

Abstract
We have cloned and characterized mouse and human variants of MONaKA, a novel protein that interacts with and modulates the plasma membrane Na,K-ATPase. MONaKA was cloned based on its sequence homology to the Drosophila Slowpoke channel-binding protein dSlob, but mouse and human MONaKA do not bind to mammalian Slowpoke channels. At least two splice variants of MONaKA exist; the splicing is conserved perfectly between mouse and human, suggesting that it serves some important function. Both splice variants of MONaKA are expressed widely throughout the CNS and peripheral nervous system, with different splice variant expression ratios in neurons and glia. A yeast two-hybrid screen with MONaKA as bait revealed that it binds tightly to the β1 and β3 subunits of the Na,K-ATPase. The association between MONaKA and Na,K-ATPase β subunits was confirmed further by coimmunoprecipitation from transfected cells, mouse brain, and cultured mouse astrocytes. A glutathione S-transferase-MONaKA fusion protein inhibits Na,K-ATPase activity from whole brain or cultured astrocytes. Furthermore, transfection of MONaKA inhibits 86Rb+ uptake via the Na,K-ATPase in intact cells. These results are consistent with the hypothesis that MONaKA modulates brain Na,K-ATPase and may thereby participate in the regulation of electrical excitability and synaptic transmission.
Keywords: sodium pump; ion transport; Na,K-ATPase; neuromodulation; alternative splicing; potassium homeostasis
Introduction
The Na,K-ATPase is a ubiquitous plasma membrane ion transporter, in fact, the first enzyme recognized to be an ion pump (Skou, 1998). It uses the hydrolysis of one molecule of ATP to drive the coupled transport of three sodium ions out of the cell and two potassium ions into the cell. Its activity is essential for maintaining membrane ion gradients responsible for membrane potential by clearing potassium from the extracellular space and removing excess sodium from the intracellular space. Cardiac glycosides, such as ouabain and strophanthidin, which are used to treat some forms of cardiac disease, produce some or all of their effects by blocking the pumping activity of the Na,K-ATPase (Smith et al., 1984).
Na,K-ATPase is a member of the family of P-type ATPases (Maeda et al., 1998). It is a heterodimeric integral membrane protein, consisting of one α and one β subunit. The α subunit, an ∼100 kDa protein with 10 predicted membrane spans, is the catalytic subunit responsible for ion translocation and ATPase activity. The β subunit, a 50–60 kDa glycosylated protein with a single membrane-spanning domain, may act as a molecular chaperone or otherwise regulate the membrane targeting and/or function of the α subunit (Kaplan, 2002). At least four distinct α subunit and three β subunit isoforms have been identified, and there appears to be no preferential association of particular α subunits with particular β subunits (Peng et al., 1997).
A recently identified gene family consisting of at least seven small single membrane-spanning domain proteins has been shown to regulate ion transport activity of the Na,K-ATPase (Sweadner and Rael, 2000). These FXYD proteins, named after an invariant amino acid sequence within the signature motif that defines the family, appear to modulate Na,K-ATPase in a tissuespecific and isoform-specific manner (Sweadner and Rael, 2000; Crambert et al., 2003; Geering et al., 2003). Findings such as these make it clear that Na,K-ATPase is highly regulated and raise the question of whether there are other still-to-be identified modulators of this critical plasma membrane ion pump.
We have been pursuing the hypothesis that many, and perhaps all, ion channel proteins exist in the plasma membrane in a regulatory complex with one or more signaling proteins that participate in the modulation of channel activity. In the course of these experiments, we identified, cloned, and characterized a protein that binds to the Drosophila Slowpoke calcium-dependent potassium channel (dSlo). We named this protein dSlob, for Drosophila Slo-binding (Schopperle et al., 1998). We describe here the cloning and characterization of mouse and human orthologs of Drosophila slob. To our surprise, we have been unable to detect binding of these orthologs to mammalian (or Drosophila) Slowpoke channels. To investigate their physiological function, we used them as bait in a yeast two-hybrid screen. Interestingly, we found that these mammalian proteins bind robustly to the β1 and β3 subunits of the Na,K-ATPase and modulate both enzymatic and ion pump activities. Accordingly, we have named the proteins MONaKA, for modulator of Na,K-ATPase.
Materials and Methods
Cloning of MONaKA. Mouse MONaKA cDNAs were amplified by PCR from a mouse brain cDNA library (Marathon-ready cDNA library; Clontech, Palo Alto, CA), using primers designed according to the sequence of GenBank accession number AK048910, which resembles Drosophila slob: 5′ primer, 5′-gcccgggatggccttcatggagaa-3′; 3′ primer, 5′-tcagccgatcttgggagcgctgtg-3′. Human MONaKA cDNAs were cloned using rapid amplification of cDNA ends (RACE) (Frohman, 1993). The template was a human brain cDNA library designed for RACE (Clontech). 3′ RACE was performed using the outer gene-specific primer 5′-tggcatgcctaccatctccggctcttaca-3′ and inner gene-specific primer 5′-cagtcccaccatggatctgaggaggaaaga-3′. 5′ RACE was performed using the outer gene-specific primer 5′-cagcccagcttaacactagccgttcc-3′ and inner gene-specific primer 5′-tatttcttccttattctccaacctatgtc-3′. The RACE products and the PCR products were sequenced and cloned into the mammalian expression vectors pcDNA3.1/V5/His-TOPO (Invitrogen, Carlsbad, CA) and pCMV-HA and pCMV-Myc (Clontech) to add either a hemagglutinin (HA) or a Myc epitope tag at the N terminus. The mouse MONaKA sequences (mMONaKA-S and mMONaKA-FL) have been deposited in GenBank with accession numbers DQ095196 (short) and DQ095197 (full-length). The human sequences (hMONaKA-S and hMONaKA-FL) have been deposited in GenBank with accession numbers DQ124708 (short) and DQ124707 (full-length).
Glutathione S-transferase-MONaKA fusion proteins. cDNAs encoding short and full-length MONaKA were fused to glutathione S-transferase (GST) in the pGEX-4T-1 vector and expressed in Escherichia coli BL21(DE3). pGEX-4T-1 with no insert was used as a control. Bacterial cultures containing the appropriate vectors were grown to a density of A600 = 0.6–0.9, induced with 1 mm isopropylthiogalactoside, and shaken for an additional 4 h. The induced bacterial cultures were pelleted, resuspended in PBS with 1 mm DTT, 1 mm EDTA, and protease inhibitors [1 mm phenylmethylsulfonyl fluoride (PMSF) plus 1 μg/ml each aprotinin, leupeptin, and pepstatin A], and then lysed by sonication. Triton X-100 (1%) was added, and the lysate was incubated with rotation for 30 min at 4°C, followed by centrifugation at 12,000 × g for 15 min at 4°C. Purification of the fusion protein was performed by adding a 50% slurry of glutathione-Sepharose 4B beads in PBS (1 μl/ml original culture volume) (Amersham Biosciences, Piscataway, NJ) for 1 h at 4°C with rotation. The beads were washed five times with PBS. GST fusion proteins were eluted with reduced glutathione elution buffer containing 50 mm Tris-HCl and 10 mm reduced glutathione (Sigma, St. Louis, MO), pH 8.0. Alternatively, the protein was retained on the washed beads and diluted to a bead/PBS slurry of 50%. Aliquots of the eluted protein or protein beads were frozen at –80°C until use.
Antibody preparation. An MONaKA antibody that recognizes both the full-length and short versions of the protein was generated by immunizing rabbits with a GST fusion protein containing amino acids 229–291 of mouse MONaKA. This sequence was cloned into the pGEX4T-1 and pMAL-P2X vectors (Amersham Biosciences, Piscataway, NJ). The maltose (pMAL) fusion protein was purified with amylose beads (New England Biolabs, Beverly, MA). The immunized rabbit serum was first passed through a GST column to remove antibodies that recognized GST and then purified with pMAL-MONaKA-conjugated beads.
