

Abstract
Type II topoisomerases regulate DNA topology by making a double-stranded break in one DNA duplex, transporting another DNA segment through this break and then resealing it. Bacterial type IIA topoisomerase inhibitors, such as fluoroquinolones and novel bacterial topoisomerase inhibitors, can trap DNA cleavage complexes with double- or single-stranded cleaved DNA. To study the mode of action of such compounds, 21 crystal structures of a “gyraseCORE” fusion truncate of Staphyloccocus aureus DNA gyrase complexed with DNA and diverse inhibitors have been published, as well as 4 structures lacking inhibitors. These structures have the DNA in various cleavage states and appear to track trajectories along the catalytic paths of the DNA cleavage/religation steps. The various conformations sampled by these multiple “gyraseCORE” structures show rigid body movements of the catalytic GyrA WHD and GyrB TOPRIM domains across the dimer interface. Conformational changes common to all compound-bound structures suggest common mechanisms for DNA cleavage-stabilizing compounds. The structures suggest that S. aureus gyrase uses a single moving-metal ion for cleavage and that the central four base pairs need to be stretched between the two catalytic sites, in order for a scissile phosphate to attract a metal ion to the A-site to catalyze cleavage, after which it is “stored” in another coordination configuration (B-site) in the vicinity. We present a simplified model for the catalytic cycle in which capture of the transported DNA segment causes conformational changes in the ATPase domain that push the DNA gate open, resulting in stretching and cleaving the gate-DNA in two steps.
Keywords: DNA gyrase, antibiotics, quinolones, NBTIs, DNA cleavage
Abbreviations: NBTIs, novel bacterial topoisomerase inhibitors; T-DNA, transported DNA segment; G-DNA, gate-DNA; WHD, winged-helix domain; GK, Greek key; LHS, left-hand side; RHS, right-hand side
Graphical Abstract
Highlights
-
•
Type II DNA topoisomerases, such as DNA gyrase, control the topological state of DNA in all cells.
-
•
As these enzymes bind, cleave and re-ligate DNA, multiple binding pockets for small compounds appear.
-
•
We discuss how crystal structures of gyrase, DNA and different compounds may be trapping different stages in the catalytic cycle of the enzyme.
-
•
We propose a model for DNA strand cleavage involving a moving metal ion.
Mechanism and Structure of Type IIA DNA Topoisomerases
Topoisomerases are essential and ubiquitous enzymes that regulate DNA topology by making temporary single- (type I) or double-strand DNA breaks (type II) [1], [2]. The DNA double helix needs to be unwound for processes such as DNA replication or transcription to take place, giving rise to the accumulation of positive supercoils ahead of transcription bubbles and replication forks, and negative supercoils or pre-catenanes behind. Topoisomerases relax supercoiled DNA and decatenate DNA to allow DNA transcription and replication to take place and are essential for genome stability [3], [4], [5]. While the creation of temporary double-strand breaks is necessary for the function of type II topoisomerases, it is potentially hazardous for the cell. Compounds that stabilize these normally transient breaks are effective therapeutics used both in cancer chemotherapy (e.g., etoposide) [6] and as anti-bacterials (e.g., fluoroquinolones) [7], [8]. Fluoroquinolones continue to represent a significant class of weapons in the physician's armory against pathogenic bacterial infections, with delafloxacin approved for use by the Food and Drug Administration in 2017 [9]. Crystal structures of many drug complexes show compounds bound in the DNA at the cleavage site physically blocking religation, in an apparently simple steric manner [10], [11], [12], [13], [14]. However, compounds such as the novel bacterial topoisomerase inhibitors (NBTIs; GSK299423) [15] and the thiophenes [16] bind away from the cleavage sites and have allosteric mechanisms for the stabilization of cleavage complexes. This points to the idea that different compounds can stabilize different conformations that the enzyme uses during the DNA cleavage and religation steps of its catalytic cycle. Two novel compounds in late-stage clinical development [17], [18], the NBTI gepotidacin and zoliflodacin (which like fluoroquinolones binds in the cleaved DNA), are active against common fluoroquinolone-resistant bacterial strains and, presumably, fluoroquinolone-resistant forms of DNA gyrase [17], [19].
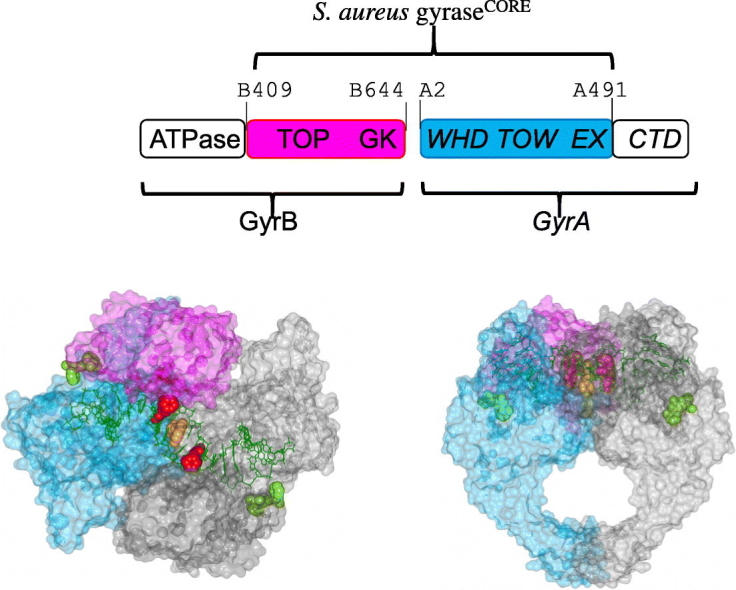
Type II DNA topoisomerases are divided into sub-types, IIA and IIB [1], [5], based on structural and evolutionary considerations. Type IIA is found in bacteria and eukaryotes, whereas IIB was discovered in archaea and more recently in plants and plasmodial parasites. Most bacteria have two type IIA topoisomerases, DNA gyrase and topoisomerase IV. DNA gyrase consists of two copies of GyrA and two copies of GyrB and functions as an A2B2 heterotetramer (Fig. 1). Topoisomerase IV has two homologous subunits, ParC and ParE, and also functions as a heterotetramer. DNA gyrase can uniquely introduce negative supercoils into DNA, while topoisomerase IV performs strand passage with two different double-stranded DNA segments and has both decatenation and relaxation activity. Eukaryotic type IIA topoisomerases are encoded as a single protein, with regions equivalent to GyrB and GyrA at the N- and C-terminus of a single subunit (Fig. 1). Residues at the DNA cleavage catalytic center are conserved between the eukaryotic and prokaryotic type IIA topoisomerases.
Fig. 1.

Schematic of type IIA topoisomerases. (A) Simplified schematic of the action of type IIA topoisomerases, such as yeast topoisomerase II (topo II), DNA gyrase and topoisomerase IV. A segment of DNA (T-DNA) is transported by capture through the ATPase domains interface and passes through a transient double-strand break established in the G-DNA (forming a DNA “gate” interface with the WHD domain). The DNA then exits the enzyme through the exit gate (EX). (B) Eukaryotic topo IIs are homodimers, and many key structures have been determined with a “core’ region (419–1180) encompassing the TOPRIM domain (TOP), the Greek key fold, the winged-helix domain (WHD), the Tower (TOW) and the exit gate (EX). (C) Fusing the C-terminal region of S. aureus GyrB (residue 409 to 644) to the N-terminal region of GyrA (residue 2 to 491) gives an “equivalent” construct. The Greek key can be deleted from this core construct in S. aureus gyrase.
Type IIA topoisomerases are able to transport a segment of DNA across a series of interfaces that form about the C2 symmetry axis (also called the dyad axis) (Fig. 1A). The first interface is formed by the two ATPase domains. ATP binding results in dimerization and the trapping of the transported DNA segment (T-DNA). The second interface is the so-called DNA gate, where the gate-DNA (G-DNA) is transiently cleaved, and residues involved in protein–protein interactions at this second interface come primarily from the winged-helix domain (WHD) as well as from the TOPRIM and Tower domains. Once the T-DNA has passed through the G-DNA (Fig. 1), it can then exit the enzyme through the “exit gate” (EX). The C-terminal domain is less conserved and is involved in substrate preference in topo IV [20] and positive DNA loop wrapping by DNA gyrase. DNA cleavage is achieved using a tyrosine residue from the WHD (Tyr123 from the GyrA subunit in Staphyloccocus aureus DNA gyrase), which forms a phosphotyrosine bond with the cleaved DNA. A catalytic metal ion (usually Mg2+) is required for cleavage and religation. Residues from the TOPRIM domain are involved in coordinating the catalytic metal. For DNA cleavage and religation to take place, the catalytic tyrosine and the scissile phosphate need to be correctly positioned with respect to the metal-binding TOPRIM domain. Type IIA topoisomerases have two active cleavage catalytic pockets making a 4-base-pair staggered break in the DNA. However, the catalytic metal ion has only been observed when phosphates from the DNA are close enough to coordinate the metal directly or indirectly via a water. The consensus is that a metal ion must move to [15], [21] or be present [22] at a position contacting the scissile phosphate for DNA cleavage to take place.
This article focuses on 25 crystal structures of S. aureus DNA gyrase, all except 2 are complexes with DNA, that have been deposited with the PDB (Table 1, Table 2). Several of these crystal structures have static disorder around the twofold axis of the complex. Such static disorder occurs when two (or more) stable configurations are observed in the crystal, which results in a density average, and are modeled by lowering the occupancy of the alternative conformation. These occupancy values reflect the frequency of the respective configurations in the crystal (see Ref. [23] pp. 373–374 and Supplementary Methods). Taking this into account, derived single biologically relevant complexes are made available online (“Research” tab at https://www.cardiff.ac.uk/people/view/1141625-bax-ben and see Table S1). To facilitate comparison of these structures, we adopt a standard BA-x numbering (for GyrB-GyrA-extended; see Supplementary Methods for details). In this S. aureus DNA gyrase BA-x numbering system, the catalytic metal is always called B5081 (or D5081), and inhibitors have CHAIN ID I and are numbered according to which pocket(s) they sit in (see below). For example, in the 1.98-Å crystal structure of GSK945237 with a 20-bp duplex, both the DNA and the compound have static disorder around the non-crystallographic axis of the complex; coordinates available are 5iwi-BA-x.pdb (crystallographic, with standard nomenclature) and 5iwi-c1a.pdb and 5iwi-c1b.pdb (“biological” single complexes derived from the crystallographic coordinates in which a single DNA and compound binding mode are present) (Table S1). We also make available coordinates for a re-refined yeast structure which sits on a crystallographic twofold axis and is complicated by static disorder, RR-3L4K, together with the originally deposited 3L4K structure (Supplementary methods, Table S4s and S3 for the refinement statistic). We also provide the DNA sequence and cleavage status for published gyraseCORE model in Table S2.
Table 1.
Four crystal structures of the S. aureus DNA gyrase GyrBA fusion truncate without inhibitors (apo and binary complexes with DNA)
| Active site 1 |
Active site 2 |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | PDB code + res. | Inhibitor | Inhibitor pockets occupied |
Metal B5081 site occupancies |
WHD Tyr C123 | DNA | DNA cleavage status | Metal D5081 site occupancies |
WHD Tyr A123 | |||||||
| 1 | 1′ | 2D | 2A | 3 | 3′ | A | B | A | B | |||||||
| 1 | 2xcqa 2.98 | None | – | – | – | – | – | – | – | Tyr | No DNA | N/A | – | – | Tyr | |
| 2 | 2xcoa 3.1 | None | – | – | – | – | – | – | – | Tyr | No DNA | N/A | – | – | Tyr | |
| 3 | 6fqv 2.6 | None | – | – | – | – | – | – | – | Tyr | 20-447 T 20-447 T |
Not cleaved |
– | – | Tyr | |
| 4 | 5cdr 2.65 | None | – | – | – | – | – | – | 1.0 | Phe | 20-12p-8 20-12p-8 |
Doubly nicked |
– | 1.0 | Phe | |
This TOPRIM-bound metal is named B5081 (or D5081) to indicate that it is coordinated by Asp 508 from GyrB, in both the A and B positions (see Fig. 6 for details). Active sites are formed across the dimer interface with the catalytic tyrosine from the DC subunit (C123) approaching the B TOPRIM domain, and the tyrosine from the BA subunit (A123) approaching the D subunit TOPRIM domain.
