

Abstract
Amyloid-β (Aβ), a peptide thought to play a crucial role in Alzheimer's disease (AD), has many targets that, in turn, activate different second-messenger cascades. Interestingly, Aβ has been found to markedly impair hippocampal long-term potentiation (LTP). To identify a new pathway that might be responsible for such impairment, we analyzed the role of the nitric oxide (NO)/soluble guanylyl cyclase (sGC)/cGMP/cGMP-dependent protein kinase (cGK)/cAMP-responsive element-binding protein (CREB) cascade because of its involvement in LTP. The use of the NO donor 2-(N,N-dethylamino)-diazenolate-2-oxide diethylammonium salt (DEA/NO), the sGC stimulator 3-(4-amino-5-cyclopropylpyrimidine-2-yl)-1-(2-fluorobenzyl)-1H-pyrazolo[3,4-b]pyridine, or the cGMP-analogs 8-bromo-cGMP and 8-(4-chlorophenylthio)-cGMP reversed the Aβ-induced impairment of CA1-LTP through cGK activation. Furthermore, these compounds reestablished the enhancement of CREB phosphorylation occurring during LTP in slices exposed to Aβ. We also found that Aβ blocks the increase in cGMP immunoreactivity occurring immediately after LTP and that DEA/NO counteracts the effect of Aβ. These results strongly suggest that, when modulating hippocampal synaptic plasticity, Aβ downregulates the NO/cGMP/cGK/CREB pathway; thus, enhancement of the NO/cGMP signaling may provide a novel approach to the treatment of AD and other neurodegenerative diseases with elevated production of Aβ.
Keywords: Alzheimer's disease, amyloid-β, hippocampus, long-term potentiation, synaptic plasticity, NO/cGMP/CREB pathway
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder, characterized by cognitive impairment, consisting of loss of memory and reasoning leading to dementia. Atrophy of specific brain areas, deposition of amyloid-β peptide (Aβ) in senile plaques and within the walls of the cerebral microvasculature, and presence of neurofibrillary tangles characterize the late stages of the disease (Selkoe, 1994). However, the earliest amnesic symptoms, occurring in the absence of any other clinical sign of brain injury, are likely to be a result of a more subtle defect, probably occurring at the level of the synapse and produced, at least in part, by Aβ (Cullen et al., 1997; Itoh et al., 1999; Vitolo et al., 2002). Low levels of pathogenic Aβ assemblies would alter mechanisms underlying excitatory neurotransmission at individual synapses, producing synaptic dysfunction before synapse loss (Selkoe, 2002). Aβ has many targets (Mattson, 1997) that, in turn, activate different second-messenger cascades. The identification of these cascades has drawn the attention of many researchers because of the potential therapeutic application of drugs aiming at limiting the adverse effects by Aβ. Interestingly, Aβ1-42 has been found to markedly inhibit long-term potentiation (LTP), a physiological correlate of synaptic plasticity that is thought to underlie learning and memory and is influenced by different second-messenger systems (Bliss and Collingridge, 1993). Therefore, several recent studies have tried to elucidate the second-messenger pathway(s) by which Aβ blocks LTP, including the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase (MAPK) as well as the metabotropic glutamate receptor type 5 (Wang et al., 2004a), the extracellular signal-regulated kinase (ERK)-MAPK cascade (Dineley et al., 2001), and the cAMP/cAMP-dependent-protein kinase (cAK)/cAMP-regulatory element-binding protein (CREB) pathway (Vitolo et al., 2002).
Both hippocampal and cortical LTP are known to involve nitric oxide (NO) (Arancio et al., 1996; Bon and Garthwaite, 2003), a membrane permeable gas that might induce CREB phosphorylation (Ohki et al., 1995; Gudi et al., 1996; Lu et al., 1999). NO is synthesized from l-arginine by the enzyme NO synthase (NOS) and induces cGMP production through the activation of the enzyme soluble guanylyl cyclase (sGC) (Garthwaite and Boulton, 1995). cGMP, in turn, activates cGMP-dependent protein-kinases (cGKs), a family of proteins with an important role as signal transduction mediators (Lohmann et al., 1997), leading to an increase of CREB phosphorylation during LTP (Lu et al., 1999). There is extensive literature suggesting that NO may be responsible for the following: (1) neuroprotection during Aβ-induced cell death (Chalimoniuk and Strosznajder, 1998; Troy et al., 2000; Wirtz-Brugger and Giovanni, 2000; Law et al., 2001; Suhara et al., 2003) and vasoactivity (Mattson, 1997; McCarty, 1998; Paris et al., 1999), and (2) Aβ toxicity in different systems ranging from in vivo animals to cell lines (Parks et al., 2001; Tran et al., 2001; Haas et al., 2002; Xie et al., 2002; Monsonego et al., 2003). We have now obtained data providing evidence that the NO/cGMP/cGK/CREB pathway might represent another crucial target through which Aβ peptides manage to induce impairment of hippocampal synaptic plasticity.
Materials and Methods
Slice preparation. All experiments were performed using 3-month-old male mice (C57BL/6; The Jackson Laboratory, Bar Harbor, ME). The protocol was approved by the Nathan Kline Institute (NKI) Institutional Animal Care and Use Committee. The animals were maintained for 1-2 months after the purchase on a 12 h light/dark cycle (with lights on at 6:00 A.M.) in temperature- and humidity-controlled rooms of the NKI Animal Facility. Food and water were available ad libitum. Animals were killed by cervical dislocation followed by decapitation. Hippocampi were quickly removed. Transverse hippocampal slices (400 μm) were cut and transferred to a recording chamber where they were maintained at 29°C and perfused with artificial CSF (ACSF) continuously bubbled with 95% O2 and 5% CO2. The ACSF composition was composed of the following (in mm): 124.0 NaCl, 4.4 KCl, 1.0 Na2HPO4, 25.0 NaHCO3, 2.0 CaCl2, 2.0 MgSO4, and 10.0 glucose.
Electrophysiological recordings. Field extracellular recordings were performed by stimulating the Schaeffer collateral fibers through a bipolar tungsten electrode and recording in CA1 stratum radiatum with a glass electrode filled with ACSF, as described previously (Vitolo et al., 2002). A 15 min baseline was recorded every minute at an intensity that evoked a response ∼35% of the maximum evoked response. LTP was induced using a theta-burst stimulation (four pulses at 100 Hz, with the bursts repeated at 5 Hz and each tetanus including three 10-burst trains separated by 15 s). In a set of experiments, LTP was induced with one 10-burst train. These experiments indicated that the same pattern of effects obtained with Aβ, DEA/NO, 8-bromo-cGMP (8-Br-cGMP), and three 10-burst train holds under a weaker tetanus (supplemental Fig. 8, available at www.jneurosci.org as supplemental material). Responses were recorded for 2 h after tetanization and measured as field-EPSP (fEPSP) slope expressed as percentage of baseline. The results were expressed as mean ± SEM.
Drugs. For the electrophysiological experiments, the following drugs were used: 2-(N,N-dethylamino)-diazenolate-2-oxide diethylammonium salt (DEA/NO), 3-(4-amino-5-cyclopropylpyrimidine-2-yl)-1-(2-fluorobenzyl)-1H-pyrazolo[3,4-b]pyridine (BAY41-2272) (Alexis Biochemicals, San Diego, CA), 1H-[1,2,4]oxadiazolo[4,3-α]quinoxalin-1-one (ODQ) (Cay-man Chemical, Ann Arbor, MI), 8-Br-cGMP, 8-(4-chlorophenylthio)-cGMP (8-pCPT-cGMP) (Biolog Life Science Institute, Bremen, Germany), (9s, 10R,12R)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylicacid hexyl ester (KT5720), (9s, 10R,12R)-2,3,9,10,11,12-hexahydro-10-methoxy-2,9-dimethyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid methyl ester (KT5823) (Calbiochem, La Jolla, CA), Aβ1-42 (American Peptide, Sunnyvale, CA), and scramble Aβ1-42 (AnaSpec, San Jose, CA). DEA/NO was required to be stored for 24 h in alkaline solution (0.01 m NaOH), diluted immediately before use, and applied for 5 min before induction of LTP at 3 μm. ODQ (10 μm), BAY41-2272 (100 μm), 8-Br-cGMP (1 μm), 8-pCPT-cGMP (1 μm), KT5823 (2 μm), and KT5720 (1 μm) were added to the bath solution for 10 min before LTP. Oligomeric Aβ1-42 was prepared as described previously (Stine et al., 2003). Briefly, the lyophilized peptide (American Peptide) was resuspended in 100% 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP; Sigma, St. Louis, MO) to 1 mm. The solution was aliquoted, and the HFIP was allowed to evaporate in the fume hood. The resulting clear peptide film was dried under vacuum in a SpeedVac and stored at -20°C. Twenty-four hours before use, the aliquots were added to dimethylsulfoxide (DMSO; Sigma) and sonicated for 10 min. Oligomeric Aβ1-42 was obtained by diluting Aβ1-42-DMSO into ACSF concentration, vortexed for 30 s, and incubated at 4°C for 24 h. Before use, this compound was added to ACSF to obtain 200 nm. Aβ1-42 with scrambled sequence was diluted in ACSF at 200 nmol immediately before use (supplemental Fig. 9, available at www.jneurosci.org as supplemental material).
Immunocytochemical experiments. Immunocytochemical measurements of phospho-CREB were performed as described previously (Lu et al., 1999). Hippocampal slices were fixed in ice-cold 4% paraformaldehyde at either 1 or 60 min after the treatment. Slices were washed three times in PBS, treated with 0.3% Triton X-100 for 60 min, washed three times in PBS again, treated with 50 mm ammonium chloride for 20 min, and incubated in 10% goat serum for 60 min. Slices were incubated with the primary antibody [rabbit polyclonal anti-phospho-CREB (Upstate Biotechnology, Lake Placid, NY) diluted 1:100 in 10% goat serum] for 36 h at 4°C. Slices were washed in PBS (six times; 2 h each time), incubated with the secondary antibody (goat anti-rabbit antibody labeled with Alexa Fluor 488; Molecular Probes, Eugene, OR), diluted 1:100 in 10% goat serum for 12 h at 4°C, and washed in PBS again (six times; 2 h each time). Slices were examined by confocal microscopy (Nikon D-Eclipse C1; Nikon, Tokyo, Japan) using a 4× and a 16× objective. Kalman averages of four scans were collected for each image. The analysis was performed using NIH Image software by an observer who was blind to the experimental treatment. The mean fluorescence intensity that exceeded a threshold set above background was determined for each slice in CA1 cell body area. The values were normalized to the values from untreated control slices from the same animal and expressed as mean percentage of control ± SEM. The specificity of the immunofluorescence was confirmed by omitting the primary antibody, which resulted in a significant reduction in fluorescence intensity. For cGMP immunofluorescence measurements, we used the same method as for phospho-CREB. In these experiments phosphodiesterase activity was blocked by adding 1 μm IBMX to the bath solution. Slices were fixed at 10 s, 5 min, and 60 min after treatment (Monfort et al., 2002). Primary antibodies consisted of rabbit polyclonal anti-cGMP (Chemicon International, Temecula, CA) diluted 1:300 in 10% goat serum.
