dmt1, is regulated by the iron regulatory protein/iron responsive element system, but in the opposite direction; an increase in intracellular iron leads to increased Ft-L protein levels. Consistent with elevated NSR labile iron levels, there was increased Ft-L immunolabeling in the NSR at all three time points in the LS-HepcKO mice (Figure 3, F, J, and N) compared with controls (Figure 3, D, H, and L). In LS-HepcKO mice, the bipolar cells are prominently labeled at all ages, and the photoreceptor inner segment label is apparent at 12 months. The pixel density analysis of Ft-L immunolabeling shows elevated signal in the NSR at all three ages (Figure 3P). No primary antibody controls show minimal red fluorescence (Figure 3, E, G, I, K, M, and O).
Previously, our laboratory demonstrated that Hepc mRNA levels in the retina are up-regulated in response to increased retinal iron levels.34 The same response was observed in the LS-HepcKO mice. There were increased Hepc mRNA levels within the NSR of the LS-HepcKO mice compared with controls at the age of 6 months (Figure 3Q). Although these results are consistent with increased labile iron in the NSR, iCP-MS showed no difference in total iron levels within the NSR of the LS-HepcKO mice compared with controls (Figure 3R) and no change in the iron/zinc ratio in the NSR of the LS-HepcKO and controls (Figure 3S).
LS-HepcKO Mice Develop Retinal Iron Overload Despite an Intact Blood-Retinal Barrier
To determine whether the elevated iron levels in the retinas of LS-HepcKO mice may have resulted, in part, from a disrupted BRB, immunolabeling for the serum protein albumin was performed. In mice with an intact BRB, albumin should be present only within the blood vessels, and it should not be able to cross into the retina freely. It was found that the albumin labeling in both the control mice (Figure 4A) and the LS-HepcKO mice (Figure 4B) at 6 months was confined to the retinal vasculature, with no indication of leakage across the inner or outer BRB in either genotype. In addition, albumin protein levels within the NSR of the LS-HepcKO versus controls at the age of 3 months were assessed using Western blot analysis. There was no difference in albumin protein levels, indicating that the inner BRB was intact in the LS-HepcKO mice at an age when there was elevated labile iron in the NSR and RPE, but no retinal degeneration (Figure 4C).
Figure 4.
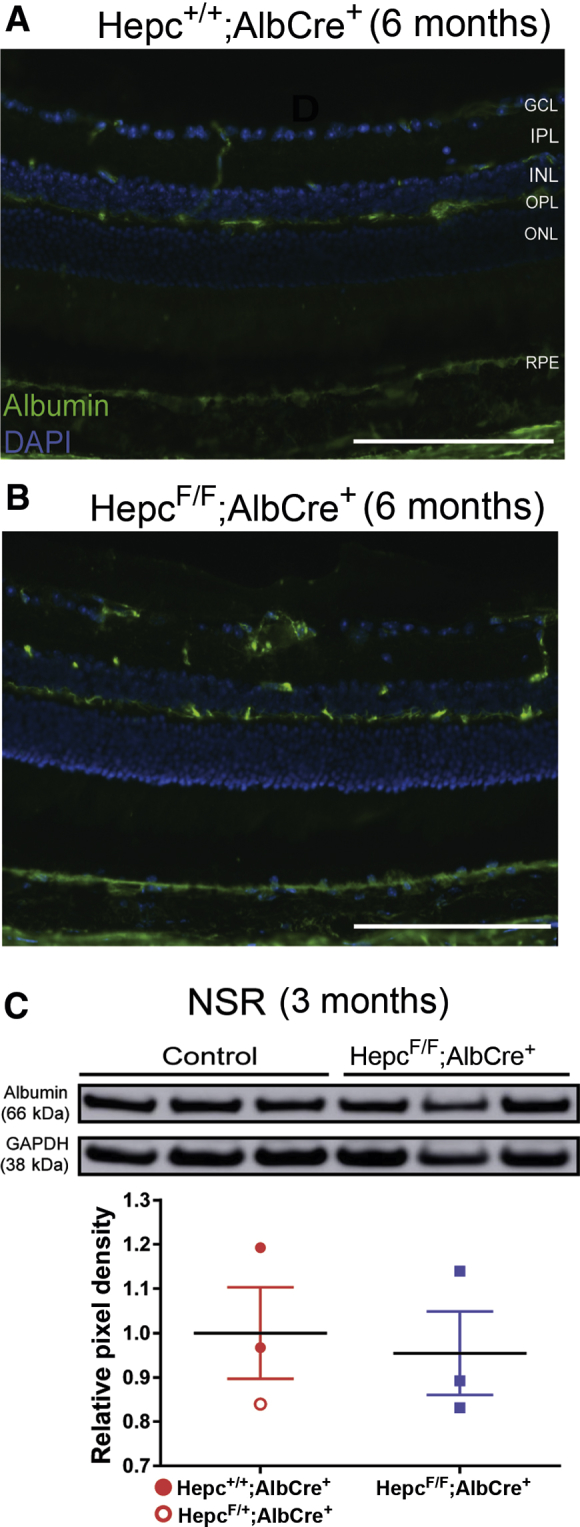
Retinal iron accumulation in the LS-HepcKO mice occurs despite an intact blood-retinal barrier (BRB). A and B: Albumin immunolabeling to test for BRB integrity in cryosections from the control (A) and LS-HepcKO (B) mice at 6 months. C: Western analysis showing albumin protein levels in the neurosensory retina (NSR) in the LS-HepcKO mice versus controls at 3 months and relative pixel density of Western analysis albumin bands corrected for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). n = 4 per genotype (A and B). Scale bars = 100 μm (A and B). GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer; RPE, retinal pigment epithelium.