Greek key present.
Table 2.
Twenty-one crystal structures of the S. aureus DNA gyrase GyrBA fusion truncate with inhibitors
| Active site 1 |
Active site 2 |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | PDB code + res. | Inhibitor | Inhibitor pockets occupied |
Metal B5081 site occupancies |
WHD Tyr C123 | DNA | DNA cleavage status | Metal D5081 site occupancies |
WHD Tyr A123 | |||||||
| 1 | 1′ | 2D | 2A | 3 | 3′ | A | B | A | B | |||||||
| 1 |
5iwi 1.98 |
‘945237 | – | – | X | X | – | – | 0.55 | 0.45 | Phe |
20–12-823 20-23cmp |
Singly nicked | 0.45 | 0.55 | Phe |
| 2 |
2xcs 2.1 Å |
‘299423 | – | – | X | X | – | – | 1.0 | – | Phe | 20–20 20–20 |
Not cleaved | 1.0 | – | Phe |
| 3 |
6qtk 2.31 Å |
gepo′ | – | – | X | X | – | – | 0.7 | 0.3 | Phe | 20–12-p8 20–12-p8 |
Doubly nicked | 1.0 | – | Phe |
| 4 |
6qtp 2.37 Å |
gepo′ | – | – | X | X | – | – | – | 0.4 | Tyr | 20-444 T 20-444 T |
Not cleaved | 0.25 | – | Tyr |
| 5 | 5iwm 2.5 | ‘945237 | – | – | X | X | – | – | 1.0 | – | Phe | 20–21 20-21cmp |
Not cleaved | 1.0 | – | Phe |
| 6 |
4bul 2.6 Å |
‘966587 | – | – | X | X | – | – | 1.0 | – | Phe | 20–23 20-23cmp |
Not cleaved | 1.0 | – | Phe |
| 7 |
5bs3 2.65 Å |
cmpd 7 | – | – | X | X | – | – | 1.0 | – | Phe | 20–20 20–20 |
Not cleaved | 1.0 | – | Phe |
| 8 |
4plb 2.69 Å |
AM8191 | – | – | X | X | – | – | 1.0 | – | Phe | 20–20 20–20 |
Not cleaved | 1.0 | – | Phe |
| 9 | 2xcra 3.5 Å | ‘299423 | – | – | X | X | – | – | – | – | Phe | 20–20 20–20 |
Not cleaved | – | – | Phe |
| 10 | 5npp 2.22 | ‘945237 + Thio2 | – | – | X | X | X | X | – | 1.0 | Tyr | 20-12p-8 20-12p-8 |
Doubly nicked | – | 1.0 | Tyr |
| 11 | 5npk 1.98 | Thio1 | – | – | – | – | X | X | – | 1.0 | Phe | 20-12p-8 20-12p-8 |
Doubly nicked | – | 1.0 | Phe |
| 12 | 6qx1 2.65 | Benzois′3 | – | – | – | – | X | X | – | 1.0 | Tyr | 20-12p-8 20-12p-8 |
Doubly nicked | – | 1.0 | Tyr |
| 13 | 6qx2 3.4 | Benzois′3 | – | – | – | – | X | X | – | – | Tyr | 20-447 T 20-447 T |
Mixed | – | – | Tyr |
| 14 | 5cdp 2.45 | Etop. | X | – | – | – | – | – | – | 1.0 | Phe | 20-12p-8 20-12p-8 |
Doubly nicked | 0.6 | 0.4 | Phe |
| 15 | 5cdm 2.5 | QPT-1 | X | X | – | – | – | – | – | 1.0 | TyrP | 20-447 T 20-447 T |
Doubly cleaved | – | 1.0 | TyrP |
| 16 | 5cdn 2.8 | Etop. | X | X | – | – | – | – | – | 1.0 | TyrP | 20–447 20–447 |
Doubly cleaved | – | 1.0 | TyrP |
| 17 | 5cdq 2.95 | Moxi. | X | X | – | – | – | – | – | 1.0 | TyrP | 20-448 T 20-448 T |
Doubly cleaved | – | 1.0 | TyrP |
| 18 | 6fqm 3.06 | IPY-t1 | X | X | – | – | – | – | – | 1.0 | TyrP | 20-448 T-U 20-448 T-U |
Doubly cleaved | – | 1.0 | TyrP |
| 19 | 6fqvs3.11 | IPY-t3 | X | X | – | – | – | – | – | 1.0 | TyrP | 20-448 T-U 20-448 T-U |
Doubly cleaved | – | 1.0 | TyrP |
| 20 | 5cdo 3.15 | QPT-1 | X | X | – | – | – | – | – | 1.0 | TyrP | 20-447 T 20-447 T |
Doubly cleaved | – | 1.0 | TyrP |
| 21 | 2xct 3.35 | Cipro. | X | X | – | – | – | – | – | 1.0 | Phe | 20–12-823 20-1223c-823c |
Doubly nicked | – | 1.0 | Phe |
The TOPRIM-bound metal is named B5081 (or D5081) to indicate that it is coordinated by Asp 508 from GyrB, in both the A and B positions (see Fig. 6 for details). Active sites are formed across the dimer interface with the catalytic tyrosine from the DC subunit (C123) approaching the B TOPRIM domain, and the tyrosine from the BA subunit (A123) approaching the D subunit TOPRIM domain. Note that “cleaved” means a phosphotyrosine bond is established, whereas “nicked” mean the 5′ phosphate is untethered.
Greek key present.
Apo and Binary DNA-Complexes of the S. aureus GyraseCORE
As described previously [15], [24], fusion truncates of the C-terminal region of GyrB (409–644) with the N-terminal region of GyrA (2–491) have been used in determining 25 published structures of the S. aureus gyraseCORE region. This construct is equivalent to the eukaryotic topo II “core” used to solve key structures of the eukaryotic enzymes (Fig. 1) [14], [25], [26]. The first structures determined for S. aureus DNA gyraseCORE were two apo structures (2XCO and 2XCQ) [15] (a rendering of 2XCQ is shown in Fig. 2A). These initial two structures were determined by molecular replacement using the Escherichia coli A59 structure (1AB4) as a search molecule [27]. Both structures are at medium-low (which we define as 2.01–3.0 Å) to low (> 3.0 Å) resolution and have a single gyraseCORE subunit in the asymmetric unit, the dimer being generated by a crystallographic twofold axis (our definitions of resolution reflect the difficulty in locating waters and having experimental data that clearly define metal coordination geometry in low- or medium-low- resolution crystal structures). The apo structures both contain a number of intrinsically disordered regions, including three α-helices at one end of the TOPRIM domain (Bα9, Bα10 and the N-terminal region of Aα1) and a number of loops that are ordered upon DNA binding (see below). Although the apo S. aureus crystals were grown in the presence of 150 mM calcium acetate (2XCO), or 85 mM magnesium formate (2XCQ), no catalytic metal was observed in either structure at the metal-binding site on the TOPRIM domain. In S. aureus gyrase, metals have only been observed at the metal-binding site on the TOPRIM domain when they interact either directly with the scissile phosphate from the DNA (the so-called “A-site” for metal binding), or when they interact with an adjacent phosphate from the backbone via a water molecule (the “B-site,” see below for the precise depiction of the metal-binding sites). This suggests that DNA binding is required to form binding sites for the catalytic metals, which involve coordinating groups from both the TOPRIM domain and the DNA. However, some DNA-containing structures do not have metals (Table 1, Table 2).
Fig. 2.

Comparison of an apo and a DNA complex of S. aureus gyrase. (A) Two orthogonal views of a 2.98-Å crystal structure (PDB code 2XCQ; [15]) of the S. aureus gyraseCORE fusion truncate (see Fig. 1). One subunit is in red (GyrB) and blue (GyrA), and the other in black (GyrB) and gray (GyrA). Note that the walls of the G-DNA binding canyon are formed by the TOPRIM (TOP) and tower (TOW) domains, while the winged helical domains (WHD) form the floor. (B) Two equivalent views of a 2.6-Å crystal structure (PDB code 6FQV; [11]) of a binary complex of the gyraseCORE-GKD (Greek key deletion) with DNA. Note that the GK domain (544–579) has been deleted from GyrB, and replaced with two residues, Thr and Gly (see text). Note also that on binding DNA, the C-terminal region of the TOPRIM domain becomes ordered (B509–B638) and (A10–A26) and interacts with the DNA (in green).
A 2.6-Å binary complex between DNA and gyraseCORE structure (6FQV), [11] (Fig. 2) does not have any compounds bound and the DNA is intact, despite the enzyme being functional for cleavage (Table 1). The DNA sits in a groove formed by the TOPRIM domain on one side and GyrA on the other side and below (Fig. 2B). Despite being obtained from crystals grown in the presence of Mn2+ ions, which can substitute Mg2+ in catalyzing phosphotransfer, no metal is observed at the catalytic site (the crystals for 6FQV were grown in conditions essentially the same as used for other structures, in which Mn2+ ions were observed at the TOPRIM domain metal-binding sites; see below and Table 1, Table 2). This is presumably due to the phosphates from the DNA backbone being too far from the TOPRIM aspartate residues involved in coordination (notably Asp508) for normal metal binding to occur. Both Mg2+ and Mn2+ favor octahedral coordination [28] with favored distances of around 2–2.3 Å, and the distance between the scissile phosphate and Asp508 is around 8 Å, precluding the formation of a coordination site. Therefore, DNA binding is essential but not sufficient for the formation of a metal-coordination site. Comparing with the apo structures (Fig. 2), DNA binding results in the ordering of the three α-helices from the TOPRIM domain (Bα9, Bα10 and the N-terminal region of Aα1). The Greek key domain (GK) is deleted in this complex, and this has been important in obtaining higher-resolution DNA-bound gyraseCORE structures (currently the only S. aureus gyrase DNA complex with the GK domain present is the 3.5-Å complex with GSK299423; 2XCR, the equivalent complex with the GK domain deleted, is at 2.1 Å, 2XCS; Table 2). In the S. aureus gyraseCORE GK deletion, residues 544–579 from GyrB are deleted and replaced with a threonine (T) and glycine (G), giving a sequence 540-AQPP-TG-YKGLGE-585. We underline the YKGLG motif as it might have some role in the control of DNA cleavage (see below for discussion of our model).
Compounds Binding at Three Different Sites Can Stabilize DNA-Cleavage Complexes
Structural studies on S. aureus DNA gyrase have identified three different sites (comprising pockets 1, 2A, 2D and 3) at or near the DNA gate where compounds (Supplementary Fig. 1 shows the chemical structures of compounds) can bind and stabilize DNA-cleavage complexes (Fig. 3); two of these pockets are largely within the DNA, and two in the protein. Site 1 (1′ for the symmetry mate) is in the cleaved DNA at the DNA-cleavage site. Compounds binding in this pocket often seem to physically block religation in a simple steric manner (Fig. 3, Fig. 4A). Such compounds often bind at both cleavage sites (four base-pairs apart) and can stabilize double-strand cleaved complexes (compounds have Chain ID I1 and I201 for the symmetry mate in the models). Pockets 2D and 2A constitute site 2 and can be occupied by the NBTIs, which sit on the twofold axis of DNA complexes. The left-hand side (LHS) of an NBTI compound, like GSK299423, occupies a pocket between the central base-pairs of the DNA (pocket 2D), while the right-hand side (RHS) occupies a pocket between the two GyrA subunits (pocket 2A) at the interface between the 2 WHDs. The LHS and RHS are linked by a central unit (Fig. 4B). Compounds in these pockets have Chain ID I2 in the models. Site 3 is a hinge pocket, between the GyrA and GyrB subunits [16]. Thiophenes that bind in the hinge pocket (Fig. 3) can stabilize both single- and double-stranded cleavage complexes [16]. Compounds in pocket 3 sit at the interface between GyrA and the TOPRIM domain on the outside of the enzyme. They do not contact the DNA (Fig. 3). Compounds in pocket 3 have Chain ID I3 and I203 in our structures (“Research” tab at https://www.cardiff.ac.uk/people/view/1141625-bax-ben and see Table S1).
Fig. 3.