Statistical analyses. Statistical analysis was performed with two-way ANOVA for LTP and CREB immunocytochemistry. Student's t test was used for cGMP immunocytochemistry. The level of significance was set for p < 0.05.
Results
DEA/NO, an NO donor, protects against Aβ-induced impairment of LTP
Electrophysiological experiments were performed by using hippocampal slices that received a tetanic stimulation to produce LTP at the Shaffer collateral-CA1 connection. In a series of experiments, we reproduced previous results showing that LTP was suppressed when hippocampal slices were exposed to 200 nm oligomeric Aβ1-42 for 20 min before tetanization (Fig. 1A) (92.26 ± 10.27 vs 225.99 ± 10.63% of baseline slope at 120 min after tetanus; n = 10/8; F(1, 16) = 98.58, p < 0.001 compared with tetanized slices treated with vehicle alone) (Vitolo et al., 2002). Aβ alone did not affect the baseline (Fig. 1A) (99.48 ± 1.43% of baseline slope at 120 min after tetanus, n = 5; F(1, 7) = 1.003, p > 0.1 compared with control slices, n = 4). As a control for non-specific effects of the peptide, we tested Aβ1-42 with a scrambled sequence (Malin et al., 2001), which did not affect the amount of potentiation (supplemental Fig. 9, available at www.jneurosci.org as supplemental material).
Figure 1.
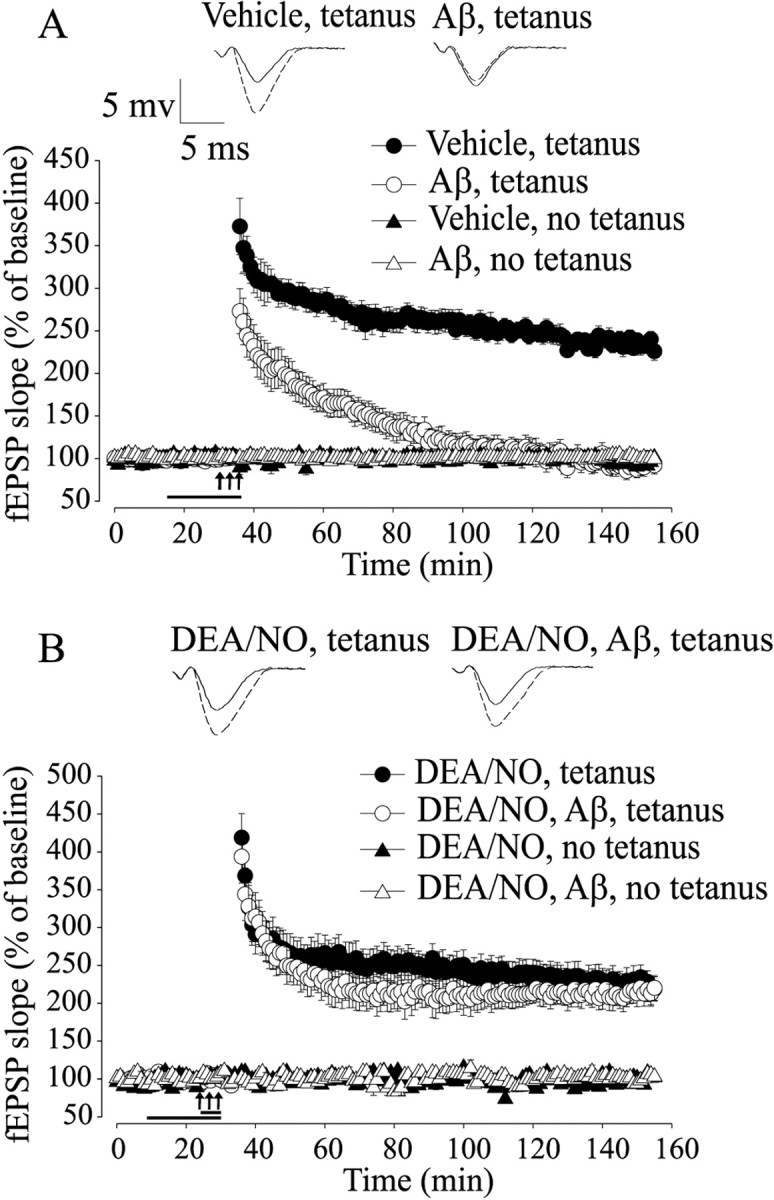
Aβ-induced LTP impairment is rescued by NO. A, LTP is strongly reduced by 200 nm oligomeric Aβ1-42 for 20 min (F(1, 16) = 98.58; p < 0.001, compared with vehicle). Aβ does not affect baseline transmission (F(1, 7) = 1.003; p > 0.1, compared with vehicle). The horizontal bar indicates the period during which Aβ was added to the bath solution. In all figures, the arrows indicate the tetanus application. The insets show representative fEPSP before (solid line) and after (dashed line) tetanus. B, DEA/NO (3 μm) reverses the Aβ-induced LTP impairment without affecting basal neurotransmission (F(1, 15) = 59.1; p <0.001, compared with Aβ plus tetanus).
In interleaved experiments, we determined whether administration of NO protects against Aβ-induced suppression of LTP. As an NO donor, we used 3 μm DEA/NO. We opted for this compound, because it delivers the NO radical and it increases the cGMP levels without affecting basal synaptic transmission (Bon and Garthwaite, 2003). A brief perfusion (5 min) with DEA/NO before induction of LTP in the presence of Aβ (200 nm) protected against LTP suppression (Fig. 1B) (220.23 ± 16.25% of baseline slope at 120 min after tetanus in slices treated with DEA/NO paired with Aβ, n = 7; F(1, 15) = 59.1, p < 0.001 compared with tetanized slices treated with Aβ). This phenomenon was not attributable to an effect of NO on baseline transmission, because perfusion with the donor alone did not affect the baseline (Fig. 1B) (102.12 ± 3.08% of baseline slope at 120 min, n = 4; F(1, 6) = 1.68, p > 0.1 compared with slices treated with vehicle). Moreover, DEA/NO alone did not induce an increase in LTP (Fig. 1B) (218.92 ± 16.04% in tetanized slices treated with DEA/NO alone, n = 6; F(1, 12) = 0.26, p > 0.5 compared with vehicle plus tetanus). Thus, these results demonstrate that NO protects against Aβ-induced inhibition of LTP.
Activation of sGC protects against Aβ-induced impairment of LTP
NO is a highly reactive molecule that can bind to many substrates, including sGC, which is its major cellular receptor (Schlossmann et al., 2003). We therefore investigated a possible role for sGC in the protection against Aβ-induced impairment of LTP. We found that a 10 min application of 10 μm ODQ, a specific irreversible sGC inhibitor (Garthwaite et al., 1995), annulled the rescue of LTP reduction by DEA/NO paired with Aβ and tetanus (Fig. 2A) (105.4 ± 7.56% of baseline slope at 120 min after tetanus in tetanized slices treated with ODQ paired with DEA/NO and Aβ, n = 5; F(1, 10) = 49.9, p < 0.001 compared with DEA/NO plus Aβ plus tetanus). Treatment with ODQ alone reduced LTP to levels equal to those obtained with Aβ (Fig. 2A) (105.11 ± 7.36% of baseline slope at 120 min after tetanus, n = 5; F(1, 13) = 0.53, p > 0.1 compared with Aβ plus tetanus).
Figure 2.

Activation of sGC protects against Aβ-induced LTP impairment. A, The sGC inhibitor ODQ (10 μm) blocks the rescue of Aβ-induced LTP suppression by DEA/NO (F(1, 10) = 49.90; p<0.001, compared with DEA/NO plus Aβ plus tetanus). ODQ alone reduces LTP to levels equal to those obtained with Aβ (F(1, 13) = 0.53; p > 0.1, compared with Aβ plus tetanus). B, The sGC stimulator BAY41-2272 (100 μm) reverses the Aβ-induced LTP impairment without affecting baseline transmission (F(1, 6) = 25.53; p < 0.001, compared with Aβ plus tetanus).
Although the fact that ODQ appears to prevent the NO-induced rescue of LTP induction in the presence of Aβ is consistent with a signaling pathway involving NO stimulation of sGC and subsequent cGMP increase, ODQ might have acted by simply disrupting the underlying mechanisms needed to support LTP. Thus, to determine whether activation of sGC plays a beneficial role in Aβ-induced synaptic dysfunction, we paired Aβ with 100 μm BAY41-2272, an sGC stimulator with no effects on phosphodiesterase (PDE) activity (Koglin et al., 2002). Perfusion of Aβ-treated slices with BAY41-2272 for 10 min rescued the LTP impairment (Fig. 2B) (211.51 ± 22.71 vs 90.51 ± 7.46% of baseline slope at 120 min after tetanus comparing tetanized slices treated with BAY41-2272 plus Aβ to Aβ; n = 4/4; F(1, 6) = 25.53; p < 0.001). BAY41-2272 with or without Aβ did not affect the baseline in slices that were not tetanized (Fig. 2B) (102.46 ± 1.97 and 98.62 ± 1.90 vs 101.78 ± 0.34% of baseline slope at 120 min; n = 3/3/3; F(1, 4) = 0.80; F(1, 4) = 0.26; p > 0.1). Finally, the sGC stimulator alone did not vary the amount of potentiation (Fig. 2B) (221.38 ± 17.21 vs 233.93 ± 12.96% of baseline slope at 120 min compared with slices treated with vehicle; n = 6/4; F(1, 8) = 0.71; p > 0.5). Together, these results indicate that stimulation of sGC might play a beneficial role against Aβ-induced LTP impairment.