Reverse transcription-PCR tissue distribution of MONaKA. Whole RNA was extracted from mouse brain, liver, kidney, spleen, lung, heart, and skeletal muscle using Ultraspec RNA Isolation System (Biotex, Houston, TX), precipitated with an equal volume of isopropanol, and washed with 75% ethanol. Two to 2.5 μg of total RNA were reverse transcribed into cDNA using oligo-dT primers (Superscript First-Strand Synthesis System for RT-PCR; Invitrogen). Outside primers that crossed the 11–12 intron/exon border to prevent amplification of contaminating genomic DNA were used initially to amplify cDNA (5′-ATCTACAAGGCAAAACCAAA-3′ and 5′-GAAATTCTGGATGGAACTGA-3′). PCR amplification of the outside primer product was performed using inside primers specific for either the full-length or short version of MONaKA. For full-length, the 5′ primer was 5′-GGTGGCCGTATTGGAGTCTA-3′, and the 3′ primer was 5′-TGAAGACAGTCCTGCTGTGG-3′. For the short version, the 5′ primer was 5′-GGTGGCCGTATTGGAGTCTA-3′, and the 3′ primer was 5′-GTTCAAAGTGGTGCATGCTC-3′. PCR was repeated three times to confirm the results.
Immunohistochemistry. Immunostaining was performed on 4% paraformaldehyde-perfused mouse tissue. Briefly, frozen, cryoprotected 20 μm sections were cut from brain, spinal cord, dorsal root ganglion, and sciatic nerve and floated in PBS. Sections were blocked with 10% normal goat serum in PBS with 0.1% Triton X-100 (PBS-T) and incubated overnight with primary antibody. Sections were rinsed in PBS-T, processed using rabbit Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA), and developed with 0.5 mg/ml 3,3′-diaminobenzidine tetrahydrochloride in PBS. Controls included preabsorbed antibody, preimmune serum, and no primary, secondary, or avidin-HRP.
Purified cell cultures. Primary hippocampal and cortical cultures were prepared from embryonic rat pups as described previously (Wilcox and Dichter, 1994) and plated on poly-l-lysine-coated plates with Neurobasal medium (Invitrogen). Cultures were allowed to grow for 5 d before harvesting for reverse transcription (RT)-PCR (see below). Purified rat oligodendrocyte cultures were prepared as described by See et al. (2004). Final purification of the cultures was done by immunopanning, which created a 94% pure oligodendrocyte culture (the remaining 6% were GFAP-positive astrocytes). Schwann cell cultures were prepared as described previously (Awatramani et al., 2002). The cultures were 95% pure, as determined by staining with p75, which specifically labels Schwann cells. Cortical astrocyte and microglial cultures were made using embryonic mouse pups (Li et al., 2003). After 7 d, flasks containing astrocytes and microglia were shaken on a horizontal clinical rotator at 260 rpm for 90 min. Microglia are loosened during this process and float into the supernatant. The supernatant containing the microglia was collected and plated into a separate flask and allowed to incubate for 1 h at 37°C in a humidified CO2 chamber. After 1 h, the medium was replaced with fresh DMEM, supplemented with 10% fetal bovine serum and penicillin and streptomycin (100 U/ml), and allowed to grow for 24 h before harvesting. Astrocyte cultures were harvested immediately after shaking. Both cultures were determined to be 98% pure by immunostaining with anti-GFAP (G-3898; Sigma), Griffonia simplicifolia lectin I isolectin B4 (L-3759; Sigma), and anti-CNPase (C5922; Sigma).
Semiquantitative RT-PCR. Semiquantitative RT-PCR was performed on cultured rat cortical and hippocampal neurons, oligodendrocytes, and Schwann cells, as well as mouse astrocytes and microglia. RNA was extracted and reverse transcribed as described above. PCR amplification was performed using primers specific for either the full-length or short version of MONaKA. For full-length, the 5′ primer was 5′-ggtggccgtattggagtcta-3′, and the 3′ primer was 5′-tgaagacagtcctgctgtgg-3′. For the short version, the 5′ primer was 5′-ggtggccgtattggagtcta-3′, and the 3′ primer was 5′-gttcaaagtggtgcatgctc-3′. Semiquantification of the PCR product was as described previously (Kendall et al., 1996) with minor modifications. Briefly, cDNA amplification of MONaKA and β-actin (5′ primer, 5′-cacggcattgtaaccaactg-3′;3′ primer, 5′-ctgggtcatcttttcacggt-3′) was performed in the same tube for each of the samples to eliminate between-sample variation and was tested to ensure that samples were in the linear phase of amplification. To test for genomic DNA contamination, MONaKA genomic primers were used as controls. Additional RT-PCR controls included primers for glial fibrillary acidic protein and neuron-specific enolase to ensure the purity of the different cultures. The semiquantitative RT-PCR was performed three times with similar results.
Yeast two-hybrid screen. The yeast two-hybrid screen was performed using MATCHMAKER System 3 (Clontech). Briefly, the entire coding sequence of the short form or full-length MONaKA from mouse were cloned into bait vector, pGBKT7, and transformed into yeast strain AH109. The baits were screened against a mouse brain cDNA library and a mouse embryo cDNA library, which were pretransformed into yeast Y187 cells. Approximately 3,000,000 mating yeast colonies were screened for MONaKA-interacting proteins. Positive clones were selected on plates lacking adenine, histidine, leucine, and tryptophan and were assayed for α-galactosidase activity. The plasmids of true positive clones were isolated from yeast cells, and the sequences of the cDNAs were determined. To confirm the phenotype, true positive clones were retested by cotransforming baits and library plasmids into yeast cells.
Na,K-ATPase cDNA constructs. Based on the yeast two-hybrid data, two cDNA constructs were created from the C terminus of mouse Na,K-ATPase β1 (amino acids 110–273) and β3 (amino acids 120–258). Each of these constructs was cloned into pCMV-Myc (Clontech), with the Myc tag located at the N terminus. These constructs were used for coimmunoprecipitation and Western blot when indicated. In addition, a full-length cDNA construct of mouse Na,K-ATPase β1 subunit was amplified by PCR from a mouse brain cDNA library. The primers, designed according to the sequence of mouse Na,K-ATPase β1 (GenBank accession number AK005071), were as follows: 5′-atggcccgcggaaaagccaaggaggaaggca-3′ and 5′-gatcagctcttaatttcaatttttacatcaaag-3′. The cDNA was cloned into pCMV-Myc and pCMV-HA (Clontech), with the tags placed at the N terminus. The construct was sequenced through the entire coding region (DNA sequencing facility, University of Pennsylvania, Philadelphia, PA). Full-length rat Na,K-ATPase α1, α2, and α3 cDNAs (ouabain sensitive) were kindly provided by Drs. Amy Moseley, Adam Redden and Jerry B. Lingrel (University of Cincinnati, Cincinnati OH) and Thomas A. Pressley (Texas Tech University Health Sciences Center, Lubbock, TX).
Coimmunoprecipitation and Western blotting. tsA201 cells, derived from HEK293 cells and characterized by Margolskee et al. (1993), were grown in minimal essential medium supplemented with 10% fetal bovine serum and penicillin and streptomycin (100 U/ml). At ∼40–50% confluency, cells were transfected with appropriate plasmids or vector only control using a calcium phosphate protocol. After 24 h, the medium was replaced with fresh supplemented medium. After an additional 24 h, the cells were washed three times in ice-cold PBS, pelleted at 3000 × g at 4°C, and resuspended in buffer containing 1% 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate and 20 mm Tris-HCl, pH 7.5, 1 mm EDTA, 120 mm NaCl, 50 mm KCl, 50 mm NaF, 2 mm DTT, and protease inhibitors (1 mm phenylmethylsulfonyl fluoride plus 1 μg/ml each aprotinin, leupeptin, and pepstatin A). The resuspended cells were incubated for 30 min at 4°C to allow solubilization. At the end of the incubation period, the lysates were spun down at 14,000 × g in a tabletop microcentrifuge at 4°C. The supernatant was removed and used for immunoprecipitation. Lysates from confluent purified astrocyte cultures were prepared in the same way.