LS-HepcKO Mice Had Evidence of RPE Degeneration Starting at 6 Months
It was next determined how exposure to high serum iron levels in LS-HepcKO mice, which resulted in RPE and NSR iron loading, affected retinal structure and function. In vivo fundus imaging was used to investigate changes in gross retinal morphology. The LS-HepcKO mice had focal regions of hypopigmentation and autofluorescence, especially around the optic nerve head, starting at 6 months (Figure 5B). In contrast, there was no evidence of hypopigmentation or autofluorescence in control mice (Figure 5A). Light microscopy of plastic sections showed focal areas of RPE hypertrophy in 6-month–old LS-HepcKO mice (Figure 5D), adjacent to areas where the RPE was relatively normal (Figure 5D), and no hypertrophy in controls (Figure 5C). There was no difference in the numbers of photoreceptor nuclei throughout the retina between the LS-HepcKO and controls at 6 months (Figure 5E). For the nuclei counts, two Hepc+/+;Alb-Cre+ mice and one Hepcflox/+;Alb-Cre+ mouse, which had similar numbers of photoreceptor nuclei, were used as controls. Both the Hepc+/+;Alb-Cre+ and Hepcflox/+;Alb-Cre+ controls had normal morphology on fundus imaging and plastic sections.
Figure 5.

Iron loading in LS-HepcKO mice leads to retinal pigment epithelium (RPE) hypertrophy and degeneration at 6 months. A and B: Representative in vivo color fundus images and green autofluorescence images. For each genotype, a representative micrograph is presented; all representative images are from male mice. C and D: Plastic sections of Hepc+/+;Alb-Cre+ control (C) and LS-HepcKO (D) retinas. D: There is RPE hypertrophy (white arrows) and adjacent regions where the RPE is normal (white arrowhead) in the LS-HepcKO retinas. For each genotype, a representative micrograph is presented; all representative images are from male mice. E: Graph of photoreceptor nuclei number per row comparing LS-HepcKO with age-matched controls. n = 8 Hepc+/+;Alb-Cre controls (A and B); n = 12 LS-HepcKO mice (A and B); n = 3 LS-HepcKO mice (C–E); n = 2 Hepc+/+;Alb-Cre+ controls (C–E); n = 1 Hepcflox/+;Alb-Cre+ control mice (C–E). Scale bars = 50 μm (C and D). GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer.
By 12 months, the LS-HepcKO mice had widespread hypopigmentation and autofluorescence on fundus imaging that covered the entirety of the retina (Figure 6B), whereas the age-matched controls still did not demonstrate any evidence of altered morphology (Figure 6A). To test whether the widespread autofluorescence observed on the fundus images in the 12-month–old LS-HepcKO mice was caused by RPE autofluorescence, cryosections of LS-HepcKO retinas were analyzed by epifluorescence microscopy. The RPE was autofluorescent on the DAPI (Figure 6C), Cy2 (Figure 6D), and Cy3 (Figure 6E) channels. There was corresponding widespread RPE degeneration evident in plastic sections of LS-HepcKO mice (Figure 6G). The RPE displayed the same histologic indicators of degeneration as observed at the age of 6 months, but in the older mice, most of the RPE was hypertrophic; the 12-month–old controls still had no evidence of RPE degeneration (Figure 6F). Other indicators of retinal degeneration were also observed in the 12-month–old LS-HepcKO mice, including focal thinning of the ONL (Figure 6H), RPE vacuolization (Figure 6H), and an undulating ONL (Figure 6I). To assess the percentage of hypertrophic RPE cells in the LS-HepcKO mice at 12 months, sagittal sections running from one edge of the retina to the other, and through the optic nerve head, were analyzed. Nearly all of the RPE cells in the surveyed 12-month LS-HepcKO retinas were hypertrophic, whereas there were no hypertrophic RPE cells throughout the retinas of the age-matched controls (Figure 6J).
Figure 6.

Iron loading in LS-HepcKO mice leads to retinal pigment epithelium (RPE) degeneration and retinal dysfunction at 12 months. A and B: Representative in vivo color fundus images and green autofluorescence images. For each genotype, a representative micrograph is presented; all representative images are from male mice. C–E: Autofluorescence imaging of cryosections on the DAPI (C), Cy2 (D), and Cy3 (E) channels in 12-month–old LS-HepcKO retinas. F and G: Plastic sections showing widespread RPE hypertrophy in LS-HepcKO mice (white arrows; G). H and I: LS-HepcKO mice also demonstrate other characteristics of retinal degeneration, including outer nuclear layer (ONL) thinning (yellow arrowheads, H), RPE vacuolization (white arrow), and undulating ONL (yellow arrowheads, I). J: Percentage hypertrophic RPE per retinal section. K: Graph of photoreceptor nuclei number comparing LS-HepcKO with age-matched controls. L–N: Electroretinography rod B (L), rod A (M), and cone B (N) amplitudes in 12-month–control and LS-HepcKO mice. n = 7 Hepc+/+;Alb-Cre controls (A and B); n = 9 LS-HepcKO mice (A and B); n = 3 LS-HepcKO mice (C–E); n = 1 Hepc+/+; Alb-Cre+ control (C–E); and n = 2 Hepcflox/+; Alb-Cre+ control mice (C–E). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗∗P < 0.0001. Scale bars = 50 μm (C–I). GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; OPL, outer plexiform layer.