8-Br-cGMP, a cGMP analog, protects against Aβ-induced impairment of LTP
Given that sGC mediates a wide range of physiological effects through elevation of intracellular cGMP levels (Schlossmann et al., 2003), we hypothesized that administration of cGMP analogs might protect against Aβ-induced block of LTP. We treated the slices with 8-Br-cGMP, a permeable cGMP analog that, at low concentrations (1 μm), specifically activates cGK (Ka, 0.01-0.21 μm for cGK, 12 μm for cAK) (Butt et al., 1992; Sekhar et al., 1992). Ten minute perfusion with 8-Br-cGMP (1 μm), before induction of LTP in the presence of Aβ, protected against LTP suppression (Fig. 3) (207.59 ± 13.76% of baseline slope at 120 min after tetanus in tetanized slices treated with 8-Br-cGMP paired with Aβ; n = 8; F(1, 16) = 50.59; p < 0.001 compared with Aβ plus tetanus). The protection was not caused by an effect of 8-Br-cGMP on baseline transmission, because perfusion with the analog alone did not modify the baseline (Fig. 3) (98.71 ± 1.04% of baseline slope at 120 min; n = 4; F(1, 6) = 0.30; p > 0.5 compared with slices treated with vehicle). Moreover, the protection was not caused by an effect of the analog on LTP, because perfusion with 8-Br-cGMP did not enhance the amounts of potentiation (Fig. 3) (211.98 ± 15.85%; n = 7; F(1, 13) = 3.60; p > 0.5 compared with slices treated with vehicle). In interleaved experiments, we reproduced the finding shown in Figure 1B that a 20 min perfusion with 200 nm oligomeric Aβ1-42 blocks LTP in the CA1 hippocampal region (98.62 ± 7.32% of baseline slope at 120 min after tetanus; n = 6 compared with control tetanized slices treated with vehicle alone, 234.21 ± 11.41%; n = 6; data not shown). These experiments indicate that the presence of a cGMP analog protects against Aβ-induced inhibition of LTP. Moreover, it is likely that the cGMP protection against Aβ-induced impairment of LTP is mediated by cGK.
Figure 3.

8-Br-cGMP, a cGMP analog, reestablishes normal LTP in Aβ-treated slices. Ten minutes of perfusion with 8-Br-cGMP (1 μm) before induction of LTP in the presence of Aβ rescues the LTP reduction (F(1, 16) = 50.59; p < 0.001, compared with Aβ plus tetanus). Basal neurotransmission is not affected in slices that are not tetanized (F(1, 6) = 0.30; p > 0.5 in 8-Br-cGMP-treated slices compared with slices treated with vehicle).
cGK is involved in the beneficial effect by cGMP analogs
The experiments reported above suggest that cGK inhibition is involved in Aβ-induced reduction of LTP. To better test the role of cGK in Aβ-induced reduction of LTP, KT5823 (2 μm), a specific cGK inhibitor (Izquierdo et al., 2000), was added to the slices treated with Aβ and 8-Br-cGMP. This compound reversed the effect of 8-Br-cGMP (Fig. 4B) (128.80 ± 9.26% of baseline slope at 120 min after tetanus; n = 5; F(1, 10) = 18.16; p < 0.05 compared with tetanized slices treated with 8-Br-cGMP plus Aβ). Treatment with KT5823 alone reduced LTP to levels equal to those obtained with Aβ (Fig. 4A) (99.3 ± 10.41% of baseline slope at 120 min after tetanus; n = 5; F(1, 13) = 0.29; p > 0.5 compared with tetanized slices treated with Aβ).
Figure 4.

cGK is involved in the beneficial effect by cGMP analogs on Aβ-induced LTP impairment. A, When KT5823 (2 μm), a specific cGK inhibitor, is added to the slices treated with Aβ and 8-Br-cGMP, the beneficial effect of 8-Br-cGMP is blocked (F(1, 10) = 18.16; p < 0.05, compared with 8-Br-cGMP plus Aβ plus tetanus). B, Similar to 8-Br-cGMP, 10 min of perfusion with 8-pCPT-cGMP (2 μm) reverses the Aβ-induced LTP impairment (F(1, 16) = 87.81; p < 0.001, compared with Aβ plus tetanus) without affecting basal neurotransmission in nontetanized slices (F(1, 5) = 0.67; p > 0.1 in slices treated with 8-pCPT-cGMP compared with vehicle-treated slices). C, Perfusion with KT5720 paired with 8-Br-cGMP and Aβ does not block LTP (F(1, 11) = 0.44; p > 0.5, compared with 8-Br-cGMP plus Aβ plus tetanus). D, KT5720 paired with 8-Br-cGMP does not produce normal LTP (F(1, 7) = 7.22; p < 0.05, compared with vehicle plus tetanus). LTP is inhibited by KT5823 plus 8-Br-cGMP (F(1, 7) = 22.72; p < 0.01, compared with vehicle plus tetanus).
Given that KT5823 might have acted by simply blocking LTP, per se, independently of 8-Br-cGMP, we also examined the effect of the specific cGK agonist 8-pCPT-cGMP. This compound has higher lipophilicity and membrane permeability than 8-Br-cGMP, and it is also selective for cGKs compared with other cGMP targets such as cGMP-stimulated PDE (Geiger et al., 1992). The Ka of 8-pCPT-cGMP is 0.04 -0.44 μm for cGK and 7.0 μm for cAK (Butt et al., 1992; Sekhar et al., 1992). Similar to 8-Br-cGMP, 10 min of perfusion with 8-pCPT-cGMP (1 μm) completely protected against the Aβ-induced LTP reduction (Fig. 4B) (225.55 ± 9.60% of baseline slope at 120 min after tetanus in tetanized slices treated with 8-pCPT-cGMP paired with Aβ; n = 8; F(1, 16) = 87.81; p < 0.001 compared with tetanized slices treated with Aβ alone), whereas 8-pCPT-cGMP alone had no effect on baseline transmission (Fig. 4B) (99.29 ± 1.32%; n = 3; F(1, 5) = 0.67; p > 0.1 compared with slices treated with vehicle), and it did not cause an increase in LTP (Fig. 4B) (217.39 ± 9.89% in tetanized slices treated with 8-pCPT-cGMP alone; n = 6; F(1, 12) = 2.59; p > 0.1 compared with tetanized slices treated with vehicle).
Given that cGMP analogs might exert their effect through cAK (Vitolo et al., 2002), the cAK inhibitor KT5720 (1 μm) was applied for 10 min in slices that were also treated with 8-Br-cGMP and Aβ, as an additional control for specificity versus cGK. This treatment did not abolish the protection against LTP impairment (Fig. 4C) (210.30 ± 15.88% of baseline slope at 120 min after tetanus; n = 6; F(1, 11) = 0.44; p > 0.5 compared with tetanized slices treated with 8-Br-cGMP plus Aβ), suggesting that the reversal of LTP impairment in Aβ-exposed slices by 8-Br-cGMP is not mediated through cAK. Similar results were obtained with simultaneous perfusion of KT5720 with 8-pCPT-cGMP and Aβ (221.52 ± 17.33% of baseline slope at 120 min after tetanus; n = 4; data not shown). KT5720 alone or paired with Aβ reduced LTP (Fig. 4C) (KT5720, 136.08 ± 3.68% of baseline slope at 120 min after tetanus, n = 5, F(1, 11) = 65.94; KT5720 plus Aβ, 115.86 ± 13.16% of baseline slope at 120 min after tetanus, n = 4, F(1, 7) = 21.39; data not shown; p < 0.05 compared with tetanized slices treated with vehicle) and did not modify baseline transmission (Fig. 4C) (96.90 ± 3.39%; n = 4; F(1, 6) = 0.96; p > 0.1). Moreover, treatment with KT5720 paired with 8-Br-cGMP failed to produce normal LTP (Fig. 4D) (143.66 ± 26.80% of baseline slope at 120 min after tetanus vs 222.92 ± 19.53% in tetanized slices that received vehicle alone; n = 4/5; F(1, 7) = 7.22; p < 0.05), whereas the same treatment did not modify baseline transmission (Fig. 4D) (98.85 ± 2.81 vs 101.46 ± 1.72%; n = 3/4; F(1, 5) = 0.34; p > 0.1). These findings are in agreement with previous data suggesting that cGK and cAK activate independent pathways merging at some later steps (Lu et al., 1999). KT5823 plus 8-Br-cGMP blocked LTP in tetanized-slices (Fig. 4D) (105.37 ± 11.98%; n = 4; F(1, 7) = 22.72; p < 0.01) without affecting baseline transmission (Fig. 4D) (101.95 ± 4.08%; n = 3; F(1, 5) = 0.21; p > 0.1). Together, these results suggest that the beneficial effect of cGMP analogs against Aβ-induced LTP impairment is mediated through the activation of their downstream target cGK.
Upregulation of the NO/cGMP/cGK cascade during Aβ-induced LTP suppression rescues the block of CREB phosphorylation
It has been shown that the NO/cGMP/cGK cascade produces LTP through CREB phosphorylation (Lu et al., 1999; Ciani et al., 2002). Thus, we investigated whether the beneficial effect of the upregulation of the NO/cGMP/cGK pathway during Aβ-induced suppression of LTP involves CREB phosphorylation. Hippocampal slices were treated as described in the electrophysiological experiments, fixed 1 or 60 min after the treatment, stained with anti-phospho-CREB antibodies at Ser-133, and viewed on a confocal microscope. We confirmed previous findings (Lu et al., 1999) showing a significant increase in the intensity of immunofluorescence in CA1 cell body area both at 1 and 60 min after application of the tetanus compared with nontetanized control slices from the same animals (Fig. 5A,E) (159.23 ± 2.86% of control vs 100.66 ± 4.04% of control at 1 min, n = 6/4, and 165.6 ± 3.57% of control vs 102.66 ± 4.04% of control at 60 min, n = 4/4; F(1, 14) = 258.80; p < 0.001). Treatment with Aβ1-42 before tetanic stimulation blocked the increase in CA1 immunofluorescence (Fig. 5A,E) (1 min, 115.18 ± 6.64% of control, n = 5; 60 min, 100.13 ± 12.89% of control, n = 5; F(1, 16) = 232.70; p < 0.001). Aβ1-42 alone without tetanization did not increase immunofluorescence (Fig. 5A,E) (103.87 ± 5.03% of control vehicle-treated slices at 1 min, n = 3; 107.04 ± 5.45% of control at 60 min, n = 3; F(1, 10) = 0.31; p > 0.5). Slices treated with scramble Aβ1-42 showed similar immunofluorescence increases as slices treated with vehicle alone (data not shown).
