

Abstract
Induction of hippocampal long-term potentiation (LTP) requires activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII), whereas maintenance of LTP additionally requires protein synthesis. We recently reported that CaMKII stimulates protein synthesis in depolarized hippocampal neurons through phosphorylation of the mRNA translation factor cytoplasmic polyadenylation element-binding protein (CPEB), and this phosphorylation is rapidly reversed by protein phosphatase 1 (PP1). Protein synthesis-dependent late-phase LTP (L-LTP) in the hippocampus requires calcium influx through the NMDA-type glutamate receptor (NMDA-R) to activate CaMKII as well as concomitant inhibition of PP1 mediated by protein kinase A. Therefore, we investigated the regulation of CPEB phosphorylation during L-LTP. Pharmacological stimulation of the NMDA-R in hippocampal slices to produce chemical long-term depression induced a brief dephosphorylation of CPEB. Modest LTP induction (once at 100 Hz), which induces a protein synthesis-independent early-phase LTP (E-LTP), resulted in a transient phosphorylation of CPEB. However, stronger stimulation (four times at 100 Hz), known to induce protein synthesis-dependent L-LTP, elicited a prolonged phosphorylation of CPEB. Furthermore, CPEB phosphorylation correlated with phosphorylation of PP1 inhibitor dopamine- and cAMP-regulated phosphoprotein, a known substrate for protein kinase A. These results evoke the hypothesis that bidirectional regulation of CPEB phosphorylation by CaMKII and protein phosphatases may serve as a mechanism to convert E-LTP into protein synthesis-dependent L-LTP by stimulating protein synthesis and thereby stabilizing synaptic enhancement.
Keywords: CaMKII, CPEB, DARPP-32, protein phosphatase, long-term potentiation, protein synthesis
Introduction
Long-lasting changes in synaptic strength, such as long-term potentiation (LTP), require intertwining biochemical cascades for their induction and maintenance. Induction of early-phase LTP (E-LTP) in the CA1 region of the hippocampus requires Ca2+ influx through the NMDA-type glutamate receptor (NMDA-R) to activate Ca2+/calmodulin-dependent protein kinase II (CaMKII) (Lisman, 2003). To convert E-LTP into late-phase LTP (L-LTP), inhibition of protein phosphatase 1 (PP1) is essential to prolong CaMKII activation and phosphorylation of downstream substrates (Blitzer et al., 1995, 1998; Brown et al., 2000; Bradshaw et al., 2003a). PP1 is inhibited during L-LTP by stimulation of Ca2+/calmodulin-dependent adenylyl cyclases, raising cAMP levels to activate protein kinase A (PKA) (Roberson and Sweatt, 1996; Nguyen and Kandel, 1997; Wong et al., 1999; Otmakhova et al., 2000). PKA phosphorylates dopamine- and cAMP-regulated phosphoprotein (DARPP-32) and inhibitor-1, potent inhibitors of PP1 when phosphorylated (Foulkes et al., 1983; Hemmings et al., 1984; Blitzer et al., 1995, 1998; Brown et al., 2000). Thus, activation of CaMKII combined with inhibition of PP1 “gates” E-LTP into L-LTP.
To maintain L-LTP, gene transcription and protein synthesis are required (Frey et al., 1988; Nguyen et al., 1994; Woo and Nguyen, 2003). Although mechanisms for initiating gene transcription during LTP are well known (Kandel, 2001), stimulation of protein synthesis is poorly understood. Recent evidence indicates that protein synthesis is stimulated in dendrites near synapses undergoing LTP (Bradshaw et al., 2003b; Klann et al., 2004). Hippocampal dendrites contain mRNAs, including CaMKII (Burgin et al., 1990), NMDA-R subunit 1 (Benson, 1997), activity-regulated cytoskeletal protein (Steward et al., 1998), and the AMPA-type glutamate receptor (AMPA-R) subunit 1 (GluR1) (Ju et al., 2004). All of the components necessary for protein synthesis are present in dendrites (Steward and Schuman, 2001), and after LTP induction, polyribosomes are recruited into spines (Ostroff et al., 2002), as is CaMKII mRNA (Havik et al., 2003). This leads to synthesis of CaMKII protein in dendrites within 5 min of LTP induction (Ouyang et al., 1999).
Regulation of dendritic protein synthesis can be accomplished by phosphorylation of several mRNA translation factors (Klann et al., 2004). For example, the translation factor cytoplasmic polyadenylation element (CPE)-binding protein (CPEB) binds the 3′-untranslated region (UTR) of mRNAs containing the CPE, which includes CaMKII mRNA (Wu et al., 1998). When phosphorylated, CPEB stimulates mRNA polyadenylation and translation initiation (Mendez et al., 2000a,b). Although originally characterized in Xenopus (Mendez et al., 2000b), CPEB is also found in hippocampal dendrites (Huang et al., 2002).
We recently demonstrated that CPEB is phosphorylated by CaMKII, but not by other protein kinases known to be activated during LTP, in cultured hippocampal neurons after depolarization (Atkins et al., 2004). CaMKII robustly phosphorylated CPEB and stimulated CPEB-dependent protein synthesis. To extend our studies, we investigated CPEB phosphorylation in hippocampal slices during LTD and LTP. Our results demonstrate that CPEB phosphorylation is bidirectionally regulated by CaMKII and PP1 in hippocampal slices, and that CPEB phosphorylation is prolonged during protein synthesis-dependent L-LTP.