To assess the degree of photoreceptor degeneration in the 12-month–old LS-HepcKO mice, the numbers of photoreceptor nuclei per column were counted in sagittal sections through the optic nerve head. Nuclei were counted at 200-μm increments to 2200 μm both superior and inferior to the optic nerve. There was a wide variation in the nuclei number across the retina in the LS-HepcKO mice, indicating that there were regions where the ONL was thinned. However, because of this large variability, there was no point where there was a significant difference in nuclei number between the LS-HepcKO mice and controls (Figure 6K). For the measurement of RPE hypertrophy and photoreceptor nuclei counts, one Hepc+/+;Alb-Cre+ mouse and two Hepcflox/+;Alb-Cre+ mice were used as controls. Both the Hepc+/+;Alb-Cre+ and Hepcflox/+;Alb-Cre+ controls had normal morphology on fundus imaging and plastic sections and similar photoreceptor nuclei counts.
The widespread RPE degeneration in the LS- HepcKO mice was associated with changes in retinal function, as measured by electroretinography. The 12-month–old LS-HepcKO mice had decreased rod b, rod a, and cone b amplitudes compared with the age-matched controls (Figure 6, L–N).
Sexual Dimorphism in LS-HepcKO Mice
LS-HepcKO mice had sex-related differences in the degree of RPE degeneration at the ages of 6 and 12 months. There was no difference in NSR or RPE Tfrc mRNA levels between the male and female LS-HepcKO mice at the ages of 3, 6, and 12 months (Figure 7, A and B). In addition, there was no difference in the intensity of Perls staining within the RPE between the 12-month–old male and female LS-HepcKO mice (Figure 7, C and D), indicating no difference in the amount of iron in the retinas of the male versus female LS-HepcKO mice. However, there was a difference between the sexes in the rate of RPE degeneration. At the age of 3 months, neither the male nor the female mice had evidence of hypopigmentation or autofluorescence on fundus imaging (Figure 7, E and F). However, at the age of 6 months, the male mice, but not the female mice, had focal regions of hypopigmentation and autofluorescence on fundus images (Figure 7, G and H). At the age of 12 months, the male LS-HepcKO mice had panretinal hypopigmentation and autofluorescence (Figure 7I), whereas there were only focal areas of hypopigmentation and autofluorescence in age-matched female mice (Figure 7J).
Figure 7.

Sexual dimorphism in retinal degeneration of 12-month–old LS-HepcKO mice. A: Graphs of relative transferrin receptor (Tfrc) mRNA levels, determined by real-time quantitative PCR (qPCR), in neurosensory retina (NSR) of LS-HepcKO male versus LS-HepcKO female mice at the indicated ages. B: Graphs of relative Tfrc mRNA levels, determined by qPCR, in retinal pigment epithelium (RPE) of LS-HepcKO male versus LS-HepcKO female mice at the indicated ages. C and D: VIP-enhanced Perls staining of plastic sections of 12-month–old male and female LS-HepcKO mice. E and F:In vivo color fundus images and green autofluorescence imaging of male and female LS-HepcKO mice at the age of 3 months. G and H: Six months. I and J: Twelve months. For each genotype, a representative micrograph is presented. Scale bars = 50 μm (C and D). GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer.
Abundant Vesicles within Hypertrophic RPE of LS-HepcKO Mice
Electron microscopy was used to further characterize the changes occurring within the iron-loaded, hypertrophic RPE of the 12-month–old LS-HepcKO mice. Compared with a 12-month–old C57BL/6J control (Figure 8A), the RPE within the LS-HepcKO mice was significantly taller (Figure 8C). A magnified image from the control (Figure 8B) is presented to demonstrate the typical composition of a healthy RPE cell. Within the RPE cell, there is a large nucleus; many melanosomes, which are localized to the apical region of the RPE; and mitochondria, which are localized to the basal region. In the LS-HepcKO RPE, there was a marked change in organelle composition. There were relatively few visible melanosomes or mitochondria. Instead, there were many vesicles, presumably endosomes/lysosomes, throughout the cytoplasm as well as phagosomes localized to the apical region (Figure 8, C and D). Both the LS-HepcKO mice and controls were euthanized in the morning, between 8 and 10 am, making it unlikely that the observed differences in RPE composition were due to documented changes associated with the circadian rhythm. There were also melanolysosomes within the RPE of the LS-HepcKO mice (Figure 8D). In addition, in several regions, there were disorganized photoreceptor inner and outer segments in the LS-HepcKO retina, with subretinal outer segment debris adjacent to the hypertrophic RPE (Figure 8E).
Figure 8.

Electron micrographs of RPE cells in the LS-HepcKO mice are hypertrophic and contain numerous lipofuscin-filled vesicles. A: Low-magnification representative image of a 12-month–old C57BL/6J control retinal pigment epithelium (RPE) demonstrates the relative size of a normal RPE cell. B: Higher-magnification image of a normal RPE cell demonstrates organelle composition in a non-hypertrophic RPE cell. C: Low-magnification image of a 12-month–old LS-HepcKO mouse demonstrates the size of the hypertrophic RPE cells. D: Higher-magnification image of 12-month–old LS-HepcKO RPE demonstrates phagosomes (Ps) in apical region, melanolysosomes (yellow arrowheads), and lysosomes (Ls). E: Disorganized outer segments adjacent to hypertrophic RPE cells. Scale bars: 10 μm (A, C, and E); 2 μm (B and D). BM, Bruch membrane; M, melanosome; OS, outer segment; V, vacuole.