Figure 5.
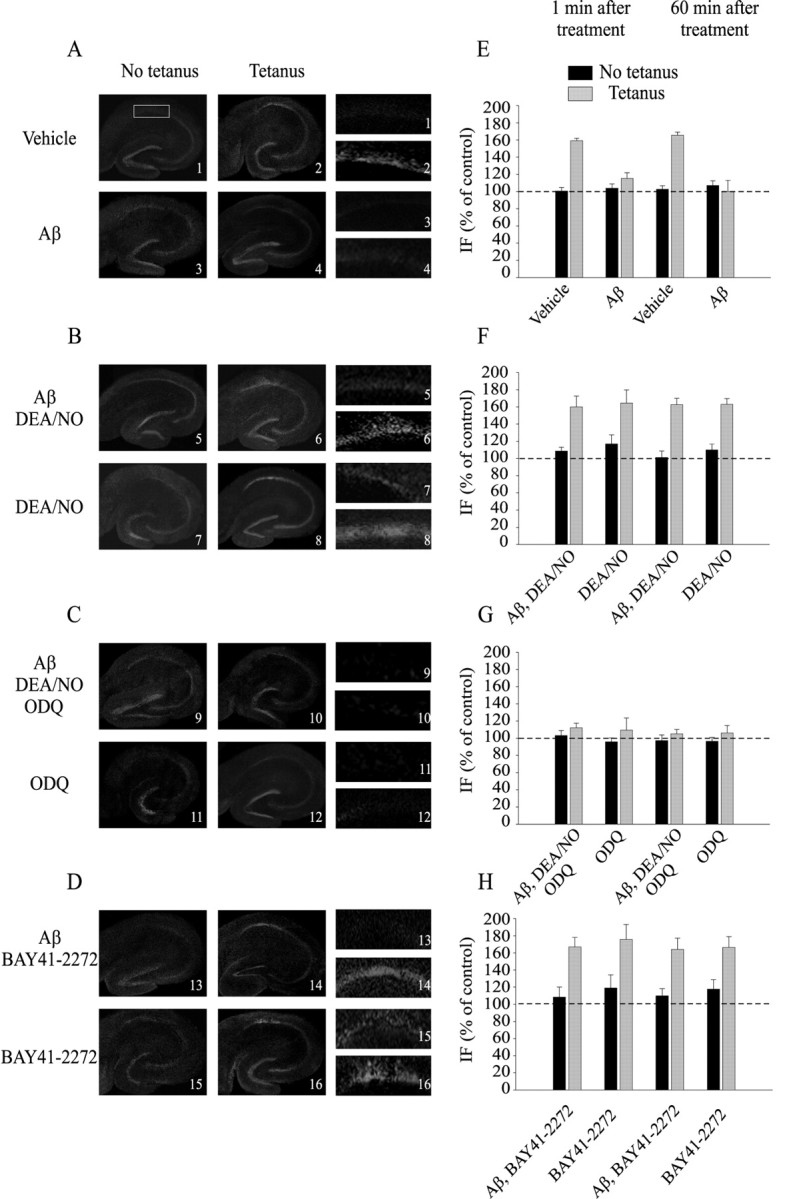
NO and sGC activation reestablish normal increase of CREB phosphorylation following Aβ paired with tetanus. A, Representative examples of hippocampal slices stained with a phospho-CREB antibody. The slices were fixed 1 min after vehicle, tetanus, Aβ, or Aβ plus tetanus. Left, Lower-power (4×) view of the entires lice. Right, Higher-power (16×) view of CA1 cell pyramidal area. B, Examples of slices fixed 1 min after either Aβ plus DEA/NO with and without tetanus or DEA/NO with and without tetanus. C, Examples of slices fixed 1 min after either Aβ plus DEA/NO plus ODQ with and without tetanus or ODQ with and without tetanus. D, Examples of slices fixed 1 min after either Aβ plus BAY41-2272 with and without tetanus or BAY41-2272 with and without tetanus. E, The increase in the intensity of immunofluorescence in the CA1 cell body area after application of the tetanus is blocked by Aβ at both 1 and 60 min after tetanus (F(1, 14) = 258.80, p < 0.001 between vehicle and vehicle plus tetanus; F(1, 16) = 232.70, p < 0.001 between vehicle plus tetanus and Aβ plus tetanus). F, DEA/NO reestablishes an increase in CREB phosphorylation in slices that are treated with Aβ at both 1 and 60 min after tetanus(F(1, 17) = 107.67; p < 0.001, compared with Aβ plus tetanus). G, Inhibition of sGC by ODQ blocks the increase in CREB phosphorylation in slices fixed 1 or 60 min after Aβ plus DEA/NO (F(1, 16) = 128.31; p < 0.001, compared with DEA/NO plus Aβ plus tetanus). H, BAY41-2272 reestablishes the phospho-CREB increase in slices that are treated with Aβ at both 1 and 60 min after tetanus (F(1,8) = 30.64; p < 0.001, compared with Aβ plus tetanus). IF, Immunofluorescence.
Addition of NO produced a significant protection against Aβ effect on phospho-CREB in tetanized slices (Fig. 5B,F) (160.01 ± 12.64% of control at 1 min, n = 6, and 162.59 ± 7.44% of control at 60 min, n = 5; F(1, 17) = 107.67; p < 0.001 compared with tetanized slices treated with Aβ). NO alone or NO paired with Aβ1-42 without tetanic stimulation did not modify immunofluorescence (Fig. 5B,F) (NO, 116.93 ± 10.61% of control at 1 min, n = 3, and 109.68 ± 6.86% of control at 60 min, n = 3; F(1, 10) = 2.91; p > 0.05 compared with vehicle; NO plus Aβ, 108.70 ± 4.48% of control at 1 min, n = 3, and 101.09 ± 7.68% of control at 60 min, n = 3; F(1, 10) = 0.16; p > 0.5 compared with vehicle).
Consistent with the electrophysiological experiments, NO was not capable of reestablishing the increase in CREB phosphorylation after tetanic stimulation in the presence of Aβ when sGC was blocked by ODQ (Fig. 5C,G) (112.13 ± 5.42% of control at 1 min, n = 5, and 105.16 ± 5.00% of control at 60 min, n = 4; F(1, 16) = 128.31; p < 0.001 compared with DEA/NO plus Aβ plus tetanus). The same treatment with no tetanus or with ODQ alone did not vary the immunofluorescence intensity (Fig. 5C,G) (ODQ plus DEA/NO plus Aβ, 103.05 ± 5.83% of control at 1 min, n = 3, and 97.25 ± 6.54% of control at 60 min, n = 3; F(1, 10) = 0.21; p > 0.5 compared with vehicle; ODQ, 95.88 ± 4.57% of control at 1 min, n = 3, and 96.53 ± 4.45% of control at 60 min, n = 3; F(1, 10) = 1.67; p > 0.1 compared with vehicle). ODQ treatment in the absence of Aβ and DEA/NO was capable of reducing tetanus-induced CREB phosphorylation to levels identical to those obtained with Aβ (Fig. 5C,G) (109.44 ± 14.20% of control at 1 min, n = 3, and 106.18 ± 8.55% of control at 60 min, n = 3; F(1, 12) = 0.001; p > 0.5 compared with Aβ plus tetanus). Treatment with BAY41-2272 plus Aβ rescued the tetanus-induced increase of phospho-CREB (Fig. 5D,H) (166.95 ± 10.96% of control at 1 min, n = 3, and 163.95 ± 13.24% of control at 60 min, n = 3; F(1, 8) = 30.64; p < 0.001 compared with tetanized slices treated with Aβ), whereas BAY41-2272 plus Aβ without tetanus did not modify immunofluorescence (Fig. 5D,H) (108.43 ± 11.61% of control at 1 min, n = 3, and 109.88 ± 8.40% of control at 60 min, n = 3; F(1, 8) = 1.93; p > 0.5 compared with vehicle). BAY41-2272 alone did not enhance tetanus-induced CREB phosphorylation (Fig. 5D,H) (166.95 ± 10.96% of control at 1 min, n = 3, and 163.95 ± 13.24% of control at 60 min, n = 3; F(1, 9) = 0.05; p > 0.5). Thus, stimulation of sGC protects against Aβ-induced block of phospho-CREB increase during LTP.
