

Abstract
Orexin A and B are neuropeptides implicated in the regulation of sleep/wakefulness and energy homeostasis. The regulatory mechanism of the activity of orexin neurons is not precisely understood. Using transgenic mice in which orexin neurons specifically express yellow cameleon 2.1, we screened for factors that affect the activity of orexin neurons (a total of 21 peptides and six other factors were examined) and found that a sulfated octapeptide form of cholecystokinin (CCK-8S), neurotensin, oxytocin, and vasopressin activate orexin neurons. The mechanisms that underlie CCK-8S-induced activation of orexin neurons were studied by both calcium imaging and slice patch-clamp recording. CCK-8S induced inward current in the orexin neurons. The CCKA receptor antagonist lorglumide inhibited CCK-8S-induced activation of orexin neurons, whereas the CCKB receptor agonists CCK-4 (a tetrapeptide form of cholecystokinin) and nonsulfated CCK-8 had little effect. The CCK-8S-induced increase in intracellular calcium concentration was eliminated by removing extracellular calcium but not by an addition of thapsigargin. Nifedipine, ω-conotoxin, ω-agatoxin, 4-ethylphenylamino-1,2-dimethyl-6-methylaminopyrimidinium chloride, and SNX-482 had little effect, but La3+, Gd3+, and 2-aminoethoxydiphenylborate inhibited CCK-8S-induced calcium influx. Additionally, the CCK-8S-induced inward current was dramatically enhanced in the calcium-free solution and was inhibited by the cation channel blocker SKF96365, suggesting an involvement of extracellular calcium-sensitive cation channels. CCK-8S did not induce an increase in intracellular calcium concentration when membrane potential was clamped at -60 mV, suggesting that the calcium increase is induced by depolarization. The evidence presented here expands our understanding of the regulation of orexin neurons and the physiological role of CCK in the CNS.
Keywords: orexin, hypocretin, patch clamp, transgenic, cholecystokinin, CCKAR
Introduction
Orexin A and orexin B (also called hypocretin-1 and hypocretin-2) are a pair of neuropeptides expressed in a specific population of neurons in the lateral hypothalamic area (LHA) (de Lecea et al., 1998; Sakurai et al., 1998). Orexin-producing neurons (orexin neurons) project throughout the brain and densely project to the monoaminergic and cholinergic nuclei in the brainstem that are implicated in the regulation of sleep/wakefulness (Peyron et al., 1998; Nambu et al., 1999). Several studies have showed that orexins activate monoaminergic neurons in vitro. Deficiency of the orexin neurons causes the sleep disorder narcolepsy, suggesting that orexin plays an important role in the maintenance of arousal (Chemelli et al., 1999; Lin et al., 1999; Peyron et al., 2000; Hara et al., 2001; Willie et al., 2003).
Recent studies have revealed that the orexin neurons receive serotonergic, noradrenergic, cholinergic, GABAergic, and glutamatergic regulation (Li et al., 2002; Yamanaka et al., 2003a; Muraki et al., 2004; Bayer et al., 2005; Grivel et al., 2005; Li and van den Pol, 2005; Sakurai et al., 2005). The activity of orexin neurons is also reported to be influenced by corticotropin-releasing factor (CRF) (Winsky-Sommerer et al., 2004), glucagon-like peptide-1 (GLP-1) (Acuna-Goycolea and van den Pol, 2004), neuropeptide Y (NPY) (Fu et al., 2004), ghrelin, leptin, and glucose (Yamanaka et al., 2003b). These results were all obtained by whole-cell patch-clamp recordings of orexin neurons. However, changes in the intracellular environment are unavoidable in electrophysiological studies, and it is impossible to record multiple orexin neurons in the same preparation simultaneously.
In this study, we established transgenic mice in which orexin neurons specifically express the calcium-sensing protein yellow cameleon 2.1 (YC2.1) (Miyawaki et al., 1999). Ca2+ imaging of orexin neurons, using hypothalamic slices prepared from these transgenic mice, revealed that several peptides including cholecystokinin (CCK) activate orexin neurons. A sulfated octapeptide form, CCK-8S, predominates in the brain and acts via two subtypes of CCK receptor, CCKA receptor (CCKAR) and CCKBR. It is known that CCK is present in many important neuronal pathways and is colocalized with classical neurotransmitters, such as dopamine in the ventral tegmental area (Hokfelt et al., 1980), GABA in the hippocampus (Gulyas et al., 1990), and CRF in the paraventricular nucleus of the hypothalamus (Mezey et al., 1985). Although CCK is believed to be involved in many functions, the physiological role of CCK in the CNS is not well understood. In the present study, we revealed that CCK-8S activates orexin neurons through the CCKAR and subsequent activation of a nonselective cation channels. The evidence presented here expands our understanding of the regulation of the orexin system and of the physiological role of CCK in the CNS.
Materials and Methods
Animal usage. All experimental procedures involving animals were approved by the University of Tsukuba Animal Care and Use Committee and were in accordance with National Institutes of Health guidelines. All efforts were made to minimize animal suffering or discomfort and to reduce the number of animals used.
Generation of orexin/YC2.1 transgenic mice. The transgenic construct was made by substituting the nLacZ gene (Sall-BamHI fragment) of the orexin/nlacZ transgenic construct (Sakurai et al., 1999) with the 1.9 kb YC2.1 fragment (a gift from Dr. A. Miyawaki, RIKEN, Wako, Japan). The transgene was excised and microinjected into pronuclei of fertilized mouse eggs (BDF1 mice) to generate transgenic founders. Founder animals were bred with BDF1 mice to produce stable orexin/YC2.1 lines. A total of 13 transgene-positive founders were obtained. However, the analysis of N1 generation shows that only one line showed a sufficiently strong expression of YC 2.1. This line was established for subsequent experiments.
Brain slice preparation. Orexin/YC2.1 mice (3-8 weeks of age) or orexin/EGFP (enhanced green fluorescent protein) mice (2-3 weeks old) were anesthetized with fluothane (Takeda, Osaka, Japan). The mice were decapitated under deep anesthesia. Brains were isolated in ice-cold cutting solution consisting of (in mm) 280 sucrose, 2 KCl, 10 HEPES, 0.5 CaCl2, 10 MgCl2, and 10 glucose, pH 7.4, bubbled with 100% O2. Brains were cut coronally into 300 μm slices with a microtome (VTA-1000S; Leica, Nussloch, Germany). Slices containing the LHA were transferred for at least 1 h to an incubation chamber at room temperature (RT) (24-26°C) filled with physiological solution containing (in mm) 135 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, pH 7.4, with NaOH.
Calcium imaging of orexin neurons. Optical recordings were performed on a fluorescence microscope (BX51WI; Olympus, Tokyo, Japan) equipped with a cooled charge-coupled device (CCD) camera (Cascade 650; Roper Scientific, Tucson, AZ) controlled by MetaFluor 5.0.7 software (Universal Imaging, West Chester, PA). YC2.1 was excited through a 440DF20 filter, and its fluorescent image was subjected to dual-emission ratio imaging through two emission filters [480DF30 for cyan fluorescent protein (CFP) and 535DF26 for yellow fluorescent protein (YFP)] controlled by a filter changer (Lambda 10-2; Sutter Instruments, Novato, CA). Images were captured at a rate of 1 Hz (300-500 ms exposure time) with 2 × 2 binning through a 20× UMPlanFI water-immersion objective (Olympus, Tokyo, Japan).
Electrophysiological recordings. Orexin/EGFP mice (Yamanaka et al., 2003a,b) were used for whole-cell recordings. Orexin/YC2.1 mice were also used for simultaneous recording of calcium imaging and slice patch clamp. The slices were transferred to a recording chamber (RC-27L; Warner Instruments, Hamden, CT) at RT on a fluorescence microscope stage (BX51WI; Olympus). Neurons that showed EGFP fluorescence were subjected to electrophysiological recording. The fluorescence microscope was equipped with an infrared camera (C2741-79; Hamamatsu Photonics, Hamamatsu, Japan) for infrared differential interference contrast (IR-DIC) imaging and a CCD camera (IK-TU51CU; Olympus) for fluorescent imaging. Each image was displayed separately on a monitor (Gawin; EIZO, Tokyo, Japan) and was saved on a Power Macintosh G4 computer (Apple Computers, Cupertino, CA) through a graphic converter (PIX-MPTV; Pixcela, Osaka, Japan).