Materials and Methods
Western blot analysis. Lysates were electrophoresed (10% SDS-PAGE) and Western blotted using the following antibodies: phospho-CPEB (pCPEB) threonine 171 (Thr171; 1:20 of hybridoma supernatant) (Atkins et al., 2004), glutathione S-transferase (1:10,000; kind gift from Dr. James W. Tracy, University of Wisconsin-Madison, Madison, WI), total CPEB (1:20 of hybridoma supernatant), phospho-GluR1 serine 831 (Ser831; 1:1000; Upstate Cell Signaling Solutions, Charlottesville, VA), GluR1 (1: 5000; Upstate Cell Signaling Solutions), phospho-CaMKII threonine 286 (Thr286; 1:1000; Affinity BioReagents, Golden, CO), CaMKII (1:2000; Affinity BioReagents), phospho-DARPP-32 threonine 34 (Thr34; 1:500; Cell Signaling Technology, Beverly, MA), DARPP-32 (1:1000; Cell Signaling Technology), and β-tubulin (1:5000; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA). Epitopes were visualized with HRP-conjugated secondary antibodies (1:1000 to 1:5000; Amersham Biosciences, Arlington Heights, IL) using ECL or ECL Plus (Amersham Biosciences) and developed on film (Phenix x-ray film BX; Phenix Research Products, Hayward, CA). Film was developed to be in a linear range and densitized using Kodak (Rochester, NY) Digital Science 1D software. Levels of phospho-protein immunoreactivity (e.g., phospho-CPEB) were normalized to total protein immunoreactivity (e.g., CPEB) and then to β-tubulin immunoreactivity to control for total protein levels. Statistical analyses were one-way ANOVAs with Tukey's t tests. Data presented are mean ± SEM values.
Hippocampal slice physiology. Transverse hippocampal slices (400 μm) were prepared from male Sprague Dawley rats (3-4 weeks of age) with a vibratome in sucrose-artificial CSF (ACSF) containing the following: 110 mm sucrose, 60 mm NaCl, 3 mm KCl, 1.25 mm NaH2PO4, 28 mm NaHCO3, 5 mm d-glucose, 500 μm CaCl2, 7 mm MgCl2, and 600 μm ascorbate, saturated with 95% O2/5% CO2. Slices were recovered in standard ACSF (in mm: 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 10 d-glucose, 2 CaCl2, and 1 MgCl2, saturated with 95% O2/5% CO2) for 30 min at 37°C and then recovered for 1-2 h at room temperature. Slices were equilibrated for 60-90 min in a Fine Science Tools (Foster City, CA) interface chamber at 31°C, except for NMDA-induced chemical long-term depression (Chem-LTD) experiments, in which recordings were done in a submersion chamber. A bipolar Teflon-coated platinum-iridium-stimulating electrode was placed in the Schaffer collateral/commissural pathway, and a borosilicate-recording micro-electrode (1-3 MΩ; filled with ACSF) was placed in the stratum radiatum of area CA1 in the hippocampus. Recordings were bandpass filtered (1-10 kHz) and then digitally sampled at 10 kHz with Molecular Devices (Union City, CA) Digidata 1200. Data were collected and analyzed using Axoscope 8.0 software. Field EPSP (fEPSP) strength was quantified by measuring the initial slope. Test stimuli were delivered and recorded at 0.0167 Hz, and stimulus intensity (5-50 μA) was adjusted to produce a field response of ∼1.5 mV, a 30-50% maximal response. Stable baselines were recorded for at least 20 min before LTP or LTD induction. LTP was induced by a single 100 Hz train 1 s long (1 × 100 Hz) with no increase in stimulus strength to ensure L-LTP was not induced or four 100 Hz trains each 1 s long separated by 5 min (4 × 100 Hz). The antagonists dl-2-amino-5-phosphonovaleric acid (APV) and 2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)amino-N-(4-chlorocinnamyl-N-methyl-benzylamine) (KN-93) were added to the ACSF solution for 30 min before baseline recording and were applied continuously throughout the recordings. For biochemical analysis, the slices were frozen in liquid nitrogen, and the CA1 region was microdissected.
Hippocampal slice biochemistry. Hippocampal slices were pharmacologically stimulated with NMDA for the indicated times in a submersion chamber containing 10 ml of ACSF at 32°C. To induce chemical LTD, slices were incubated with 25 μm NMDA in ACSF for 3 min at 32°C. Slices were then washed in 10 ml of ACSF and transferred to a new chamber with ACSF to assay CPEB phosphorylation at 20 and 60 min after the initial 3 min of NMDA treatment. After stimulation, the slices were transferred to a nylon screen and frozen on liquid nitrogen. For pharmacological experiments, two slices were pooled and homogenized for each condition. For physiology experiments, the CA1 region was microdissected, and one CA1 region was homogenized and assayed for each condition. Samples were homogenized using a Dounce homogenizer in lysis buffer containing the following: 50 mm HEPES, pH 7.5, 10 mm Na4P2O7, 100 mm NaF, 1 mm Na3VO4, 20 mm EDTA, 20 mm EGTA, 100 mm NaCl, 1 mm benzamidine, 20 μg/ml aprotinin, 20 μg/ml leupeptin, 1 μm microcystin-lr, 1 μm PMSF, 1× phosphatase inhibitor cocktail set I (Calbiochem, La Jolla, CA), and 1× complete protease inhibitor cocktail set (Roche Applied Science, Indianapolis, IN). Samples were boiled with 2× sample buffer supplemented with 1 μm microcystin-lr, 100 mm NaF, 100 mm β-glycerol phosphate, 20 mm EDTA, 20 mm EGTA, 1 μm PMSF, 25 μg/ml aprotinin, and 25 μg/ml leupeptin.
Materials. NMDA was obtained from Sigma (St. Louis, MO). KN-93 was obtained from Calbiochem. Tetrodotoxin (TTX) and APV were obtained from Tocris Cookson (Ellisville, MO).
Results
Regulation of CPEB phosphorylation by NMDA treatment of hippocampal slices
Depolarization of cultured hippocampal neurons produces a robust, but transient (maximal in <1 min), phosphorylation of CPEB at its regulatory site, Thr171, by CaMKII (Atkins et al., 2004). This is accompanied by a significant increase in CPEB-dependent protein synthesis measured 3 h later. Because CaMKII-mediated phosphorylation of CPEB is potently enhanced by inhibition of PP1 in cultured neurons, we speculated that the transient nature of CPEB phosphorylation after neuronal depolarization was a result of strong phosphatase activity. Thus, sustained phosphorylation of CPEB would require simultaneous activation of CaMKII and inhibition of PP1, conditions demonstrated to be essential for induction of protein synthesis-dependent L-LTP (Blitzer et al., 1995, 1998; Brown et al., 2000; Bradshaw et al., 2003a).