Similar to NO, either 8-Br-cGMP or 8-pCPT-cGMP reestablished the increase in phospho-CREB following tetanus and Aβ (Fig. 6A,B) (8-Br-cGMP plus Aβ plus tetanus, 165.73 ± 9.93% of control at 1 min, n = 4, and 156.91 ± 8.13% of control at 60 min, n = 4; F(1, 14) = 141.50; p < 0.001 compared with Aβ plus tetanus; 8-pCPT-cGMP plus Aβ plus tetanus, 163.14 ± 5.55% of control after 1 min, n = 3, and 166.06 ± 8.75% of control after 60 min, n = 3; F(1, 12) = 96.98; p < 0.001 compared with Aβ plus tetanus). Perfusion with 8-pCPT-cGMP paired with Aβ without tetanic stimulation showed a slight increase in immunofluorescence at 1 min and 60 min, whereas this increase was found to a less extent in slices treated with 8-Br-cGMP plus Aβ (Fig. 6A,B) (8-pCPT-cGMP plus Aβ, 118.59 ± 9.03% of control at 1 min, n = 3, and 116.25 ± 11.98% of control at 60 min, n = 3; F(1, 10) = 5.68; p < 0.05; 8-Br-cGMP plus Aβ, 109.64 ± 12.47% of control at 1 min, n = 3, and 99.94 ± 10.94% of control at 60 min, n = 3; F(1, 10) = 0.09; p > 0.5 compared with vehicle). The protection was not caused by enhancement of LTP by either NO or cGMP analogs that was maintained in the presence of Aβ, because perfusion with these compounds alone did not enhance tetanus-induced CREB phosphorylation [(Fig. 5B,E) (NO plus tetanus, 164.36 ± 15.34% of control at 1 min, n = 4, and 162.89 ± 6.58% of control at 60 min, n = 4; F(1, 14) = 0.072; p > 0.5)] [(Fig. 6A,B) (8-Br-cGMP plus tetanus, 169.84 ± 15.62% of control at 1 min, n = 4, and 164.83 ± 12.44% of control at 60 min, n = 4; F(1, 14) = 1.57; p > 0.05)] [(Fig. 6A,B) (8-pCPT-cGMP plus tetanus, 164.11 ± 11.49% of control at 1 min, n = 4, and 163.75 ± 13.48% of control at 60 min, n = 4; F(1, 14) = 0.07; p > 0.5 compared with vehicle plus tetanus)]. Interestingly, BAY41-2272, 8-Br-cGMP, or 8-pCPT-cGMP alone without Aβ and tetanus produced a slight increase in immunofluorescence [(Fig. 5D,H) (BAY41-2272, 118.98 ± 15.13% of control at 1 min, n = 4, and 117.71 ± 10.97% of control at 60 min, n = 3; F(1, 9) = 4.25; p < 0.05 compared with vehicle)] [(Fig. 6A,B) (8-Br-cGMP, 120.14 ± 9.3% of control at 1 min, n = 3, and 119.72 ± 6.26% of control at 60 min, n = 4; F(1, 11) = 9.03; p < 0.05 compared with vehicle; 8-pCPT-cGMP, 127.41 ± 11.58% of control at 1 min, n = 3, and 122.07 ± 7.15% of control at 60 min, n = 3; F(1, 10) = 13.17; p < 0.001 compared with vehicle)]. This result is consistent with the slight potentiation observed in cultured hippocampal neurons after treatment with cGMP analogs (Arancio et al., 1995). When the cGK inhibitor KT5823 (2 μm) was added to the slices treated with Aβ and 8-Br-cGMP before tetanization, the increase in phospho-CREB was reduced at 1 and 60 min (Fig. 6C,D) (111.95 ± 8.9% of control, n = 3, and 114.38 ± 11.34% of control, n = 3; F(1, 10) = 79.89; p < 0.001). The same treatment with no tetanus or with KT5823 alone did not vary the immunofluorescence intensity [(Fig. 6C,D) (100.24 ± 5.62% of control at 1 min, n = 3, and 110.11 ± 4.51% of control at 60 min, n = 3; F(1, 10) = 0.25; p > 0.5 in slices that received KT5823 paired with 8-Br-cGMP and Aβ compared with vehicle-treated slices)] [(Fig. 6C,D) (101.57 ± 6.19% of control at 1 min, n = 3, and 108.26 ± 3.93% of control at 60 min, n = 3; F(1, 10) = 0.19; p > 0.5 in slices that were treated with KT5823 alone compared with vehicle)]. KT5823 treatment alone or paired with 8-Br-cGMP was capable of reducing tetanus-induced CREB phosphorylation (Fig. 6C,D) (KT5823, 109.25 ± 7.20% of control at 1 min, n = 3, and 107.79 ± 8.01% of control at 60 min, n = 3; F(1, 12) = 209.61; p < 0.001; KT5823 plus 8-Br-cGMP, 107.58 ± 14.12% of control at 1 min, n = 3, and 105.99 ± 12.54% of control at 60 min, n = 3; F(1, 9) = 86.71; p < 0.001 compared with vehicle plus tetanus). Together, these results indicate that an increase in NO and cGMP levels can protect against Aβ suppression of enhancement of CREB phosphorylation during LTP through cGK activation.
Figure 6.
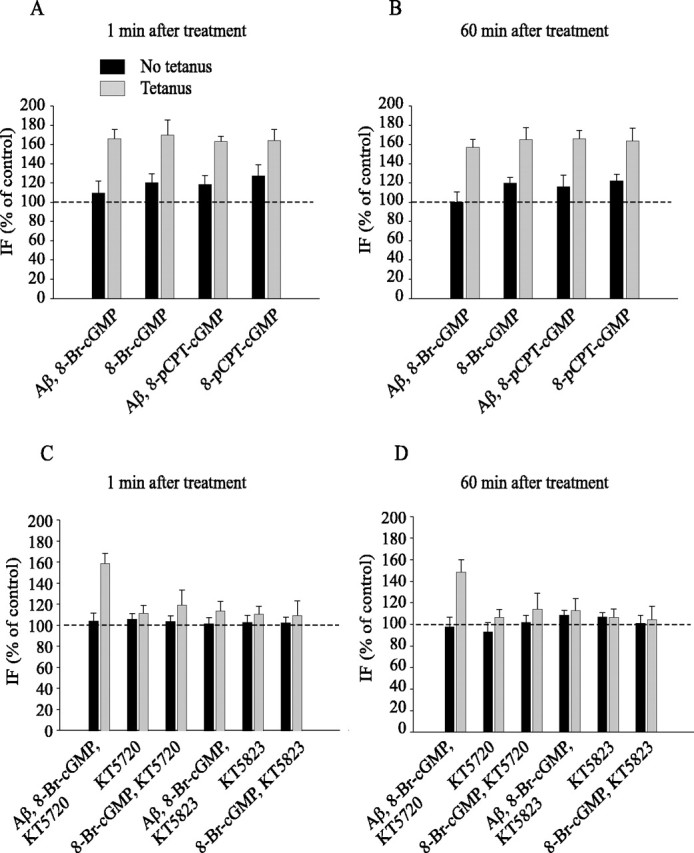
cGK activation protects against Aβ suppression of enhancement of CREB phosphorylation during LTP. A, The cGMP analogs 8-Br-cGMP and 8-pCPT-cGMP reestablish normal increase of CREB phosphorylation in slices that are treated with Aβ plus tetanus at both 1 min (A) and 60 min (B) after tetanus (F(1, 14) = 141.50, p < 0.001 in slices treated with 8-Br-cGMP plus Aβ plus tetanus compared with slices treated with Aβ plus tetanus; F(1, 12) = 96.98, p < 0.001 in slices treated with 8-pCPT-cGMP plus Aβ plus tetanus compared with slices treated with Aβ plus tetanus). C, The protection against suppression of CREB phosphorylation by cGMP analogs is not reversed by inhibiting cAK activity with KT5720 at 1 and 60 min (D) after the tetanus (F(1, 12) = 3.58; p > 0.05, compared with tetanized slices perfused with 8-Br-cGMP plus Aβ). KT5720 is still capable of reducing tetanus-induced CREB phosphorylation (F(1,12) = 266.39; p < 0.001, compared with tetanized slices perfused with vehicle). Tetanus-induced immunofluorescence is still low after treatment with KT5720 paired with 8-Br-cGMP (F(1, 8) = 3.17; p > 0.1, compared with vehicle).Similar to the electrophysiological experiments, the increase in phospho-CREB is reduced when the cGK inhibitor KT5823 (2 μm) is added to the slices treated with Aβ and 8-Br-cGMP before tetanization (F(1, 10) = 79.89; p < 0.001, compared with slices treated with Aβ plus 8-Br-cGMP). KT5823 treatment in the absence of Aβ and 8-Br-cGMP is capable of reducing tetanus-induced CREB phosphorylation (F(1, 12) = 209.61; p < 0.001, compared with vehicle plus tetanus). IF, Immunofluorescence. Error bars represent SEM.
As in the electrophysiological experiments, the protection against suppression of CREB phosphorylation by 8-Br-cGMP was not reversed by inhibiting cAK activity with KT5720 (Fig. 6C,D) (156.15 ± 9.89% of control at 1 min, n = 4, and 150.80 ± 11.38% of control at 60 min, n = 4; F(1, 12) = 3.58; p > 0.05 compared with 8-Br-cGMP plus tetanus), even if KT5720 was still capable of reducing tetanus-induced CREB phosphorylation (Fig. 6C,D) (110.06 ± 7.31% of control at 1 min, n = 3, and 107.71 ± 7.51% of control at 60 min, n = 3; F(1, 12) = 266.39; p < 0.001 compared with tetanized-slices perfused with vehicle). KT5720 paired with 8-Br-cGMP and Aβ without tetanus did not affect immunofluorescence intensity (Fig. 6C,D) (102.90 ± 7.30% of control at 1 min, n = 3, and 98.96 ± 9.02% of control at 60 min, n = 3; F(1, 10) = 1.4; p > 0.1 compared with vehicle). Similarly, KT5720 alone did not vary immunofluorescence intensity (Fig. 6C,D) (104.45 ± 5.42% of control at 1 min, n = 3, and 94.30 ± 8.65% of control at 60 min, n = 3; F(1, 10) = 0.33; p > 0.5). Tetanus-induced immunofluorescence was still low after treatment with KT5720 paired with 8-Br-cGMP or Aβ compared with vehicle-treated slices (Fig. 6C,D) (8-Br-cGMP plus KT5720, 117.70 ± 13.76% of control vehicle-treated slices at 1 min, n = 3, and 115.40 ± 15.03% of control at 60 min, n = 3; F(1, 8) = 3.17; p > 0.1; Aβ plus KT5720, 101.95 ± 11.51% of control at 1 min, n = 3, and 105.94 ± 12.30% of control at 60 min, n = 3; F(1, 8) = 0.33; data not shown; p > 0.1). KT5720 plus 8-Br-cGMP or KT5720 plus Aβ did not modify CREB immunofluorescence in slices that did not receive tetanic stimulation (Fig. 6C,D) (8-Br-cGMP plus KT5720, 102.11 ± 5.56% of control at 1 min, n = 3, and 103.26 ± 6.49% of control at 60 min, n = 3; F(1, 8) = 0.67; KT5720 plus Aβ, 100.81 ± 13.52% of control at 1 min, n = 3, and 102.92 ± 15.40% of control at 60 min, n = 3; F(1, 8) = 88.78; data not shown; p > 0.1). Thus, cAK is not involved in the effects of cGMP analogs on CREB phosphorylation during Aβ treatment.