Loss of Retina-Produced Hepc Does Not Affect Retinal Iron Levels or Retinal Health in Aged Mice
To determine whether retina-produced Hepc plays a significant role in maintaining retinal iron homeostasis during periods when the systemic iron levels are normal, an RS-HepcKO model was developed. To delete Hepc from the retina, the Hepcflox/flox strain was crossed with a strain expressing mRx-Cre, which leads to the expression of Cre-recombinase in most cells within the NSR and some RPE cells.22 The RS-HepcKO and controls were aged to 6 and 12 months. This model was validated by measuring the Hepc mRNA levels in both the retina and the liver of RS-HepcKO (Hepcflox/flox;mRx-Cre+) and control (Hepc+/+;mRx-Cre+) mice. As expected, there was a significant reduction in Hepc mRNA levels within the NSR of the RS-HepcKO mice compared with controls (Figure 9A). There was also a reduction in Hepc mRNA levels in the RPE of RS-HepcKO mice compared with controls (Figure 9B), although to a lesser extent than the reduction seen in the RS-HepcKO NSR. As expected, there was no difference in liver levels of Hepc mRNA between the RS-HepcKO mice and controls (Figure 9C). Consistent with this, the retina-specific HepcKO did not affect the serum iron levels, as both the serum iron concentration (Figure 9D) and serum Tf saturation (Figure 9E) were the same in both the control and RS-HepcKO mice.
Figure 9.

Analysis of RS-HepcKO model at 6 and 12 months in mice. A: Graph of Hepc mRNA levels in neurosensory retina (NSR), measured by real-time quantitative PCR (qPCR), in RS-HepcKO versus controls at 6 months. B: Hepc mRNA levels in retinal pigment epithelium (RPE) of RS-HepcKO versus controls at 6 months. C: Hepc mRNA levels in liver of RS-HepcKO versus controls at 6 months. D: Serum iron concentration of RS-HepcKO mice versus controls at 6 months. E: Serum transferrin saturation of RS-HepcKO mice versus controls at 6 months. F: Graph of Tfrc mRNA levels, measured by qPCR, in NSR of RS-HepcKO versus controls at 6 months. G: Graph of Tfrc mRNA levels, measured by qPCR, in RPE of RS-HepcKO versus controls at 6 months. H: Graph of Dmt1 mRNA levels, measured by qPCR, in NSR of RS-HepcKO versus controls at 6 months. I: Graph of Dmt1 mRNA levels, measured by qPCR, in RPE of RS-HepcKO versus controls at 6 months. J and K:In vivo fundus images and autofluorescence images of 6-month–old RS-HepcKO and control mice. L: VIP-enhanced Perls iron staining of plastic sections of 12-month–old RS-HepcKO retina. M and N:In vivo fundus images and autofluorescence images of 12-month–old RS-HepcKO and control mice. Statistical analysis was performed using two-group, two-sided t-test. ∗P < 0.05, ∗∗∗∗P < 0.0001. Scale bar = 50 μm (L). GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer.
To determine whether loss of retina-produced Hepc affects retinal iron homeostasis under normal serum iron conditions, retinal iron levels were assessed using several of the indirect and direct methods discussed above. There was no evidence of altered retinal iron levels at the age of 6 months in RS-HepcKO mice compared with age-matched controls. Specifically, there was no difference in Tfrc mRNA levels between the RS-HepcKO and controls in either the NSR (Figure 9F) or the RPE (Figure 9G). Similarly, there was no change in Dmt1 mRNA levels in either the NSR (Figure 9H) or the RPE (Figure 9I) between the RS-HepcKO mice and age-matched controls. Fundus images of the 6-month–old RS-HepcKO mice and age-matched controls did not demonstrate any retinal changes (Figure 9, J and K). In retina sections from 12-month–old mice, there was no Perls staining in the NSR or RPE in the RS-HepcKO mice (Figure 9L) and no changes in retinal morphology on fundus images from the 12-month–old RS-HepcKO mice compared with controls (Figure 9, M and N).
Discussion
In this study, liver- and retina-specific HepcKOs were used to determine whether Hepc produced in either of these locations is important for retinal iron regulation and health. The LS-HepcKO, but not the RS-HepcKO, had retinal iron accumulation and degeneration. The LS-HepcKO had high plasma iron levels due to unregulated iron absorption from the diet, indicating that high plasma iron levels lead to retinal iron overload and subsequent retinal degeneration despite the presence (and up-regulation) of retina-produced Hepc. The LS-HepcKO mice had elevated labile iron in NSR and RPE and elevated total iron in the RPE. The RPE iron loading was progressive, with the strongest Perls staining in the 12-month–old LS-HepcKO mice. These elevated iron levels caused retinal degeneration, most prominently affecting the RPE.
Our laboratory has previously demonstrated that systemic dysregulation of the Hepc/Fpn axis leads to retinal iron accumulation and degeneration.19, 20 However, these models did not demonstrate whether the retinal iron accumulation occurred because of long-term exposure to high plasma iron levels or, alternatively, loss of Fpn regulation by Hepc within the retina. Other studies have shown that Hepc, produced by the heart, has a significant role in regulating local cardiac iron levels even when systemic iron levels are unchanged,15 hinting that retina-produced Hepc may play a similar critical role in retinal iron homeostasis. However, the normal retinal iron levels and morphology in RS-HepcKO mice refute this possibility.