Inhibitors (I) can stabilize the cleaved state through binding at different positions. (A) View of the un-cleaved binary complex equivalent to Fig. 2c. (B) Closer view of the binary complex (6FQV) showing the proximity of the two Asp83 residues at the WHDs interface. (C) Global view of compound binding sites on the S. aureus gyraseCORE illustrated by moxifloxacin (in sites 1 and 1′), GSK299423 (in site 2) and thiophene 2 (in sites 3 and 3″). The structure represented is a composite between a thiophene 2 and GSK945237 bound structure (PDB code: 5NPP) and a moxifloxacin-bound structure (PDB code: 5CDQ). (D) Closer view of the moxifloxacin structure (5CDQ) showing moxifloxacin (I1 and I201), Mn2+ (yellow sphere) and the separation of the Asp83 residues. (E) Alternative view of the composite representation shown in panel C. (F) Closer view of the NBTIs/thiophene structure (5NPP) showing GSK945237 (2), and thiophene 2 (3 and 3′), Mn2+ (yellow sphere) and the separation of the two Asp83 residues.
Fig. 4.

Comparison of cleaved and uncleaved DNA complexes. (A) The 2.5-Å structure of QPT1 (I1 and I201 in coordinates avalailable in our website, see text) with cleaved DNA (PDB code 5CDM; [10]). One subunit has carbons in magenta (GyrB) and slate-blue (GyrA), and the other in black (GyrB) and gray (GyrA); carbons in DNA are green. Oxygens are red; nitrogen, blue; and sulfur and phosphorus, orange. Residues contacting the catalytic metal and the catalytic tyrosine (TyrP A123 and TyrP C123) are shown as fatter sticks, and most residues are in thinner line representation. Carbons in inhibitors (I1 and I201) are yellow. The phosphorus in the scissile phosphate is also highlighted in yellow and arrowed (black arrows). Dotted lines between the 3’-OHs (red arrows) and phosphorus atoms illustrate that after cleavage in this complex, these atoms are some 6.5 Å apart. (B) The 2.1-Å structure of GSK299423 (I 2) with uncleaved DNA (PDB code 2XCS; [15]). The black arrows point at the phosphorous atom, and the red arrows point at the 3′-oxygen of the bond to be cleaved. The catalytic tyrosine has been mutated to a phenylalanine (Phe A123 and Phe C123).
Site 1
Compounds binding in site 1 tend to be slightly wedge-shaped compounds that intercalate in the DNA at each cleavage site contacting bases from the DNA. Compounds sitting in this pocket can make contacts with the GyrA WHD and/or the GyrB TOPRIM domain. We have obtained crystal structures with this pocket occupied by fluoroquinolones (FQ), the quinoline pyrimidinetrione (QPT-1), etoposide [10] and imidazopyrazinones (IPYs) [11] (Fig. 3 and Table 2). Anti-bacterial quinazolinediones (QZ) also occupy this pocket; a structure with a QZ bound to the same pocket has also been obtained [21]. The standard fluoroquinolone-binding mode involves interactions with the GyrA subunit via the now well-characterized water–metal ion bridge (Fig. 5) [13], [29], [30], [31]. The conformational flexibility of the water–metal ion bridge may be important. Blower et al. [30] report that, for Mycobacterium tuberculosis gyrase complexes with DNA and fluoroquinolones, complex stability correlates with antimicrobial activity. Structure-guided drug design has recently produced some more potent fluoroquinolones [32], [33], [34]. Fluoroquinolones modified at the N1 position, which are gyrase inhibitors but do not promote DNA cleavage, have also been reported [35]. The water–metal ion bridge is important for the stability of the complex, and residues involved in coordinating the metal are frequently mutated in bacterial strains that are resistant to the FQs [36]. This and safety concerns with fluoroquinolones (https://www.ema.europa.eu/en/medicines/human/referrals/quinolone-fluoroquinolone-containing-medicinal-products) are spurs to investigate the antibacterial therapeutic potential of other chemotypes binding in this pocket. The QZs are such compounds and rely on other interactions for the stability of the complex. The IPYs are a new class of bacterial type IIA topoisomerase inhibitor that bind at site 1 that do not form a water–metal-ion bridge but do establish contact with the serine involved in its formation, leading to some cross-resistance [11], [37]. They also establish contact with the arginine located just before the catalytic tyrosine in the primary sequence [11]. Interestingly, this arginine is essential for cleavage and therefore cannot be acquired as a resistance mutation by pathogens.
Fig. 6.

A-configuration and B-configuration metal coordination. (A) Left panel: The 2.1-Å structure of GSK299423 (PDB code: 2XCS) has a Mn2+ ion at the A (or 3′) site. For clarity, only the DNA, compound, catalytic tyrosine (mutated to Phe), metal and coordinating residues and waters (small red spheres) are shown. View is similar to Fig. 4B. The dotted lines show approximate positions of Asp 508 and Mn2+ ion for B-configuration metal. Right panel: Density around the modeled Mn2+ in 2XCS (blue cage, contoured at 8.4 rmsd). The coordinating oxygens were omitted, and the difference map (light green cage, contoured at 5.4 rmsd) is shown around the modeled coordinating oxygen. (B) Left panel: The 2.5-Å structure of QPT-1 (PDB code: 5CDM) has a Mn2+ ion in the B-configuration (similar view to Fig. 4A). The DNA has been cleaved by the catalytic tyrosine. Carbon atoms in DNA covalently attached to the catalytic tyrosines are colored as in tyrosine A123 (slate-blue) or C123 (gray). The position of an A-configuration metal is shown (broken circle) as in panel A, left panel. Right panel: Density around the modeled B-configuration Mn2+ for 5NPP (the highest-resolution B-configuration metal structure) is shown as in panel A, right panel (contoured at 5.1 rmsd). The omit map for coordinating oxygen is shown as above in a lighter-green cage (contoured at 4.1 rmsd).
Fig. 5.

Simplified schematics of binding modes of moxifloxacin and QPT-1 in site 1.
Simplified schematic binding modes derived from S. aureus gyraseCORE crystal structures with DNA [16] are shown for the following: (A) moxifloxacin and (B) QPT-1. In panel B, zoliflodacin, a derivative of QPT-1, is shown faintly (see Supplementary Fig. 1 for chemical structures). Compounds binding in pocket 1 (largely within the DNA—green rectangle) can extend out of the DNA into a broad open pocket on GyrB.
Currently, the most advanced novel chemotype site 1 binder is zoliflodacin, a derivative of QPT-1 [18]. Structural studies suggest that the ability of the barbituric acid moiety on QPT-1 to adopt different tautomers may be important in allowing compounds to remain bound as pocket 1 changes shape [10]. The barbituric acid does not definitely adopt a pyrimidinetrione tautomer when inhibiting the enzyme; therefore, we suggest the term quinoline barbituric acids (QBAs) rather than quinolinepyrimidinetriones (QPTs) for this class of inhibitor [10]. However, we stick to the accepted terminology throughout this review to avoid confusion. The amino acid residues that QPT-1 interacts with on GyrB are conserved in human type IIA topoisomerases. Therefore, the specificity of QPT-1 for bacterial topoisomerases is difficult to explain but may be due to small differences in the size and shape of pocket 1 in the bacterial and human enzymes, or to differences in the energetics of the DNA gate (see below). Having a totally different scaffold in the same pocket as fluoroquinolones offers many opportunities for “scaffold hopping” (Fig. 5), and it will be exciting to see how the QPT class of compounds progress in phase III clinical trials and beyond.
Imidazopyrazinones are a new class of bacterial type IIA topoisomerase inhibitor that bind in site 1 [11]. Interestingly, although one crystal structure (with two complexes in the asymmetric unit) was observed to have doubly cleaved DNA at both cleavage pockets (IPY, 6FQM, Table 2), the other IPY crystal structure contain both a doubly cleaved complex and a cleavage complex with two compounds, but only one strand cleaved (IPY-t3, 6FQS, Table 2; Supplementary Fig. 2). In the complex with one strand cleaved and one strand uncleaved, the compound has a different binding mode at the pocket with the uncleaved strand. This suggests that transitioning to the cleaved state is accompanied by a flipping of the compound. It is known that fluoroquinolones can bind the enzyme without cleavage occurring [38]. It would therefore be interesting to try to determine a crystal structure of a fluoroquinolone in complex with uncleaved DNA and the YtoF mutant, to see if a similar flip in binding mode occurs on cleavage with fluoroquinolones to that we have observed with IPY-t3 (Supplementary Fig. 6).
Etoposide is an anti-cancer drug that also has antibacterial activity [10]. Crystal structures show that the way etoposide binds to human topoisomerase II [14] and S. aureus gyrase [10] is very similar. The ability of etoposide to stabilize single-stranded DNA cleavage (as well as double-stranded DNA cleavage) at a range of concentrations with both bacterial [10] and human [39] type IIA topoisomerases is interesting and constitutes an exception among the pocket 1 binders, which generally stabilize double-strand cleavage. It is not yet clear, when one etoposide is bound, if the DNA is cleaved where the etoposide is bound, or if it is cleaved four base-pairs away as some data suggest [40].
Site 2
Novel (or non-fluoroquinolone) bacterial topoisomerase inhibitors (NBTIs) were discovered in SmithKline Beecham in the 1990s and developed at GSK to produce gepotidacin, an NBTI that is currently going through phase II clinical trials [19]. The NBTIs were also developed simultaneously by Rhone-Poulenc Rorer (now part of Sanofi); see Ref. [41] for a historical perspective on the development of these compounds. The name NBTI [15] has caused some confusion in the literature. In this review, we apply the name NBTIs to the compounds that sit in the two pockets (forming site 2, see below) on the twofold symmetry axis. This is in accordance with accepted terminology. However, we suggest that a better name for this class of compounds might be taps-NBTIs (for twofold axis pockets binding NBTIs) to clearly distinguish them from the many other novel (non-fluoroquinolone) bacterial topoisomerase inhibitors that continue to emerge.
NBTIs do not have a single chemical scaffold; however, they have some features in common that that allow them to bind on the twofold axis of the complex as shown in structures solved at GSK [15], [16], [42], [43] and by others [44], [45], namely, a planar LHS that intercalates on the twofold axis of the complex in a pocket in the DNA midway between the two cleavage sites (pocket 2D) and an RHS that sits in a pocket between the two GyrA subunits on the twofold (pocket 2A), and a central linker that usually has a basic nitrogen adjacent to Asp83 (in S. aureus DNA gyrase; Fig. 4). Although pocket 2A (the GyrA pocket) is conserved in bacterial gyrases and topo IVs, it is not conserved in human topo IIs, accounting for the specificity of NBTIs [15].
The initial S. aureus gyraseCORE crystallization system was developed in GSK to support the development of NBTIs. The optimization of the DNA sequence and identification of a more stable complex with Mn2+ (cf Mg2+ or Ca2+) and the catalytic Y123F mutant were critical in producing initially, a 3.5-Å crystal structure with GSK299423, then with the deletion of the GK domain, a 2.1-Å structure was generated [15]. This structure produced a clear view of the A (or 3′) metal coordination at the DNA cleavage sites, including the waters coordinating the catalytic metal ion. However, because GSK299423 sits on the twofold axis, the electron density for the compound is averaged around the non-crystallographic twofold axis of the complex and the water structure around the compound is not so clear (Supplementary Fig. 8). A 2-year campaign in GSK to try and produce NBTI crystal structures in this P61 space group with the compounds in a single orientation, trying to exploit the fact that the ends of the DNA pack differently in this space group, produced structures with asymmetric DNAs disordered around the non-crystallographic twofold symmetry axis [42], [43]. The highest resolution (1.98-Å) structure, with GSK945237, had an artificial nick in one strand and the electron density maps for the active sites were clear, although there was static disorder around the non-crystallographic twofold symmetry axis. The conclusion from this 1.98-Å GSK945237 structure [43] was that the metal coordination geometry for the A (3′) and B (Y) metal binding sites are incompatible with two metals being bound at the same time.