Immunoprecipitation was performed by preclearing the lysate with protein A/G PLUS-Agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA) for 2 h at 4°C. The beads were removed by centrifugation at 3000 × g for 5 min, and an aliquot of lysate was removed to confirm protein expression by Western blot (positive control). Proteins were immunoprecipitated from lysates by preincubating the precleared lysates with appropriate antibodies for 1 h before adding the protein A/G beads. The entire mixture was incubated overnight at 4°C with rotation. The next day, the beads were rinsed three times with PBS, and the protein was eluted with 2× loading buffer. Precleared lysates or immunoprecipitates were then subjected to 4–15% gradient SDS-PAGE (Bio-Rad, Hercules, CA).
After SDS-PAGE, proteins were transferred to nitrocellulose membranes (Bio-Rad). The membranes were blocked with 5% nonfat milk in TBST (10 mm Tris-HCl, pH 7.5, 150 mm NaCl, and 0.1% Tween 20) and then incubated with appropriate primary antibodies in blocking buffer at 4°C overnight (see below). After three washes with TBST, the blots were incubated with horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG (anti-mouse NA931 and anti-rabbit NA934; Amersham Biosciences, Arlington Heights, IL) for 1 h at room temperature. Proteins were detected with an enhanced ECL Western blotting detection reagent (Amersham Biosciences, Arlington Heights, IL). Molecular Analyst software (Bio-Rad) was used to quantify Western films.
Antibodies used for Western blot and coimmunoprecipitation were purchased from Upstate Biotechnology (Lake Placid, NY; anti-Na,K-ATPase β1 06–170, β3 06–817, and α1 05–369), Sigma (anti-Myc M5546, anti-HA H9658, and anti-GST A7340), and Invitrogen (anti-V5 SC138).
Mouse brain membrane preparation. Mice were anesthetized and decapitated, and brains were immediately removed and snap frozen in liquid nitrogen. Individual brains were pulverized and then homogenized in a buffer containing 2.5 mm KCl, 250 mm sucrose, and 25 mm HEPES, pH 7.4, protease inhibitors (1 mm PMSF plus 1 μg/ml each aprotinin, leupeptin, and pepstatin A), and 2 mm DTT. The homogenate was centrifuged at 1000 × g for 10 min to remove excess debris. The supernatant was removed and centrifuged for 1 h at 150,000 × g. After this centrifugation, the supernatant was collected as the cytoplasmic fraction and the pellet as the crude membranes. The pellet was washed with the same buffer and centrifuged again for 1 h at 150,000 × g. After the second centrifugation, the membrane pellet was resuspended in a buffer containing 1% Triton X-100, 20 mm Tris-HCl, pH 7.5, 10 mm EDTA, 120 mm NaCl, 50 mm KCl, 50 mm NaF, protease inhibitors, and 2 mm DTT to the same final volume as the cytoplasmic fraction.
GST-MONaKA pull-down of Na,K-ATPase β subunits from native cells and tissue. A total of 0.5 μg (500 μl) of crude mouse brain membranes or purified cultured astrocyte lysates were incubated with 50 μl of a 50% slurry of GST or GST-MONaKA beads at 4°C for 2 h (n = 3). The beads were washed with PBS five times, resuspended in 2× SDS loading buffer, and subjected to 4–15% gradient SDS-PAGE. Na,K-ATPase β1 and β3 subunits were detected by Western blot.
Deglycosylation. Mouse brain membranes were prepared as described above, and proteins were deglycosylated using peptide N-glycosidase F (PNGase F; New England Biolabs), following the protocol provided with the enzyme and buffers. Briefly, a 25 μg sample of the membrane preparation was denatured in 50 μl of glycoprotein denaturing buffer (final concentration of 0.5% SDS with 1% β-mercaptoethanol) at 100°C for 10 min, followed by the addition of a final concentration of 50 mm sodium phosphate buffer, pH 7.5, 1% NP-40, and 150 U of PNGase. The entire reaction mixture was incubated for 2 h at 37°C and separated by SDS-PAGE. Western blots were developed as described above, using Na,K-ATPase β1or β3 antibodies (Upstate Biotechnology).
Cell surface biotinylation. Cell surface biotinylation was performed as described previously (Wen and Levitan, 2002). Briefly, tsA201 cells were transfected with either HA-MONaKA alone or Myc-Na,K-ATPase β1 subunit alone or were cotransfected with HA-MONaKA and Myc-β1 subunit together. Forty-eight hours later, the cells were washed three times with PBS and incubated with 2 ml of 0.5 mg/ml Sulfo-NHS-LC-Biotin (Pierce; Rockford, IL) in PBS, pH 8.0, at room temperature for 30 min with gentle agitation. The biotin reagent was removed by washing the cells three times with PBS. Cells were harvested and resuspended in Western blot lysis buffer (described above) containing 20 μl of ImmunoPure immobilized streptavidin beads (Pierce) to isolate the biotinylated proteins. Protein in total cell lysates and streptavidin precipitates was detected by Western blotting with anti-HA and anti-Myc antibodies. Control cells were transfected with Myc-14-3-3, an intracellular protein that served as a negative control, as described previously (Zhou et al., 1999). The experiment was repeated three times.
Preparation of Na,K-ATPase from mouse brain. Na,K-ATPase from mouse brain was prepared using a scaled-down protocol for the purification of Na,K-ATPase from rat brain (Mayrand et al., 1982). All steps, unless otherwise specified, were performed at 4°C. The meninges were removed from 25 mouse brains, and the brains were minced with scissors in medium A (0.25 m sucrose, 1 mm Tris-EDTA, and 10 mm imidazole-HCl, pH 7.7), homogenized with a tissue homogenizer at 6000 rpm, and centrifuged for 5 min in a Beckman Instruments (Fullerton, CA) J2-HC rotor at 1100 × g, followed by a 20 min and then 5 min spin at 7700 × g, each time retaining and recentrifuging the supernatant. The supernatant was centrifuged using a 50.2Ti rotor in a Beckman Instruments XL-90 Ultracentrifuge at 55,000 × g for 30 min. The white membrane portion of the pellet was retained, discarding the brown pellet. The white membrane pellet was homogenized in medium A using six strokes in a Dounce homogenizer and then frozen at –20°C until use.
Final purification of Na,K-ATPase was performed by thawing and continuously stirring the membrane preparation at 25°C for 30 min with 3mm ATP, 50 mm imidazole-HCl, pH 7.7, and 2 mm Tris-EDTA, with a protein (milligrams per milliliter) to SDS (milligrams per milliliter) ratio of 1.9. All subsequent centrifugation steps were performed with a 50.2Ti rotor in a Beckman Instruments XL-90 Ultracentrifuge. The resuspended membrane mixture was added to ice-cold medium B (2.1 m sucrose, 5 mm Tris-EDTA, and 20 mm imidazole-HCl, pH 7.7) and centrifuged at 250,000 × g for 90 min. The floating protein band was removed and homogenized using six strokes of a Dounce homogenizer, resuspended in medium A, and centrifuged at 150,000 × g for 45 min. The resulting pellet was resuspended in medium C (20% glycerol v/v, 5.4% sucrose w/v, 1 mm Tris-EDTA, and 25 mm imidazole-HCl, pH 7.4), homogenized with six strokes in a Dounce homogenizer, and centrifuged for an additional 150,000 × g. The pellet was then resuspended and homogenized with a Dounce homogenizer in medium A plus 10% v/v glycerol and centrifuged at 150,000 × g for 30 min. The final pellet was diluted to 1 mg/ml in medium A with 10% glycerol and stored at –20°C until use. SDS-PAGE followed by Coomassie blue staining was performed to check for the appropriate size bands and the purity of the protein. All protein quantification was performed using a Bio-Rad Protein Assay kit, based on the method of Bradford (Bio-Rad).