As a first step toward understanding the regulation of CPEB phosphorylation in hippocampal slices, we tested whether NMDA-R stimulation, which is required for hippocampal LTP, would modulate CPEB phosphorylation. NMDA-R stimulation triggers the activation of CaMKII or protein phosphatases depending on the level of receptor activation and resulting Ca2+ influx intensity and duration (Schulman, 1995). CPEB was modestly and transiently phosphorylated between 10 s and 1 min during a 10 min NMDA stimulation (Fig. 1A,B). This paralleled activation of CaMKII, as measured by autophosphorylation at Thr286 (Fig. 1A,C). However, after 10 min of NMDA-R stimulation, CaMKII activation returned to near basal levels, whereas CPEB was strongly dephosphorylated. To determine whether differential stimulation of the NMDA-R could prolong CPEB phosphorylation, an NMDA dose-response curve was performed. At all concentrations tested, NMDA treatment resulted in significant dephosphorylation of CPEB at 10 min (Fig. 1D,E). This dephosphorylation of CPEB was blocked by the NMDA-R antagonist APV, whereas an AMPA-R antagonist (DNQX) or blockade of action potential firing by the sodium channel blocker TTX was ineffective (Fig. 1F).
Figure 1.

CPEB and CaMKII phosphorylation and dephosphorylation during NMDA-R activation. A, Representative Western blots of pCPEB, total CPEB (CPEB), autophosphorylated CaMKII (pCaMKII), and total CaMKII from hippocampal slices stimulated with NMDA (25 μm) for the indicated time points. B, Densitized results of Western blot analysis for pCPEB indicated that pCPEB modestly and significantly increased only after 30 s or 1 min of NMDA stimulation (n = 16; *p < 0.05 for each). Ten minutes of NMDA stimulation led to a significant dephosphorylation of CPEB (n = 12; **p < 0.01). C, Densitized results of phospho-Thr286 CaMKII (pCaMKII) revealed a significant increase at 10 s (n = 15; *p < 0.05), 30 s (n = 16; **p < 0.01), and 1 min (n = 16; **p < 0.01). Although CPEB was dephosphorylated at 10 min of stimulation, phospho-CaMKII was still nonsignificantly elevated over baseline at 10 min (n = 12). D, Representative Western blots of phospho-Thr171 CPEB (pCPEB), total CPEB (CPEB), and a total protein control (Tubulin) during an NMDA dose-response curve. E, At all concentrations tested, NMDA application for 10 min resulted in dephosphorylation of CPEB (pCPEB; n = 8 for each concentration; ***p < 0.001). No changes in total CPEB levels were seen (CPEB). F, Dephosphorylation of CPEB with NMDA (n = 8; ***p < 0.001) was blocked by the NMDA-R antagonist APV (50 μm; n = 4) but not by the AMPA-R antagonist DNQX (20 μm; n = 4; **p < 0.01) or the sodium channel antagonist TTX (1 μm; n = 4; ***p < 0.001).
In the context of hippocampal slice physiology, brief bath application of NMDA to hippocampal slices induces Chem-LTD. The mechanisms underlying Chem-LTD are not well known, but it occludes induction of low-frequency-induced LTD (LFS-LTD), suggesting that they use common mechanisms. LFS-LTD in region CA1 of the hippocampus is mediated by dephosphorylation of Ser845 in GluR1 (Lee et al., 1998) and endocytosis of AMPA-Rs (Carroll et al., 2001) but not protein synthesis (Huber et al., 2000). Hippocampal slices were incubated for 3 min with 25 μm NMDA, and NMDA was then washed out. Bath application of NMDA transiently abolished synaptic transmission because of depolarization of the neurons, followed by a short recovery period and then a depression of evoked synaptic responses (Chem-LTD) between 40 and 70 min (Fig. 2A), essentially identical to that reported previously (Lee et al., 1998). Chem-LTD resulted in a net dephosphorylation of CPEB Thr171 at 3 min followed by a slow recovery to basal levels between 20 and 60 min (Fig. 2B). Thus, decreasing synaptic strength in the hippocampus resulted in a transient dephosphorylation of CPEB.
Figure 2.
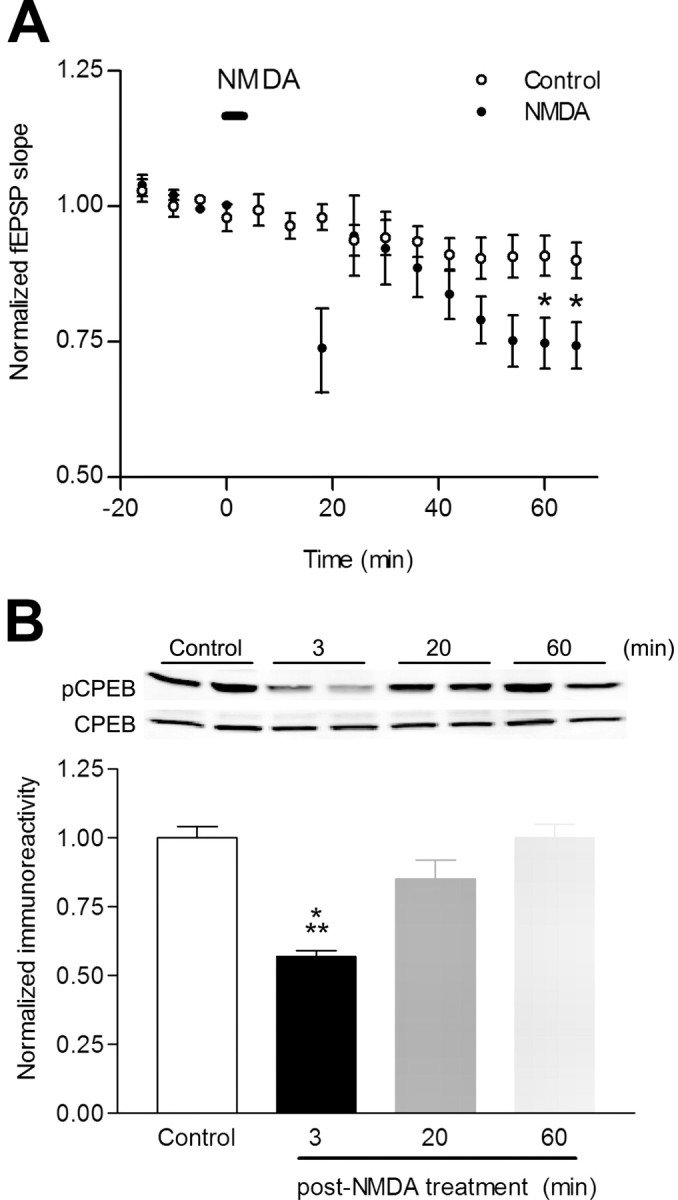
NMDA-induced Chem-LTD dephosphorylates CPEB. A, Control slices treated with vehicle (DMSO; 0.01%) for 3 min had modest rundown, whereas slices treated with NMDA (25 μm) for 3 min showed prolonged depression between 40 and 70 min after washout (t = 66 min; control fEPSP slope, 0.90 ± 0.3, n = 6; NMDA fEPSP slope, 0.74 ± 0.4, n = 12; p < 0.05). The bar indicates when NMDA was applied. B, Representative Western blots of hippocampal slices probed with phospho-CPEB and total CPEB treated with either vehicle or NMDA. Three minutes of NMDA treatment (n = 5; ***p < 0.001) significantly decreased CPEB phosphorylation with a slow recovery back to baseline levels at 20 min (n = 5) and 60 min (n = 7) after drug treatment.