Site 3
This site is formed by a pocket situated between the GyrA and GyrB subunits (Fig. 3), and compounds binding in this pocket do not contact the DNA. However, they can stabilize both double- and single-stranded DNA-cleavage complexes [16]. The “floor” of the pocket is partly formed by the Aα1 helix (from GyrA). This Aα1 helix is inserted into the TOPRIM domain and bends at its C-terminal end (proximal to pocket 3) when the apo and DNA-bound S. aureus gyrasecore complexes are compared, hence arriving at the name “hinge pocket.” However, sequence and structural differences suggest that it does not do this in eukaryotes. Although most S. aureus gyrasecore complexes with DNA and compounds have the GyrA and GyrB subunits within a covalently fused gyrasecore subunit in the same orientation [10], the 2.6-Å binary complex (6FQV, Table 1) shows a distinct movement between the TOPRIM domain (which includes the N-terminal end of the Aα1 helix) and the WHD (which includes the C-terminal end of the Aα1 helix) within a covalently fused subunit (compare Fig. 3A and C). Pocket 3 assumes a different shape in crystal structures of gyrasecore DNA-cleavage complexes compared to the binary complex with uncleaved DNA [11], [16]. Superposing the 1.98-Å crystal structure of thiophene 1 with the 2.6-Å binary structure shows clashes of the compound with the protein in the binary structure conformation (Supplementary Fig. 7). Therefore, we conclude that compounds such as thiophene 1 inserting into the pocket prevent DNA cleavage complexes from relaxing into the binary state.
The 1.98-Å complex with thiophene 1 is the highest-resolution structure we have obtained containing B-configuration metal geometry, which appears conserved between bacterial and human type IIA topoisomerases. The highest-resolution structure we have obtained with an A-site metal is the 2.1-Å structure with GSK299423 (Table 2).
The Role of S. aureus GyraseCORE Crystallization System in Compound Development
Although much of drug development is concerned with optimizing the ADMET properties of compounds, target potency cannot be ignored. The solving of structures showing compounds bound to their target thus remains an essential part of pre-clinical compound development, helping the chemistry effort in maintaining or improving specificity and potency.
The S. aureus gyraseCORE crystallization system, developed by GSK to support the development of NBTIs, had relatively little impact on the chemical development of gepotidacin, which is similar to important progenitor compounds GSK966587 [42] and GSK945237 [43], which were synthesized before the first S. aureus gyraseCORE structure with an NBTI was determined (see Supplementary Fig. 1 for chemical structures). Although many potent NBTIs with good antibacterial activity have been reported, a major challenge remains hERG toxicity [42], [46], for which target-bound structure are not particularly helpful.
The successful structure determination of the first structure of a fluoroquinolone complex showing the water–metal ion bridge in GSK, the 3.25-Å moxifloxacin complex with Acinetobacter baumannii topo IV [13], used a DNA sequence identified by Arnoldi and co-workers [47] (Table S2) and followed the deposition of a quinazolinedione structure in the PDB [21]. The identification of the water–metal ion bridge was essential in illuminating target-based resistance to fluoroquinolones and helped the development of new compounds overcoming this resistance. Early attempts to use the S. aureus gyraseCORE system for fluoroquinolone structure determinations at GSK were not successful, tending to produce limited resolution ambiguous data sets such as the 3.35-Å twinned ciprofloxacin structure, which we have now re-refined and re-deposited with the PDB (2XCT; see Supplementary Materials). Therefore, the determination of the first medium-high-resolution structure of a fluoroquinolone complex [30] was an impressive feat. Fluoroquinolone crystals do not tend to diffract to very high resolution, and Blower et al. [30] reported that with the M. tuberculosis gyrase crystals they used, “the diffraction quality varied greatly not only between crystals but also between different regions of a single crystal.”
The deletion of the GK domain in the S. aureus gyraseCORE produced a dramatic improvement in resolution in complexes with the NBTI GSK299423, from 3.5 to 2.1 Å (Table 2), as well as a clear view of an A active-site metal. Attempts to reproduce this GK deletion strategy in about 10 Gram-negative gyrases and topo IVs were not successful.
The initial 1.98-Å thiophene hinge binder complex facilitated the building of higher activity into the initial compounds through an added methyl group, filling a cavity in the hinge pocket [16]. This illustrates the usefulness of the gyraseCORE in building potency, especially when higher resolution can be achieved. More recently, structure-guided drug design has produced a second class of hinge binder compounds [48].
Finally, the use of the S. aureus gyraseCORE crystal system to investigate other antibacterial gyrase “poisons” has produced some interesting structures [10], [11], [16]. Consideration of these structures has allowed us to develop two different single-moving-metal mechanisms for DNA cleavage by S. aureus gyrase (one of these is presented in Fig. 7, Fig. 8 and below, and an alternative model is presented in the Supplementary discussion and Supplementary Figs. 9 and 10). It is difficult to prove mechanisms, but it is hoped that by considering different possibilities, experiments can be devised that can distinguish between them. The two single-moving-metal mechanisms we present have many aspects in common (below and in Supplementary discussions) but are different.
Fig. 7.

Dynamic model for the cleavage–religation cycle catalyzed by a single metal ion. (1) Relative movement of the GyrA subunit (see Fig. 3), extension of the DNA and tilting of the TOPRIM domain create a coordination environment for a single metal ion, the A-configuration, involving Asp508 residue from the TOPRIM domain, water molecules, and two oxygens from the scissile phosphate (see inset). (2) Through its contacts with the scissile phosphate, the metal catalyzes phosphotransfer to the catalytic tyrosine. The phospho-tyrosine then moves away after cleavage, disrupting the A-configuration, which favors the transfer of the metal to the B-configuration (inset) initially filled by a water molecule contacting the A-configuration metal. (3) Opening of the DNA gate obliterates the A-configuration but does not affect the B-configuration, allowing the metal ion to be stored in the B-configuration during strand passage. Keeping the catalytic metal away from the phospho-tyrosine can prevent detrimental hydrolysis of the bond during strand passage. (4) Closure of the DNA gate brings the phospho-tyrosine closer to the catalytic metal in the B-configuration. (5) We hypothesize that phospho-tyrosine is being brought even closer to the B-configuration coordination cage and can disrupt a coordinating water molecule, resulting in metal recapture by the phosphate and the catalysis of phosphotransfer to the 3´-OH (which remains close to the B-configuration during the cycle). This re-creates the A-configuration and panel 6. Relaxation into the binary state can then ensure irreversibility by disrupting the A-configuration. We envision that controlling the motion of the DNA and the position of the scissile phosphate allows the enzyme to control cleavage and religation. This control is presumably coupled to the ATP hydrolysis and exchange cycle. Alternatively, a compound binding the enzyme might also influence the position of the scissile phosphate.
Fig. 8.

A simplified model for DNA cleavage by S. aureus DNA gyrase. (A) A G-DNA segment is shown in a binary complex, based on binary structure (6FQV). No metal is bound. The scissile phosphate (red) is outlined on the DNA backbone (green). D83 and D83’ are close to each other at the GyrA/GyrA’ dimer interface (red lines). (B) A T-DNA segment has been captured (not shown) and the domains at the DNA-gate start to move, stretching the substrate gate DNA. (C) A single metal (yellow sphere) is captured in the A-configuration, on one side. (D) The bottom strand has been cut, the metal is in the B-configuration, the gate is pushed further open and a metal is captured in the A-configuration on the other side. (E) Both DNA strands are now cleaved. Metals are in B-configurations. (F) The T-DNA is outlined passing through the DNA gate. (G) The above model is broadly consistent with the scheme published by Zechiedrich et al. [49]; E is the enzyme and S is the substrate DNA, which is either uncleaved (E.S—A,B), single cleaved (E-S1—C,D) or doubly cleaved (E-S11—E,F).
Why Different Compounds Stabilize Different Amounts of Single- and Double-Strand Cleavage
We have mentioned that each class of compounds presents a characteristic ratio of double- to single-strand cleavage. The above considerations fail to explain this. It is important to note that each strand of DNA is involved in the formation of its own catalytic cleavage pocket through involvement (directly or via a water molecule) in the coordination of the catalytic metal.
Interestingly, although compounds binding in pocket 3 stabilize both double- and single-stranded DNA-cleavage complexes, they do so without changing the preference for cleavage sites seen (at a much lower level) in the absence of compounds [16]. This contrasts with fluoroquinolones, which do change cleavage site preferences, presumably through their interaction with DNA [16]. This suggests that interaction with the DNA near the cleavage site can influence cleavage.
We have mentioned that two binary complexes have been solved with S. aureus gyrase (6FQV and 5CDR). The relative positions of the domains within a gyraseCORE subunit in 6FQV are different from those in any other S. aureus gyrase core complex with DNA. 6FQV has domains in positions intermediate between the apo structures and the other S. aureus complexes with DNA and traps an “uncleaved” conformation (see below). In 5CDR, introducing artificial “nicks” in the DNA at both cleavage sites, by annealing a 12mer with a phosphate at the 5´ end with an 8mer, gives a 20-bp duplex which crystallizes well (20-12p-8; Table 1, Table 2) [24]. However, in the binary complex with this artificially doubly nicked DNA (5CDR), the central four base pairs (where the four base-pair overhangs should anneal) of the DNA are largely disordered. Interestingly, a similar observation is made with the 1.98-Å structure, which has a compound bound in the allosteric hinge pocket but no compound in the DNA (5NPK; Table 2) [16]. This disorder has been similarly observed in a human topo II structure [25]. On the other hand, crystal structures obtained with the same doubly nicked DNA and compounds bound to the DNA show ordered central base pairs (e.g., the etoposide-bound structure [10]). This again points toward the idea that compounds that interact with DNA in the central region can influence its structure and therefore its cleavage status. We surmise that when the cleaved conformation is adopted by the enzyme, the DNA has some flexibility and can oscillate between single-, double-strand cleaved and uncleaved, unless a compound(s) is bound to the DNA and inhibits this flexibility, thereby favoring single- or double-strand cleavage. In biochemical assays, complexes stabilized by the hinge binders are a mixture of uncleaved, single- and double-strand cleaved [16]. When the full-length enzyme is not bound by compounds, the addition of ATP also results in a mixture of trappable single- and double-strand cleavage [11], [16]. This suggests that, during the catalytic cycle, the enzyme naturally and transiently can adopt cleaved conformations.
In that view, the symmetrical binding of FQs to pocket 1 would favor double-strand cleavage by favoring the formation of a catalytic cleavage pocket on both sides. The NBTIs on the other hand stabilize only single-strand cleavage, which is consistent with them having an asymmetrical binding mode. In the 2.1-Å crystal structure of GSK299423 with S. aureus gyrase and DNA (2XCS), the compound sits on the twofold axis and is not C2 symmetric (see above). Since such asymmetric compounds interact with DNA, it is likely that they can asymmetrically influence DNA conformation and thereby stabilize only single-strand cleavage. However, the exact conformation of DNA in this asymmetric complex is difficult to ascertain due to the lack of crystal structure with compound and DNA in a single orientation. More specifically, it has not yet been possible to correlate a compound orientation to a DNA orientation.
General Mechanistic and Energetic Features of DNA Gyrase Poisoning
The ability of compounds to stabilize DNA gyrase-DNA cleavage complexes is dubbed “poisoning” as it converts an essential enzyme into a deleterious lesion. These have, to date, been much more successful in the clinic than the “catalytic” ATPase inhibitors [50]. The mechanism described above for the allosteric hinge binders reflects general features of the mechanism of poisoning by a variety of compounds binding to a variety of pockets. Compound binding to any of the pockets is generally associated with a “sliding” of the WHDs along the DNA gate interface. This is accompanied by a “tilting” of each TOPRIM domain toward the DNA (Fig. 3A, C), accompanying the C-terminal bending of the Aα1 helix described above. These motions were also observed by Wendorff et al. [51] with an etoposide complex with human topo II, suggesting that they are conserved across type IIA topoisomerases. The DNA itself is extended compared to the binary complex and, as a result, the distance between the scissile phosphate and the metal-coordinating residues is reduced and allows the capture of a catalytic metal (usually Mn2+ in S. aureus gyraseCORE structures). Indeed, nearly all the compound-bound structures have a single metal bound at the catalytic site, whereas the binary uncleaved complex and the apo structures do not. These movements are small (< 5 Å) but relatively consistent across compound-bound structures and represent a transition toward the cleaved state, which is stabilized by compound binding. The sliding of the two WHDs opens up a cavity (pocket 2A) into which GSK299423 can bind, as evidenced by the separation of the two D83 residues from the WHDs, which are close enough to establish contact at the interface in the uncleaved binary complex (Fig. 3B). Consistently, mutating D83 to an N residue disfavors the transition to the cleaved state, presumably by allowing the establishment of hydrogen bonds at the WHDs interface in the binary complex [11]. GSK299423 can be viewed as a “locking pin” that slides into a cavity formed on proper alignment of the WHDs.