ATPase assay. The ATPase assay was performed based on a standard protocol (Jorgensen, 1988). GST fusion protein beads were washed with reaction buffer (130 mm NaCl, 20 mm KCl, 3 mm MgCl2, and 25 mm imidazole-HCl, pH 7.5) three times to get rid of any residual PBS and resuspended to a 50% bead/buffer slurry. GST fusion protein bead slurry (10 μl) was preincubated for 10 min at 37°C with 10 μl (10 μg) of semipurified Na,K-ATPase with the addition of 30 μl of reaction buffer. After the incubation, the entire 50 μl was transferred to test tubes containing 1 ml of prewarmed incubation buffer. Half of the samples included 5.0 mm ouabain. The reaction was initiated by the addition of 3 mm ATP, and the samples were incubated for 5 min at 37°C. The reaction was stopped with 1 ml of ice-cold 0.5 m HCl containing 170 mm ascorbic acid, 4 mm ammonium heptamolybdate, and 1% SDS, on ice. For color development, 1.5 ml of a solution containing 150 mm sodium metaarsenite, 70 mm sodium citrate, and 2% acetic acid was added, and the tubes were incubated at 37°C for 10 min. The tubes were centrifuged at 3500 × g for 5 min to concentrate the beads at the bottom. A 1 ml aliquot was taken from the tubes and transferred to disposable cuvettes for analysis. The absorbance was read at 850 nm. Alternatively, eluted GST fusion protein was used in the assay, and the assay was performed in a similar manner as with the beads with similar results. Controls included GST-dSlob, no membrane, washed and unbound glutathione-Sepharose beads, GST only, Na,K-ATPase without ATP, and all assay components added with stop buffer on ice to determine baseline inorganic phosphate levels. All assays were performed in triplicate. Statistical significance was assessed by a paired t test.
86Rb+ uptake assay. 86Rb+ uptake was measured by standard methods (Munzer et al., 1994; Garty et al., 2002) with slight modification. tsA201 cells were transfected with vector with or without MONaKA, or with Na,K-ATPase α1β1 subunits with or without MONaKA, using a standard calcium phosphate protocol. Forty-eight hours later, the cells were washed twice with 10 ml of 140 mm NaCl, 5 mm KCl, 1 mm MgCl2,1mm CaCl2, 5 mm glucose, and 5 mm HEPES-Tris, pH 7.4, and resuspended into 1 ml with gentle pipetting. A 200 μl aliquot of the resuspension was removed for Western blot and protein concentration analysis. The remaining cells were split into four tubes, centrifuged at 3000 × g, and resuspended in the buffer above, with the addition of 0.1 mm furosemide, 0.01 mm monensin, with or without 5 mm ouabain. The tubes were preincubated for 10 min at 37°C, and then 2.5 μCi of 86RbCl (Amersham Biosciences, Piscataway, NJ) diluted in buffer with furosemide, monensin, and/or ouabain, as above, was added to each tube for a final reaction mixture of 500 μl. After an additional 0–15 min at 37°C, the cells were placed on ice with the addition of 500 μl of ice-cold PBS containing 5 mm BaCl2 to stop the reaction. The tubes were centrifuged at 3000 × g and washed three times with cold PBS containing 5 mm BaCl2. Cells were lysed with 500 μl of 1 m NaOH, and the radioactivity in the lysate was determined on a Wallac 1410 liquid scintillation counter (Amersham Biosciences, Piscataway, NJ). Radioactivity was normalized to protein concentration, determined by Bio-Rad Protein Assay kit (Bio-Rad). The expression of the α and β subunits and MONaKA was examined by Western blotting to confirm equivalent expression in each plate. Statistical significance was assessed by a paired t test.
Results
Sequence and splice variants of MONaKA
We cloned mouse and human variants of MONaKA based on their sequence similarity to Drosophila Slob. Mouse and human MONaKA exhibit >90% amino acid sequence identity and >97% similarity to one another (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). They also are identical in all properties that we have tested, and so we will not distinguish between them here. MONaKA is most similar to Drosophila Slob in a central domain constituting approximately half of the protein but exhibits little similarity in the N- and C-terminal domains (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). There are at least two splice variants of MONaKA (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material), depending on the absence or presence of a 33 bp cassette that is inserted at the site marked by the vertical arrow in supplemental Figure 1A (available at www.jneurosci.org as supplemental material). This cassette contains a stop codon that produces a truncated form of the protein (515 amino acids in human; 514 amino acids in mouse). In the absence of this cassette, a 578 amino acid human protein (581 amino acids in mouse) that we call full-length MONaKA is produced. The alternative splicing is conserved perfectly between mouse and human MONaKAs (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material).
A conserved domain search reveals that MONaKA contains a PX-like domain near its N terminus (amino acids 32–122), of the kind that might bind to phosphoinositides (Wishart et al., 2001). It also contains a putative protein kinase domain in the central portion of the protein (amino acids 198–349). This is the region in which MONaKA most closely resembles Drosophila Slob, and we found recently that Drosophila Slob exhibits protein kinase activity that can be modulated by the cAMP-dependent protein kinase (Zeng et al., 2004). An examination of the MONaKA sequence also reveals an unusually large number of proline residues, often clustered together to form polyproline motifs (supplemental Fig. 1A, gray box, available at www.jneurosci.org as supplemental material). The longest of these polyproline motifs is absent from the short splice variant of MONaKA. The functional significance of these multiple proline residues remains to be determined.
Distribution of MONaKA
Using RT-PCR, we examined the mRNA distribution of both full-length and short MONaKA in various mouse tissues. mRNA for full-length MONaKA was found in all tissues examined, with the short form found in all tissues except skeletal muscle and very low levels in spleen (Fig. 1). Focusing on the nervous system, we used a polyclonal antibody that recognizes both splice variants of the protein and immunohistochemical techniques to verify MONaKA localization. We found MONaKA staining throughout the nervous system, in both neurons and glia (Figs. 2, 3). Within neurons, staining is strong in the cytoplasm of the cell body and axon in both the CNS and peripheral nervous system. Staining in the CNS is seen in cortical and thalamic neurons, neurons of the hippocampal formation, cerebellum and brainstem (Fig. 2), and spinal cord α motoneurons (Fig. 3A,C). The most intense immunoreactivity is observed in peripheral dorsal root ganglion neurons (Fig. 3D,E). Figure 3E shows the clear cytoplasmic distribution of MONaKA in both the cell body and axon of a single dorsal root ganglion neuron. In fact, the axonal staining is so intense that the dorsal root entry zone and superficial lamina of the spinal cord are also strongly immunopositive (Fig. 3A).
Figure 1.

MONaKA mRNA distribution in various mouse tissues. All tissues examined contain mRNA for the full-length (FL) version of the protein, as determined by RT-PCR. The short (S) version is also found in most tissues, with a low amount in spleen and none in skeletal muscle. FL product size, 439 bp; S product size, 450 bp. RT-PCR was performed three times in different animals with similar results.
Figure 2.
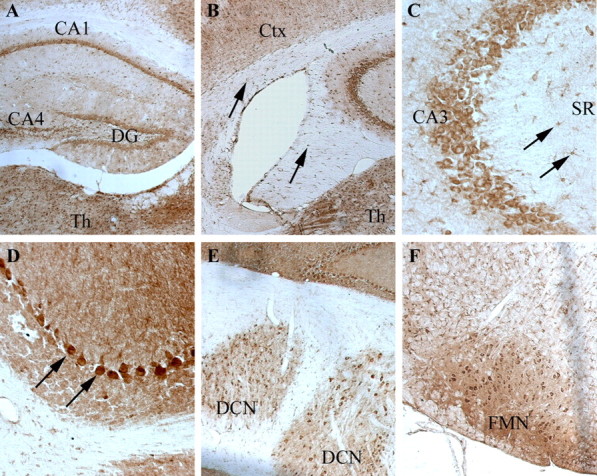
Distribution of MONaKA in mouse brain. MONaKA immunostaining in the cortex and hippocampus (A–C), cerebellum (D, E), and brainstem (F). There is a widespread distribution of MONaKA protein within the cytoplasm of both neurons and glia. Immunopositive neurons are found in dentate gyrus granule cells (DG), as well as in CA pyramidal cells of the hippocampal formation (A). At higher magnification (C), an example of the cytoplasmic distribution of MONaKA is seen within pyramidal cells of the CA3 region of the hippocampus. In addition, astrocytes within the stratum radiatum (SR) (C, arrows) are labeled. Thalamic (Th) and cortical (Ctx) neuronal staining is noted in all areas examined (A, B). Oligodendrocyte labeling is demonstrated by punctate staining in the fornix and corpus callosum (B, arrows). In the cerebellum, the most intense staining is found in the Purkinje cell bodies (D, arrows) and deep cerebellar nuclei (E, DCN), with less immunoreactivity present in the molecular and granule cell layers (D). In the brainstem (F), facial motor nucleus (FMN) neurons projecting out to the peripheral nervous system demonstrate strong cytoplasmic staining. Staining was performed three times in different animals with similar results.