Recordings were performed with an Axopatch 200B amplifier (Molecular Devices, Union City, CA) using a borosilicate pipette (GC150-10; Harvard Apparatus, Holliston, MA) prepared by a micropipette puller (P-97; Sutter Instruments, Pangbourne, UK) filled with intracellular solution (4-10 MΩ) consisting of (in mm) 145 KCl, 1 MgCl2, 10 HEPES, 1.1 EGTA-Na3, 2 MgATP, and 0.5 Na2-GTP, pH 7.3, with KOH. Osmolarity of the solution was checked by a vapor pressure osmometer (model 5520; Wescor, Logan, UT). The osmolarity of the internal and external solutions was 280-290 and 320-330 mOsm/L, respectively. The liquid junction potential of the patch pipette and perfused extracellular solution was estimated to be 3.9 mV and was applied to the data. Recording pipettes were under positive pressure while advanced toward individual cells in the slice. Tight seals on the order of 0.5-1.0 GΩ were made by negative pressure. The membrane patch was then ruptured by suction. The series resistance during recording was 10-25 MΩ and was compensated. The reference electrode was an Ag-AgCl pellet immersed in bath solution. During recordings, cells were superfused with extracellular solution at a rate of 1.6 ml/min using a peristaltic pump (Dynamax; Rainin, Oakland, CA) at RT. To measure the membrane resistance, depolarizing and hyperpolarizing current pulses were applied to cells at durations of 200 ms at 20 pA steps at 2 s intervals from the resting membrane potential (-60 mV) set by varying the intensity of a constantly injected current. Spontaneous EPSCs (sEPSCs) and spontaneous IPSCs (sIPSCs) were recorded in orexin neurons under whole-cell voltage-clamp mode at a holding potential of -60 mV. sEPSCs were recorded using KCl-based pipette solution containing the sodium channel blocker N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide (QX-314) (1 mm) to inhibit action potentials in the recording neuron and in the presence of picrotoxin (100 μm) in the bath solution. sIPSCs were recorded using KCl-based pipette solution containing QX-314 (1 mm) in the presence of dl-2-amino-5-phosphono-pentanoic acid (AP-5) (50 μm) and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (20 μm) in the bath solution. The frequency of sEPSCs or sIPSCs was measured using pClamp software; only those events with amplitudes >10 pA were used. Frequency and amplitude were represented as a mean of 200 s duration.
The output signal was low-pass filtered at 5 kHz and digitized at 10 kHz. Data were recorded on a computer through a Digidata 1322A analog-to-digital converter using pClamp software version 8.2 (Molecular Devices). The trace was processed for presentation using Origin 6.1 (OriginLab, Northampton, MA) and Canvas 9.0.5 software (ACD Systems of America, Miami, FL).
Drugs. CCK-8S, nonsulfated CCK-8 (CCK-8NS), a tetrapeptide form of CCK (CCK-4), ω-conotoxin GVIA, ω-agatoxin IVA, the peptide toxin SNX-482 (Peptide Institute, Osaka, Japan), 2-aminoethoxydiphenylborate (2-APB), SKF96365 (Calbiochem, La Jolla, CA), nifedipine, thapsigargin, LaCl3, lorglumide, CNQX, AP-5 (Sigma, St. Louis, MO), 4-ethylphenylamino-1,2-dimethyl-6-methylaminopyrimidinium chloride (ZD7288; Tocris Cookson, Ellisville, MO), picrotoxin, tetrodotoxin (TTX), and GdCl3 (Wako, Osaka, Japan) were dissolved in extracellular solution and applied by bath application.
Immunohistochemistry. Mice were anesthetized deeply with diethyl ether and perfused sequentially with 20 ml of chilled saline and 20 ml of chilled 4% paraformaldehyde in 0.1 m phosphate buffer. The brains were removed and immersed in the same fixative solution for 24 h at 4°C and then immersed in the 30% sucrose solution for at least 2 d. The brains were quickly frozen in embedding solution (Sakura, Tokyo, Japan). For orexin immunoreactivity, coronal sections of orexin/YC2.1 mice brains were incubated with rabbit anti-orexin antiserum (1:2000) (Nambu et al., 1999) for 24 h at 4°C. These sections were incubated with Alexa 594-labeled goat anti-rabbit IgG antibody (1:800; Molecular Probes, Eugene, OR) for 1 h at RT. For orexin and CCKAR double staining, coronal sections of C57BL/6J mice brains were incubated with rabbit anti-CCKAR antiserum (1:2000; American Research Products, Belmont, MA) for 48 h at 4°C. These sections were incubated with Alexa 594-labeled anti-rabbit IgG (1:1000; Molecular Probes) for 1 h at RT. These sections were then incubated with guinea pig anti-orexin antiserum (1:1000) for 48 h at 4°C and incubated with Alexa 488-labeled goat anti-guinea pig IgG (1:800; Molecular Probes) for 1 h at RT. The sections were mounted and examined with a fluorescence microscope (AX-70; Olympus). To confirm the specificity of antibodies, incubations without primary antibody were conducted as a negative control in each experiment and no signal was observed. Numbers of YFP-positive or orexin-immunoreactive (ir) cells were counted in every four brain slices (40 μm thickness, -1.34 to -2.54 mm from bregma) under fluorescent microscopy.
Statistical analysis. Data were analyzed by two-way ANOVA followed by post hoc analysis of significance with the Fisher's protected least significant difference test or Dunnett's test using the StatView 5.0 software package for Macintosh (Abacus Concepts, Berkeley, CA). p values < 0.05 were considered statistically significant.
Results
Specific expression of YC2.1 in orexin neurons
We made transgenic mice in which the YC2.1 is expressed under control of the human prepro-orexin promoter (orexin/YC2.1 transgenic mice). Specific expression of YC2.1 in orexin neurons was confirmed by immunohistochemistry. In the orexin/YC2.1 transgenic mouse brain, both YFP and CFP fluorescence were observed in the same neuron, but no such fluorescence was observed in wild-type littermate brains (data not shown). Immunoreactivity for orexin (red; Alexa-594) was also readily detected in the LHA. A merged picture (YFP fluorescence and orexin-ir) revealed that YFP fluorescence was exclusively observed in the orexin neurons: no ectopic expression of YC2.1 was observed throughout the brain by extensive observation of the transgenic mice brains (Fig. 1A). In this line of transgenic mice, ∼50% of orexin neurons express YC2.1 (a total of five male and female transgenic mouse brains were counted). Transgenic mice derived from this line were used for all subsequent experiments.
Figure 1.

A, The Ca2+ indicator YC2.1 is expressed in the orexin neurons in the orexin/YC2.1 mouse brain. YFP and CFP fluorescence were observed in the same neurons. The merged picture (orexin-ir and YFP) shows that the expression of YC2.1 is restricted to orexin-immunoreactive neurons in the LHA. Approximately 50% of orexin neurons expressed YC2.1 (n = 5). B, C, The function of YC2.1 was confirmed by glutamate application. B, A pseudocolor ratio image of orexin neuron during application of glutamate. The images were captured at the times indicated in the graph. Glutamate (300 μm) was applied by bath application during the period represented by the bar in the graph. Color represents a ratio of 0.85 (blue) to 1.05 (red). C, Glutamate application increased the YFP/CFP ratio in a concentration-dependent manner. The Δ ratio is normalized to a high concentration of glutamate application (1000 μm). D-F, Simultaneous recording of calcium imaging and slice patch clamp. The neuron subjected to calcium imaging was whole-cell patch clamped, and depolarizing pulses were applied through patch pipette for 10 s (60-90 pA; duration of 5 ms; 20, 50, and 100 Hz). D, Bottom trace shows electrical stimulation through pipette, top trace shows evoked action potentials in the recording neuron. E, Intracellular calcium concentration increased in a firing frequency-dependent manner. Blue and green lines at the top of the graph show CFP and YFP intensity, respectively. F, Bar graph summarizes the data in E (n = 6). Values are represented by mean ± SEM.
Verification of YC2.1 as an indicator of neuronal activation
To confirm the function of expressed YC2.1 in orexin neurons, glutamate was applied to orexin neurons in a hypothalamic slice preparation (300 μm thickness). Previously, we reported that glutamate strongly depolarized orexin neurons through ionotropic glutamate receptors (Yamanaka et al., 2003a). Glutamate application increased the YFP/CFP ratio in a concentration-dependent manner: CFP fluorescence intensity decreased while YFP fluorescence intensity reciprocally increased simultaneously by fluorescence resonance energy transfer (n = 10) (Fig. 1B,C). Application of a high concentration of potassium (45 mm) also increased the YFP/CFP ratio to 108.1 ± 0.8% of the basal level (n = 11). These responses were reversible; the ratio returned to the basal level after glutamate washout. The same concentration of glutamate induced almost the same ratio change at the second application (data not shown).