These domain movements are not simply a result of DNA cleavage since they are observed in the GSK299423-bound structure (2XCS) in which the catalytic tyrosines are mutated to phenylalanines and the DNA is therefore intact [15]. Moreover, data suggest that a significant proportion of thiophene–gyrase–DNA complexes do not have the DNA cleaved [16]. Similarly a significant proportion of gepotidicin complexes do not have the DNA cleaved [52]. In the case of pocket 1, compounds intercalate and we speculate that it favors the extension of the DNA. Moreover, the transition to the cleaved state favors the alignment of residues involved in contacting the compound and the cavity between the two bases. It is known that fluoroquinolones can bind the enzyme without cleavage occurring [38], again suggesting that the transition to a different conformation precedes, and determines cleavage. It is possible that the steric impediment to religation also plays a role in the efficiency of cleavage for these compounds. Indeed, they are usually more efficient than the allosteric compounds (NBTIs and thiophene).
We therefore suggest a model in which the enzyme needs to undergo conformational changes resulting in DNA extension and the formation of a favorable coordination site for a single metal ion, which can then catalyze the phosphotransfer reaction to achieve DNA cleavage. The primary mode of action of compounds is to stabilize this otherwise unfavorable state (as low or no cleavage is observed in their absence). The only structure of type II topoisomerase with the DNA gate opened was recently published [25]. In this structure (with human topo II), the WHDs separate seemingly by a sliding motion similar to the one we describe, only more extensive. We therefore speculate that the transition to the cleaved state would constitute a “prelude” to the opening of the DNA gate by a continuation of the sliding motion. However, no bacterial type IIA topoisomerase structure with DNA bound and the DNA gate opened has yet been obtained.
This suggests that in order to cleave the DNA, gyrase has to stretch the central four base pairs. This increases the distance between the scissile phosphates and reduces the stacking interactions between the central bases. DNA is a very deformable molecule, but this stretching will have some energetic cost. It may also explain the vulnerability of the DNA-gate to intercalating compounds, which can also stretch the DNA. According to this model, the energy of compound binding must compensate the energetic cost of transitioning to the cleaved state. This energetic cost is presumably due to the loosening of the protein interface in addition to the stretching of the DNA. Hence, we predict that affecting either the compound binding energy or the energy of transition to the cleaved state could affect the efficiency of poisoning. Consistently, mutations affecting the residues contacting compounds result in resistance to poisoning. For instance, mutating the residues involved in the water–metal ion bridge results in resistance to the FQs [29], [53], [54]. Moreover, mutations that affect the transition to the cleaved state (like D83, above) confer resistance to several different classes of gyrase poison despite not establishing contact with them [11]. We predict that other residues might have this property, notably at the interface.
This model can also help to understand other facts such as the bacterial specificity of QPT-1. Some data suggest that the interface might be tighter in the eukaryotic enzyme. Namely, gyrase can relax DNA without ATP pointing to a “loose” interface. This suggests that the energy of transition to the cleaved state is higher for the eukaryotic enzyme and the binding energy of QPT-1 might not be sufficient to compensate and will not be able to poison. In other words, when the DNA-gate closes in human type IIA enzymes, the wedge-shaped QPT-1 can be squeezed out of the DNA-cleavage site pocket, whereas with bacterial type IIA topoisomerases, QPT-1 remains trapped [10].
Role of the Metal Ion at the Cleavage Site: Dynamic Re-configuring of the Catalytic Core
There is consensus in the literature that there are two metal-binding sites (per catalytic pocket) in the TOPRIM domain, about 3 Å apart. Biochemical [55], [56] and structural data [12], [15], [22] suggest that in order for DNA cleavage to take place, a metal must be making interactions with the 3′-oxygen of the scissile phosphate. Problems in obtaining well-diffracting crystals with clear views are not uncommon with type IIA topoisomerases, which can make ascertaining the position and coordination of the catalytic metal difficult. Therefore, we focus on relatively high-resolution structures, which show clear views of the coordination mode of the catalytic metals.
As mentioned above, only gyraseCORE structures with compounds bound and/or DNA cleaved have a metal at the catalytic pocket. Only a single metal at each catalytic pocket is consistently observed. The coordination geometry is found to correlate with the cleavage status of the DNA. In the 2.1-Å crystal structure of GSK299423 with DNA and S. aureus DNA gyrase (2XCS), the DNA is uncleaved at both catalytic pockets, and a single metal ion establishes contact with the scissile phosphate (Fig. 4, Supplementary Figs. 2 and 3). The catalytic tyrosine mutated to a phenylalanine is observed in the vicinity pointing toward the scissile phosphate. We surmise that this configuration is “poised” for cleavage. This coordination configuration for the metal is dubbed the “A-configuration” or “A-site.” Metal in the A-configuration is coordinated by two oxygens from the scissile phosphate, Asp508 and Glu435 from the TOPRIM domain, and two water molecules forming an octahedral geometry favored by the Mn2+ ion (Fig. 5, the difference map is shown for the coordinating oxygen, showing octahedral geometry). Note that in the following discussion, we use the term “configuration” to describe metal status instead of “site” because the metal-binding site is observed in different configurations depending on whether the metal is at the A- or B-site. Note that a single metal, seemingly close to the A position, is observed in a cleaved structure (3KSA [21]). However, the resolution being lower, it is difficult to ascertain the coordination geometry in this case. Therefore, the status of this metal is unclear. The A-configuration is incompatible with metal binding at the B-site and vice versa. Super-imposing the two configurations using as a reference the coordinating residues induces a clash between a coordinating water molecule from the A-configuration and the metal in the B-configuration.
When the DNA is cleaved, the coordination of the metal is altered and the metal is observed in the so-called “B-configuration” or “B-site.” This is consistently observed across all cleaved structures. Figure 4 shows the metal status on the highest resolution S. aureus DNA cleavage complex structure (5CDM), which has QPT-1 bound at pockets 1. When the DNA is cleaved, the scissile phosphate moves away from the metal and the metal moves to the B-site, where it is coordinated by D508 (as in the A-configuration), D510 and four water molecules, one of which contacts the adjacent phosphate on the 3′-OH side. Figure 5B shows the density of the metal and coordinating oxygens as above. Superimposing the two structures with D508 as a reference (more accurately the C-Cα bond of the residual chain) shows a rotation of the D508 residual chain that seems to accompany the metal from the A-configuration to the B-configuration. See also Supplementary Figs. 2 and 4.
The case of the 1.98-Å GSK945237 structure [43] is interesting. In this structure, one DNA strand is uncleaved, while the other strand has an artificial nick (5IWI). The electron density shows two positions for metal ions (Mn2+), one at the A-configuration and one at the B-configuration, but the occupancy for each metal is about 0.5. The 1.98-Å electron density at the two active catalytic pockets clearly shows an average of the structure seen in the 2XCS structure (with uncleaved DNA) and a second conformation with the nicked DNA (like the 5CDM structure, see Supplementary Fig. 2). In the crystal structure, the asymmetric nicked DNA is bound with a random orientation with respect to the nick, resulting in each cleavage catalytic pocket being an average of the cleaved and uncleaved configuration in the solved structure. The 1.98-Å density clearly shows two different configurations: a single metal in the B-configuration when the DNA is cleaved and a single metal in the A-configuration when the DNA is uncleaved, averaging resulting in the modeling of two metals with half occupancy (Supplementary Fig. 2). The coordination geometry of metal ion at the B configuration is consistent with other structures with a metal at the B-configuration, which do not display such static disorder (see Supplementary Methods for details).
Considering all of the above, we propose a dynamic model for the cleavage-religation cycle of DNA gyrase (and possibly all type IIA topoisomerase) in which conformational changes undergone by the enzyme re-configure the catalytic metal coordination site in the catalytic pocket to control cleavage and religation. Figure 7 depicts such a model. After DNA binding, the sliding of the WHDs and the tilting of the TOPRIM domains create a favorable coordination environment for a single metal per catalytic pocket (the A-configuration, step 1). Contact with the scissile phosphate allows the A-configuration metal to catalyze phosphotransfer to the catalytic tyrosine, the scissile phosphate moves away from the metal, obliterating the A-configuration. The metal is then transferred to the B-configuration (accompanied by a rotation of Asp508, step 2) which is now more favorable and importantly is not affected by the opening of the DNA gate (step 3). The closure of the DNA gate (step 4) brings the scissile phosphate in the vicinity of the B-configuration metal and allows the recapture of the metal by the scissile phosphate (step 5). Irreversibility is achieved by a conformational transition to the uncleaved conformation (step 6). In this model, the enzyme controls the movement of the scissile phosphate which, in turn, controls the metal status and ultimately cleavage and religation. Moving the scissile phosphate away from the metal, which is “stored” in the B-configuration, has the advantage of making hydrolysis of the phosphotyrosine bond impossible since no metal can be stably coordinated by the enzyme around the scissile phosphate when the DNA gate is opened. Consistently in the open-gate structure obtained with human topo II, a single metal is observed in the B-configuration [25]. Moreover, an etoposide-bound structure of the human enzyme also shows a single metal in the B-configuration per catalytic pocket, further suggesting that a single metal might be conserved among type IIA topoisomerases.
Therefore, we surmise that what controls the configuration of the metal coordination sphere is the position of the scissile phosphate and preceding phosphate. When the enzyme assumes the “cleaved” configuration, both configurations are possible, albeit not at the same time (see below for further discussion). The fact that metal is observed in the A-configuration when the DNA is not cleaved (2XCS) suggests that the A-configuration has more affinity than the B-configuration when the tyrosine is mutated to a phenylalanine. When phospho-transfer has occurred, the scissile phosphate (now bonded to the catalytic tyrosine) moves away from D508 and the metal moves to the B-configured site. In that view, religation would only require the scissile phosphate to come nearer the B-configured site metal to recapture it by re-forming the A-configured site. This would be consistent with the idea that the A-configured site has more affinity. There is little structural evidence on how this recapture would occur. However, we suggest that the recapture can occur through limited motion afforded by the enzyme flexibility in this “cleaved” conformation. This is supported by the observation that the thiophene–gyrase–DNA complexes are a mixture of single-, double-strand cleaved and uncleaved. This indicates that the enzyme can religate even when the “cleaved” conformation is stabilized by compounds. There are also data suggesting that it is possible to religate cleavage complexes while compounds are still bound. The above considerations do not preclude further structural controls on religation. Indeed, it is highly likely that such control exists (see below in the description of our model).
It is difficult to distinguish whether the metal initially binds in the A-configuration directly or binds in the B-configuration first and is subsequently “recruited” by the scissile phosphate. It is expected that the A-configuration is very transient since it leads to cleavage. Therefore, the stable A-configuration observed when the tyrosine is mutated by a phenylalanine might not necessarily reflect the true situation and is only reached when catalysis is not possible. An alternative model might have the scissile phosphate coming in close enough proximity to the B-configuration metal to disrupt its water coordination, resulting in the true catalytic configuration (see Supplementary Figs. 9 and 10 and Supplementary discussion).
Two-Metal versus One-Metal Cleavage
Whether two metals binding to one catalytic pocket is necessary for cleavage remains a matter of debate. Biochemical studies showing two different metals can synergistically enhance cleavage rates have been interpreted as favoring the two-metal per catalytic pocket hypothesis [56], [57]. However, we suggest that they do not achieve this by both binding to the same catalytic pocket [22] but rather by cooperating in “trans,” with one in the A-configuration on the first TOPRIM domain and the other in the B-configuration on the second TOPRIM domain [15]. We favor the single-metal model, at least for bacterial type IIA topoisomerases.
The electron density maps for several early low-resolution (> 3 Å) and medium-low-resolution (2.5–3 Å) structures are not very clear [12], [15], [21], [22], [26], and allow alternative interpretations for ligand-binding modes and/or metal coordination geometry at the DNA-cleavage sites to the published structure. We have re-refined two of these structures (see Supplementary Methods) to include the water–metal ion bridge (for 2XCT) and chemically reasonable catalytic metal coordination geometry consistent with later higher-resolution structures: the 3.35-Å ciprofloxacin structure 2XCT [15] and the 2.98-Å yeast topo II structure (3L4K) [22]. With this re-refinement, all structures, to the best of our knowledge, are consistent with a single metal ion in either the B-configuration (as in the 2.16-Å crystal structure of etoposide with a human topo II [14]) or the A-configuration (as in the 2.1-Å structure of S. aureus DNA gyrase with GSK299423 [15]). 3L4K is the only structure in the PDB interpreted as having two metals at a single catalytic site at the same time. This structure is complicated by static disorder around a crystallographic twofold axis. Single “biological” complexes derived from the crystal structures for both the original (3L4K) and re-refined (RR-3L4K) coordinates are made available (Table S4). The re-refined coordinates of 3L4K are consistent with a single moving metal mechanism for DNA cleavage by type IIA topoisomerases (Supplementary Fig. 5).