Aβ blocks the increase in cGMP immunofluorescence occurring immediately after LTP induction
To further investigate the beneficial effects of NO donors and cGMP analogs on Aβ-induced LTP suppression, we next asked whether Aβ interferes directly with the NO/cGMP/cGK pathway by blocking the cGMP increase normally occurring immediately after LTP induction (Monfort et al., 2002). Hippocampal slices were treated with vehicle or 200 nm oligomeric Aβ1-42 for 20 min, fixed 10 s, 5 min, or 60 min after the treatment, and stained with anti-cGMP antibodies. As demonstrated previously (Monfort et al., 2002), tetanic stimulation was capable of increasing the cGMP immunofluorescence at 10 s after the application of the tetanus (140.87 ± 9.53% of control untreated slices from the same animals, n = 5; t(8) = 3.35; p < 0.01 compared with vehicle), whereas cGMP immunofluorescence was similar to control values after 5 or 60 min (Fig. 7A,B) (5 min, 102.15 ± 6.61% of control, n = 4, t(6) = 0.26; 60 min, 95.27 ± 12.78% of control, n = 4, t(6) = 0.35, p > 0.5). However, Aβ treatment abolished the tetanus-induced increase in cGMP immunofluorescence at 10 s. In fact, no significant differences were detected at 10 s, 5 min, and 60 min comparing slices treated with Aβ plus tetanus to control slices (Fig. 7A,B) (10 s, 105.21 ± 9.39% of control, n = 4, t(7) = 0.40; 5 min, 99.88 ± 11.18% of control, n = 4, t(6) = 0.25; 60 min, 92.47 ± 10.29% of control, n = 4, t(6) = 0.19; p > 0.5). Moreover, addition of DEA/NO reestablished the increase of cGMP immunofluorescence in slices that were tetanized in the presence of Aβ (Fig. 7C) (141.21 ± 10.11% of control at 10 s; n = 5; t(8) = 0.022; p > 0.5 compared with tetanized slices at 10 s) without affecting immunofluorescence at 5 and 60 min (Fig. 7C) (5 min, 102.37 ± 11.41% of control, n = 4, t(6) = 0.30; 60 min, 94.99 ± 12.84% of control, n = 4, t(6) = 0.33; p > 0.5 compared with vehicle). The effect of DEA/NO was not caused by an effect of the donor on the increase in cGMP immunofluorescence, because DEA/NO alone did not enhance the tetanus-induced cGMP increase (Fig. 7C) (10 s, 142.68 ± 10.64% of control, n = 5, t(8) = 0.11; 5 min, 101.24 ± 11.52% of control, n = 4, t(6) = 0.05; 60 min, 98.52 ± 14.27% of control, n = 4, t(6) = 0.17; p > 0.5 compared with tetanus). These results demonstrate that Aβ blocks a cGMP increase occurring during LTP induction. Moreover, the fact that DEA/NO reestablishes normal cGMP production by sGC during LTP induced in the presence of Aβ suggests that Aβ downregulates the NO/cGMP/cGK/CREB pathway during synaptic plasticity.
Figure 7.
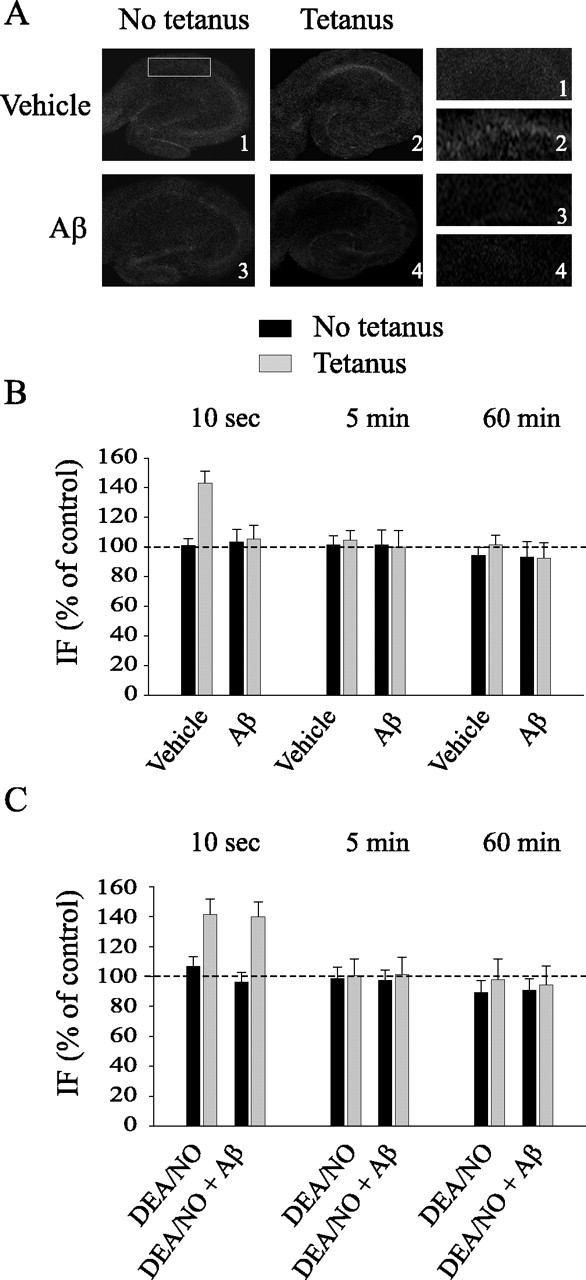
Aβ blocks the increase in cGMP immunofluorescence occurring immediately after LTP. A, Representative examples of hippocampal slices stained with a cGMP antibody and fixed 10 s after either vehicle plus tetanus and Aβ or Aβ plus tetanus. Left, Lower-power (4×) view of the entire slice. Right, Higher-power (16×) view of CA1 cell pyramidal area. B, Hippocampal slices are fixed 10 s, 5 min, or 60 min after vehicle or Aβ both in basal conditions and after tetanus. Aβ blocks the increase of cGMP immunofluorescence in tetanus-treated slices at 10 s (t(7) = 0.40; p > 0.5 between vehicle-treated slices and slices treated with Aβ plus tetanus). cGMP immunofluorescence does not vary at 5 or 60 min after the tetanus (t(6) = 0.25; t(6) = 0.19; p > 0.5). C, DEA/NO reestablishes the increase of cGMP immunofluorescence in tetanized slices exposed to Aβ at 10 s (t(8) = 0.022; p > 0.5 compared with tetanized slices). cGMP immunofluorescence does not vary at 5 or 60 min after the tetanus (t(6) = 0.30; t(6) = 0.33; p > 0.5). IF, Immunofluorescence. Error bars represent SEM.
Discussion
The present study was inspired by the observation that hippocampal LTP, a model of synaptic plasticity that is thought to underlie learning and memory (Bliss and Collingridge, 1993), is suppressed after exposure to sublethal doses of Aβ (Cullen et al., 1997; Itoh et al., 1999). Because our study and other studies have provided evidence indicating that activation of the NO/cGMP/cGK/CREB pathway is involved in hippocampal plasticity (O'Dell et al., 1991; Schuman and Madison, 1991; Arancio et al., 1995, 1996, 2001; Lu et al., 1999; Antonova et al., 2001), we decided to assess whether this cascade might play a role in Aβ-induced LTP suppression. To determine whether NO plays a beneficial effect on Aβ-induced reduction of LTP, we used the NO donor DEA/NO and found that it rescues LTP impairment in hippocampal slices perfused with Aβ. This result is in agreement with studies suggesting that NO has a protective effect on Aβ-induced damage of the nervous system (McCarty, 1998; Troy et al., 2000; Wirtz-Brugger and Giovanni, 2000). Moreover, this finding is consistent with studies performed on PC12 cells, sympathetic neurons, and hippocampal neurons, showing that inhibition of NO synthesis by N-nitro-l-arginine methyl ester does not protect against Aβ-induced neurotoxicity, whereas treatment with the NO generator S-nitroso penicillamine exerts a neuroprotective effect because of the inhibition of the proapoptotic factor caspase-2 by nitrosylation (Troy et al., 2000). Finally, this result is consistent with the observations that Aβ impairs NO generation by decreasing NMDA receptor signal transduction (McCarty, 1998) by subtracting NADPH availability to NO synthase (Venturini et al., 2002) or by inhibiting the phosphorylation of the serine-threonine kinase Akt (Suhara et al., 2003). However, a large body of evidence indicates that NO plays a neurotoxic role in Aβ-induced synaptic dysfunction and cell death through the generation of peroxinitrite. Interestingly, in most of these studies, NO neurotoxicity seems to be attributable to the stimulation of microglial inducible NOS (iNOS) (McCann, 1997; Tran et al., 2001; Wong et al., 2001; Haas et al., 2002; Xie et al., 2002; Monsonego et al., 2003; Wang et al., 2004b), instead of the constitutive NOS, which includes the neuronal (nNOS) and the endothelial isoforms. Moreover, an increase in hippocampal iNOS and a decrease in nNOS have been shown in aged rats (Law et al., 2002). Thus, the dual and apparently contrasting effect of NO might be explained through a possible differential role of these enzymes causing the following: (1) synaptic dysfunction and cell death in case of activation of iNOS and (2) lack of synaptic plasticity for downregulation of NO production, as in the present studies.
To explore steps downstream of NO production, we hypothesized that sGC, the main mediator of NO signal transduction, is involved in the beneficial effect by DEA/NO (Schlossmann et al., 2003). In agreement with this hypothesis, pairing of Aβ with NO and ODQ, an sGC inhibitor, blocked the neuroprotective effect by DEA/NO. Moreover, BAY41-2272 rescued the Aβ-induced LTP impairment, and DEA/NO reestablished the cGMP immunofluorescence increase in tetanized slices exposed to Aβ. Interestingly, Aβ decreases sGC expression in brain astroglial cells (Baltrons et al., 2002), and sGC activity is reduced in the superior temporal cortex of AD patients (Bonkale et al., 1995). Therefore, it would be interesting to test in future experiments whether Aβ reduces sGC expression and/or activity in our preparation.
Given that sGC produces cGMP (Schlossmann et al., 2003), we hypothesized that increasing levels of cGMP could rescue the impairment of synaptic plasticity caused by Aβ. Indeed, both 8-Br-cGMP and 8-pCPT-cGMP reversed the inhibition of LTP by Aβ. Consistent with these results, the increase of cGMP levels has been found to reduce both Aβ-induced cell death (Wirtz-Brugger and Giovanni, 2000) and vasoactivity (Paris et al., 2000). Moreover, a decrease in cGMP levels has been detected either in vessels and neurons after Aβ treatment or in aged brains (Bonkale et al., 1995; Vallebuona and Raiteri, 1995; Chalimoniuk and Strosznajder, 1998; Paris et al., 1999; Baltrons et al., 2002).