Protein synthesis-dependent LTP increases CPEB phosphorylation
To determine whether increasing synaptic strength also regulates CPEB phosphorylation, we investigated the effect of LTP induction on CPEB phosphorylation. Protein synthesis-dependent L-LTP is “gated” by PKA phosphorylation of inhibitor-1/DARPP-32 to inhibit PP1 and thereby prolong CaMKII autophosphorylation and downstream substrate phosphorylation (Blitzer et al., 1995, 1998; Brown et al., 2000; Bradshaw et al., 2003a). If hippocampal slices are tetanized with modest tetanic stimulation (e.g., 1 × 100 Hz), only protein kinase-dependent E-LTP, which does not require protein synthesis, is induced and lasts for 1-3 h (Huang and Kandel, 1994). However, if multiple tetanic stimulation (e.g., 4 × 100 Hz) is used, both protein kinase-dependent E-LTP and protein synthesis-dependent L-LTP that lasts beyond 3 h are induced (Huang and Kandel, 1994; Nguyen et al., 1994; Tang et al., 2002; Kelleher et al., 2004). E-LTP activates CaMKII (Fukunaga et al., 1993; Barria et al., 1997), and L-LTP stimulates dendritic protein synthesis (Ouyang et al., 1999; Miller et al., 2002; Bradshaw et al., 2003b). Thus, prolonging CaMKII-mediated phosphorylation of CPEB by concomitant PKA-mediated phosphorylation of DARPP-32 to inhibit PP1 would be an attractive model of how protein synthesis is selectively stimulated in L-LTP.
LTP was induced with either a single 100 Hz tetanus, 1 s long, or four 100 Hz tetani, each 1 s long spaced by 5 min (Fig. 3A). E-LTP induction (1 × 100 Hz) produced a modest but transient CPEB phosphorylation that was significant at 5 min and returned to baseline levels at 10 min (Fig. 3B,C). There was no detectable increase in DARPP-32 phosphorylation with this protocol. However, an L-LTP induction protocol (4 × 100 Hz) produced a prolonged CPEB phosphorylation that lasted ≥30 min, and this paralleled phosphorylation of DARPP-32 (Fig. 3B,D).
Figure 3.

CPEB phosphorylation depends on the LTP induction paradigm. A, Hippocampal slice physiology of slices tetanized with either 1 × 100 Hz or 4 × 100 Hz stimulation. Both paradigms induced hippocampal LTP as measured by the fEPSP slope (t = 45 min; 1 × 100 Hz LTP fEPSP slope, 1.42 ± 0.1, n = 4; 4 × 100 Hz LTP fEPSP slope, 1.71 ± 0.11, n = 9). Arrows indicate when the tetanization was applied. B, Representative Western blots of hippocampal CA1 regions probed with phospho-CPEB, total CPEB, phospho-DARPP-32, total DARPP-32, and tubulin antibodies. Control slices (C) versus LTP slices (L) are compared. C, Densitized results of Western blots from slices stimulated with 1 × 100 Hz. CPEB phosphorylation significantly increased only at 5 min (n = 11; p < 0.001) and returned to baseline by 10 min (n = 7). DARPP-32 phosphorylation did not increase at any time point. D, Densitized results of Western blots from slices stimulated with 4 × 100 Hz. This protocol elicited significant increases in CPEB phosphorylation at 5 min (n = 10; ***p < 0.001), 10 min (n = 7; ***p < 0.001), and 30 min (n = 11; *p < 0.05) after tetanization. DARPP-32 phosphorylation also increased with this protocol at 5 min (n = 9; ***p < 0.001) and 10 min (n = 7; **p < 0.01) after LTP induction.
To test whether the increase in CPEB phosphorylation during L-LTP was mediated by NMDA-R activation, we tetanized hippocampal slices (4 × 100 Hz) in the presence of the NMDA-R antagonist APV and then assayed for CPEB phosphorylation. APV blocked LTP (Fig. 4A) and the increases in both CPEB and DARPP-32 phosphorylation (Fig. 4B). The general CaM kinase inhibitor KN-93 strongly reduced, but did not completely attenuate, the potentiation in fEPSP response with tetanic stimulation (Fig. 4A), but it did block the increase in CPEB phosphorylation as well as CaMKII autophosphorylation (Fig. 4B). We have shown previously that KN-93, but not its inactive analog KN-92, attenuated CPEB phosphorylation and CaMKII autophosphorylation in response to neuronal depolarization (Atkins et al., 2004). KN-93 treatment also inhibited phosphorylation of DARPP-32 surprisingly so, because CaMKII does not phosphorylate DARPP-32. However, stimulation of NMDA-R during LTP can also activate CaM-dependent phosphodiesterases (PDEs) to lower cAMP. We have shown previously that activated CaMKII phosphorylates and inhibits CaM-dependent PDEs (Hashimoto et al., 1989), which would result in elevated PKA activity and, therefore, increased phosphorylation of DARPP-32. Inhibition of CaMKII by KN-93 would have the opposite effect, thereby suppressing phosphorylation of DARPP-32, as we observed. These results suggest that CPEB phosphorylation during LTP is mediated through NMDA-R activation and CaMKII activity. Furthermore, maintenance of CPEB phosphorylation during protein synthesis-dependent L-LTP correlated with phosphorylation of the PP1 inhibitor DARPP-32.