This being said, there are a series of arguments that can be raised against the one-metal interpretation. First of all, the above interpretation of the two-metal model as averaging of the two one-metal configurations must be qualified. It is difficult to assign occupancy unambiguously to atoms in medium- to low-resolution structures. The hypothesis of half occupancy (or for that matter full occupancy) is therefore a choice guiding the refinement. The reason we have made that choice is that one metal only has been unambiguously observed in a high-resolution structure (1.98 Å) with static disorder (5IWI). The geometry modeled in these cases is in accordance with what is known of coordination chemistry for divalent metals. We argue that it is therefore justified to use it as a guide to refine low-resolution structure. However, we would like to stress that we do not think that our refinement of such structure settles the debate, merely that such data could be compatible with the one-metal model.
Moreover, while our interpretation of the 5IWI apparent two-metal density is relatively straightforward, due to the nicked DNA adopting both orientations, the case of 5CDP is not so simple. In this structure, a doubly nicked DNA is crystallized along with the gyraseCORE and etoposide. Only one etoposide molecule is observed in pocket 1, while the symmetric pocket 1′ is free of compound. As one would expect, according to our model, one metal in the B-configuration is observed on the etoposide site. However, two metals are observed on the other site, despite the DNA being nicked on that side as well. Our interpretation is that it is due to the two configurations coexisting in the crystal (i.e., static disorder) independently of the symmetry axis, the relative occupancies reflecting the respective frequency of each configuration. When no compound is present, the scissile phosphate being flexible, it can sometimes adopt a conformation in which it can coordinate an A-configuration metal but sometimes it does not; this results in the metal being observed in the B-configuration. In 5CDP, the presence of an etoposide on the other side restricts the flexibility of the untethered DNA, thereby allowing an A-configuration metal at least some of the time. This is our favored interpretation, but because the structure has static disorder and the data only extend to 2.45-Å resolution, other interpretations might be possible. It should be noted, however, that the geometry of metal coordination is different in the case of 5CDP compared to others, the presumed “A-configuration” metal being coordinated mainly by water molecules, which are difficult to resolve at this resolution (Supplementary Fig. 11). The scissile phosphate is modeled by two conformations suggesting limited flexibility. 5CDP refinement is difficult, and therefore, any interpretation would be subject to caution. The main point is that, in high-resolution structures, the one-metal interpretation allows clear and unambiguous coordination geometry (like in 5IWI).
The case of 5CDP also illustrates an argument made by Wendorff et al. [51]. In this work, only one metal is observed in an etoposide-bound complex. It was proposed that this deviation from the two-metal model is due to the compound reconfiguring the active site and preventing the A-configuration and thereby preventing religation. This would be a consistent interpretation of 5CDP. In fact, our interpretation is not that different. The idea that removing the A-configuration metal prevents religation is not controversial and actually compatible with both models. The only difference between the two models is the source of the metal filling the A-configuration when religation occurs. If it comes from the solution, two metals will be the catalytic configuration; if it is mobilized from the B-configuration, you get the one-metal mechanism. We think it is unlikely that the one-metal observation results from compound reconfiguring the active site. Several, unrelated compounds show one metal, including the allosteric hinge binders that do not interact with DNA. Moreover, 5CDR does not have any compound bound and shows only one metal. Nevertheless, crystallography always samples out stable conformations, and it is possible that the true catalytic configuration has not been observed, only pre- and post-catalytic configurations are likely to be observed. It is worth noting, however, that from existing knowledge of phospho-transfer reactions, only one metal is absolutely required for catalysis: the A-configuration metal. It is not at all clear what a B-configuration metal would do to make itself essential for catalysis, in addition to the A-configuration metal, in such a way that the only catalytic configuration would be with two metals bound. We argue that the one-metal configuration is the minimal required for activity. The two sites having different affinity, the configuration could also be different depending on the concentration of divalent metal in the solution (or the intracellular media).
We have used the 20-12p-8 DNA in many unpublished crystal structures with NBTIs. When the resolution is sufficiently high (as in the 1.98-Å 5IWI), two conformations are clearly visible in electron density maps. However, as the resolution becomes lower (as in the 2.45-Å 5CDP structure), the two conformations are not always so clearly seen in electron density maps. We have refined all medium-high and medium-low structures consistently with our unambiguous high-resolution structures. However, given the poor quality of the data, we cannot be 100% certain that such structures could not be open to other interpretations.
As a direction for future studies, we would like to suggest trying to sample out the true catalytic configuration using fluoride compounds of divalent metal standing in for the scissile phosphate. This might settle the debate, although the requirement for high resolution might make it challenging. It is also expected that real-time crystallography data would shed light on the catalytic mechanism.
An Integrated Model for the Strand-Passage Reaction
In this simplified model, the catalytic cycle starts when the G-DNA is captured. To achieve cleavage, the DNA is then bent [26], resulting in the central four base pairs between the two DNA-cleavage states adopting an A-DNA like conformation. At this stage, the distance between the two scissile phosphates is about 21 Å (as in the binary complex, 6FQV). With the DNA bound and the protein in this conformation, there is no metal bound. Interestingly for yeast topo II, Mueller-Planitz and Herschlag [58] identified two conformations that can have G-DNA bound: the first conformation efficiently “binds DNA at a diffusion limited rate but cannot efficiently cleave DNA,” while the second conformation binds (and releases) DNA very slowly but allows DNA cleavage. Presumably, the first conformation has the G-DNA bound but not bent, precluding cleavage, while the second conformation has the DNA bent, allowing cleavage. However, it is also possible that the first conformation is captured by the binary complex 6FQV. There is to date no structure with the DNA unbent. It is expected that such a structure would be informative.
When the T-DNA has been captured by the ATPase domains, the captured T-DNA and ATPase domains start to try and push open the DNA-gate (Fig. 8B–F; see also Fig. 1). Initially, this moves a TOPRIM domain and starts to stretch the G-DNA segment allowing a single metal to be captured (Fig. 8B). We then envision that if metals are bound, they either are recruited directly to the A-configuration (Fig. 7, Fig. 8) or normally reside in the B-configuration, unless transiently attracted to interact with the scissile phosphate if it comes close enough (resulting in an A-configuration metal, Supplementary Figs. 9 and 10). When the metal is in the A-configuration, DNA cleavage occurs (steps B–C Fig. 8), and the scissile phosphate from the now cleaved DNA (attached to Tyr A123) relaxes away from the cleavage site (Fig. 8C), and the metal returns to the B-configuration. Once one strand has been cleaved and as the T-DNA pushes the DNA-gate open, the DNA-gate will adopt an asymmetric conformation [10] (Fig. 8D) and then the second cleavage step will occur (Fig. 8D–E). Deweese and Osheroff [59] state that for topo II “it is believed that cleavage of the first stand introduces flexibility in the DNA that allows the substrate to attain an acutely bent transition state that is required for efficient cleavage,” and that the rate for cleavage of the second strand is faster than that for the first strand. The idea of an asymmetric orientation of the T-segment during passage is supported by some cryo-EM work [60].
The simplified model suggests that once the T-DNA has passed through the DNA-gate, at least one of the DNA strands must be religated before the exit gate can open. Mutation at the C-gate favors cleavage in yeast topoisomerase II, and Schmidt et al. [22] have proposed a structural model for the coupling of cleavage to C-gate closure, which controls the position of the tyrosine. We suggest that, when the tyrosine is bonded to the scissile phosphate, such control could in turn influence the position of the scissile phosphate and recapture of the metal to achieve religation. Another layer of control could involve the GK domain (as mentioned above). Interestingly, in both S. aureus binary structures (Table 1, 6FQV and 5CDR), the main-chain N-H group of Leu583 (the L from the YKGLG motif) is within hydrogen-bonding distance of one of the side-chain oxygens of Asp508. However, in 5CDR (where the DNA is cleaved), the other side-chain oxygen from Asp508 coordinates a Mn2+ ion (whereas no metal is observed in 6FQV). This raises the possibility of an involvement of the Greek key fold in controlling metal coordination. We have previously proposed [10] that the function of the GK domain is to interact with the T-DNA and prevent the catalytic metal in the B-configuration moving to re-cleave the G-DNA, as the exit gate opens and the T-DNA is released. If the exit gate opened while the G-DNA contained a double-stranded break, this might be potentially hazardous for the cell. We suggest that the Greek key could exert control of the metal position through the YKGLG motif, to which it is directly adjacent. Whether the Greek key is ordered or not might be associated with such control.
The presence of a single-strand cleaved intermediate during this cycle is evidenced by the mixture of single- and double-strand cleavage induced by the enzyme in the presence of ATP [16], also suggesting that the ATP hydrolysis cycle favors the transition to the cleaved state. The exact nature of this singly cleaved intermediate is difficult to ascertain. Biochemical and structural data show that some asymmetry can be transiently or permanently assumed by the enzyme. As discussed above, many compounds stabilize a significant amount of single-strand breaks as opposed to double-strand breaks only. Etoposide and the thiophenes stabilize a mixture of single- and double-strand breaks [10], [16], and the NBTIs only stabilize single-strand breaks [15], [52]. A single-strand intermediate is observed during cleavage induced by the fluoroquinolone ciprofloxacin and also during religation of the cleavage complex [11], [61]. In addition, some structures with asymmetry were solved (Casym class of structure [10]). In those structures, however, the DNA is doubly cleaved so it is difficult to correlate the cleavage status with the structural conformation on each site of the C2 axis. As mentioned previously, an asymmetric DNA can be bound both ways in the crystal, resulting in the symmetric averaging of a potential asymmetric structure. Despite several years of effort, we failed to obtain crystals in which an asymmetric DNA is bound in a single orientation [24]. To date, all structures with S. aureus DNA gyrase with asymmetric DNAs [43] have the DNA in two orientations related by the C2 symmetry of the complex. The structural characterization of the asymmetric state(s) with singly cleaved DNA remains to be done, The Casym structures suggest that asymmetry in cleavage could be coupled to an asymmetry in protein domains conformation.
Taken together, our data suggest the following simple model for the strand passage reaction (Fig. 8):
-
1.
Capture of G-DNA segment
-
2.
G-DNA segment bending, no metal bound (binary complex structure 6FQV)
-
3.
Capture of T-DNA- segment by ATPase domains.
This causes DNA at the gate to be stretched through a conformational transition to the cleaved state. The exact mechanism is unclear. This is asymmetric since the T-segment probably does not enter orthogonally to the G-segment [25] and causes one metal to bind at the A-configuration on one side only (but see Supplementary Fig. 10).
-
4.
The metal at the first cleaved site now moves to the B-configuration and, as the T-DNA continues to push the DNA-gate open, the second scissile phosphate moves close to the TOPRIM domain, allowing cleavage of the second DNA strand through capture of a second metal.
-
5.
The DNA-gate now opens as the T-DNA is pushed downward and through.
-
6.
Once the T-DNA has moved through, the DNA-gate swings closed (like a swinging-door).
-
7.
The T-DNA is now between the DNA-gate and exit gate. The T-segment segment has then the potential to bind one or two of the GK domains. We hypothesize that interaction with only one of the GK domains might underlie the stepwise religation. This binding might influence the cleavage status through the YKGLG motif being moved to hold the catalytic metal in the B-configuration and to prevent the catalytic metal moving back to the A-configuration while the exit gate is opening. The exact nature of this coupling is unclear.
-
8.
The opening of the exit gate releases the T-segment and forces religation, presumably through the motion described in Ref. [22].