It is surprising that the BRB does not prevent retinal iron excess in the LS-HepcKO mice. The BRB consists of two components: the inner BRB, which is composed of tight junctions between the retinal vascular endothelial cells; and the outer BRB, which is formed by tight junctions between RPE cells. Transferrin-bound iron may enter the retina through the iron importer, TfR, which is localized to both the luminal membrane of the retinal vascular endothelial cells and the basal membrane of the RPE. The RPE Tfrc mRNA levels in LS-HepcKO mice are greatly reduced, even at the early 3-month time point, indicating that iron accumulation within the RPE is not likely by import of transferrin-bound iron from the choriocapillaris through the basal membrane. Instead, the RPE may become iron loaded because of daily phagocytosis of iron-laden photoreceptor outer segments.35 When retinal iron levels are normal, the RPE can properly digest the photoreceptor outer segments and the iron within the phagolysosome is exported into the RPE cytoplasm for use, storage, or export through the basal membrane into the choriocapillaris. However, in the LS-HepcKO mice, the RPE may have difficulty exporting the outer segment–derived iron from iron-damaged lysosomes. This may be exacerbated by the observed reduction in Dmt1, which exports iron from endosomes/lysosomes. Iron export from the RPE basolateral membrane may also be impaired because there is no unsaturated apotransferrin in the blood available to accept it.
This still leaves open the question of how iron crosses the BRB when Tfr levels are diminished. There was no difference in NSR or RPE Tfrc mRNA levels between the LS-HepcKO mice and controls at the 15-day time point. At this early time point, the BRB is just closing and, therefore, the lack of iron accumulation at this early time point indicates that the iron overload seen at subsequent time points occurs by transport of iron across the intact outer and/or inner BRB. It is possible that the non–transferrin-bound iron present in the serum of LS-HepcKO mice is imported into retinal vascular endothelial cells and/or RPE by divalent metal transporters Zip8, Zip14, and/or Dmt1. These proteins are expressed in vascular endothelial cells and RPE, and can be up-regulated by iron overload,25 potentially leading to continued iron import from plasma into these cells. Support for this concept comes from the finding that Zip14 is necessary for liver iron overload in several mouse models where plasma iron levels are high.36 Another possibility is that iron damages the cells constituting the BRB, leading to leakage. This possibility was tested by immunolabeling and performing Western blot analysis on NSRs from LS-HepcKO retinas for the serum protein albumin, and evidence of albumin leakage into the retina was not detected.
In addition to determining the role of retina-produced Hepc, another objective of this study was to help define how iron overload affects retinal health to better understand the pathophysiology of retinal degeneration that involves iron dysregulation. The microscopic appearance of the iron-laden RPE cells in the 12-month–old LS-HepcKO mice provides insight into the pathways that ultimately lead to RPE degeneration under chronic iron overload conditions. The RPE of the 12-month–old LS-HepcKO mice had many of the histologic features observed in aged retinas. These include the formation of lipofuscin granules and complex granules, including melanolysosomes and melanolipofusin, as well as a decrease in the amount of cytosolic free space.37, 38 The RPE lipofuscin was observed as autofluorescence on both fundus imaging and in cryosections. On electron microscopy, there was a dramatic shift in the composition of the LS-HepcKO RPE cells compared with age-matched controls, with a large increase in the number of vesicles, presumably consisting of endosomes/lysosomes throughout the RPE cells and an increase in the number of phagosomes containing undigested photoreceptor outer segments in the apical RPE in some sections. It was also observed that the photoreceptor outer segments were highly disorganized, with accumulation of membranous material in the subretinal space adjacent to some of the hypertrophic RPE cells.
A prior cell culture study suggests potential mechanisms for the morphologic changes in the RPE. Chronic iron exposure in the medium caused increased lysosome biogenesis, but impaired lysosome function, as measured by failure to acidify and diminished production of the active cathepsin D protein needed for proper lysosome function.39 In LS-HepcKO mice, RPE lysosomes may display the same dysfunction after long-term exposure to the iron-laden contents of the phagosomes, with an increase in lysosome number, but a decrease in the ability of the lysosome to properly digest phagosomes. The RPE continues to phagocytize the photoreceptor outer segments, but the failing lysosomes are unable to accept more material, leading to the increase in the number of phagosomes observed in the LS-HepcKO mice. Ultimately, as the mouse ages, the RPE fails to phagocytize the photoreceptor outer segments entirely, leading to the observed buildup of photoreceptor-derived membranous debris in the subretinal space. An increase in the number of complex granules within the RPE of the LS-HepcKO mice, including the formation of melanolysosomes, was also observed. Previous studies from our laboratory suggest that iron accumulation within the RPE may increase melanogenesis and may also lead to melanosome degradation.40, 41 The formation of the melanolysosomes in our model suggests that melanosomes, which have antioxidant activity, may be attempting to protect the RPE by fusing with the dysfunctional lysosomes.42
The histologic features observed in the LS-HepcKO mice provide insight into the mechanisms of iron-induced RPE degeneration and may represent some features of age-related macular degeneration. The hypertrophic, autofluorescent RPE cells in the LS-HepcKO mice resemble RPE cells located on the edges of regions of geographic atrophy.43, 44 In the 12-month–old LS-HepcKO mice, RPE hypertrophy, RPE vacuolization, and photoreceptor inner and outer segment disorganization were observed. These results, together with the prior findings of increased RPE iron levels in age-related macular degeneration eyes,4 suggest that the RPE hypertrophy in geographic atrophy may be due, in part, to RPE iron accumulation.
It was also observed that the LS-HepcKO mice displayed sexual dimorphism in response to retinal iron loading. Male mice had more severe retinal degeneration, as evidenced by fundus imaging (Figure 5). However, there was no evidence that there was any difference in RPE iron levels between the male and female LS-HepcKO mice. These data indicate that sex did not affect the ability of iron to get into the RPE but affected the response of the retina to prolonged iron loading. Estrogen has previously been demonstrated to have an inverse association with progression of age-related macular degeneration.45, 46 In addition, in the rodent light damage model, estrogen is retina protective.45 The difference in phenotype between the male and female LS-HepcKO mice could also be due to retinal protection by estrogen.