Figure 4.
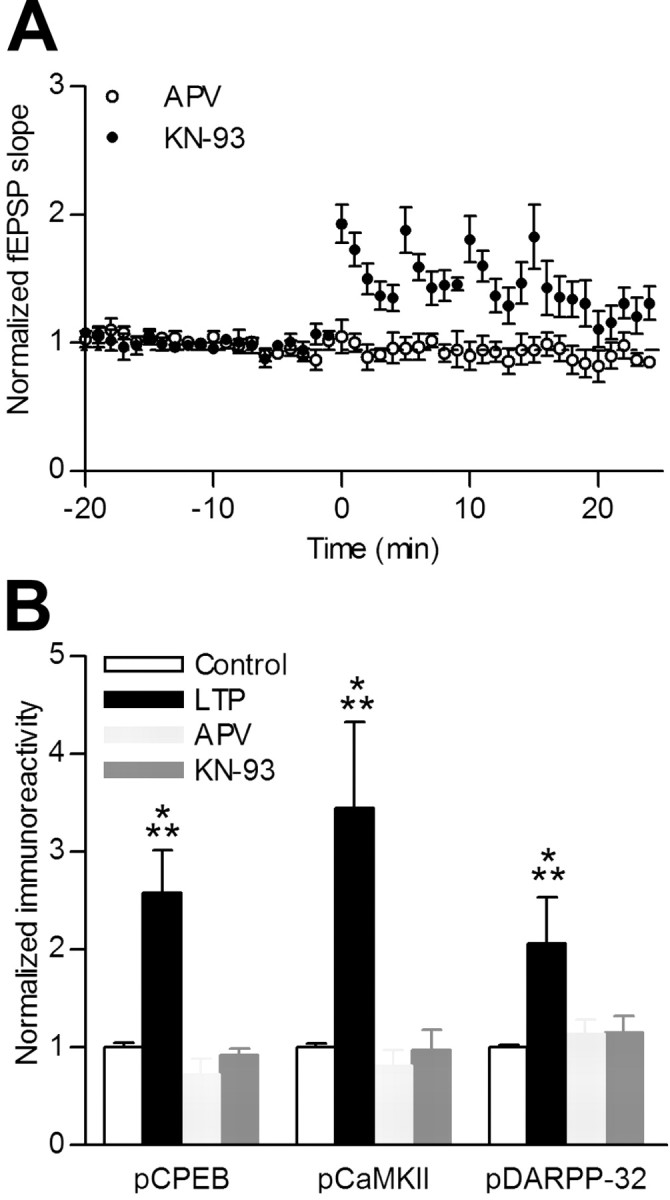
Increased CPEB phosphorylation during hippocampal LTP was blocked by the NMDA-R and CaM-kinase antagonists APV and KN-93, respectively. Arrows indicate when the tetanization was applied. A, Hippocampal slice recordings with 4 × 100 Hz tetanization in the presence of APV (50 μm; t = 10 min; normalized LTP fEPSP slope, 2.03 ± 0.38, n = 7; normalized LTP+APV fEPSP slope, 0.93 ± 0.1, n = 6; p < 0.05) or KN-93 (10 μm; normalized LTP+KN-93 fEPSP slope, 1.17 ± 0.13; n = 7). B, Densitized results from Western blots of slices stimulated with 4 × 100 Hz with APV (n = 6) or KN-93 (n = 7). Neither CPEB nor DARPP-32 phosphorylation increased 10 min after LTP tetanization when APV or KN-93 were applied. ***p < 0.001.
Discussion
Tightly controlling the phosphorylation status of substrates is integral for decoding synaptic inputs and tuning into the appropriate second-messenger signaling cascades. We have shown previously that the dendritic mRNA translation factor CPEB is phosphorylated at its regulatory site, Thr171, by CaMKII and dephosphorylated by PP1 in cultured hippocampal neurons (Atkins et al., 2004). These observations prompted the hypothesis that CaMKII-mediated phosphorylation of CPEB may regulate dendritic protein synthesis during NMDA-R-dependent LTP in the hippocampus. Our current results demonstrate that bath application of NMDA to hippocampal slices under conditions that induce Chem-LTD produced a rapid but transient dephosphorylation of CPEB. Stimulation of hippocampal slices with a paradigm that induced E-LTP (1 × 100 Hz) that is protein synthesis independent only transiently increased CPEB phosphorylation. However, induction of protein synthesis-dependent LTP (4 × 100 Hz) resulted in a prolonged CPEB phosphorylation that correlated with phosphorylation of DARPP-32, a PP1 inhibitor. Thus, our current data support the model that CaMKII-mediated phosphorylation of CPEB, coupled with inhibition of PP1 by PKA phosphorylation of DARPP-32/inhibitor 1, may be involved in regulating dendritic protein synthesis during L-LTP (Fig. 5). In support of this model, downregulation of PP1 activity during LTP has been observed previously (Mulkey et al., 1994).
Figure 5.

Model for bidirectional regulation by CaMKII and PP1 of CPEB-mediated protein synthesis. Modest LTP induction (1 × 100 Hz) stimulates Ca2+ influx through the NMDA-R (1). Ca2+ binds calmodulin (CaM) to activate CaMKII (2) through autophosphorylation. Activated CaMKII phosphorylates CPEB (3) to stimulate protein synthesis (4). The elevated Ca2+ also activates PP2B (5), which dephosphorylates DARPP-32, thereby activating PP1 to dephosphorylate CPEB (6) and limiting protein synthesis. However, robust LTP (4 × 100 Hz) further elevates Ca2+, stimulating adenylyl cyclase (AC) (7) to generate cAMP. This activates PKA (8), which phosphorylates DARPP-32, thereby inhibiting PP1 to prolong CPEB phosphorylation and protein synthesis. Green represents events that enhance protein synthesis, and red denotes inhibitory events.