Conclusions and Future Directions
The main subject of this article is the interpretation of x-ray crystal structures of topoisomerase–DNA–drug complexes. However, it remains difficult to obtain high-resolution (< 2-Å) x-ray crystal structures of DNA complexes of the DNA-cleavage gate of type IIA topoisomerases. As mentioned above, several low- and medium-low-resolution structures with potentially ambiguous interpretations of ligand binding and/or metal coordination geometry at the DNA-cleavage sites have been published [12], [15], [21], [22]. Two of these have now been re-refined and the coordinates made available.
Overall, the most parsimonious interpretation of the current structural data is that only a single metal ion is ever present at any one DNA-cleavage active site at any one time. Although the possibility that two metals could bind at one active site at the same time cannot be ruled out, in our opinion, convincing crystallographic evidence of this two-metal possibility has yet to be demonstrated. Despite the fact that coordination geometry for many metal ions has been reasonably well defined by small-molecule crystal structures, there are a number of structures in the PDB where the coordination geometry of the metal binding sites does not match what has been established with small molecules. Our single-metal model for the cleavage–religation cycle preserves an acceptable coordination geometry for the catalytic metal. However, it remains to be directly tested and the dynamics of the metal at the catalytic pocket remain to be explored.
Based on both the crystallographic and biochemical data, we have proposed two different models in which type IIA topoisomerases control DNA cleavage and religation prior to and after strand passage, respectively, through a number of conformational changes (Fig. 7, Fig. 8 and Supplementary Figs. 9 and 10). These changes result in changes of DNA conformation at the cleavage catalytic pocket, which favor the capture of a single catalytic metal ion per catalytic pocket. The metal status is determined by the position of the scissile phosphate and the preceding phosphate, which form part of the coordination site responsible for the catalysis of the phosphotransfer reaction (the A-configuration). The scissile phosphate moves away when the DNA is cleaved (notably by the opening of the DNA gate), and the metal can be transferred to the B-configuration which indirectly interacts with the 3´-OH side of the cleavage, thereby keeping the catalyst with the nucleophile needed for the reverse phophotransfer reaction and away from any other nucleophile (like water for instance). The enzyme controls religation by controlling the position of the scissile phosphate, which approaches the B-configuration metal after closure of the DNA gate and presumably recaptures the catalytic metal.
In this model, the action of cleavage complex-stabilizing compounds is twofold. First, they bind to pockets that are only present when the enzyme has the ability to cleave DNA. They therefore stabilize a conformation of the enzyme in which cleavage is rendered possible by shortening the distance between the scissile phosphates and their cognate catalytic pockets. This constitutes the “allosteric” mode of action. Second, compounds can also interact directly with the DNA in the central region, thereby influencing the position of the scissile phosphate and the cleavage status of the DNA. This explain why some compounds stabilize various proportion of single- and double-strand breaks.
Structural biology is yet to precisely define the domain movements associated with T-DNA segment movement, although a number of studies have helped define and develop current understanding of DNA gyrase dynamics [60], [62], [63], [64], [65], [66]. Of particular interest is how the structural transition assumed by the enzyme as it progresses along its DNA binding, cleavage and religation cycle, are coupled to the transport of the T-DNA segment across the enzyme interface and ultimately ATP binding and hydrolysis. It is well established that strand passage is coupled to ATP binding and hydrolysis, although the precise mode of this coupling is yet to be understood [67]. Consistent with the idea that the ATP binding and hydrolysis cycle influence the conformational changes involved in cleavage, it was found that ATP accelerates the appearance of cleavage induced by ciprofloxacin and the IPY compounds [11], [61]. It should be noted that T-DNA capture is asymmetric and a sequential ATP hydrolysis model has been proposed for yeast topo II in which strand passage is coupled to the hydrolysis of the first ATP [68], [69], [70]. It is tempting to speculate that the asymmetry in cleavage reflects the intrinsic, and transient, asymmetry of the strand-passage reaction.
Acknowledgments
This work was supported by a Wellcome Trust Investigator Award (110072/Z/15/Z to A.M.), by the BBSRC Institute Strategic Programme Grants BB/J004561/1 and BB/P012523/1 (A.M.) and by funding from the Innovative Medicines Initiative Joint Undertaking under grant agreement no. 115583, resources of which are composed of financial contribution from the European Union's Seventh Framework Programme (FP7/2007-2013) and EFPIA companies' in-kind contribution. The ENABLE project is also financially supported by contributions from Academic and SME partners.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmb.2019.07.008.
Contributor Information
Benjamin D. Bax, Email: baxb@cardiff.ac.uk.
Thomas Germe, Email: Thomas.Germe@jic.ac.uk.
Appendix A. Supplementary data
Supplementary material
Supplementary figures
References
- 1.Bush N.G., Evans-Roberts K., Maxwell A. DNA topoisomerases. EcoSal Plus. 2015;6 doi: 10.1128/ecosalplus.ESP-0010-2014. [DOI] [PubMed] [Google Scholar]
- 2.Collin F., Karkare S., Maxwell A. Exploiting bacterial DNA gyrase as a drug target: current state and perspectives. Appl. Microbiol. Biotechnol. 2011;92:479–497. doi: 10.1007/s00253-011-3557-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Espeli O., Marians K.J. Untangling intracellular DNA topology. Mol. Microbiol. 2004;52:925–931. doi: 10.1111/j.1365-2958.2004.04047.x. [DOI] [PubMed] [Google Scholar]
- 4.Pommier Y., Sun Y., Huang S.-Y.N., Nitiss J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016 Nov;17(11):703–721. doi: 10.1038/nrm.2016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat. Rev. Mol. Cell Biol. 2002;3:430–40. doi: 10.1038/nrm831. [DOI] [PubMed]
- 6.Nitiss J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer. 2009;9:338–350. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan P.F., Huang J., Bax B.D., Gwynn M.N. Recent developments in inhibitors of bacterial type IIA topoisomerases. In: Gualerzi C.O., Brandi L., Fabbretti A., Pon C.L., editors. Antibiotics. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2013. pp. 263–297. [Google Scholar]
- 8.Drlica K., Malik M. Fluoroquinolones: action and resistance. Curr. Top. Med. Chem. 2003;3:249–282. doi: 10.2174/1568026033452537. [DOI] [PubMed] [Google Scholar]
- 9.Markham A. Delafloxacin: first global approval. Drugs. 2017;77:1481–1486. doi: 10.1007/s40265-017-0790-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan P.F., Srikannathasan V., Huang J., Cui H., Fosberry A.P., Gu M. Structural basis of DNA gyrase inhibition by antibacterial QPT-1, anticancer drug etoposide and moxifloxacin. Nat. Commun. 2015;6:10048. doi: 10.1038/ncomms10048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Germe T., Voros J., Jeannot F., Taillier T., Stavenger R.A., Bacque E. A new class of antibacterials, the imidazopyrazinones, reveal structural transitions involved in DNA gyrase poisoning and mechanisms of resistance. Nucleic Acids Res. 2018 May 4;46(8):4114–4128. doi: 10.1093/nar/gky181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laponogov I., Sohi M.K., Veselkov D.A., Pan X.-S., Sawhney R., Thompson A.W. Structural insight into the quinolone–DNA cleavage complex of type IIA topoisomerases. Nat. Struct. Mol. Biol. 2009;16:667–669. doi: 10.1038/nsmb.1604. [DOI] [PubMed] [Google Scholar]
- 13.Wohlkonig A., Chan P.F., Fosberry A.P., Homes P., Huang J., Kranz M. Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat. Struct. Mol. Biol. 2010;17:1152–1153. doi: 10.1038/nsmb.1892. [DOI] [PubMed] [Google Scholar]
- 14.Wu C.C., Li T.K., Farh L., Lin L.Y., Lin T.S., Yu Y.J. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science. 2011;333:459–462. doi: 10.1126/science.1204117. [DOI] [PubMed] [Google Scholar]
- 15.Bax B.D., Chan P.F., Eggleston D.S., Fosberry A., Gentry D.R., Gorrec F. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature. 2010;466:935–940. doi: 10.1038/nature09197. [DOI] [PubMed] [Google Scholar]
- 16.Chan PF, Germe T, Bax BD, Huang J, Thalji RK, Bacque E, et al. Thiophene antibacterials that allosterically stabilize DNA-cleavage complexes with DNA gyrase. Proc. Natl. Acad. Sci. U. S. A. 2017;114:E4492-E500. [DOI] [PMC free article] [PubMed]
- 17.Biedenbach D.J., Bouchillon S.K., Hackel M., Miller L.A., Scangarella-Oman N.E., Jakielaszek C. In vitro activity of gepotidacin, a novel triazaacenaphthylene bacterial topoisomerase inhibitor, against a broad spectrum of bacterial pathogens. Antimicrob. Agents Chemother. 2016;60:1918–1923. doi: 10.1128/AAC.02820-15. https://doi.org/10.128/AAC.02820–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacobsson S., Golparian D., Alm R.A., Huband M., Mueller J., Jensen J.S. High in vitro activity of the novel spiropyrimidinetrione AZD0914, a DNA gyrase inhibitor, against multidrug-resistant Neisseria gonorrhoeae isolates suggests a new effective option for oral treatment of gonorrhea. Antimicrob. Agents Chemother. 2014;58:5585–5588. doi: 10.1128/AAC.03090-14. (Epub 2014 Jun 30) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Riordan W, Tiffany C, Scangarella-Oman N, Perry C, Hossain M, Ashton T, et al. Efficacy, safety, and tolerability of gepotidacin (GSK2140944) in the treatment of patients with suspected or confirmed gram-positive acute bacterial skin and skin structure infections. Antimicrob. Agents Chemother. 2017;61(6).AAC.02095–16. doi: 10.1128/AAC.-16. Print 2017 Jun. [DOI] [PMC free article] [PubMed]
- 20.Vos S.M., Lee I., Berger M. Distinct regions of the Escherichia coli ParC C-terminal domain are required for substrate discrimination by topoisomerase IV. J. Mol. Biol. 2013;425:3029–3045. doi: 10.1016/j.jmb.2013.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laponogov I., Pan X.S., Veselkov D.A., McAuley K.E., Fisher L.M., Sanderson M.R. Structural basis of gate-DNA breakage and resealing by type II topoisomerases. PLoS One. 2010;5 doi: 10.1371/journal.pone.0011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmidt B.H., Burgin A.B., Deweese J.E., Osheroff N., Berger J.M. A novel and unified two-metal mechanism for DNA cleavage by type II and IA topoisomerases. Nature. 2010;465:641–644. doi: 10.1038/nature08974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giacovazzo C. Oxford University Press; New York: 2002. Fundamentals of Crystallography. [Google Scholar]
- 24.Srikannathasan V., Wohlkonig A., Shillings A., Singh O., Chan P.F., Huang J. Crystallization and initial crystallographic analysis of covalent DNA-cleavage complexes of Staphyloccocus aureus DNA gyrase with QPT-1, moxifloxacin and etoposide. Acta crystallographica Section F, Structural biology communications. 2015;71:1242–1246. doi: 10.1107/S2053230X15015290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen S.F., Huang N.L., Lin J.H., Wu C.C., Wang Y.R., Yu Y.J. Structural insights into the gating of DNA passage by the topoisomerase II DNA-gate. Nat. Commun. 2018;9:3085. doi: 10.1038/s41467-018-05406-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong K.C., Berger J.M. Structural basis for gate-DNA recognition and bending by type IIA topoisomerases. Nature. 2007;450:1201–1205. doi: 10.1038/nature06396. [DOI] [PubMed] [Google Scholar]