To examine the relationship between firing rate and intracellular calcium concentration, we performed simultaneous recordings of calcium imaging and slice patch clamp. When a depolarizing pulse (20-100 Hz, 60-90 pA, duration of 5 ms) was applied through the pipette electrode for 10 s, neurons generated action potentials. The YFP/CFP ratio increased in an applied frequency-dependent manner, suggesting that the YFP/CFP ratio increase reflects an increase in firing rate and neuronal activity (Fig. 1D). Twenty, 50, and 100 Hz firing increased the YFP/CFP ratio to 105.2 ± 1.5, 108.6 ± 1.6, and 112.0 ± 1.8% of the basal level, respectively (Fig. 1E,F) (n = 6). These results suggest that functional YC2.1 is expressed in the orexin neurons and that these orexin/YC2.1 transgenic mice are useful for monitoring the activity of orexin neurons. To test whether the physiological properties of orexin neurons are altered by YC2.1 expression, the membrane characteristics of YC2.1-expressing orexin neurons were compared with those of EGFP-expressing orexin neurons. Input resistance of YC2.1-expressing orexin neurons and EGFP-expressing orexin neurons prepared from the orexin/EGFP mice are 408 ± 53 MΩ (n = 11) and 399 ± 23 MΩ (n = 11), respectively (p = 0.87; not significantly different). Input resistance was calculated from the slope of the current-voltage relationship obtained by step current injection in current-clamp mode. Additionally, membrane capacitance and after hyperpolarization of YC2.1-expressing neurons were 25.2 ± 5.7 pF (n = 6) and -56.1 ± 1.4 mV (n = 6), respectively. These values are similar to our previous report, obtained by using orexin/EGFP mice (30.5 ± 8.9 pF and -59.3 ± 6.2 mV, respectively) (Yamanaka et al., 2003a), suggesting that basic membrane properties of orexin neurons are not affected by the expression of YC2.1 protein.
Muscimol, a GABAA receptor agonist, decreased the YFP/CFP ratio in a concentration-dependent manner (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The YFP/CFP ratio was also decreased by serotonin application in a concentration-dependent manner (data not shown). We reported previously that orexin neurons are strongly hyperpolarized by serotonin (Muraki et al., 2004). It may be difficult to lower baseline calcium in neurons. We speculate that the intracellular calcium concentrations of neurons were increased during the slice preparation and assay procedure. Under these conditions, the intracellular calcium concentration might be lowered by adding 5-HT or muscimol. It is unlikely that an increase in intracellular calcium concentration in orexin neurons is caused by a high-frequency excitatory input, because TTX application did not affect YFP/CFP ratio (percentage alteration of YFP/CFP ratio after TTX application was 98.9 ± 0.3%; n = 30). However, these data suggest that orexin/YC2.1 mice are also useful in screening for substances that inhibit the activity of orexin neurons.
CCK-8S activates orexin neurons
To identify endogenous peptides that affect the activity of orexin neurons, we applied various peptides known to be involved in the regulation of sleep/wakefulness and/or feeding onto the orexin neurons using hypothalamic slices prepared from orexin/YC2.1 transgenic mice. Screening was performed in the presence of TTX (1 μm). Among these peptides, neurotensin, CCK-8S, oxytocin, and vasopressin induced an increase in [Ca2+]i in orexin neurons (Table 1). Figure 2A shows that NPY and galanin-like peptide had no effect, but CCK-8S induced a robust increase in the YFP/CFP ratio in the orexin neurons. The CCK-8S-induced response peaked within 1 min when applied and returned to basal level 5-10 min after washout. Figure 2B shows that CCK-8S induced an increase in [Ca2+]i in orexin neurons in a concentration-dependent manner (EC50 = 8.2 ± 2.6 nm; n = 20). Neurotensin and vasopressin also induced an increase in [Ca2+]i in orexin neurons in a concentration-dependent manner (supplemental Figs. 2, 3, available at www.jneurosci.org as supplemental material).
Table 1.
The screening using orexin/YC2.1 transgenic mice
|
Substances |
Concentration |
n |
Response |
|---|---|---|---|
| Peptides | |||
| Neuropeptide Y | 100 nm | 20 | ND |
| Orexin A | 1 μm | 27 | ND |
| Galanin | 1 μm | 17 | ND |
| QRFP | 300 nm | 13 | ND |
| Melanin-concentrating hormone | 1 μm | 10 | ND |
| Galanin-like peptide | 100 nm | 20 | ND |
| Neurotensin | 100 nm | 8 | Activation |
| Calcitonin gene-related peptide | 100 nm | 8 | ND |
| Insulin | 300 nm | 11 | ND |
| Urocortin | 300 nm | 21 | ND |
| Cocaine-and amphetamine-regulated transcript | 300 nm | 18 | ND |
| CCK-8S | 10 nm | 20 | Activation |
| α-Melanocyte-stimulating hormone | 1 μm | 13 | ND |
| Agouti-related protein | 1 μm | 19 | ND |
| Neuropeptide B | 1 μm | 25 | ND |
| Substance P | 300 nm | 16 | ND |
| Neuropeptide S | 1 μm | 10 | ND |
| Prolactin-releasing peptide | 1 μm | 13 | ND |
| Vasoactive intestinal peptide | 1 μm | 17 | ND |
| Vasopressin | 300 nm | 13 | Activation |
| Oxytocin | 300 nm | 13 | Activation |
| Nonpeptides | |||
| Serotonin | 100 μm | 13 | Inhibition |
| Noradrenaline | 100 μm | 10 | Inhibition |
| Dopamine | 100 μm | 15 | Inhibition |
| Muscimol | 30 μm | 13 | Inhibition |
| Adenosine | 100 μm | 17 | ND |
| Histamine |
100 μm
|
17 |
ND |
All substances were applied by bath application for 2 min. ND, Not detected.
Figure 2.

CCK-8S activates orexin neurons. A, In the presence of TTX, many peptides were screened by bath application (Table 1). NPY (100 nm) and galanin-like peptide (GALP; 100 nm) had no effect (n = 20), although CCK-8S (100 nm) increased intracellular calcium concentration in the orexin neurons. Lines at the top of the graph show CFP and YFP intensity, respectively. Intensity of CFP fluorescence decreased while intensity of YFP fluorescence increased simultaneously when CCK-8S was applied. B, CCK-8S induced an increase in intracellular calcium concentration in a concentration-dependent manner. EC50 was 8.2 ± 2.6 nm (n = 20). C, D, Whole-cell current-clamp recording of orexin neurons showing that CCK-8S (30 nm) depolarized orexin neurons in the presence (C, bottom) or absence (C, top) of TTX (1 μm). D, Concentration dependency of the CCK-8S-induced depolarization in the presence of TTX. The EC50 and Emax were 7.7 ± 2.7 nm and 12.7 ± 1.7 mV, respectively (n = 10-15). E, Typical trace of CCK-8S-induced inward current under voltage-clamp recording at a holding potential of -60 mV. F, Concentration dependency of CCK-8S-induced inward current. EC50 and Emax were 3.3 ± 0.7 nm and 17.9 ± 1.4 pA, respectively (n = 10-15). Peptides were applied by bath application during the period represented by bars. Values are represented by mean ± SEM.
We examined the detailed mechanism of CCK-8S-induced response by slice patch clamp using orexin/EGFP transgenic mice (Yamanaka et al., 2003a,b). Under whole-cell current-clamp mode recording conditions, CCK-8S (30 nm) bath application depolarized and increased firing frequency in the orexin neurons (n = 20) (Fig. 2C, top). CCK-8S (30 nm) also induced depolarization in the orexin neurons in the presence of TTX (from -53.4 ± 1.8 to -42.2 ± 1.8 mV; n = 18) (Fig. 2C, bottom), suggesting that CCK-8S directly depolarized orexin neurons. The time course of CCK-8S-induced response showed good agreement with that obtained in calcium imaging. The response peaked within 1 min when applied, and membrane potential returned to the basal level 3-5 min after washout. Almost all orexin neurons tested were depolarized by CCK-8S (96%; 247 of 256). A small number of orexin neurons showed no response or faint depolarization (4%; 9 of 256). Twenty-six percent (22 of 86) of EGFP-negative neurons (non-orexin neurons and possibly a few orexin neurons, because EGFP fluorescence of 20% of orexin neurons is under the detection limit) in the same area showed a depolarization, and 74% (64 of 86) showed no detectable effect. Figure 2D demonstrates that CCK-8S depolarized orexin neurons in the presence of TTX in a concentration-dependent manner; EC50 and maximum effect (Emax) were 7.7 ± 2.7 nm and 12.7 ± 1.7 mV, respectively (n = 8-27). At a holding potential of -60 mV under voltage clamp, CCK-8S (100 nm) induced an inward current in orexin neurons in the presence of TTX (19.7 ± 2.5 pA; n = 16) (Fig. 2E). Figure 2F demonstrates that CCK-8S induced inward current in orexin neurons in a concentration-dependent manner; EC50 and Emax were 3.3 ± 0.7 nm and 17.9 ± 1.4 pA, respectively (n = 11-43). In addition to synaptic transmission blocked by TTX to exclude indirect action of CCK-8S on orexin neurons, glutamatergic synaptic transmission was blocked by the ionotropic glutamate receptor antagonists. CCK-8S-induced depolarization was not blocked by coapplication of the AMPA-type ionotropic glutamate receptor antagonist CNQX (25 μm), the NMDA-type ionotropic glutamate receptor antagonist AP-5 (25 μm), and TTX (n = 5 and data not shown). The depolarizing effect of CCK-8S on orexin neurons via CCK receptors was confirmed using the nonhydrolyzable GDP analog GDPβS in the recording pipette solution. Although the amplitude of the CCK-8S (100 nm)-induced inward current just after patch membrane rupture was comparable with that obtained using normal pipette solution (26.5 ± 3.8 pA; n = 5), CCK-8S-induced inward current was significantly depressed 10-15 min after the membrane was ruptured [5.6 ± 2.0 pA; n = 5; p = 0.0017; paired t test (vs just after rupture)]. These results suggest that CCK-8S depolarizes the membrane potential of orexin neurons in a GTP-dependent manner, acting via G-protein.