During hippocampal LTP, CPEB and DARPP-32 phosphorylation were blocked by APV, an inhibitor of the NMDA-R, and by KN-93, a general CaM-kinase inhibitor. Although DARPP-32 is phosphorylated by PKA (Hemmings et al., 1984), its phosphorylation can be indirectly modulated by CaM-kinases. During hippocampal LTP, elevated intracellular Ca2+ can activate CaM-dependent PDEs to lower cAMP levels. However, this negative-feedback loop is countered by activated CaMKII, which phosphorylates and inhibits CaM-dependent PDEs (Hashimoto et al., 1989), thereby elevating PKA activity and increasing phosphorylated DARPP-32 levels. Thus, inhibition of CaMKII by KN-93 would have the opposite effect to suppress pDARPP-32, as we observed during L-LTP.
Bidirectional regulation of CPEB phosphorylation has important physiological implications, in that the ratios of key kinase/phosphatase activities determine downstream cellular responses (Fig. 5). For example, the phosphorylation status of DARPP-32 and inhibitor-1 is determined by the ratio of PKA/PP2B activities, thus gating the expression of L-LTP. L-LTP suppressed by a PKA inhibitor can also be rescued by postsynaptic injection of phosphorylated inhibitor-1 (Blitzer et al., 1998). Furthermore, L-LTP is impaired in mice lacking CaM-dependent adenylyl cyclase types 1 and 8 (Wong et al., 1999), whereas in mice overexpressing type 1 adenylyl cyclase, a 1 × 100 Hz stimulus induces sustained LTP that is blocked by a PKA inhibitor (Wang et al., 2004). Moreover, in mice expressing a PP2B inhibitor, transient suppression of PP2B activity facilitates LTP in vitro and in vivo (Malleret et al., 2001), showing that PP2B exerts an inhibitory constraint on LTP. Thus, activation of PKA overcomes PP2B activity to gate E-LTP into L-LTP. In our previous study (Atkins et al., 2004), the PKA inhibitor H-89 did not affect pCPEB induced by depolarization, but because CPEB was only transiently phosphorylated, it is unlikely that PKA was activated under those conditions.
Similarly, the ratio of CaMKII/PP1 activities determines the phosphorylation state of CPEB and, thereby, CPEB-dependent protein synthesis. Thus, induction of E-LTP produced only a transient phosphorylation of CPEB (Fig. 5). In contrast, a protocol known to induce protein synthesis-dependent L-LTP gave a prolonged CPEB phosphorylation that was sustained beyond 30 min. Moreover, the phosphorylation of CPEB during L-LTP correlated temporally with increased phosphorylation of DARPP-32 (Fig. 3). Thus, CPEB-dependent protein synthesis during L-LTP may require simultaneous activation of CaMKII and inhibition of PP1 through PKA-mediated phosphorylation of DARPP-32 or inhibitor-1. This suggests that the ratio of CaMKII/PP1 activities in LTP regulates the magnitude and duration of CPEB phosphorylation, which may in turn control the level of dendritic protein synthesis.
Although it is well established that CPEB phosphorylation stimulates protein synthesis, the role of CPEB in synaptic plasticity is not well defined. In a model of NMDA-dependent synaptic plasticity in the visual cortex, experience-induced synthesis of αCaMKII, the mRNA of which contains two CPEs (Wu et al., 1998), was dependent on mRNA polyadenylation and translation (Wu et al., 1998; Wells et al., 2001), strongly implicating the CPEB pathway. Consistent with this interpretation, translation in hippocampal neurons of transfected reporter constructs containing the 3′-UTR of CaMKII with its two CPEs was stimulated by depolarization with KCl (Atkins et al., 2004). In mice lacking the CPEB-1 gene, which encodes for the only CPEB isoform that is regulated by Thr171 phosphorylation (Theis et al., 2003), LTP induced by a single tetanus was reduced in CPEB-1 knock-out mice (Alarcon et al., 2004). However, a multiple tetanization protocol was able to essentially obviate this deficit, suggesting that other CPEB isoforms may have compensated for the lack of CPEB-1 (Theis et al., 2003). Alternatively, there are multiple mechanisms in addition to CPEB that regulate protein synthesis during LTP. For example, CPEB forms a complex with maskin, a translation inhibitory molecule; this complex sequesters the 5′-cap mRNA-binding protein, elongation initiation factor 4E (eIF4E), until CPEB is phosphorylated (Stebbins-Boaz et al., 1999; Mendez et al., 2000a; Cao and Richter, 2002). However, eIF4E is not only regulated by binding to the CPEB/maskin complex but also through the eIF4E-binding protein (4EBP1) (Matthews et al., 2000). During LTP, phosphorylation of 4EBP1 releases sequestered eIF4E to stimulate dendritic protein synthesis (Kelleher et al., 2004). Additionally, phosphorylation of eukaryotic elongation factor 2, another translation factor that binds the 5′-UTR, suppresses general protein synthesis but selectively increases CaMKII mRNA translation in synaptosomes during NMDA-R stimulation (Scheetz et al., 2000). These pathways are among several that could stimulate translation of dendritic mRNAs and compensate for the lack of CPEB-1 in the knock-out mouse (Klann et al., 2004).
Phosphorylation of CPEB Thr171 is one mechanism that regulates CPEB; however, the three other mammalian CPEB isoforms do not contain a homologous regulatory Thr171 site, suggesting that they have alternative regulatory mechanisms (Theis et al., 2003). These other mammalian CPEB isoforms do contain several predicted phosphorylation sites. For example, PKA-mediated phosphorylation of Aplysia CPEB, which lacks a homologous Thr171 site, increased actin mRNA polyadenylation and protein synthesis during long-term facilitation (Liu and Schwartz, 2003). Additionally, a tantalizing observation is that one of the Aplysia neuronal CPEB isoforms exists in two conformations; one is a prion-like state (Si et al., 2003). When in this state, CPEB-mediated protein translation is activated, possibly generating a self-perpetuating molecular memory to increase dendritic protein synthesis. This has led to the speculation that perhaps this mechanism produces a very stable change in synaptic efficacy (Si et al., 2003). It remains to be determined whether mammalian CPEB isoforms form prion-like states, and whether this occurs during hippocampal synaptic plasticity.
Initiating dendritic protein translation through CPEB phosphorylation allows for rapid, local increases in protein levels at the synapses undergoing potentiation. Although CaMKII is activated during both E-LTP and L-LTP, we found that coincident activation of CaMKII and inhibition of PP1 was necessary to sustain the CaMKII-mediated phosphorylation of CPEB during LTP (Fig. 5). Having CPEB coupled to PP1 may provide the neuron a means to tightly regulate dendritic protein synthesis during different forms of synaptic plasticity.