Another important finding of our experiments is that the increase in cGMP levels, normally occurring immediately after tetanic stimulation (Monfort et al., 2002), is blocked in Aβ-treated slices. This finding indicates that Aβ directly interferes with the NO/cGMP/cGK/CREB pathway to suppress LTP. Optical, fluorescence, and NMR spectroscopy studies have demonstrated that the cofactor NADPH strongly complexes soluble Aβ with a fast kinetic forming a binary adduct (Venturini et al., 2002). This results in diminished availability of the cofactor for NO-synthase functioning (Venturini et al., 2002). Thus, a particularly attractive hypothesis that could explain our findings on Aβ-induced block of synaptic plasticity by linking directly Aβ with the NO/cGMP/cGK/CREB pathway is that Aβ functions as a sink for NADPH, preventing the production of NO by NO synthase and the following cascade of events, including cGMP production, until CREB phosphorylation. An alternative possibility is that the lack of cGMP fluorescence increase after Aβ application is attributable to an increase of phosphodiesterase activity and/or a decrease in sGC activity and/or expression. Phosphodiesterase activity increase has been demonstrated on both isolated blood vessels and cultured microglia, in which phosphodiesterase type V inhibition reestablishes normal vasoactivity and blocks inflammatory response caused by Aβ (Paris et al., 1999). A reduction in sGC activity has been shown in the superior temporal cortex of AD patients (Bonkale et al., 1995). Aβ has been shown to reduce sGC expression in astroglial cells (Baltrons et al., 2002). The answers to these questions remain to be explored. Nonetheless, our findings have identified a novel signaling pathway inactivated by amyloid peptides during synaptic plasticity, the NO/cGMP/cGK/CREB cascade.
Another finding of our studies was that Aβ blocks an increase in CREB phosphorylation during LTP. CREB is a transcription factor involved in several types of learning and memory in different species ranging from invertebrates to mammals (Bourtchuladze et al., 1994; Yin et al., 1994; Lonze and Ginty, 2002). It is known that both activation of NMDA receptors and membrane depolarization induce CREB phosphorylation at Ser-133, a crucial step for its resulting transcriptional activity during synaptic plasticity (Montminy, 1997; Shieh et al., 1998; Tao et al., 1998; Hu et al., 1999). Suppression of CREB phosphorylation results in inhibition of late-LTP and long-term memory (Bourtchuladze et al., 1994; Tully, 1997; Lonze and Ginty, 2002). The finding that Aβ induces block of CREB phosphorylation is consistent with the role of CREB in synaptic plasticity. More importantly, in agreement with our results, Aβ has been shown to suppress CREB phosphorylation in cultured cortical neurons (Tong et al., 2001) as well as in cultured hippocampal neurons after glutamate stimulation (Vitolo et al., 2002). Therefore, the present study extends findings from cultured neurons to the slice preparation, a system in which neuronal circuits are intact.
Application of the NO donor DEA/NO, the sGC stimulator BAY41-2272, or the cGMP analogs 8-Br-cGMP and 8-pCPT-cGMP, together with Aβ1-42, reestablished normal levels of phospho-CREB during potentiation. This finding is consistent with the observation that the inhibition of one of the components of the NO/cGMP/cGK pathway suppresses CREB phosphorylation (Lu et al., 1999; Gudi et al., 2000; Ciani et al., 2002). Most importantly, the effect of cGMP analogs on CREB phosphorylation and LTP was specifically mediated through cGK, because treatment with the cAK inhibitor KT57230 did not suppress the beneficial effect of cGMP analogs, as occurred with the cGK inhibitor KT5823. Considering that cAMP/cAK and NO/cGMP/cGK pathways are likely to converge onto CREB to maintain LTP (Lu et al., 1999), it was important to define whether the NO/cGMP/cGK/CREB system is also involved in Aβ-induced suppression of synaptic plasticity in addition to the cAMP/cAK/CREB pathway (Vitolo et al., 2002).
Recent studies have provided evidence that the Aβ-mediated inhibition of LTP induction involves stimulation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase pathway after the activation of an unidentified Aβ receptor(s) by Aβ and activation of the metabotropic glutamate receptor type 5 by glutamate (Wang et al., 2004a). Aβ has been found to bind and activate the α-7 nicotinic receptor with downregulation of the ERK/MAPK/CREB cascade in APP(K670N:M671L) mice (Dineley et al., 2001, 2002). A decrease in PKA basal activity was found in cultured hippocampal neurons exposed to sublethal doses of Aβ (Vitolo et al., 2002). With the present study, we have added a new pathway that is downregulated by Aβ, the NO/cGMP/cGK/CREB cascade. Together, these findings suggest a model in which different mechanisms mediate the multifaceted action of Aβ. Until we find a way to block amyloid production, our findings might be therapeutically relevant, because the most efficacious way to counteract symptoms caused by the abnormal production of Aβ might be to interfere with Aβ different targets and downstream pathways. Our study suggests that intervention using NO donors or drugs enhancing cGMP levels may turn out to ameliorate cognition in patients affected by AD and other neurodegenerative diseases characterized by abnormal production of Aβ.
Footnotes
This work was supported by the Alzheimer's Association and by National Institutes of Health Grants NS15076, NS049442, and AG17490. We thank Michael Shelanski for helpful comments.
Correspondence should be addressed to Ottavio Arancio, Department of Pathology, Columbia University, 630W 168th Street, New York, NY 10032. E-mail: oa1@columbia.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/256887-11$15.00/0
References
- Antonova I, Arancio O, Trillat AC, Zablow L, Kandel ER, Hawkins RD (2001) Rapid increase in immunoreactive presynaptic terminals during long-lasting potentiation. Science 294: 1547-1550. [DOI] [PubMed] [Google Scholar]
- Arancio O, Kandel ER, Hawkins RD (1995) Activity-dependent long-term enhancement of transmitter release by presynaptic 3′, 5′-cyclic GMP in cultured hippocampal neurons. Nature 376: 74-80. [DOI] [PubMed] [Google Scholar]
- Arancio O, Kiebler M, Lee CJ, Lev-Ram V, Tsien RY, Kandel ER, Hawkins RD (1996) Nitric oxide acts directly in the presynaptic neuron to produce long-term potentiation in cultured hippocampal neurons. Cell 87: 1025-1035. [DOI] [PubMed] [Google Scholar]
- Arancio O, Antonova I, Gambaryan S, Lohmann SM, Wood J, Lawrence D, Hawkins RD (2001) Presynaptic cGMP-dependent protein kinase contributes to long-lasting potentiation in cultured hippocampal neurons. J Neurosci 21: 143-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltrons MA, Pedraza CE, Heneka MT, Garcia A (2002) β-Amyloid peptides decrease guanylyl cyclase expression in astroglial cells. Neurobiol Dis 10: 139-149. [DOI] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31-39. [DOI] [PubMed] [Google Scholar]
- Bon CL, Garthwaite J (2003) On the role of nitric oxide in hippocampal long-term potentiation. J Neurosci 5: 1941-1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonkale WL, Winblad B, Ravid R, Cowburn RF (1995) Reduced nitric oxide responsive guanylyl cyclase activity in the superior temporal cortex of patients with Alzheimer's disease. Neurosci Lett 187: 5-8. [DOI] [PubMed] [Google Scholar]
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ (1994) Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 79: 59-68. [DOI] [PubMed] [Google Scholar]
- Butt E, Nolte C, Schulz S, Beltman J, Beavo JA, Jastorff B, Walter U (1992) Analysis of the functional role of cGMP-dependent protein kinase in intact human platelets using a specific activator 8-para-chlorophenylthio-cGMP. Biochem Pharmacol 43: 2591-2600. [DOI] [PubMed] [Google Scholar]
- Chalimoniuk M, Strosznajder JB (1998) Aging modulates nitric oxide synthesis and cGMP levels in hippocampus and cerebellum. Effects of amyloid beta peptide. Mol Chem Neuropathol 35: 77-95. [DOI] [PubMed] [Google Scholar]
- Ciani E, Guidi S, Bartesaghi R, Contestabile A (2002) Nitric oxide regulates cGMP-dependent cAMP-responsive element binding protein phosphorylation and Bcl-2 expression in cerebellar neurons: implication for a survival role of nitric oxide. J Neurochem 82: 1282-1289. [DOI] [PubMed] [Google Scholar]
- Cullen WK, Suh YH, Anwyl R, Rowan MJ (1997) Block of LTP in rat hippocampus in vivo by beta-amyloid precursor protein fragments. Neuro-Report 8: 3213-3217. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD (2001) β-Amyloid activates the mitogen-activated protein kinase cascade via hippocampal α7 nicotinic acetylcholine receptors: in vitro and in vivo mechanisms related to Alzheimer's disease. J Neurosci 21: 4125-4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KT, Xia X, Bui D, Sweatt JD, Zheng H (2002) Accelerated plaque accumulation, associative learning deficits, and up-regulation of alpha7 nicotinic receptor protein in transgenic mice co-expressing mutant human presenilin 1 and amyloid precursor proteins. J Biol Chem 277: 22768-22780. [DOI] [PubMed] [Google Scholar]
- Garthwaite J, Boulton CL (1995) Nitric oxide signaling in the central nervous system. Annu Rev Physiol 57: 683-706. [DOI] [PubMed] [Google Scholar]
- Garthwaite J, Southam E, Boulton CL, Nielson EB, Schmidt K, Mayer B (1995) Potent and selective inhibition of nitric oxide sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3α]quinoxalin-1-one. Mol Pharmacol 48: 184-188. [PubMed] [Google Scholar]
- Geiger J, Nolte C, Butt E, Sage SO, Walter U (1992) Role of cGMP and cGMP-dependent protein kinase in nitrovasodilator inhibition of agonist-evoked calcium elevation in human platelets. Proc Natl Acad Sci USA 89: 1031-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudi T, Huvar I, Meinecke M, Lohmann SM, Boss GR, Pilz RB (1996) Regulation of gene expression by cGMP-dependent protein kinase. J Biol Chem 271: 4597-4600. [DOI] [PubMed] [Google Scholar]