CCK-8S activates orexin neurons via the CCKAR
To identify the subtype of CCK receptor involved in the CCK-8S-induced depolarization of orexin neurons, preferential CCK receptor agonists and antagonists were used. Two subtypes of CCK receptors are known in mammals, CCKA (also known as CCK1) and CCKB (also known as CCK2) receptors. CCKAR is mainly expressed in peripheral tissues, whereas CCKBR is abundant in the brain. Although CCK-8S binds to both CCKA and CCKB receptors with similar affinities, CCK-8NS and CCK-4 selectively bind to CCKBR with an affinity 50-70 times higher than to CCKAR (Fossa et al., 1997). These CCKBR selective agonists induced very weak inward current in the orexin neurons even at a 100 times higher concentration than when CCK-8S was used (Fig. 3A,B) (CCK-4, n = 5, p = 0.0005; CCK-8NS, n = 16, p = 0.0126; ANOVA). Subsequently, we tested the effects of the CCKAR selective antagonist lorglumide on this response. Sequentially applied CCK-8S induced almost the same amplitude of inward current (85.6 ± 9.2%; n = 6; p > 0.05; not significantly different; ANOVA followed by a Dunnett's procedure for multiple comparisons). However, CCK-8S-induced inward current was attenuated by pretreatment of lorglumide in a concentration-dependent manner (Fig. 3C,D). Pretreatment of slices with 0.01 and 1 μm lorglumide for 2 min inhibited 10 nm CCK-8S-induced inward current to 57.1 ± 14.9% (n = 9; p < 0.05; ANOVA, followed by Dunnett's) and 9.4 ± 4.9% (n = 6; p < 0.05; ANOVA, followed by Dunnett's), respectively, compared with before antagonist treatment. The effect of lorglumide on CCK-8S-induced response was also confirmed by calcium imaging of orexin neurons using orexin/YC2.1 mice brain slices in the presence of TTX (1 μm). Lorglumide pretreatment for 5 min significantly inhibited a CCK-8S-induced increase in [Ca2+]i by 41.0 ± 8.8% (0.1 μm; n = 6; p < 0.0001; ANOVA), 31.9 ± 5.3% (1 μm; n = 15; p < 0.0001; ANOVA) and 5.9 ± 5.2% (10 μm; n = 6; p < 0.0001; ANOVA) (Figs. 3E,F). These results suggest that CCKAR is involved in CCK-8S-induced depolarization of orexin neurons.
Figure 3.

CCK-8S activates orexin neuron through the CCKAR. A, B, The effects of CCKAR or CCKBR agonists on the orexin neurons. CCK-8S (0.01 μm, n = 6), a CCKAR and CCKBR nonselective agonist, induced an inward current in orexin neurons. However, CCK-4 (1 μm, n = 5) and CCK-8NS (1 μm, n = 16), CCKBR preferential agonists, induced weak inward current even at a high concentration. CCK-4 and CCK-8NS were dissolved in DMSO and added to extracellular solution. Final DMSO concentration in the extracellular solution was 0.1%. The extracellular solution, which contained 0.1% DMSO alone was used as vehicle control and had no effect. C, D, The effect of CCKAR selective antagonist, lorglumide, on the CCK-8S-induced inward current. Sequentially applied CCK-8S induced almost the same amplitude of inward current. Pretreatment with lorglumide (1 μm) for 2 min inhibited CCK-8S (10 nm)-induced inward current. Δ Current was normalized to CCK-8S (10 nm) before experiments. E, F, The effect of lorglumide on CCK-8S (30 nm)-induced increase in [Ca2+]i. Orexin/YC2.1 mice brain slices were used for calcium imaging of orexin neurons. CCK-8S and lorglumide were applied by bath application during the period indicated by the bar. The experiment was performed in the presence of TTX (1 μm). Lorglumide (0.1 μm, n = 6; 1 μm, n = 15; 10 μm, n = 6) inhibited CCK-8S-induced increase in [Ca2+]i in a concentration-dependent manner. Values are mean ± SEM. *p<0.05.
CCKAR immunoreactivity in orexin neurons
To confirm the expression of CCKAR in orexin neurons, double-label immunofluorescence analysis was performed. The merged picture indicates that almost all orexin-ir neurons showed CCKAR immunoreactivity (Fig. 4C, arrowheads). We also observed orexin-negative neurons in the hypothalamus that express CCKAR (Fig. 4C, arrow). This CCKAR antibody showed strong immunoreactivities in the paraventricular nuclei, amygdala, and preoptic area (data not shown), areas that are known to densely express CCKAR (Mercer and Beart, 2004).
Figure 4.

Orexin neurons express CCKAR. A, Orexin-ir neurons were located in the lateral hypothalamic area (Alexa 488, green). B, CCKAR-ir was observed in the same area (Alexa 594, red). CCKAR-ir was observed on the somata and dendrites. C, Immunoreactivity for orexin and CCKAR overlaps in the merged image. Arrowheads indicate that orexin neurons express CCKAR. The arrow indicates that a non-orexin neuron in the same area expressed CCKAR as well. Scale bar, 50 μm.
The effect of CCK-8S on sEPSC and sIPSC
It has been reported that the neuropeptide GLP-1 has both indirect and direct effects on orexin neurons to modulate synaptic transmission (Acuna-Goycolea and van den Pol, 2004). To examine the possibility that CCK-8S also affects synaptic inputs to orexin neurons, sEPSCs and sIPSCs were recorded in orexin neurons under whole-cell voltage-clamp mode at a holding potential of -60 mV. CCK-8S (100 nm) had little effect on sEPSC or sIPSC frequency (Fig. 5) or amplitude [sEPSC: control, 35.1 ± 3.6 pA, CCK-8S (100 nm), 35.4 ± 3.0 pA, n = 6, p = 0.76 (not significantly different); sIPSC: control, 63.0 ± 7.5 pA, CCK-8S (100 nm), 61.7 ± 6.0 pA, n = 6, p = 0.75 (not significantly different)]. These results suggest that CCK-8S does not modulate glutamatergic or GABAergic synaptic transmission to orexin neurons.
Figure 5.

CCK-8S had no effect on glutamatergic or GABAergic synaptic transmission. sEPSCs and sIPSCs were recorded by whole-cell voltage clamp at a holding potential of -60 mV. sEPSCs were recorded in the presence of the GABAA receptor antagonist picrotoxin (100 μm), whereas sIPSCs were recorded in the presence of the ionotropic glutamate receptor antagonists AP-5 (50 μm) and CNQX (20 μm). A, sEPSC frequency (n = 6). B, sIPSC frequency (n = 6). PSC frequency was represented as a mean of 200 s. Values are mean ± SEM. Wash, Washout.