Figure 3.
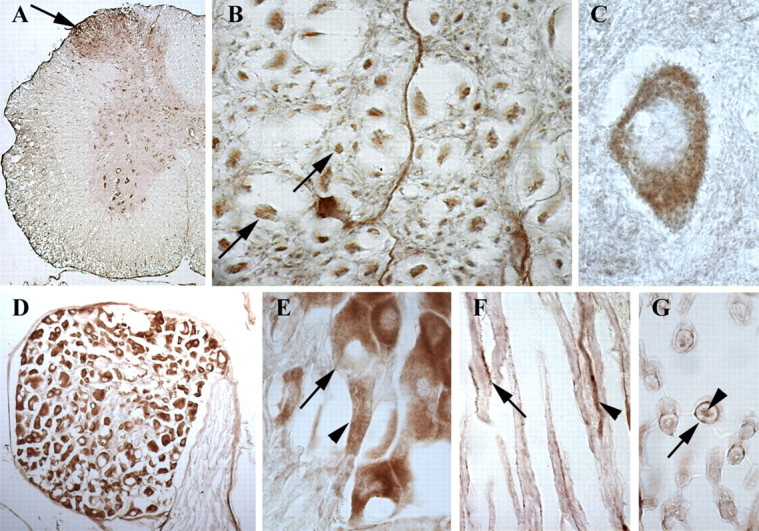
Distribution of MONaKA in mouse spinal cord and peripheral nervous system. In a low-magnification cross-section of lumbar spinal cord (A), note the intense staining in the dorsal root entry zone and superficial lamina (arrow). This staining is most likely in the axons of the dorsal root ganglion (D; E, arrowhead), which shows the most intense staining in the CNS or peripheral nervous system. At higher magnification, axonal staining is seen in spinal cord white matter (B, arrows), dorsal root ganglion neurons (E, arrow), and sciatic nerve (F, arrowhead; G). Within the ventral horn of the spinal cord, cytoplasmic staining is seen in α motoneurons (A, C) with a lack of staining in the nucleus or nucleolus (C). A longitudinal section of sciatic nerve demonstrates Schwann cell immunoreactivity on the membrane of the Schwann cell but not within the myelin itself (F, arrow; G, arrow), as well as in sciatic nerve axons (F, arrowhead; G, arrowhead).
Glial cells of the peripheral nervous system and CNS also demonstrate MONaKA immunoreactivity. Within the CNS, oligodendrocytes (Fig. 2B), astrocytes (Fig. 2C), and microglia (data not shown) all are immunopositive for MONaKA. In the peripheral nervous system, Schwann cells also express MONaKA (Fig. 3F,G); however, the expression pattern appears to be on the Schwann cell membrane, not within the myelin or, more interestingly, not at the nodes of Ranvier. This widespread expression pattern in both neurons and glia suggests that MONaKA may play an important role in the nervous system.
Differential expression of full-length and short versions of MONaKA in neurons and glia
Because the immunohistochemical staining could not differentiate between the short and full-length versions of MONaKA, we performed semiquantitative RT-PCR on purified cultured cells with primers specific to each splice variant. We found both full-length and short version mRNA in all cells examined but in different ratios depending on cell type. Purified hippocampal and cortical neurons produce more short version than full-length MONaKA mRNA (Fig. 4A,B, compare lanes 1, 3 with 2, 4). Glial cells, conversely, have more full-length than short version mRNA (Fig. 4A,B, compare lanes 5, 7, 9 with 6, 8, 10). The findings that both the full-length and short forms of MONaKA are present in the nervous system, and that they exhibit differential expression ratios in neurons and glia, are intriguing, and suggest that the two forms of MONaKA may play different roles in different cell types.
Figure 4.

Semiquantitative RT-PCR analysis of MONaKA transcripts in cultured cells. A, Both hippocampal and cortical neurons (lanes 1–4) have an intense band for the short version of MONaKA (MO) and have full-length message as well, whereas astrocytes, oligodendrocytes, and microglia (lanes 5–10) show a more intense band for full-length than short MONaKA. The left lane shows molecular weight markers. The expected PCR product size is 439 bp for the full-length transcript and 450 bp for the short transcript. Lanes: 1, cortical neurons, full-length; 2, cortical neurons, short; 3, hippocampal neurons, full-length; 4, hippocampal neurons, short; 5, astrocytes, full-length; 6, astrocytes, short; 7, oligodendrocytes, full-length; 8, oligodendrocytes, short; 9, microglia, full-length; 10, microglia, short. B, Densitometric quantitation of the relative amounts of MONaKA mRNA. Each bar represents results from cultures prepared from 8–14 pooled animals, normalized to an internal β-actin standard (146 bp band), and RT-PCR was performed three times to ensure reproducibility. Lane labeling is the same as in A.
MONaKA binds to Na,K-ATPase β subunits
To explore the physiological role of MONaKA, both the short and full-length versions were used as bait in a yeast two-hybrid screen. These baits were screened against a mouse brain cDNA library and a mouse embryo cDNA library. Approximately 3 million mating yeast colonies were screened for MONaKA-interacting proteins. Among the positive clones, 12 exhibited sequence identity to the C-terminal domain of the β1 subunit of the Na,K-ATPase. One of the 12 clones encoded amino acid residues 110–277 of this 304 amino acid protein, and the remaining 11 encoded amino acids 110–273 (Fig. 5A). Another nine positive clones were found to encode amino acids 120–258 of the 278 amino acid β3 subunit of the Na,K-ATPase (Fig. 5A). Approximately half of these positive clones were isolated using full-length MONaKA and the other half using short MONaKA as bait. These results are consistent with the possibility that both the short and full-length versions of MONaKA interact with the plasma membrane Na,K-ATPase.
Figure 5.

MONaKA binds to C-terminal fragments of Na,K-ATPase β subunits. A, Results from the yeast two-hybrid screen. Both the short and full-length MONaKA sequences were used as baits in yeast two-hybrid screens. Of 21 positive clones, nine encoded amino acids 120–258 of the β3 subunit of the Na,K-ATPase. The other 12 clones encoded a portion of the C-terminal domain of the β1 subunit of the Na,K-ATPase. n indicates number of clones. B, Mouse MONaKA (MO) coimmunoprecipitates with the β1 and β3 subunit C-terminal fragments identified in the yeast two-hybrid screen (23 and 22 kDa, respectively). tsA201 cells were transfected with either the short or full-length versions of HA-tagged MONaKA, together with Myc-tagged Na,K-ATPase β1 or β3 subunit C-terminal fragments. Identical results were obtained for β1 (left panels) and β3 (right panels) subunit fragments. Western blot demonstrates that the β subunit fragment expression (top panels) and immunoprecipitation (IP)(middle panels) are similar in the absence or presence of MONaKA. A β subunit fragment band is observed in the MONaKA immunoprecipitate (bottom panels) only when β subunit fragment and MONaKA are transfected together. No bands were seen when vector only was used as control (data not shown).