Dysregulation of retinal iron homeostasis has been implicated in various retinal diseases,47, 48, 49, 50, 51 although the causes of retinal iron overload are not yet clear. Therefore, it is necessary to probe the basic mechanisms that govern how the retina processes iron under normal and high plasma iron levels. In this study, it was demonstrated that the retina is susceptible to iron accumulation when plasma iron levels are high, despite the presence of an intact BRB. In addition, it was demonstrated that iron accumulation within the RPE can promote degeneration and retinal dysfunction. It was also shown that although the retina produces its own Hepc, retinal Hepc up-regulation in response to iron overload does not prevent retinal iron accumulation. Together, these data indicate that high plasma iron levels that exceed serum transferrin binding capacity may represent a danger to the retina.
Footnotes
Supported by NIH/National Eye InstituteEY015240 (J.L.D.); the UPenn Vision Science Training grant 5T32EY007035-37 (B.H.B.); Research to Prevent Blindness (J.L.D.); the F.M. Kirby Foundation (J.L.D.); a gift in memory of Lee F. Mauger, M.D. (J.L.D.); the Paul and Evanina Bell Mackall Foundation Trust (J.L.D.); and Institute of Molecular Genetics of the Academy of Sciences of the Czech Republic grant MEYSLO1419 (Z.K.).
Disclosures: None declared.
References
- 1.Beard J.L. Iron biology in immune function, muscle metabolism and neuronal functioning. J Nutr. 2001;131:568S–579S. doi: 10.1093/jn/131.2.568S. discussion 580S. [DOI] [PubMed] [Google Scholar]
- 2.Aisen P., Enns C., Wessling-Resnick M. Chemistry and biology of eukaryotic iron metabolism. Int J Biochem Cell Biol. 2001;33:940–959. doi: 10.1016/s1357-2725(01)00063-2. [DOI] [PubMed] [Google Scholar]
- 3.Rogers B.S., Symons R.C.A., Komeima K., Shen J., Xiao W., Swaim M.E., Yuan Y.G., Kachi S., Campochiaro P.A. Differential sensitivity of cones to iron-mediated oxidative damage. Invest Ophthalmol Vis Sci. 2007;48:438–445. doi: 10.1167/iovs.06-0528. [DOI] [PubMed] [Google Scholar]
- 4.Hahn P., Milam A.H., Dunaief J.L. Maculas affected by age-related macular degeneration contain increased chelatable iron in the retinal pigment epithelium and Bruch's membrane. Arch Ophthalmol. 2003;121:1099–1105. doi: 10.1001/archopht.121.8.1099. [DOI] [PubMed] [Google Scholar]
- 5.Rouault T.A. Iron metabolism in the CNS: implications for neurodegenerative diseases. Nat Rev Neurosci. 2013;14:551–564. doi: 10.1038/nrn3453. [DOI] [PubMed] [Google Scholar]
- 6.Gröger A., Berg D. Does structural neuroimaging reveal a disturbance of iron metabolism in Parkinson's disease? implications from MRI and TCS studies. J Neural Transm. 2012;119:1523–1528. doi: 10.1007/s00702-012-0873-0. [DOI] [PubMed] [Google Scholar]
- 7.Smith M.A., Zhu X., Tabaton M., Liu G., McKeel D.W., Cohen M.L., Wang X., Siedlak S.L., Dwyer B.E., Hayashi T., Nakamura M., Nunomura A., Perry G. Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J Alzheimers Dis. 2010;19:353–372. doi: 10.3233/JAD-2010-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pietrangelo A. Hereditary hemochromatosis: a new look at an old disease. N Engl J Med. 2004;350:2383–2397. doi: 10.1056/NEJMra031573. [DOI] [PubMed] [Google Scholar]
- 9.Roth A.M., Foos R.Y. Ocular pathologic changes in primary hemochromatosis. Arch Ophthalmol. 1972;87:507–514. doi: 10.1001/archopht.1972.01000020509003. [DOI] [PubMed] [Google Scholar]
- 10.Smith S.B., Liu K., Thangaraju M., Ha Y., Ganapathy V., Martin P.M., Gnana-Prakasam J.P. Absence of iron-regulatory protein Hfe results in hyperproliferation of retinal pigment epithelium: role of cystine/glutamate exchanger. Biochem J. 2009;424:243–252. doi: 10.1042/BJ20090424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song D., Kanu L.N., Li Y., Kelly K.L., Bhuyan R.K., Aleman T., Morgan J.I.W., Dunaief J.L. AMD-like retinopathy associated with intravenous iron. Exp Eye Res. 2016;151:122–133. doi: 10.1016/j.exer.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nemeth E., Tuttle M.S., Powelson J., Vaughn M.D., Donovan A., Ward D.M.V., Ganz T., Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 13.Ganz T. Hepcidin in iron metabolism. Curr Opin Hematol. 2004;11:251–254. doi: 10.1097/00062752-200407000-00004. [DOI] [PubMed] [Google Scholar]
- 14.McCarthy R.C., Kosman D.J. Ferroportin and exocytoplasmic ferroxidase activity are required for brain microvascular endothelial cell iron efflux. J Biol Chem. 2013;288:17932–17940. doi: 10.1074/jbc.M113.455428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lakhal-Littleton S., Wolna M., Chung Y.J., Christian H.C., Heather L.C., Brescia M., Ball V., Diaz R., Santos A., Biggs D., Clarke K., Davies B., Robbins P.A. An essential cell-autonomous role for hepcidin in cardiac iron homeostasis. Elife. 2016;5:e19804. doi: 10.7554/eLife.19804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cetin Y., Gehrke S.G., Kulaksiz H., Bachmann S., Stremmel W., Theilig F., Rost D., Janetzko A. The iron-regulatory peptide hormone hepcidin: expression and cellular localization in the mammalian kidney. J Endocrinol. 2005;184:361–370. doi: 10.1677/joe.1.05729. [DOI] [PubMed] [Google Scholar]