Footnotes
This work was supported by National Institutes of Health Grant NS 27037 (T.R.S.). We thank Sean Nygaard for technical assistance, Dr. Anthony Oliva for critical reading of this manuscript, and members of the Soderling laboratory for helpful discussions.
Correspondence should be addressed to Dr. Thomas R. Soderling, Vollum Institute, Oregon Health and Science University, 3181 Southwest Sam Jackson Park Road, Portland, OR 97239. E-mail: soderlit@ohsu.edu.
C. M. Atkins' present address: The Miami Project to Cure Paralysis, University of Miami, Miami, FL 33136.
Copyright © 2005 Society for Neuroscience 0270-6474/05/255604-07$15.00/0
References
- Alarcon JM, Hodgman R, Theis M, Huang YS, Kandel ER, Richter JD (2004) Selective modulation of some forms of schaffer collateral-CA1 synaptic plasticity in mice with a disruption of the CPEB-1 gene. Learn Mem 11: 318-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins CM, Nozaki N, Shigeri Y, Soderling TR (2004) Cytoplasmic polyadenylation element binding protein-dependent protein synthesis is regulated by calcium/calmodulin-dependent protein kinase II. J Neurosci 24: 5193-5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A, Muller D, Derkach V, Griffith LC, Soderling TR (1997) Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science 276: 2042-2045. [DOI] [PubMed] [Google Scholar]
- Benson DL (1997) Dendritic compartmentation of NMDA receptor mRNA in cultured hippocampal neurons. NeuroReport 8: 823-828. [DOI] [PubMed] [Google Scholar]
- Blitzer RD, Wong T, Nouranifar R, Iyengar R, Landau EM (1995) Postsynaptic cAMP pathway gates early LTP in hippocampal CA1 region. Neuron 15: 1403-1414. [DOI] [PubMed] [Google Scholar]
- Blitzer RD, Connor JH, Brown GP, Wong T, Shenolikar S, Iyengar R, Landau EM (1998) Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science 280: 1940-1942. [DOI] [PubMed] [Google Scholar]
- Bradshaw JM, Kubota Y, Meyer T, Schulman H (2003a) An ultrasensitive Ca2+/calmodulin-dependent protein kinase II-protein phosphatase 1 switch facilitates specificity in postsynaptic calcium signaling. Proc Natl Acad Sci USA 100: 10512-10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw KD, Emptage NJ, Bliss TV (2003b) A role for dendritic protein synthesis in hippocampal late LTP. Eur J Neurosci 18: 3150-3152. [DOI] [PubMed] [Google Scholar]
- Brown GP, Blitzer RD, Connor JH, Wong T, Shenolikar S, Iyengar R, Landau EM (2000) Long-term potentiation induced by theta frequency stimulation is regulated by a protein phosphatase-1-operated gate. J Neurosci 20: 7880-7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgin KE, Waxham MN, Rickling S, Westgate SA, Mobley WC, Kelly PT (1990) In situ hybridization histochemistry of Ca2+/calmodulin-dependent protein kinase in developing rat brain. J Neurosci 10: 1788-1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Q, Richter JD (2002) Dissolution of the maskin-eIF4E complex by cytoplasmic polyadenylation and poly(A)-binding protein controls cyclin B1 mRNA translation and oocyte maturation. EMBO J 21: 3852-3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll RC, Beattie EC, von Zastrow M, Malenka RC (2001) Role of AMPA receptor endocytosis in synaptic plasticity. Nat Rev Neurosci 2: 315-324. [DOI] [PubMed] [Google Scholar]
- Foulkes JG, Strada SJ, Henderson PJ, Cohen P (1983) A kinetic analysis of the effects of inhibitor-1 and inhibitor-2 on the activity of protein phosphatase-1. Eur J Biochem 132: 309-313. [DOI] [PubMed] [Google Scholar]
- Frey U, Krug M, Reymann KG, Matthies H (1988) Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res 452: 57-65. [DOI] [PubMed] [Google Scholar]
- Fukunaga K, Stoppini L, Miyamoto E, Muller D (1993) Long-term potentiation is associated with an increased activity of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem 268: 7863-7867. [PubMed] [Google Scholar]
- Hashimoto Y, Sharma RK, Soderling TR (1989) Regulation of Ca2+/calmodulin-dependent cyclic nucleotide phosphodiesterase by the autophosphorylated form of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem 264: 10884-10887. [PubMed] [Google Scholar]
- Havik B, Rokke H, Bardsen K, Davanger S, Bramham CR (2003) Bursts of high-frequency stimulation trigger rapid delivery of pre-existing alpha-CaMKII mRNA to synapses: a mechanism in dendritic protein synthesis during long-term potentiation in adult awake rats. Eur J Neurosci 17: 2679-2689. [DOI] [PubMed] [Google Scholar]
- Hemmings Jr HC, Greengard P, Tung HY, Cohen P (1984) DARPP-32, a dopamine-regulated neuronal phosphoprotein, is a potent inhibitor of protein phosphatase-1. Nature 310: 503-505. [DOI] [PubMed] [Google Scholar]
- Huang YS, Jung MY, Sarkissian M, Richter JD (2002) N-methyl-d-aspartate receptor signaling results in Aurora kinase-catalyzed CPEB phosphorylation and alpha CaMKII mRNA polyadenylation at synapses. EMBO J 21: 2139-2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Kandel ER (1994) Recruitment of long-lasting and protein kinase A-dependent long-term potentiation in the CA1 region of hippocampus requires repeated tetanization. Learn Mem 1: 74-82. [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF (2000) Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288: 1254-1257. [DOI] [PubMed] [Google Scholar]
- Ju W, Morishita W, Tsui J, Gaietta G, Deerinck TJ, Adams SR, Garner CC, Tsien RY, Ellisman MH, Malenka RC (2004) Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat Neurosci 7: 244-253. [DOI] [PubMed] [Google Scholar]
- Kandel ER (2001) The molecular biology of memory storage: a dialogue between genes and synapses. Science 294: 1030-1038. [DOI] [PubMed] [Google Scholar]
- Kelleher III RJ, Govindarajan A, Jung HY, Kang H, Tonegawa S (2004) Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell 116: 467-479. [DOI] [PubMed] [Google Scholar]
- Klann E, Antion MD, Banko JL, Hou L (2004) Synaptic plasticity and translation initiation. Learn Mem 11: 365-372. [DOI] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF (1998) NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron 21: 1151-1162. [DOI] [PubMed] [Google Scholar]
- Lisman J (2003) Long-term potentiation: outstanding questions and attempted synthesis. Philos Trans R Soc Lond B Biol Sci 358: 829-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Schwartz JH (2003) The cytoplasmic polyadenylation element binding protein and polyadenylation of messenger RNA in Aplysia neurons. Brain Res 959: 68-76. [DOI] [PubMed] [Google Scholar]
- Malleret G, Haditsch U, Genoux D, Jones MW, Bliss TV, Vanhoose AM, Weitlauf C, Kandel ER, Winder DG, Mansuy IM (2001) Inducible and reversible enhancement of learning, memory, and long-term potentiation by genetic inhibition of calcineurin. Cell 104: 675-686. [DOI] [PubMed] [Google Scholar]
- Matthews MB, Sonenberg N, Hershey JWB (2000) Origins and principles of translational control. In: Translational control of gene expression (Sonenberg N, Hershey JWB, Matthews MB, eds), pp 1-31. Cold Spring Harbor, NY: Cold Spring Harbor.