To confirm the results of the yeast two-hybrid screen, the C-terminal fragments of the Na,K-ATPase β1 and β3 subunits isolated from the screen were cloned into mammalian cell expression vectors with epitope tags. As shown in Figure 5B, both the β1 subunit fragment (left) and the β3 subunit fragment (right) coimmunoprecipitate with MONaKA. Coimmunoprecipitation in the other direction gave similar results (data not shown). This finding confirms that these C-terminal fragments of the β1 and β3 subunits bind to MONaKA under coimmunoprecipitation conditions. Using a similar approach, we also tested the interaction of full-length Na,K-ATPase β1 subunit with MONaKA. As shown in Figure 6, immunoprecipitation in both directions results in the coimmunoprecipitation of the β1 subunit and MONaKA. The results shown in Figures 5 and 6 were obtained using full-length MONaKA, but identical results were obtained with the short splice variant (data not shown). Together, these results demonstrate clearly that MONaKA is a binding partner for the Na,K-ATPase β subunits in mammalian cells.
Figure 6.

Coimmunoprecipitation of MONaKA and β1 subunit. HA-tagged full-length MONaKA was cotransfected with Myc-tagged Na,K-ATPase β1 subunit. Western blot of cell lysates demonstrates that both the glycosylated (48 kDa) and nonglycosylated (38 kDa) forms of the β1 subunit (A, lysates) as well as MONaKA (B, lysates) are both expressed. The β1 subunit can be detected in a MONaKA immunoprecipitate (A), and, conversely, MONaKA can be detected in a β1 subunit immunoprecipitate (IP) (B), when the β1 subunit and MONaKA are transfected together. Vector transfection alone showed no bands in either direction (data not shown). Immunoprecipitation antibodies were either anti-Myc (β1) or anti-HA (MONaKA), with detection antibodies either anti-Myc or anti-MONaKA. The prominent 48 kDa band in the β1 and MO IP lanes in A is IgG. The 65 kDa band in the MO IP lane in B is probably MO coimmunoprecipitating with endogenous β subunit.
MONaKA is associated with Na,K-ATPase β1 and β3 subunits in native cells and tissue
To determine whether MONaKA interacts with Na,K-ATPase β subunits in native cells and tissue, we constructed GST-MONaKA fusion proteins using both the short and full-length splice variants. As shown in Figure 7A, GST-MONaKA can pull down both the glycosylated and nonglycosylated forms of β subunit protein from purified cultured mouse astrocytes (left) and from whole mouse brain (right). Both full-length and short GST-MONaKA are effective in pulling down the β subunits (lanes 3 and 4), whereas GST alone is without effect (lane 2). Similarly, as shown in Figure 7B, MONaKA coimmunoprecipitates with β subunits from cultured astrocytes (top) and whole mouse brain (bottom). Thus, the interaction between MONaKA and Na,K-ATPase β subunits occurs not only in transfected cells but also in native tissues.
Figure 7.
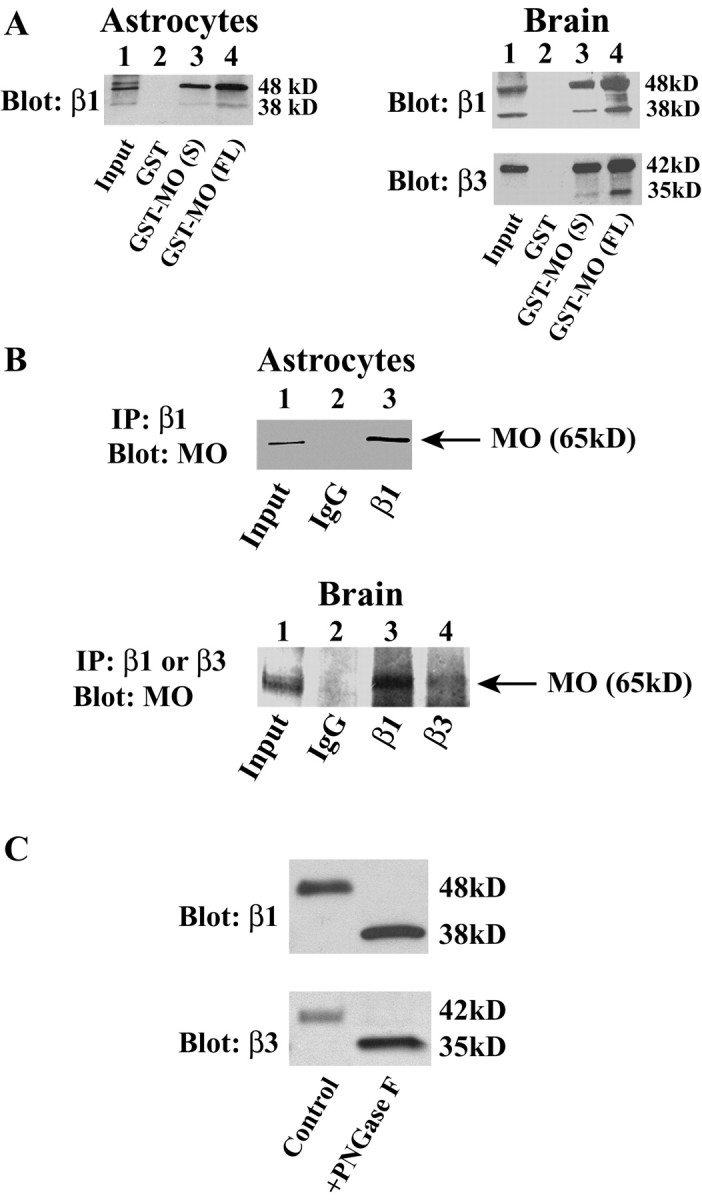
MONaKA interacts with native Na,K-ATPase β1 and β3 subunits. A, GST pull-down. Native β subunits of Na,K-ATPase can be pulled down from lysates of purified cultured astrocytes (left) or whole mouse brain crude membranes (right) by both the short (S) and full-length (FL) versions of GST-MONaKA (MO) but not by GST alone. B, Immunoprecipitation (IP) with an anti-β subunit antibody of Na,K-ATPase can coimmunoprecipitate MONaKA from lysates of cultured astrocytes (top) or whole mouse brain crude membranes (bottom). C, The specificity of the antibodies used to immunoprecipitate and blot for the β1 and β3 subunits was confirmed by deglycosylation of brain membrane samples; the antibodies recognize both the glycosylated and deglycosylated forms of the native β1 (top panel) and β3 (bottom panel) subunits.
To confirm the specificity of the β antibodies used, we deglycosylated mouse brain membrane preparations and compared them with untreated samples using Western blot analysis. Figure 7C confirms that both the β1 and β3 subunits are recognized by the antibodies in both their glycosylated and deglycosylated forms.
MONaKA is distributed in both plasma membrane and cytoplasm in the brain
Although our immunohistochemical analysis suggested that much of MONaKA in neurons and glia is localized in the cytoplasm (Figs. 2, 3), the resolution of this method is not sufficient to determine whether any of the protein is associated with the plasma membrane in which the Na,K-ATPase resides. To examine this, we used two different methods: (1) Western blot analysis of separated brain membranes and cytoplasm, and (2) cell surface biotinylation. For Western blot analysis, we isolated mouse brain membranes and compared them with samples of brain cytoplasm. As shown in Figure 8A, both the brain cytoplasm and brain membrane fractions contain MONaKA protein. Using gel densitometric quantitation, we found that approximately two-thirds of MONaKA protein is associated with the membrane, with the remaining one-third found in the cytoplasm.
Figure 8.

MONaKA is associated with the plasma membrane. A, Brain cytoplasmic and membrane fractions were analyzed by Western blot to determine the distribution of MONaKA (MO). MONaKA protein is found in both the brain cytoplasm and membrane fractions, with approximately two-thirds of the protein associated with the membrane based on densitometric analysis. B, Cell surface biotinylation. tsA201 cells were transfected with Myc-Na,K-ATPase β1 subunit (lane 1) or HA-MONaKA (lane 2) or both together (lane 3). After surface biotinylation (see Materials and Methods), both MONaKA and the β1 subunit (positive control) can be detected in avid in precipitates (bottom panels) using anti-HA or -Myc antibodies, respectively. The cytoplasmic protein Myc-14-3-3 is not present in the avidin precipitate under these experimental conditions (lane 4).