- 17.Kulaksiz H., Fein E., Redecker P., Stremmel W., Adler G., Cetin Y. Pancreatic β-cells express hepcidin, an iron-uptake regulatory peptide. J Endocrinol. 2008;197:241–249. doi: 10.1677/JOE-07-0528. [DOI] [PubMed] [Google Scholar]
- 18.Gnana-Prakasam J.P., Martin P.M., Smith S.B., Ganapathy V. Expression and function of iron-regulatory proteins in retina. IUBMB Life. 2010;62:363–370. doi: 10.1002/iub.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hadziahmetovic M., Song Y., Ponnuru P., Iacovelli J., Hunter A., Haddad N., Beard J., Connor J.R., Vaulont S., Dunaief J.L. Age-dependent retinal iron accumulation and degeneration in hepcidin knockout mice. Invest Ophthalmol Vis Sci. 2011;52:109–118. doi: 10.1167/iovs.10-6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Theurl M., Song D., Clark E., Sterling J., Grieco S., Altamura S., Galy B., Hentze M., Muckenthaler M.U., Dunaief J.L. Mice with hepcidin-resistant ferroportin accumulate iron in the retina. FASEB J. 2016;30:813–823. doi: 10.1096/fj.15-276758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Postic C., Shiota M., Niswender K.D., Jetton T.L., Chen Y., Moates J.M., Shelton K.D., Lindner J., Cherrington A.D., Magnuson M.A. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic β cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 1999;274:305–315. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- 22.Klimova L., Lachova J., Machon O., Sedlacek R., Kozmik Z. Generation of mRx-Cre transgenic mouse line for efficient conditional gene deletion in early retinal progenitors. PLoS One. 2013;8:e63029. doi: 10.1371/journal.pone.0063029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolkow N., Song D., Song Y., Chu S., Hadziahmetovic M., Lee J.C., Iacovelli J., Grieco S., Dunaief J.L. Ferroxidase hephaestin's cell-autonomous role in the retinal pigment epithelium. Am J Pathol. 2012;180:1614–1624. doi: 10.1016/j.ajpath.2011.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J.-Y., White D.J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sterling J., Guttha S., Song Y., Song D., Hadziahmetovic M., Dunaief J.L. Iron importers Zip8 and Zip14 are expressed in retina and regulated by retinal iron levels. Exp Eye Res. 2017;155:15–23. doi: 10.1016/j.exer.2016.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dunaief J.L., Zhong Y., Song Y., Hadziahmetovic M., Song D. Systemic administration of the iron chelator deferiprone protects against light-induced photoreceptor degeneration in the mouse retina. Free Radic Biol Med. 2012;53:64–71. doi: 10.1016/j.freeradbiomed.2012.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hadziahmetovic M., Dentchev T., Song Y., Haddad N., He X., Hahn P., Pratico D., Wen R., Harris Z.L., Lambris J.D., Beard J., Dunaief J.L. Ceruloplasmin/hephaestin knockout mice model morphologic and molecular features of AMD. Invest Ophthalmol Vis Sci. 2008;49:2728–2736. doi: 10.1167/iovs.07-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lyubarsky A.L., Falsini B., Pennesi M.E., Valentini P., Pugh E.N. UV- and midwave-sensitive cone-driven retinal responses of the mouse: a possible phenotype for coexpression of cone photopigments. J Neurosci. 1999;19:442–455. doi: 10.1523/JNEUROSCI.19-01-00442.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lyubarsky A.L., Lem J., Chen J., Falsini B., Iannaccone A., Pugh E.N. Functionally rodless mice: transgenic models for the investigation of cone function in retinal disease and therapy. Vision Res. 2002;42:401–415. doi: 10.1016/s0042-6989(01)00214-0. [DOI] [PubMed] [Google Scholar]
- 30.Zumerle S., Mathieu J.R.R., Delga S., Heinis M., Viatte L., Vaulont S., Peyssonnaux C. Targeted disruption of hepcidin in the liver recapitulates the hemochromatotic phenotype. Blood. 2014;123:3646–3650. doi: 10.1182/blood-2014-01-550467. [DOI] [PubMed] [Google Scholar]
- 31.Harford J.B., Klausner R.D., McBride O.W., Samaniego F., Burgess W.H., Tang C.K., Kaptain S., Rouault T.A., Haile D.J. Cloning of the cDNA encoding an RNA regulatory protein--the human iron-responsive element-binding protein. Proc Natl Acad Sci U S A. 2006;87:7958–7962. doi: 10.1073/pnas.87.20.7958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rouault T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat Chem Biol. 2006;2:406–414. doi: 10.1038/nchembio807. [DOI] [PubMed] [Google Scholar]