- Mendez R, Murthy KG, Ryan K, Manley JL, Richter JD (2000a) Phosphorylation of CPEB by Eg2 mediates the recruitment of CPSF into an active cytoplasmic polyadenylation complex. Mol Cell 6: 1253-1259. [DOI] [PubMed] [Google Scholar]
- Mendez R, Hake LE, Andresson T, Littlepage LE, Ruderman JV, Richter JD (2000b) Phosphorylation of CPE binding factor by Eg2 regulates translation of c-mos mRNA. Nature 404: 302-307. [DOI] [PubMed] [Google Scholar]
- Miller S, Yasuda M, Coats JK, Jones Y, Martone ME, Mayford M (2002) Disruption of dendritic translation of CaMKIIalpha impairs stabilization of synaptic plasticity and memory consolidation. Neuron 36: 507-519. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Endo S, Shenolikar S, Malenka RC (1994) Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature 369: 486-488. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Kandel ER (1997) Brief theta-burst stimulation induces a transcription-dependent late phase of LTP requiring cAMP in area CA1 of the mouse hippocampus. Learn Mem 4: 230-243. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Abel T, Kandel ER (1994) Requirement of a critical period of transcription for induction of a late phase of LTP. Science 265: 1104-1107. [DOI] [PubMed] [Google Scholar]
- Ostroff LE, Fiala JC, Allwardt B, Harris KM (2002) Polyribosomes redistribute from dendritic shafts into spines with enlarged synapses during LTP in developing rat hippocampal slices. Neuron 35: 535-545. [DOI] [PubMed] [Google Scholar]
- Otmakhova NA, Otmakhov N, Mortenson LH, Lisman JE (2000) Inhibition of the cAMP pathway decreases early long-term potentiation at CA1 hippocampal synapses. J Neurosci 20: 4446-4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Y, Rosenstein A, Kreiman G, Schuman EM, Kennedy MB (1999) Tetanic stimulation leads to increased accumulation of Ca2+/calmodulin-dependent protein kinase II via dendritic protein synthesis in hippocampal neurons. J Neurosci 19: 7823-7833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Sweatt JD (1996) Transient activation of cyclic AMP-dependent protein kinase during hippocampal long-term potentiation. J Biol Chem 271: 30436-30441. [DOI] [PubMed] [Google Scholar]
- Scheetz AJ, Nairn AC, Constantine-Paton M (2000) NMDA receptor-mediated control of protein synthesis at developing synapses. Nat Neurosci 3: 211-216. [DOI] [PubMed] [Google Scholar]
- Schulman H (1995) Protein phosphorylation in neuronal plasticity and gene expression. Curr Opin Neurobiol 5: 375-381. [DOI] [PubMed] [Google Scholar]
- Si K, Lindquist S, Kandel ER (2003) A neuronal isoform of the Aplysia CPEB has prion-like properties. Cell 115: 879-891. [DOI] [PubMed] [Google Scholar]
- Stebbins-Boaz B, Cao Q, de Moor CH, Mendez R, Richter JD (1999) Maskin is a CPEB-associated factor that transiently interacts with elF-4E. Mol Cell 4: 1017-1027. [DOI] [PubMed] [Google Scholar]
- Steward O, Schuman EM (2001) Protein synthesis at synaptic sites on dendrites. Annu Rev Neurosci 24: 299-325. [DOI] [PubMed] [Google Scholar]
- Steward O, Wallace CS, Lyford GL, Worley PF (1998) Synaptic activation causes the mRNA for the IEG Arc to localize selectively near activated postsynaptic sites on dendrites. Neuron 21: 741-751. [DOI] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM (2002) A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci USA 99: 467-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis M, Si K, Kandel ER (2003) Two previously undescribed members of the mouse CPEB family of genes and their inducible expression in the principal cell layers of the hippocampus. Proc Natl Acad Sci USA 100: 9602-9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Ferguson GD, Pineda VV, Cundiff PE, Storm DR (2004) Overexpression of type-1 adenylyl cyclase in mouse forebrain enhances recognition memory and LTP. Nat Neurosci 7: 635-642. [DOI] [PubMed] [Google Scholar]
- Wells DG, Dong X, Quinlan EM, Huang YS, Bear MF, Richter JD, Fallon JR (2001) A role for the cytoplasmic polyadenylation element in NMDA receptor-regulated mRNA translation in neurons. J Neurosci 21: 9541-9548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC, Muglia LJ, Storm DR (1999) Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron 23: 787-798. [DOI] [PubMed] [Google Scholar]
- Woo NH, Nguyen PV (2003) Protein synthesis is required for synaptic immunity to depotentiation. J Neurosci 23: 1125-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Wells D, Tay J, Mendis D, Abbott MA, Barnitt A, Quinlan E, Heynen A, Fallon JR, Richter JD (1998) CPEB-mediated cytoplasmic polyadenylation and the regulation of experience-dependent translation of alpha-CaMKII mRNA at synapses. Neuron 21: 1129-1139. [DOI] [PubMed] [Google Scholar]