As a secondary confirmation, we used a surface biotinylation reagent to label with biotin all proteins that have a portion of their sequence outside the plasma membrane. After the removal of the biotinylation reagent, biotinylated proteins were isolated by precipitation with avidin beads. As shown in Figure 8B, MONaKA is present in the avidin precipitate from tsA201 cells, whether it was transfected alone (lane 2) or together with the β1 subunit of the Na,K-ATPase (lane 3). In contrast, when a similar experiment was performed in cells transfected with the ζ isoform of 14-3-3, which is known to be exclusively a cytoplasmic protein, no 14-3-3 is found in the avidin precipitate (lane 4). To test the possibility that MONaKA is not itself an intrinsic membrane protein but rather binds to some other membrane protein that can react with biotin, we washed the avidin precipitate with SDS at concentrations ranging from 2 to 10% before separation of the proteins on polyacrylamide gels. This treatment disrupts protein–protein interactions such that only the biotinylated proteins themselves would remain present in the avidin precipitate. MONaKA is still present in the avidin precipitate after this stringent treatment (data not shown), suggesting that at least some proportion of the protein behaves as an intrinsic membrane protein in these heterologous cells. In addition, the β1 subunit of Na,K-ATPase, which is known to be membrane bound, is also present after the SDS wash (data not shown), confirming that the avidin–biotin interaction survives this stringent treatment.
MONaKA modulates the Na,K-ATPase
To determine whether the binding of MONaKA to Na,K-ATPase β subunits can modulate the properties of the protein, we tested whether MONaKA can influence the ATPase activity in vitro. A standard protocol was used for the partial purification of mouse brain Na,K-ATPase. The generation of inorganic phosphate from ATP by this membrane fraction was measured with a colorimetric assay, and the ouabain-sensitive fraction (∼90%) of the total ATPase activity was taken as the activity attributable to the plasma membrane Na,K-ATPase. When purified GST-MONaKA fusion protein (either short or full-length) is added to this in vitro assay, the ATPase activity is inhibited by 30–38% (Fig. 9A). In contrast, neither GST-Drosophila Slob nor GST alone inhibits the ATPase activity (data not shown). These data are consistent with the possibility that MONaKA binding to Na,K-ATPase β subunit can acutely influence the enzymatic activity of the Na,K-ATPase. Similar results were also obtained when crude membranes were prepared from cultured astrocytes (data not shown).
Figure 9.

GST MONaKA inhibits Na,K-ATPase activity in vitro and 86Rb+ uptake in intact cells. A, The production of inorganic phosphate from ATP by a partially purified Na,K-ATPase preparation was measured by a colorimetric assay. The ouabain-sensitive portion of the total ATPase activity was taken as the basal Na,K-ATPase activity (1.2 μmol Pi/mgprotein/min). This activity is inhibited by both short (S) and full-length (FL) GST-MONaKA (MO) but not by GST alone or GST-Drosophila Slob (data not shown) (*p < 0.05, significantly different from control). Similar results were obtained with Na,K-ATPase from purified cultured astrocytes (data not shown). B, tsA201 cells were transfected with vector (Mock) with or without full-length MONaKA (left two lanes) or with α1β1 Na,K-ATPase subunits with or without full-length MONaKA (right two lanes). The ouabain-sensitive portion of total 86Rb+ uptake was taken as that attributable to the Na,K-ATPase. Identical results were obtained with the short version of MONaKA (data not shown). Each data point is the mean ± SE of three experiments (*p < 0.05, significantly different from transfection without MONaKA). Both endogenous and α1β1-mediated 86Rb+ uptake are inhibited by MONaKA.
To determine whether MONaKA can also influence the ion transport activity of the Na,K-ATPase, we examined 86Rb+ uptake into intact cells via the Na,K-ATPase. tsA201 cells were transfected, and 86Rb+ uptake was measured as described in Materials and Methods. As shown in Figure 9B, endogenous ouabain-sensitive (∼90% of the total) 86Rb+ uptake is reduced 19% in MONaKA transfected cells (Fig. 9B, left two lanes). The tsA201 cells are derived from HEK296 cells, which contain endogenous Na,K-ATPase as well as MONaKA (confirmed by RT-PCR; data not shown). In addition, cotransfection of MONaKA together with Na,K-ATPase α1 and β1 subunits also decreases ouabain-sensitive 86Rb+ uptake to approximately the same extent when compared with α1β1 transfected controls (Fig. 9B, right two lanes). MONaKA cotransfection does not alter the expression level of the Na,K-ATPase α1 and β1 subunits (data not shown).
Discussion
There is much interest in the possibility that the properties of membrane ion channels are subject to long-term modulation by a variety of molecular mechanisms (Levitan, 1999). Our laboratory has been studying the Drosophila protein dSlob, which binds to and modulates the dSlo voltage and calcium-activated potassium channel. Based on sequence homology, we cloned mouse and human orthologs of Drosophila Slob. The mammalian protein, however, does not bind to mammalian Slowpoke channels. Rather, it binds to and modulates Na,K-ATPase, and thus we named it MONaKA to reflect its functional properties.
MONaKA exists in at least two splice variants, and the splicing is conserved perfectly between mouse and human. The protein contains a putative protein kinase domain in its central region that is most similar to Drosophila Slob, and we found recently that Drosophila Slob does indeed exhibit a regulated protein kinase activity (Zeng et al., 2004). MONaKA also contains a PX motif of the sort that has been shown to bind phosphoinositides (Wishart et al., 2001), although we have not yet tested the possibility that such binding does indeed occur. Another striking feature of the MONaKA sequence is the presence of multiple proline residues organized in polyproline motifs, most notably in the C-terminal domain. Such motifs often participate in protein–protein interactions (Pawson and Nash, 2003). Of particular interest is the fact that the longest polyproline motif is present only in the full-length splice variant of MONaKA. The expression of both splice variants is widespread throughout the CNS and peripheral nervous system, in neurons and glia, but the relative amount of the two splice variants varies from one cell type to another. The general pattern is that glial cells of various types tend to express more full-length than short MONaKA, whereas the opposite is true for neurons. The functional significance of this differential expression of the splice variants remains to be determined.
When we first isolated MONaKA, we were surprised to find that it does not bind to mammalian Slowpoke channels. However, it is now known that the region of Drosophila Slob necessary for its binding to the dSlo channel lies outside the central domain in which Drosophila slob most resembles MONaKA (Zhou et al., 2003). Instead, a yeast two-hybrid screen identified the β1 and β3 subunits of the Na,K-ATPase as MONaKA binding partners. We did not identify β2 subunit isoforms in the screen, but it is possible that β2 subunit cDNA is not well represented in the libraries we used. The interaction of MONaKA with the β subunits is robust because it survives coimmunoprecipitation in a detergent-containing solution. A combination of such coimmunoprecipitation and GST fusion protein pull-down experiments makes it clear that MONaKA interacts with the Na,K-ATPase β subunits both in vitro and in vivo and in native cells as well as in a mammalian cell expression system. Although Drosophila Slob binds to a channel and mammalian MONaKA to an ion pump, one common feature between them is their interaction with membrane proteins involved in transmembrane potassium flux.
Another surprise was the finding that MONaKA appears to be tightly associated with the plasma membrane, even under conditions that should dissociate it from any of its binding partners. The presence of a PX-like phosphoinositide binding motif (Wishart et al., 2001) in the MONaKA sequence is consistent with membrane association but cannot account for the surface biotinylation results. Although there are no predicted membranespanning domains in the MONaKA sequence, the biotinylation data suggest that at least a portion of the protein is outside the plasma membrane. The C-terminal portion of the Na,K-ATPase β subunits, to which MONaKA binds, is also thought to be extracellular (Crambert and Geering, 2003). We are exploring the possibility that MONaKA may be secreted from neurons or glia, perhaps via an exocytotic mechanism.