Role of extracellular calcium in CCK-8S-induced response
To determine the source of CCK-8S-induced calcium mobilization, calcium imaging of orexin neurons using brain slices of orexin/YC2.1 mice was performed in the presence of TTX (1 μm). First, to deplete intracellular calcium stores, slices were pretreated with thapsigargin (1 μm) for 5 min. Thapsigargin pretreatment did not affect the CCK-8S-induced intracellular calcium increase. CCK-8S-induced increment of YFP/CFP ratio was 94.2 ± 9.0% (vehicle control, n = 7) and 97.4 ± 6.6% (1 μm thapsigargin, n = 13) of that induced with CCK-8S application in the absence of thapsigargin (Fig. 6A). In contrast, removing extracellular calcium significantly decreased the CCK-8S-induced [Ca2+]i increase to 12.1 ± 4.6% (n = 9; p < 0.0001; ANOVA) of the control, suggesting that calcium influx is the main source of the CCK-8S-induced [Ca2+]i increase (Fig. 6A). Subsequently, to reveal which channel is involved in the CCK-8S-induced calcium influx, various channel blockers were tested. To examine any contribution of the voltage-dependent calcium channel in this response, nifedipine, ω-conotoxin, ω-agatoxin, and SNX-482, which are L-type, N-type, P-type, and R-type calcium channel blockers, respectively, were tested. Nifedipine, ω-conotoxin, ω-agatoxin, and SNX-482 did not have any effect on the CCK-8S-induced [Ca2+]i increase (Fig. 6B). The relative increments of YFP/CFP ratio were 94.2 ± 9.0% (vehicle control, 0.1% ethanol, n = 7), 107.9 ± 13.4% (10 μm nifedipine, n = 5), 82.1 ± 7.0% (30 μm nifedipine, n = 8, p = 0.32, ANOVA), 90.8 ± 5.1% (10 μm ω-conotoxin, n = 7), 94.2 ± 3.9% (10 nm ω-agatoxin, n = 5), and 91.6 ± 4.6% (100 nm SNX-482, n = 8). ZD7288, a blocker of hyperpolarization-activated and cyclic nucleotide-gated (HCN) channel and the T-type calcium channel (Felix et al., 2003), also had no effect on the CCK-8S-induced [Ca2+]i increase. The relative change of YFP/CFP ratio was 89.9 ± 3.2% (100 μm ZD7288, n = 6). In contrast, the nonselective cation channel blockers La3+, Gd3+, and 2-APB significantly inhibited the CCK-8S-induced [Ca2+]i increase in a concentration-dependent manner (Fig. 6C). The relative changes of YFP/CFP ratio were 69.7 ± 10.3% (10 μm La3+, n = 8, p < 0.0001, ANOVA), 2.5 ± 2.2% (100 μm La3+, n = 7, p < 0.001, ANOVA), 65.9 ± 8.2% (10 μm Gd3+, n = 7, p < 0.001, ANOVA), 4.0 ± 4.3% (100 μm Gd3+, n = 6, p < 0.0001, ANOVA), 71.5 ± 3.8% (10 μm 2-APB, n = 5, p = 0.0014, ANOVA), 23.3 ± 6.4% (30 μm 2-APB, n = 6, p < 0.0001, ANOVA), and 9.5 ± 2.3% (100 μm 2-APB, n = 5, p < 0.0001, ANOVA). These results suggest that calcium influx through the nonselective cation channel is involved in the CCK-8S-induced [Ca2+]i increase.
Figure 6.
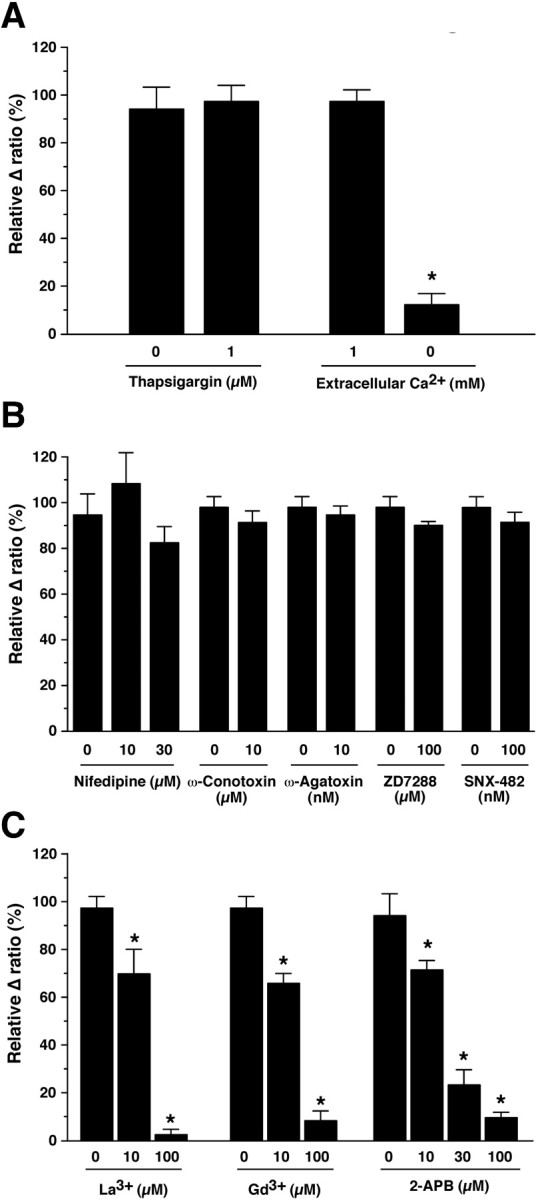
Ca2+ imaging of orexin neurons using brain slices prepared from orexin/YC2.1 mice revealed that CCK-8S activates nonselective cation channels on the orexin neurons. The experiments were performed in the presence of TTX (1 μm). A, CCK-8S-induced increase in [Ca2+]i was significantly inhibited by removal of extracellular calcium (n = 9) but was not inhibited by thapsigargin (1 μm) treatment (n = 13). B, The effect of voltage-dependent calcium channel blockers on CCK-8S-induced increase in [Ca2+]i. Nifedipine (10 μm, n = 5; 30 μm, n = 8), ω-conotoxin (10 μm, n = 7), ω-agatoxin (10 nm, n = 5), ZD7288 (100 μm, n = 6), and SNX-482 (100 nm, n = 8) did not inhibit CCK-8S-induced increase in [Ca2+]i. C, The effect of nonselective cation channel blockers on CCK-8S-induced increase in [Ca2+]i. La3+ (10 μm, n = 8; 100 μm, n = 7), Gd3+ (10 μm, n = 7; 100 μm, n = 6), and 2-APB (10 μm, n = 5; 30 μm, n = 6; 100 μm, n = 5) inhibited CCK-8S-induced increase in [Ca2+]i in a concentration-dependent manner. Data are normalized by Δ ratio obtained by CCK-8S (30 nm) application before experiments. Drugs were dissolved in the extracellular solution and were applied by bath application for 5 min before the experiments. Thapsigargin, nifedipine, and 2-APB were dissolved in ethanol and were compared with vehicle control (0.1% ethanol). Values are mean ± SEM. *p < 0.05.
CCK-8S-induced current are potentiated by removal of extracellular Ca2+
To examine the properties of the CCK-8S-induced current in more detail, additional electrophysiological experiments were performed. First, the contribution of Na+ to this CCK-8S-induced depolarization was studied. Experiments were done in which NaCl in the extracellular solution was replaced by an equimolar concentration of choline chloride in the presence of TTX (1 μm). When choline was substituted for sodium, the CCK-8S-induced inward current was strongly depressed in all of the cells tested. In Na+-free solution, CCK-8S (100 nm)-induced inward current was only 2.2 ± 0.7 pA (n = 5; p < 0.001; ANOVA), suggesting that CCK-8S-induced depolarization was primarily dependent on extracellular Na+ (Fig. 7A). Subsequently, to examine the contribution of calcium ions in this response, calcium was removed from extracellular solution. Although the removal of divalent cations from extracellular solution itself induced inward current, the detailed mechanism was not elucidated. As shown in Figure 7B, removal of extracellular calcium markedly potentiated the CCK-8S-induced inward current. In the presence or absence of calcium ions in the extracellular solution, the CCK-8S (5 nm)-induced inward currents were 14.8 ± 5.5 or 201.0 ± 48.0 pA, respectively (n = 7; p < 0.002; ANOVA). The CCK-8S-induced inward current increased ∼14-fold in calcium-free solution, suggesting that the CCK-8S-induced inward current was suppressed by extracellular calcium ions. Reversal potential of the CCK-8S-induced current in the calcium-free extracellular solution (in mm: 140 NaCl, 2 CsCl, 1 MgCl2, 1 EGTA, 10 HEPES, and 10 glucose) was near 0 mV (-2.3 ± 2.1 mV; n = 5) when measured using a CsCl pipette solution (in mm: 145 CsCl, 1 MgCl2, 10 HEPES, 1.1 EGTA, and 0.5 Na2-GTP) (Fig. 7C,D). This reversal potential is midway between negative Cs+ and positive Na+, suggesting the involvement of an activation of the nonselective cation channels in CCK-8S-induced depolarization of orexin neurons. The nonselective cation channel blocker La3+ significantly inhibited the CCK-8S-induced inward current, also supporting this idea. In the absence of extracellular calcium ions, the CCK-8S-induced inward current before and after La3+ (10 μm) application was 201.0 ± 48.0 and 67.0 ± 17.8 pA, respectively (Fig. 7F) (n = 5; p < 0.05; ANOVA).
Figure 7.