- 27.Morais Cabral J.H., Jackson A.P., Smith C.V., Shikotra N., Maxwell A., Liddington R.C. Crystal structure of the breakage-reunion domain of DNA gyrase. Nature. 1997;388:903–906. doi: 10.1038/42294. [DOI] [PubMed] [Google Scholar]
- 28.Bock C.W., Katz A.K., Markham G.D., Glusker J.P. Manganese as a replacement for magnesium and zinc: functional comparison of the divalent ions. J. Am. Chem. Soc. 1999;121:7360–7372. [Google Scholar]
- 29.Aldred K.J., Kerns R.J., Osheroff N. Mechanism of quinolone action and resistance. Biochemistry. 2014;53:1565–1574. doi: 10.1021/bi5000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blower T.R., Williamson B.H., Kerns R.J., Berger J.M. Crystal structure and stability of gyrase-fluoroquinolone cleaved complexes from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 2016;113:1706–1713. doi: 10.1073/pnas.1525047113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aldred K.J., McPherson S.A., Turnbough C.L., Jr., Kerns R.J., Osheroff N. Topoisomerase IV-quinolone interactions are mediated through a water–metal ion bridge: mechanistic basis of quinolone resistance. Nucleic Acids Res. 2013;41:4628–4639. doi: 10.1093/nar/gkt124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aldred K.J., Blower T.R., Kerns R.J., Berger J.M., Osheroff N. Fluoroquinolone interactions with Mycobacterium tuberculosis gyrase: enhancing drug activity against wild-type and resistant gyrase. Proc. Natl. Acad. Sci. U. S. A. 2016;113:E839–E846. doi: 10.1073/pnas.1525055113. (Epub 2016 Jan 20) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laponogov I, Pan XS, Veselkov DA, Cirz RT, Wagman A, Moser HE, et al. Exploring the active site of the Streptococcus pneumoniae topoisomerase IV-DNA cleavage complex with novel 7,8-bridged fluoroquinolones. Open Biol. 2016;6(9).rsob.160157. doi: 10.1098/rsob. [DOI] [PMC free article] [PubMed]
- 34.Wagman A.S., Cirz R., McEnroe G., Aggen J., Linsell M.S., Goldblum A.A. Synthesis and microbiological evaluation of novel tetracyclic fluoroquinolones. ChemMedChem. 2017;12:1687–1692. doi: 10.1002/cmdc.201700426. (Epub 2017 Oct 2) [DOI] [PubMed] [Google Scholar]
- 35.Towle TR, Kulkarni CA, Oppegard LM, Williams BP, Picha TA, Hiasa H, et al. Design, synthesis, and evaluation of novel N-1 fluoroquinolone derivatives: probing for binding contact with the active site tyrosine of gyrase. Bioorg. Med. Chem. Lett. 2018;28:1903–10. doi: 10.016/j.bmcl.2018.03.085. Epub Mar 30. [DOI] [PMC free article] [PubMed]
- 36.Redgrave L.S., Sutton S.B., Webber M.A., Piddock L.J.V. Fluoroquinolone resistance: mechanisms, impact on bacteria, and role in evolutionary success. Trends Microbiol. 2014;22:438–445. doi: 10.1016/j.tim.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 37.Jeannot F., Taillier T., Despeyroux P., Renard S., Rey A., Mourez M. Imidazopyrazinones (IPYs): non-quinolone bacterial topoisomerase inhibitors showing partial cross-resistance with quinolones. J. Med. Chem. 2018;61:3565–3581. doi: 10.1021/acs.jmedchem.7b01892. [DOI] [PubMed] [Google Scholar]
- 38.Critchlow S.E., Maxwell A. DNA cleavage is not required for the binding of quinolone drugs to the DNA gyrase–DNA complex. Biochemistry. 1996;35:7387–7393. doi: 10.1021/bi9603175. [DOI] [PubMed] [Google Scholar]
- 39.Bromberg K.D., Burgin A.B., Osheroff N. A two-drug model for etoposide action against human topoisomerase IIalpha. J. Biol. Chem. 2003;278:7406–7412. doi: 10.1074/jbc.M212056200. (Epub 2002 Dec 8) [DOI] [PubMed] [Google Scholar]
- 40.Infante Lara L., Fenner S., Ratcliffe S., Isidro-Llobet A., Hann M., Bax B. Coupling the core of the anticancer drug etoposide to an oligonucleotide induces topoisomerase II-mediated cleavage at specific DNA sequences. Nucleic Acids Res. 2018;46:2218–2233. doi: 10.1093/nar/gky072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lesuisse D., Tabart M. 1.2 Importance of chirality in the field of anti-infective agents. In: Carreira E.M., Yamamoto H., editors. Comprehensive Chirality. Elsevier; Amsterdam: 2012. pp. 8–29. [Google Scholar]
- 42.Miles T.J., Hennessy A.J., Bax B., Brooks G., Brown B.S., Brown P. Novel hydroxyl tricyclics (e.g., GSK966587) as potent inhibitors of bacterial type IIA topoisomerases. Bioorg. Med. Chem. Lett. 2013;23:5437–5441. doi: 10.1016/j.bmcl.2013.07.013. [DOI] [PubMed] [Google Scholar]
- 43.Miles T.J., Hennessy A.J., Bax B., Brooks G., Brown B.S., Brown P. Novel tricyclics (e.g., GSK945237) as potent inhibitors of bacterial type IIA topoisomerases. Bioorg. Med. Chem. Lett. 2016;26:2464–2469. doi: 10.1016/j.bmcl.2016.03.106. [DOI] [PubMed] [Google Scholar]
- 44.Singh S.B., Kaelin D.E., Wu J., Miesel L., Tan C.M., Meinke P.T. Oxabicyclooctane-linked novel bacterial topoisomerase inhibitors as broad spectrum antibacterial agents. ACS Med. Chem. Lett. 2014;5:609–614. doi: 10.1021/ml500069w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singh SB, Kaelin DE, Wu J, Miesel L, Tan CM, Black T, et al. Tricyclic 1,5-naphthyridinone oxabicyclooctane-linked novel bacterial topoisomerase inhibitors as broad-spectrum antibacterial agents-SAR of left-hand-side moiety (Part-2). Bioorg Med Chem Lett. 2015;25:1831–5. doi: 10.016/j.bmcl.2015.03.044. Epub Mar 24. [DOI] [PubMed]
- 46.Kolaric A., Minovski N. Novel bacterial topoisomerase inhibitors: challenges and perspectives in reducing hERG toxicity. Future Med. Chem. 2018;10:2241–2244. doi: 10.4155/fmc-2018-0272. (Epub 2018 Sep 14) [DOI] [PubMed] [Google Scholar]
- 47.Arnoldi E., Pan X.S., Fisher L.M. Functional determinants of gate-DNA selection and cleavage by bacterial type II topoisomerases. Nucleic Acids Res. 2013;41:9411–9423. doi: 10.1093/nar/gkt696. (Epub 2013 Aug 12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thalji RK, Raha K, Andreotti D, Checchia A, Cui H, Meneghelli G, et al. Structure-guided design of antibacterials that allosterically inhibit DNA gyrase. Bioorg. Med. Chem. Lett. 2019;22:30168–4. [DOI] [PubMed]
- 49.Zechiedrich E.L., Christiansen K., Andersen A.H., Westergaard O., Osheroff N. Double-stranded DNA cleavage/religation reaction of eukaryotic topoisomerase II: evidence for a nicked DNA intermediate. Biochemistry. 1989;28:6229–6236. doi: 10.1021/bi00441a014. [DOI] [PubMed] [Google Scholar]
- 50.Maxwell A., Bush N.G., Germe T., SJ McKie. Non-quinolone topoisomerase inhibitors. In: Fong I.W., Shlaes D., Drlica K., editors. Antimicrobial Resistance in the 21st Century. Springer; New York: 2018. pp. 593–618. [Google Scholar]
- 51.Wendorff T.J., Schmidt B.H., Heslop P., Austin C.A., Berger J.M. The structure of DNA-bound human topoisomerase II alpha: conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J. Mol. Biol. 2012;424:109–124. doi: 10.1016/j.jmb.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gibson E.G., Bax B., Chan P.F., Osheroff N. Mechanistic and structural basis for the actions of the antibacterial gepotidacin against Staphylococcus aureus gyrase. ACS Infect Dis. 2019;28 doi: 10.1021/acsinfecdis.8b00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hooper D.C., Jacoby G.A. Topoisomerase inhibitors: fluoroquinolone mechanisms of action and resistance. Cold Spring Harbor pers in med. 2016;6 doi: 10.1101/cshperspect.a025320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aldred K.J., McPherson S.A., Wang P., Kerns R.J., Graves D.E., Turnbough C.L., Jr. Drug interactions with Bacillus anthracis topoisomerase IV: biochemical basis for quinolone action and resistance. Biochemistry. 2012;51:370–381. doi: 10.1021/bi2013905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deweese J.E., Burgin A.B., Osheroff N. Using 3′-bridging phosphorothiolates to isolate the forward DNA cleavage reaction of human topoisomerase IIalpha. Biochemistry. 2008;47:4129–4140. doi: 10.1021/bi702194x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deweese J.E., Burgin A.B., Osheroff N. Human topoisomerase IIalpha uses a two-metal-ion mechanism for DNA cleavage. Nucleic Acids Res. 2008;36:4883–4893. doi: 10.1093/nar/gkn466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Noble C.G., Maxwell A. The role of GyrB in the DNA cleavage–religation reaction of DNA gyrase: a proposed two metal-ion mechanism. J. Mol. Biol. 2002;318:361–371. doi: 10.1016/S0022-2836(02)00049-9. [DOI] [PubMed] [Google Scholar]
- 58.Mueller-Planitz F., Herschlag D. DNA topoisomerase II selects DNA cleavage sites based on reactivity rather than binding affinity. Nucleic Acids Res. 2007;35:3764–3773. doi: 10.1093/nar/gkm335. (Epub 2007 May 21) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Deweese J.E., Osheroff N. The use of divalent metal ions by type II topoisomerases. Metallomics. 2010;2:450–459. doi: 10.1039/c003759a. (Epub 2010 May 21) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Soczek K.M., Grant T., Rosenthal P.B., Mondragon A. CryoEM structures of open dimers of gyrase A in complex with DNA illuminate mechanism of strand passage. Elife. 2018;7:41215. doi: 10.7554/eLife.41215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kampranis S.C., Maxwell A. The DNA gyrase–quinolone complex. ATP hydrolysis and the mechanism of DNA cleavage. J. Biol. Chem. 1998;273:22615–22626. doi: 10.1074/jbc.273.35.22615. [DOI] [PubMed] [Google Scholar]
- 62.Gubaev A., Klostermeier D. DNA-induced narrowing of the gyrase N-gate coordinates T-segment capture and strand passage. Proc. Natl. Acad. Sci. U. S. A. 2011;108:14085–14090. doi: 10.1073/pnas.1102100108. (Epub 2011 Aug 4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lanz M.A., Klostermeier D. Guiding strand passage: DNA-induced movement of the gyrase C-terminal domains defines an early step in the supercoiling cycle. Nucleic Acids Res. 2011;39:9681–9694. doi: 10.1093/nar/gkr680. (Epub 2011 Aug 31) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Basu A., Schoeffler A.J., Berger J.M., Bryant Z. ATP binding controls distinct structural transitions of Escherichia coli DNA gyrase in complex with DNA. Nature structural & molecular biology. 2012;19:538–546. doi: 10.1038/nsmb.2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rudolph M.G., Klostermeier D. Mapping the spectrum of conformational states of the DNA- and C-gates in Bacillus subtilis gyrase. J. Mol. Biol. 2013;425:2632–2640. doi: 10.1016/j.jmb.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 66.Basu A, Parente AC, Bryant Z. Structural dynamics and mechanochemical coupling in DNA gyrase. J Mol Biol. 2016;428:1833–45. doi: 10.016/j.jmb.2016.03.016. Epub Mar 22. [DOI] [PMC free article] [PubMed]
- 67.Bates A.D., Maxwell A. Energy coupling in type II topoisomerases: why do they hydrolyze ATP? Biochemistry. 2007;46:7929–7941. doi: 10.1021/bi700789g. (Epub 2007 Jun 20) [DOI] [PubMed] [Google Scholar]
- 68.Harkins T.T., Lewis T.J., Lindsley J.E. Pre-steady-state analysis of ATP hydrolysis by Saccharomyces cerevisiae DNA topoisomerase II. 2. Kinetic mechanism for the sequential hydrolysis of two ATP. Biochemistry. 1998;37:7299–7312. doi: 10.1021/bi9729108. [DOI] [PubMed] [Google Scholar]
- 69.Harkins T.T., Lindsley J.E. Pre-steady-state analysis of ATP hydrolysis by Saccharomyces cerevisiae DNA topoisomerase II. 1. A DNA-dependent burst in ATP hydrolysis. Biochemistry. 1998;37:7292–7298. doi: 10.1021/bi9729099. [DOI] [PubMed] [Google Scholar]
- 70.Baird C.L., Harkins T.T., Morris S.K., Lindsley J.E. Topoisomerase II drives DNA transport by hydrolyzing one ATP. Proc. Natl. Acad. Sci. U. S. A. 1999;96:13685–13690. doi: 10.1073/pnas.96.24.13685. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material
Supplementary figures