- Gudi T, Casteel DE, Vinson C, Boss GR, Pilz RB (2000) NO activation of fos promoter elements requires nuclear translocation of G-kinase I and CREB phosphorylation but is independent of MAP kinase activation. Oncogene 19: 6324-6333. [DOI] [PubMed] [Google Scholar]
- Haas J, Storch-Hagenlocher B, Biessmann A, Wildemann B (2002) Inducible nitric oxide synthase and argininosuccinate synthetase: co-induction in brain tissue of patients with Alzheimer's dementia and following stimulation with beta-amyloid 1-42 in vitro. Neurosci Lett 322: 121-125. [DOI] [PubMed] [Google Scholar]
- Hu SC, Chrivia J, Ghosh A (1999) Regulation of CBP-mediated transcription by neuronal calcium signaling. Neuron 22: 799-808. [DOI] [PubMed] [Google Scholar]
- Itoh A, Akaike T, Sokabe M, Nitta A, Iida R, Olariu A, Yamada K, Nabeshima T (1999) Impairments of long-term potentiation in hippocampal slices of beta-amyloid-infused rats. Eur J Pharmacol 382: 167-175. [DOI] [PubMed] [Google Scholar]
- Izquierdo LA, Vianna M, Barrosw DM, Souza TM, Arenghi P, Sant'Anna MK, Rodrigues C, Medina JH, Izquierdo I (2000) Short- and long-term memory are differentially affected by metabolic inhibitors given into hippocampus and enthorinal cortex. Neurobiol Learn Mem 73: 141-149. [DOI] [PubMed] [Google Scholar]
- Koglin M, Stasch JP, Behrends S (2002) BAY 41-2272 activates two isoforms of nitric oxide-sensitive guanylyl cyclase. Biochem Biophys Res Commun 292: 1057-1062. [DOI] [PubMed] [Google Scholar]
- Law A, Gauthier S, Quirion R (2001) Neuroprotective and neurorescuing effects of isoform-specific nitric oxide synthase inhibitors, nitric oxide scavenger, and antioxidant against beta-amyloid toxicity. Br J Pharmacol 133: 1114-1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law A, O'Donnel J, Gauthier S, Quirion R (2002) Neuronal and inducible nitric oxide synthase expressions and activities in the hippocampi and cortices of young adult, aged cognitively unimpaired, and impaired Long-Evans rats. Neuroscience 112: 267-275. [DOI] [PubMed] [Google Scholar]
- Lohmann SM, Vaandrager AB, Smolenski A, Walter U, De Jonge HG (1997) Distinct and specific functions of cGMP-dependent protein kinases. TIBS 22: 307-312. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35: 605-623. [DOI] [PubMed] [Google Scholar]
- Lu YF, Kandel ER, Hawkins RD (1999) Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J Neurosci 19: 10250-10261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin DH, Crothers MK, Lake JR, Goyarzu P, Plotner RE, Garcia SA, Spell SH, Tomsic BJ (2001) Hippocampal injections of amyloid β-peptide 1-40 impair subsequent one-trial/day reward learning. Neurobiol Learn Mem 76: 125-137. [DOI] [PubMed] [Google Scholar]
- Mattson MP (1997) Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev 77: 1081-1132. [DOI] [PubMed] [Google Scholar]
- McCann SM (1997) The nitric oxide hypothesis of brain aging. Exp Gerontol 32: 431-440. [DOI] [PubMed] [Google Scholar]
- McCarty MF (1998) Vascular nitric oxide may lessen Alzheimer's risk. Med Hypotheses 51: 465-476. [DOI] [PubMed] [Google Scholar]
- Monfort P, Munoz MD, Koseko E, Felipo V (2002) Long-term potentiation in hippocampus involves sequential activation of soluble guanylate cyclase, cGMP-dependent protein kinase, and cGMP-degrading phosphodiesterase. J Neurosci 22: 10116-10122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsonego A, Imitola J, Zota V, Oida T, Weiner HL (2003) Microglia-mediated nitric oxide cytotoxicity of T cells following amyloid beta-peptide presentation to Th1 cells. J Immunol 171: 2216-2224. [DOI] [PubMed] [Google Scholar]
- Montminy M (1997) Transcriptional regulation by cyclic AMP. Annu Rev Biochem 66: 807-822. [DOI] [PubMed] [Google Scholar]
- O'Dell TJ, Hawkins RD, Kandel ER, Arancio O (1991) Tests of the roles of two diffusible substances in long-term potentiation: evidence for nitric oxide as a possible early retrograde messenger. Proc Natl Acad Sci USA 88: 11285-11289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohki K, Yoshida K, Hagiwara M, Harada T, Takamura M, Ohashi T, Matsuda H, Imaki J (1995) Nitric oxide induces c-fos gene expression via cyclic AMP response element binding protein (CREB) phosphorylation in rat retinal pigment epithelium. Brain Res 696: 140-144. [DOI] [PubMed] [Google Scholar]
- Paris D, Town T, Parker TA, Tan J, Humphrey J, Crawford F, Mullan M (1999) Inhibition of Alzheimer's beta-amyloid induced vasoactivity and proinflammatory response in microglia by a cGMP-dependent mechanism. Exp Neurol 157: 211-221. [DOI] [PubMed] [Google Scholar]
- Paris D, Town T, Parker T, Humphrey J, Mullan M (2000) Beta-amyloid vasoactivity and proinflammation in microglia can be blocked by cGMP-elevating agents. Ann NY Acad Sci 903: 446-450. [DOI] [PubMed] [Google Scholar]
- Parks JK, Smith TS, Trimmer PA, Bennett Jr JP, Parker Jr WD (2001) Neurotoxic Abeta peptides increase oxidative stress in vivo through NMDA-receptor and nitric-oxide-synthase mechanisms, and inhibit complex IV activity and induce a mitochondrial permeability transition in vitro. J Neurochem 76: 1050-1056. [DOI] [PubMed] [Google Scholar]
- Schlossmann J, Feil R, Hofmann F (2003) Signaling through NO and cGMP-dependent protein kinases. Ann Med 35: 21-27. [DOI] [PubMed] [Google Scholar]
- Schuman EM, Madison DV (1991) A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science 254: 1503-1506. [DOI] [PubMed] [Google Scholar]
- Sekhar KR, Hatchett RJ, Shabb JB, Wolfe L, Francis SH, Wells JN, Jastorff B, Butt E, Chakinala MM, Corbin JD (1992) Relaxation of pig coronary arteries by new and potent cGMP analogs that selectively activate type I alpha, compared with type I beta, cGMP-dependent protein kinase. Mol Pharmacol 42: 103-108. [PubMed] [Google Scholar]
- Selkoe DJ (1994) Normal and abnormal biology of the beta-amyloid precursor protein. Annu Rev Neurosci 17: 489-517. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ (2002) Alzheimer's disease is a synaptic failure. Science 298: 789-791. [DOI] [PubMed] [Google Scholar]
- Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A (1998) Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron 20: 727-740. [DOI] [PubMed] [Google Scholar]
- Stine Jr WB, Dahlgren KN, Krafft GA, LaDu MJ (2003) In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J Biol Chem 278: 11612-11622. [DOI] [PubMed] [Google Scholar]
- Suhara T, Magrane J, Rosen K, Christensen R, Kim HS, Zheng B, McPhie DL, Walsh K, Querfurth H (2003) Abeta42 generation is toxic to endothelial cells and inhibits eNOS function through an Akt/GSK-3beta signaling-dependent mechanism. Neurobiol Aging 24: 437-451. [DOI] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME (1998) Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20: 709-726. [DOI] [PubMed] [Google Scholar]
- Tong L, Thornton PL, Balazs R, Cotman CW (2001) Beta-amyloid-(1-42) impairs activity-dependent cAMP-response element-binding protein signaling in neurons at concentrations in which cell survival is not compromised. J Biol Chem 276: 17301-17306. [DOI] [PubMed] [Google Scholar]
- Tran MH, Yamada K, Olariu A, Mizuno M, Ren XA, Nabeshima T (2001) Amyloid-β-peptide induces nitric oxide production in rat hippocampus: association with cholinergic dysfunction and amelioration by inducible nitric oxide synthase inhibitors. FASEB J 15: 1407-1409. [DOI] [PubMed] [Google Scholar]
- Troy CM, Rabacchi SA, Friedman WJ, Frappier TF, Brown K, Shelanski ML (2000) Caspase-2 mediates neuronal cell death induced by β-amyloid. J Neurosci 20: 1386-1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully T (1997) Regulation of gene expression and its role in long-term memory and synaptic plasticity. Proc Natl Acad Sci USA 94: 4239-4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallebuona F, Raiteri M (1995) Age-related changes in NMDA receptor/nitric oxide/cGMP pathway in the hippocampus and cerebellum of freely moving rats subjected to transcerebral microdialysis. Eur J Neurosci 7: 694-701. [DOI] [PubMed] [Google Scholar]
- Venturini G, Colasanti M, Persichini T, Fioravanti E, Ascensi P, Palomba L, Cantoni O, Musci G (2002) β-Amyloid inhibits NOS activity by subtracting NADPH availability. FASEB J 16: 1970-1972. [DOI] [PubMed] [Google Scholar]
- Vitolo OV, Sant'Angelo A, Costanzo V, Battaglia F, Arancio O, Shelanski M (2002) Amyloid β peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci USA 99: 13217-13221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R (2004a) Block of long-term potentiation by naturally secreted and synthetic amyloid-β-peptide in hippocampal slices is mediated via activation of the kinases c-jun n-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci 24: 3370-3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Rowan MJ, Anwyl R (2004b) β-Amyloid-mediated inhibition of NMDA receptor-dependent long-term potentiation induction involves activation of microglia and stimulation of inducible nitric oxide synthase and superoxide. J Neurosci 24: 6049-6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtz-Brugger F, Giovanni A (2000) Guanosine 3′,5′-cyclic monophosphate mediated inhibition of cell death induced by nerve growth factor withdrawal and beta-amyloid: protective effects of propentofylline. Neuroscience 99: 737-750. [DOI] [PubMed] [Google Scholar]
- Wong A, Luth HJ, Deuther-Conrad W, Dukic-Stefanovic S, Gasic-Milenkovic J, Arendt T, Munch G (2001) Advanced glycation endproducts co-localize with inducible nitric oxide synthase in Alzheimer's disease. Brain Res 920: 32-40. [DOI] [PubMed] [Google Scholar]
- Xie Z, Wei M, Morgan TE, Fabrizio P, Han D, Finch CE, Longo VD (2002) Peroxynitrite mediates neurotoxicity of amyloid β-peptide 1-42- and lipopolysaccharide-activated microglia. J Neurosci 22: 3484-3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin JC, Wallach JS, Del Vecchio M, Wilder EL, Zhou H, Quinn WG, Tully T (1994) Induction of a dominant negative CREB transgene specifically blocks long-term memory in Drosophila Cell 79: 49-58. [DOI] [PubMed] [Google Scholar]