A, The effect of extracellular Na+ on the CCK-8S-induced inward current. NaCl was replaced by an equimolar concentration of choline chloride in the presence of TTX (1 μm). CCK-8S (10 nm)-induced inward current was dramatically decreased in the Na+-free solution (n = 7). B, The effect of extracellular Ca2+ on the CCK-8S-induced inward current. CCK-8S (5 nm) induced weak inward current in the presence of extracellular calcium concentration. This inward current was dramatically increased in the Ca2+-free solution (n = 7). The bar graph in B shows a fold increase in inward current in the Ca2+-free solution. C, D, The current-voltage relationship obtained by voltage step protocol using a CsCl pipette in the Ca2+-free extracellular solution. The steady-state current, at the end of the voltage step (marked by circle in C), is plotted in current-voltage relationship. I-V curve shows that the reversal potential of the CCK-8S (100 nm)-induced current was -2.3 ± 2.1 mV (n = 5). Neurons were voltage clamped at 0 mV for 2 s, and the membrane potential was stepped from -100 to +100 mV with an increment of 20 mV at a duration of 40 ms. Open circles indicate control, and filled circles indicate CCK-8S (100 nm) application. E, F, The effects of the nonselective cation channel blocker SKF96365 and La3+. E, SKF96365 (10 μm) inhibited CCK-8S-induced inward current in the Ca2+-free extracellular solution. F, Bar graph indicates that SKF96365 inhibited CCK-induced current in a concentration-dependent manner (n = 5) in the Ca2+-free extracellular solution. La3+ (10 μm) also significantly inhibited CCK-8S-induced inward current (n = 5). Values are mean ± SEM. *p < 0.05.
Several recent reports suggest that the transient receptor potential (TRP) channel family plays an important role in the receptor-operated influx of cations (Spassova et al., 2004; Takai et al., 2004). In addition, the current through TRP channels are known to be suppressed by the presence of extracellular calcium ions (Lintschinger et al., 2000; Hill, 2001). To examine whether TRP channels are involved in the CCK-induced response, the effect of the SKF96365, which is often used as a TRP channel blocker (Halaszovich et al., 2000), on the CCK-8S-induced inward current was tested. In the absence of extracellular calcium, CCK-8S induced a 201.0 ± 48.0 pA (n = 7) inward current. Pretreatment of SKF96365 inhibited the CCK-8S-induced inward current in a concentration-dependent manner (Fig. 7E,F). SKF96365 (1 and 10 μm) inhibited the CCK-8S-induced inward current by 85.1 ± 24.1 pA (n = 4; p = 0.034; ANOVA) and 47.2 ± 4.7 pA (n = 5; p = 0.004; ANOVA), respectively. Together, these results indicate that CCK-8S depolarizes orexin neurons by activating the extracellular calcium ion-sensitive nonselective cation channels, which are possibly TRP channels, through the CCKAR.
Simultaneous recording of calcium imaging and whole-cell voltage clamp on the same neuron
To study whether the increase in intracellular calcium concentration induced by CCK-8S is caused by depolarization, simultaneous recording of calcium imaging and whole-cell voltage clamp on the same neuron was performed. Neurons subjected to calcium imaging were whole-cell patch clamped at -60 mV. Under this condition, CCK-8S (100 nm) was applied by bath application in the presence of TTX (1 μm). Amplitude of CCK-8S-induced inward current was comparable with that obtained by patch-clamp recording using orexin/EGFP mice [19.7 ± 2.5 pA (n = 17) vs 20.1 ± 3.0 pA (n = 10); p = 0.92; not significantly different]. However, an increase in intracellular calcium, measured by YFP/CFP ratio recording, was not observed under the voltage-clamp condition (Fig. 8). Increments of YFP/CFP ratio were 0.015 ± 0.003 (n = 10) when neurons were voltage-clamped. This value is markedly smaller than that observed in the normal conditions (0.085 ± 0.01; n = 28; p = 0.0003; ANOVA) (Fig. 8B,C). CCK-8S (100 nm) induced an increase in intracellular calcium when clamp mode was switched to current clamp from voltage clamp (data not shown). These results suggest that an increase in intracellular calcium occurred secondary to depolarization.
Figure 8.

Simultaneous recording of calcium imaging and slice patch clamp. A, The neuron subjected to calcium imaging was whole-cell patch clamped. Membrane potential was held at -60 mV. CCK-8S (100 nm) was applied by bath application during the period represented by the bar. The experiment was performed in the presence of TTX (1 μm). B and C summarize an inward current and Δ ratio alteration obtained in A. Values are mean ± SEM. *p < 0.05.
Discussion
In the present study, we showed that orexin/YC2.1 transgenic mice are useful for the screening of substances that affect the activity of orexin neurons. Calcium imaging using these mice revealed that CCK-8S, neurotensin, and vasopressin activate orexin neurons. The mechanisms underlying CCK-8S-induced activation of orexin neurons were analyzed in detail by both calcium imaging using orexin/YC2.1 and slice patch clamp using orexin/EGFP transgenic mice. These experiments revealed that CCK-8S induces activation of orexin neurons through an activation of the extracellular calcium-sensitive cation channel via a CCKAR.
Calcium imaging of orexin neurons using orexin/YC2.1 transgenic mice brain slice
The genetically encoded calcium sensor YC2.1 was specifically expressed in the orexin neurons. The advantages of this method are as follows: (1) the images obtained were of a high signal-to-noise ratio, because the expression of YC2.1 is restricted to the orexin neurons. Additionally, YFP/CFP ratio imaging reduces the noise. (2) This system minimizes artificial damage because it is not necessary to load a calcium-sensing dye, such as fura-2 and fluo-3. (3) Adult mice (6-8 weeks of age) can be used for experiments as well as young mice. (4) The activity of several orexin neurons can be simultaneously recorded in the same field of view. Although orexin neurons are widely distributed, we could measure 5-10 orexin neurons simultaneously in one experiment. (5) This system is suitable for long time recording. Although we did not show data from long time recordings here, we could continuously record the same neurons for 5-6 h if bleaching of fluorescent proteins was avoided by minimizing exposure time.
CCK-8S activates orexin neurons
Calcium imaging of orexin neurons using orexin/YC2.1 mice revealed that CCK-8S activates orexin neurons. The fact that CCK-8S had little effect on sPSCs and CCK-8S-induced depolarization or inward current in orexin neurons even in the presence of TTX suggest direct activation through CCK receptor(s) on the orexin neurons. The CCKB receptor agonists CCK-4 and CCK-8NS had little effect on orexin neurons, whereas the CCKA receptor antagonist lorglumide inhibited both the CCK-8S-induced inward current and the increase in intracellular calcium concentration, strongly suggesting an involvement of CCKA receptor in this response. CCKAR-like immunoreactivity observed on all orexin-like immunoreactive neurons also supports this idea. CCKAR-like immunoreactivity was observed not only on orexin neurons but also on non-orexin neurons in the LHA. Patch-clamp electrophysiology using orexin/EGFP mice showed that 26% of non-EGFP-positive neurons in the LHA are also activated by CCK-8S.
CCKAR is expressed on the neurons in the CNS as well as in peripheral tissues (Noble et al., 1999; Mercer and Beart, 2004). CCKAR is reported to activate neurons through decreased potassium conductance in rat dorsal motor nucleus (Zheng et al., 2004) or activation of the L-type calcium channel in cultured myenteric neurons (Zhang et al., 2002) or the nonselective cation channel in nodose ganglion (Dun et al., 1991). In the present study, we showed that the CCK-8S-induced increase in [Ca2+]i is dependent on calcium influx. The fact that removing extracellular calcium ions enhanced the CCK-8S-induced inward current 14-fold suggests the possible involvement of a voltage-dependent calcium channel or of TRP channel families in this response, because these channels are reported to become highly permeable to monovalent ions when divalent ions are removed from extracellular environment (Hill, 2001). The CCK-8S-induced increase in [Ca2+]i was inhibited by nonselective cation channel blockers, La3+, Gd3+, and 2-APB, but was not inhibited by voltage-dependent calcium channel blockers, nifedipine, ω-conotoxin, ω-agatoxin, and SNX-482, also suggesting an involvement of the nonselective cation channel in this response. Additionally, the CCK-8S-induced inward current was almost abolished by replacement of extracellular sodium with choline, and the reversal potential of the CCK-8S-induced current shows good agreement with a nonselective cation channel, also supporting this idea. ZD7288, an HCN channel blocker, had no effect, excluding an involvement of an HCN channel (Fig. 6). The CCK-8S-induced inward current in the absence of extracellular calcium ions was inhibited by SKF96365, which is often used as a TRP channel blocker (Halaszovich et al., 2000). These results show that the use of channel blockers demonstrate a profile that most closely fits members of the TRP family. Ishibashi et al. (2003) reported a similar result using acutely isolated rat cardiac synaptic ganglia, showing that noradrenaline evoked an extracellular Ca2+- or Mg2+-sensitive inward current, which was inhibited by La3+, Gd3+, and SKF96365, and they claimed possible involvement of a TRP channel in this response. Several types of TRP channels might be involved, because intracellular calcium concentration was not increased by CCK-8S under the voltage-clamped condition held at -60 mV (Fig. 8). Most likely, the channels primarily opened by CCK-8S have a preference for monovalent ions, and induced depolarization. The secondarily opened channels opened by depolarization have a preference for divalent ions. In the present study, although we did not elucidate the detailed intracellular mechanism of an activation of the nonselective cation channel through the CCKAR, the phospholipase C or protein kinase C (PKC) pathway might be involved in this response, because the CCKA receptor is believed to couple with the Gq and Gs subclass of G-proteins (Pangalos and Davies, 2002). TRP receptors, especially the TRPC family, are reported to be activated by diacylglycerol or by PKC-mediated phosphorylation (Hofmann et al., 1999; Okada et al., 1999; Spassova et al., 2004). Thus, it is possible that TRP channels are activated downstream of CCKAR via the Gq-PKC pathway.