- 33.Hahn P., Song Y., Ying G.S., He X., Beard J., Dunaief J.L. Age-dependent and gender-specific changes in mouse tissue iron by strain. Exp Gerontol. 2009;44:594–600. doi: 10.1016/j.exger.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao L., Li Y., Song D., Song Y., Theurl M., Wang C., Cwanger A., Su G., Dunaief J.L. A high serum iron level causes mouse retinal iron accumulation despite an intact blood-retinal barrier. Am J Pathol. 2014;184:2862–2867. doi: 10.1016/j.ajpath.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song D., Dunaief J.L. Retinal iron homeostasis in health and disease. Front Aging Neurosci. 2013;5:24. doi: 10.3389/fnagi.2013.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coffey R., Knutson M.D., Chan A., Jenkitkasemwong S., Zhang W., Wang C.-Y., Fukada T., Kim J.-S., Biel T., Hojyo S. SLC39A14 is required for the development of hepatocellular iron overload in murine models of hereditary hemochromatosis. Cell Metab. 2015;22:138–150. doi: 10.1016/j.cmet.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feeney-Burns L. Lipofuscin of human retinal pigment epithelium. Am J Ophthalmol. 1980;90:783–787. doi: 10.1016/s0002-9394(14)75193-1. [DOI] [PubMed] [Google Scholar]
- 38.Feeney-Burns L., Hilderbrand E.S., Eldridge S. Aging human RPE: morphometric analysis of macular, equatorial, and peripheral cells. Invest Ophthalmol Vis Sci. 1984;25:195–200. [PubMed] [Google Scholar]
- 39.Ke Y.-Y., Wu C.-Y., Wang J.-Y., Yang R.-C., Huang S.-W., Chang S.-H., Kao J.-K., Wang S.-C., Shieh J.-J., Ho L.-W. Chronic iron overload results in impaired bacterial killing of THP-1 derived macrophage through the inhibition of lysosomal acidification. PLoS One. 2016;11:e0156713. doi: 10.1371/journal.pone.0156713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wolkow N., Song Y., Wu T.-D., Qian J., Guerquin-Kern J.-L., Dunaief J.L. Aceruloplasminemia: retinal histopathologic manifestations and iron-mediated melanosome degradation. Arch Ophthalmol. 2011;129:1466–1474. doi: 10.1001/archophthalmol.2011.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song Y., Iacovelli J., Li Y., Lee J.C., Song D., Wolkow N., Maminishkis A., Alekseev O., Dunaief J.L. Iron upregulates melanogenesis in cultured retinal pigment epithelial cells. Exp Eye Res. 2014;128:92–101. doi: 10.1016/j.exer.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rózanowski B., Burke J.M., Boulton M.E., Sarna T., Rózanowska M. Human RPE melanosomes protect from photosensitized and iron-mediated oxidation but become pro-oxidant in the presence of iron upon photodegradation. Invest Ophthalmol Vis Sci. 2008;49:2838–2847. doi: 10.1167/iovs.08-1700. [DOI] [PubMed] [Google Scholar]
- 43.Rudolf M., Vogt S.D., Curcio C.A., Huisingh C., McGwin G., Wagner A., Grisanti S., Read R.W. Histologic basis of variations in retinal pigment epithelium autofluorescence in eyes with geographic atrophy. Ophthalmology. 2013;120:821–828. doi: 10.1016/j.ophtha.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ach T., Freund K.B., Zanzottera E.C., Curcio C.A., Huisingh C., Messinger J.D. Visualizing retinal pigment epithelium phenotypes in the transition to atrophy in neovascular age-related macular degeneration. Retina. 2016;36 Suppl 1:S26–S39. doi: 10.1097/IAE.0000000000001330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Snow K.K., Cote J., Yang W., Davis N.J., Seddon J.M. Association between reproductive and hormonal factors and age-related maculopathy in postmenopausal women. Am J Ophthalmol. 2002;134:842–848. doi: 10.1016/s0002-9394(02)01755-5. [DOI] [PubMed] [Google Scholar]
- 46.Edwards D.R.V., Gallins P., Polk M., Ayala-Haedo J., Schwartz S.G., Kovach J.L., Spencer K., Wang G., Agarwal A., Postel E.A., Haines J.L., Pericak-Vance M., Scott W.K. Inverse association of female hormone replacement therapy with age-related macular degeneration and interactions with ARMS2 polymorphisms. Invest Ophthalmol Vis Sci. 2010;51:1873–1879. doi: 10.1167/iovs.09-4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim H.J., Zhao J., Sparrow J.R., Ueda K., Dunaief J.L., Song Y. Iron promotes oxidative cell death caused by bisretinoids of retina. Proc Natl Acad Sci U S A. 2018;115:4963–4968. doi: 10.1073/pnas.1722601115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith S.B., Arjunan P., Tawfik A., Promsote W., Martin P., Gnana-Prakasam J.P., Thangaraju M., Kisselev O., Ganapathy V., Chaudhary K., Veeranan-Karmegam R., Ananth S. Iron overload accelerates the progression of diabetic retinopathy in association with increased retinal renin expression. Sci Rep. 2018;8:3025. doi: 10.1038/s41598-018-21276-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shindler K.S., Steenstra C., Chen Y., Khan R.S., Song Y., Cheah J.H., Cwanger A., Snyder S.H., Kim S.F., Bang S., Dunaief J. Dexras1, a small GTPase, is required for glutamate-NMDA neurotoxicity. J Neurosci. 2013;33:3582–3587. doi: 10.1523/JNEUROSCI.1497-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yefimova M.G., Jeanny J.C., Keller N., Sergeant C., Guillonneau X., Beaumont C., Courtois Y. Impaired retinal iron homeostasis associated with defective phagocytosis in Royal College of Surgeons rats. Invest Ophthalmol Vis Sci. 2002;43:537–545. [PubMed] [Google Scholar]
- 51.Deleon E., Lederman M., Berenstein E., Meir T., Chevion M., Chowers I. Alteration in iron metabolism during retinal degeneration in rd10 mouse. Invest Ophthalmol Vis Sci. 2009;50:1360–1365. doi: 10.1167/iovs.08-1856. [DOI] [PubMed] [Google Scholar]