Physiological significance of CCK-8S-mediated activation of orexin neurons
What is the consequence of the activating actions of CCK for orexin neuron activity? In addition to orexins being implicated in the regulation of sleep/wakefulness states, they increase food intake and locomotor activity when administered centrally to animals (Sakurai et al., 1998; Chemelli et al., 1999; Yamanaka et al., 1999; Nakamura et al., 2000; Hara et al., 2001). Thus, the activation of CCK to activate orexin neurons appears paradoxical because CCK suppresses food intake (Gibbs et al., 1973; Schick et al., 1986; Kopin et al., 1999) and locomotor activity (Hirosue et al., 1992) when administered intraperitoneally or intracerebroventricularly. It has also been reported that a CCKAR antagonist, L-364,718, suppresses feeding-induced sleep in rats, suggesting that CCKAR activation might participate in postprandial hyper-somnolence and that CCK is a somnogen (Shemyakin and Kapas, 2001).
In contrast, the Otsuka-Long-Evans-Tokushima Fatty rats, which lack expression of the CCKAR gene, show a reduced amount of large movements during the dark phase before the manifestation of non-insulin-dependent diabetes mellitus compared with control rats (Sei et al., 1999). This study demonstrates that the CCKAR might play a role in maintaining the level of motor activity. These discrepancies might stem from the wide distribution of the CCK receptors. Because CCK receptors are widely distributed throughout the brain, the pharmacological effects of CCK are the consequence of simultaneous stimulation of CCK receptors in various brain regions. Activation of orexin neurons by CCK might be counterbalanced by the effects of CCK on other brain regions. Somnogenic and anorexic actions of CCK might be partly reduced by the activation of orexin neurons by CCK. This mechanism might be apparent in supporting the vigilance level after food intake, when the CCK level is elevated. The report in which human male volunteers having high-fat meals received an intravenous infusion of the CCKAR receptor antagonist loxiglumide supports this idea; subjects who were infused with loxiglumide on their first test day felt significantly more fatigued, sleepy, and tense and less vigorous, less efficient, and had lower energetic arousal during the loxiglumide infusion than during the saline infusion (Wells et al., 1997). Additional studies using conditional knock-out mice that lack CCKAR in orexin neurons are needed to clarify the physiological role of CCK on orexin neurons.
Where is the origin of putative CCK input to the orexin neurons? Recent analysis of afferents to orexin neurons using transgenic mice, which specifically express a genetically encoded retrograde tracer in orexin neurons, provides valuable information to address this issue. This study demonstrated that orexin neurons receive innervations from various brain areas. Among these areas, the basolateral amygdala, medial preoptic area, and paraventricular nucleus overlap with previously reported localization of CCK-containing neurons (Mezey et al., 1985; Fox et al., 1990; Mascagni and McDonald, 2003). These brain areas are thought to be involved in stress response, fear, anxiety, cognition, sexual behavior, and memory and in the generation of sensations such as fear. If these neurons innervate orexin neurons, CCKergic activation of orexin neurons might be involved in the generation of these brain functions.
In this report, we show that the expression of a genetically encoded calcium sensor in specific neurons using a specific promoter is useful to search for substances that affect the activity of those neurons. CCK-8S activates orexin neurons, suggesting that CCK may have a role in enhancing arousal through orexin neurons in the hypothalamus.
Footnotes
This work was supported by grants-in-aid for scientific research (S) and (B) and for young scientists (B), the 21st Century Center of Excellence Program from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, the Kanae Foundation, and National Institutes of Health Grants RO1MH61755 and RO1AG020584. We thank Charles Jones for proofreading.
Correspondence should be addressed to Dr. Akihiro Yamanaka, Department of Molecular Pharmacology, Graduate School of Comprehensive Human Sciences, University of Tsukuba, Tsukuba, Ibaraki 305-8575, Japan. E-mail: yamank@md.tsukuba.ac.jp.
Copyright © 2005 Society for Neuroscience 0270-6474/05/257459-11$15.00/0
N.T. and A.Y. contributed equally to this work.
References
- Acuna-Goycolea C, van den Pol A (2004) Glucagon-like peptide 1 excites hypocretin/orexin neurons by direct and indirect mechanisms: implications for viscera-mediated arousal. J Neurosci 24: 8141-8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer L, Eggermann E, Serafin M, Grivel J, Machard D, Muhlethaler M, Jones BE (2005) Opposite effects of noradrenaline and acetylcholine upon hypocretin/orexin versus melanin concentrating hormone neurons in rat hypothalamic slices. Neuroscience 130: 807-811. [DOI] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB, Yanagisawa M (1999) Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell 98: 437-451. [DOI] [PubMed] [Google Scholar]
- de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett II FS, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG (1998) The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci USA 95: 322-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dun NJ, Wu SY, Lin CW (1991) Excitatory effects of cholecystokinin octapeptide on rat nodose ganglion cells in vitro. Brain Res 556: 161-164. [DOI] [PubMed] [Google Scholar]
- Felix R, Sandoval A, Sanchez D, Gomora JC, De la Vega-Beltran JL, Trevino CL, Darszon A (2003) ZD7288 inhibits low-threshold Ca(2+) channel activity and regulates sperm function. Biochem Biophys Res Commun 311: 187-192. [DOI] [PubMed] [Google Scholar]
- Fossa AA, DePasquale MJ, Morrone J, Zorn SH, Bryce D, Lowe JA, McLean S (1997) Cardiovascular effects of cholecystokinin-4 are mediated by the cholecystokinin-B receptor subtype in the conscious guinea pig and dog. J Pharmacol Exp Ther 281: 180-187. [PubMed] [Google Scholar]
- Fox CA, Adam DE, Watson Jr R, Hoffman GE, Jacobson CD (1990) Immunohistochemical localization of cholecystokinin in the medial preoptic area and anterior hypothalamus of the Brazilian gray short-tailed opossum: a sex difference. J Neurobiol 21: 705-718. [DOI] [PubMed] [Google Scholar]
- Fu LY, Acuna-Goycolea C, van den Pol AN (2004) Neuropeptide Y inhibits hypocretin/orexin neurons by multiple presynaptic and postsynaptic mechanisms: tonic depression of the hypothalamic arousal system. J Neurosci 24: 8741-8751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs J, Young RC, Smith GP (1973) Cholecystokinin decreases food intake in rats. J Comp Physiol Psychol 84: 488-495. [DOI] [PubMed] [Google Scholar]
- Grivel J, Cvetkovic V, Bayer L, Machard D, Tobler I, Muhlethaler M, Serafin M (2005) The wake-promoting hypocretin/orexin neurons change their response to noradrenaline after sleep deprivation. J Neurosci 25: 4127-4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyas AI, Gorcs TJ, Freund TF (1990) Innervation of different peptide-containing neurons in the hippocampus by GABAergic septal afferents. Neuroscience 37: 31-44. [DOI] [PubMed] [Google Scholar]
- Halaszovich CR, Zitt C, Jungling E, Luckhoff A (2000) Inhibition of TRP3 channels by lanthanides. Block from the cytosolic side of the plasma membrane. J Biol Chem 275: 37423-37428. [DOI] [PubMed] [Google Scholar]
- Hara J, Beuckmann CT, Nambu T, Willie JT, Chemelli RM, Sinton CM, Sugiyama F, Yagami K, Goto K, Yanagisawa M, Sakurai T (2001) Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron 30: 345-354. [DOI] [PubMed] [Google Scholar]
- Hill B (2001) Ion channels of excitable membranes. In: Calcium dynamics, epithelial transport, and intercellular coupling (Hill B, ed), pp 290-291. Sunderland, MA: Sinauer.
- Hirosue Y, Inui A, Miura M, Nakajima M, Okita M, Himori N, Baba S, Kasuga M (1992) Effects of CCK antagonists on CCK-induced suppression of locomotor activity in mice. Peptides 13: 155-157. [DOI] [PubMed] [Google Scholar]
- Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G (1999) Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397: 259-263. [DOI] [PubMed] [Google Scholar]
- Hokfelt T, Skirboll L, Rehfeld JF, Goldstein M, Markey K, Dann O (1980) A subpopulation of mesencephalic dopamine neurons projecting to limbic areas contains a cholecystokinin-like peptide: evidence from immunohistochemistry combined with retrograde tracing. Neuroscience 5: 2093-2124. [DOI] [PubMed] [Google Scholar]
- Ishibashi H, Umezu M, Jang IS, Ito Y, Akaike N (2003) Alpha 1-adrenoceptor-activated cation currents in neurones acutely isolated from rat cardiac parasympathetic ganglia. J Physiol (Lond) 548: 111-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopin AS, Mathes WF, McBride EW, Nguyen M, Al-Haider W, Schmitz F, Bonner-Weir S, Kanarek R, Beinborn M (1999) The cholecystokinin-A receptor mediates inhibition of food intake yet is not essential for the maintenance of body weight. J Clin Invest 103: 383-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, van den Pol AN (2005) Direct and indirect inhibition by catecholamines of hypocretin/orexin neurons. J Neurosci 25: 173-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Gao XB, Sakurai T, van den Pol AN (2002) Hypocretin/orexin excites hypocretin neurons via a local glutamate neuron-A potential mechanism for orchestrating the hypothalamic arousal system. Neuron 36: 1169-1181. [DOI] [PubMed] [Google Scholar]
- Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, Qiu X, de Jong PJ, Nishino S, Mignot E (1999) The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell 98: 365-376. [DOI] [PubMed] [Google Scholar]
- Lintschinger B, Balzer-Geldsetzer M, Baskaran T, Graier WF, Romanin C, Zhu MX, Groschner K (2000) Coassembly of Trp1 and Trp3 proteins generates diacylglycerol- and Ca2+-sensitive cation channels. J Biol Chem 275: 27799-27805. [DOI] [PubMed] [Google Scholar]
- Mascagni F, McDonald AJ (2003) Immunohistochemical characterization of cholecystokinin containing neurons in the rat basolateral amygdala. Brain Res 976: 171-184. [DOI] [PubMed] [Google Scholar]
- Mercer LD, Beart PM (2004) Immunolocalization of CCK1R in rat brain using a new anti-peptide antibody. Neurosci Lett 359: 109-113. [DOI] [PubMed] [Google Scholar]
- Mezey E, Reisine TD, Skirboll L, Beinfeld M, Kiss JZ (1985) Cholecystokinin in the medial parvocellular subdivision of the paraventricular nucleus. Co-existence with corticotropin-releasing hormone. Ann NY Acad Sci 448: 152-156. [DOI] [PubMed] [Google Scholar]
- Miyawaki A, Griesbeck O, Heim R, Tsien RY (1999) Dynamic and quantitative Ca2+ measurements using improved cameleons. Proc Natl Acad Sci USA 96: 2135-2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraki Y, Yamanaka A, Tsujino N, Kilduff TS, Goto K, Sakurai T (2004) Serotonergic regulation of the orexin/hypocretin neurons through the 5-HT1A receptor. J Neurosci 24: 7159-7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Uramura K, Nambu T, Yada T, Goto K, Yanagisawa M, Sakurai T (2000) Orexin-induced hyperlocomotion and stereotypy are mediated by the dopaminergic system. Brain Res 873: 181-187. [DOI] [PubMed] [Google Scholar]
- Nambu T, Sakurai T, Mizukami K, Hosoya Y, Yanagisawa M, Goto K (1999) Distribution of orexin neurons in the adult rat brain. Brain Res 827: 243-260. [DOI] [PubMed] [Google Scholar]
- Noble F, Wank SA, Crawley JN, Bradwejn J, Seroogy KB, Hamon M, Roques BP (1999) International Union of Pharmacology. XXI. Structure, distribution, and functions of cholecystokinin receptors. Pharmacol Rev 51: 745-781. [PubMed] [Google Scholar]
- Okada T, Inoue R, Yamazaki K, Maeda A, Kurosaki T, Yamakuni T, Tanaka I, Shimizu S, Ikenaka K, Imoto K, Mori Y (1999) Molecular and functional characterization of a novel mouse transient receptor potential protein homologue TRP7. Ca2+-permeable cation channel that is constitutively activated and enhanced by stimulation of G protein-coupled receptor. J Biol Chem 274: 27359-27370. [DOI] [PubMed] [Google Scholar]
- Pangalos MN, Davies CH (2002) Understanding G protein-coupled receptors and their role in the CNS. In: Cholecystokinin receptors (Nobel F, Roques BP, eds), pp 242-263. New York: Oxford UP.
- Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS (1998) Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci 18: 9996-10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, Charnay Y, Nevsimalova S, Aldrich M, Reynolds D, Albin R, Li R, Hungs M, Pedrazzoli M, Padigaru M, Kucherlapati M, Fan J, Maki R, Lammers GJ, Bouras C, Kucherlapati R, Nishino S, Mignot E (2000) A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med 6: 991-997. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M (1998) Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 92: 573-585. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Moriguchi T, Furuya K, Kajiwara N, Nakamura T, Yanagisawa M, Goto K (1999) Structure and function of human prepro-orexin gene. J Biol Chem 274: 17771-17776. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Nagata R, Yamanaka A, Kawamura H, Tsujino N, Muraki Y, Kageyama H, Kunita S, Takahashi S, Goto K, Koyama Y, Shioda S, Yanagisawa M (2005) Input of orexin/hypocretin neurons revealed by a genetically encoded tracer in mice. Neuron 46: 297-308. [DOI] [PubMed] [Google Scholar]
- Schick RR, Yaksh TL, Go VL (1986) Intracerebroventricular injections of cholecystokinin octapeptide suppress feeding in rats—pharmacological characterization of this action. Regul Pept 14: 277-291. [DOI] [PubMed] [Google Scholar]
- Sei M, Sei H, Shima K (1999) Spontaneous activity, sleep, and body temperature in rats lacking the CCK-A receptor. Physiol Behav 68: 25-29. [DOI] [PubMed] [Google Scholar]
- Shemyakin A, Kapas L (2001) L-364,718, a cholecystokinin-A receptor antagonist, suppresses feeding-induced sleep in rats. Am J Physiol Regul Integr Comp Physiol 280: R1420-R1426. [DOI] [PubMed] [Google Scholar]
- Spassova MA, Soboloff J, He LP, Hewavitharana T, Xu W, Venkatachalam K, van Rossum DB, Patterson RL, Gill DL (2004) Calcium entry mediated by SOCs and TRP channels: variations and enigma. Biochim Biophys Acta 1742: 9-20. [DOI] [PubMed] [Google Scholar]
- Takai Y, Sugawara R, Ohinata H, Takai A (2004) Two types of non-selective cation channel opened by muscarinic stimulation with carbachol in bovine ciliary muscle cells. J Physiol (Lond) 559: 899-922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells AS, Read NW, Fried M, Borovicka J, D'Amato M (1997) Effects of a specific CCK-A antagonist, loxiglumide, on postprandial mood and sleepiness. J Psychopharmacol 11: 241-246. [DOI] [PubMed] [Google Scholar]
- Willie JT, Chemelli RM, Sinton CM, Tokita S, Williams SC, Kisanuki YY, Marcus JN, Lee C, Elmquist JK, Kohlmeier KA, Leonard CS, Richardson JA, Hammer RE, Yanagisawa M (2003) Distinct narcolepsy syndromes in orexin receptor-2 and orexin null mice: molecular genetic dissection of non-REM and REM sleep regulatory processes. Neuron 38: 715-730. [DOI] [PubMed] [Google Scholar]
- Winsky-Sommerer R, Yamanaka A, Diano S, Borok E, Roberts AJ, Sakurai T, Kilduff TS, Horvath TL, de Lecea L (2004) Interaction between the corticotropin-releasing factor system and hypocretins (orexins): a novel circuit mediating stress response. J Neurosci 24: 11439-11448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka A, Sakurai T, Katsumoto T, Yanagisawa M, Goto K (1999) Chronic intracerebroventricular administration of orexin-A to rats increases food intake in daytime, but has no effect on body weight. Brain Res 849: 248-252. [DOI] [PubMed] [Google Scholar]
- Yamanaka A, Muraki Y, Tsujino N, Goto K, Sakurai T (2003a) Regulation of orexin neurons by the monoaminergic and cholinergic systems. Biochem Biophys Res Commun 303: 120-129. [DOI] [PubMed] [Google Scholar]
- Yamanaka A, Beuckmann CT, Willie JT, Hara J, Tsujino N, Mieda M, Tominaga M, Yagami K, Sugiyama F, Goto K, Yanagisawa M, Sakurai T (2003b) Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron 38: 701-713. [DOI] [PubMed] [Google Scholar]
- Zhang W, Segura BJ, Mulholland MW (2002) Cholecystokinin-8 induces intracellular calcium signaling in cultured myenteric neurons from neonatal guinea pigs. Peptides 23: 1793-1801. [DOI] [PubMed] [Google Scholar]
- Zheng Z, Lewis MW, Travagli RA (2004) In vitro analysis of the effects of cholecystokinin (CCK) on rat brainstem motorneurons. Am J Physiol Gastrointest Liver Physiol 288: G1066-G1073. [DOI] [PMC free article] [PubMed] [Google Scholar]