Most interesting is the finding that MONaKA can modulate the ATPase and ion transport activities of the Na,K-ATPase. The ATPase activity from whole brain or cultured astrocytes is inhibited by the addition of exogenous MONaKA, suggesting that the Na,K-ATPase is not fully saturated with endogenous MONaKA in these cells. It has been known for a long time that the Na,K-ATPase can be regulated by ion concentration gradients, and more recently it has become evident that endogenous modulators, most notably members of the FXYD protein family, exist (Sweadner and Rael, 2000). Our findings that the Na,K-ATPase can be modulated by both the short and full-length splice variants of MONaKA add to the emerging concept that the Na,K-ATPase is subject to complex modulatory influences.
Prolonged or intense neuronal firing can lead to the accumulation of extracellular potassium ions and the disruption of the normal plasma membrane ion gradients. Elevated extracellular potassium, in turn, can cause membrane depolarization, leading to excitotoxicity as a result of the excessive release of the neurotransmitter glutamate from astrocytes or synaptic terminals (Albensi and Janigro, 2003). Because potassium accumulates to high concentrations in the relatively restricted extracellular space bounded by the presynaptic and postsynaptic neuronal elements and the associated astrocyte (Cholet et al., 2002), this may be a particularly acute problem at CNS synapses. It has long been thought that the strategic location of astrocytes allows them to play a critical role in potassium homeostasis at the synapse (Hertz, 1978) via activation of their prominent Na,K-ATPase as well as other mechanisms (Amedee et al., 1997). Thus, it is particularly interesting that MONaKA is expressed at high levels in astrocytes (as well as other glial cells) and modulates the astrocyte Na,K-ATPase.
An additional role for the astrocyte Na,K-ATPase at the synapse is to help with the uptake of neurotransmitter released into the synaptic cleft. The glutamate transporters responsible for uptake into astrocytes are powered by the plasma membrane sodium gradient and mediate the coupled transport of sodium and glutamate into the cell. The resulting elevated intracellular sodium activates the Na,K-ATPase, resulting in the restoration of the normal ionic gradients (Magistretti and Pellerin, 1999). Interestingly, the two major astrocyte sodium-dependent glutamate transporters are tightly colocalized with the Na,K-ATPase in perisynaptic regions (Cholet et al., 2002). Consistent with this close association is the finding that glutamate uptake by cultured astrocytes stimulates Na,K-ATPase activity, possibly by the recruitment of a specific isoform that is mobilized in response to synaptic activity (Pellerin and Magistretti, 1997). It will be interesting to examine the role of MONaKA in the regulation of neuronal excitability and synaptic transmission via its modulation of the Na,K-ATPase.
Footnotes
This work was supported by a grant to I.B.L. from the National Institutes of Health. We thank Amy Moseley, Adam Redden, Jerry B. Lingrel, and Thomas Pressley for Na,K-ATPase α and β subunit cDNAs. We are grateful to Steve Scherer, Marc Dichter, Margie Maronski, Judy Grinspan, Phil Haydon, and Eric Burlingame for providing us with cultures of neurons and glia and to members of the Levitan laboratory for helpful comments and suggestions.
Correspondence should be addressed to Irwin B. Levitan, Department of Neuroscience, University of Pennsylvania School of Medicine, 3450 Hamilton Walk, Philadelphia, PA 19104-6074. E-mail: levitani@mail.med.upenn.edu.
S. M. Cibulsky's present address: Division of Cardiovascular Research, Children's Hospital, Boston, MA 02115.
C. Ding's present address: Room 207, 2/F, Centre for Infectious Diseases, Postgraduate Education Centre, School of Public Health, Prince of Wales Hospital, Shatin, New Territories, Hong Kong.
M. Holmqvist's present address: Synta Pharmaceuticals, 45 Hartwell Avenue, Lexington, MA 02421.
H. Mao's present address: Department of Radiation Oncology, University of North Carolina at Chapel Hill, Manning Drive 101, Chapel Hill, NC 27599.
DOI:10.1523/JNEUROSCI.0635-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/257934-10$15.00/0
H.M. and T.S.F. contributed equally to this work.
References
- Albensi BC, Janigro D (2003) Traumatic brain injury and its effects on synaptic plasticity. Brain Inj 17: 653–663. [DOI] [PubMed] [Google Scholar]
- Amedee T, Robert A, Coles JA (1997) Potassium homeostasis and glial energy metabolism. Glia 21: 46–55. [DOI] [PubMed] [Google Scholar]
- Awatramani R, Shumas S, Kamholz J, Scherer SS (2002) TGFbeta1 modulates the phenotype of Schwann cells at the transcriptional level. Mol Cell Neurosci 19: 307–319. [DOI] [PubMed] [Google Scholar]
- Cholet N, Pellerin L, Magistretti PJ, Hamel E (2002) Similar perisynaptic glial localization for the Na+,K+-ATPase alpha 2 subunit and the glutamate transporters GLAST and GLT-1 in the rat somatosensory cortex. Cereb Cortex 12: 515–525. [DOI] [PubMed] [Google Scholar]
- Crambert G, Geering K (2003) FXYD proteins: new tissue-specific regulators of the ubiquitous Na,K-ATPase. Sci STKE 2003: RE1. [DOI] [PubMed] [Google Scholar]
- Crambert G, Beguin P, Uldry M, Monnet-Tschudi F, Horisberger JD, Garty H, Geering K (2003) FXYD7, the first brain- and isoform-specific regulator of Na,K-ATPase: biosynthesis and function of its posttranslational modifications. Ann NY Acad Sci 986: 444–448. [DOI] [PubMed] [Google Scholar]
- Frohman MA (1993) Rapid amplification of complementary-DNA ends for generation of full-length complementary DNAs: thermal race. Methods Enzymol 218: 340–356. [DOI] [PubMed] [Google Scholar]
- Garty H, Lindzen M, Scanzano R, Aizman R, Fuzesi M, Goldshleger R, Farman N, Blostein R, Karlish SJ (2002) A functional interaction between CHIF and Na-K-ATPase: implication for regulation by FXYD proteins. Am J Physiol Renal Physiol 283: F607–F615. [DOI] [PubMed] [Google Scholar]
- Geering K, Beguin P, Garty H, Karlish S, Fuzesi M, Horisberger JD, Crambert G (2003) FXYD proteins: new tissue- and isoform-specific regulators of Na,K-ATPase. Ann NY Acad Sci 986: 388–394. [DOI] [PubMed] [Google Scholar]
- Hertz L (1978) An intense potassium uptake into astrocytes, its further enhancement by high concentrations of potassium, and its possible involvement in potassium homeostasis at the cellular level. Brain Res 145: 202–208. [DOI] [PubMed] [Google Scholar]
- Jorgensen PL (1988) Purification of Na+,K+-ATPase: enzyme sources, preparative problems, and preparation from mammalian kidney. Methods Enzymol 156: 29–43. [DOI] [PubMed] [Google Scholar]
- Kaplan JH (2002) Biochemistry of Na,K-ATPase. Annu Rev Biochem 71: 511–535. [DOI] [PubMed] [Google Scholar]
- Kendall G, Crankson H, Ensor E, Lublin DM, Latchman DS (1996) Activation of the gene encoding decay accelerating factor following nerve growth factor treatment of sensory neurons is mediated by promoter sequences within 206 bases of the transcriptional start site. J Neurosci Res 45: 96–103. [DOI] [PubMed] [Google Scholar]
- Levitan IB (1999) Modulation of ion channels by protein phosphorylation. How the brain works. Adv Second Messenger Phosphoprotein Res 33: 3–22. [DOI] [PubMed] [Google Scholar]
- Li N, Sul JY, Haydon PG (2003) A calcium-induced calcium influx factor, nitric oxide, modulates the refilling of calcium stores in astrocytes. J Neurosci 23: 10302–10310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda M, Hamano K, Hirano Y, Suzuki M, Takahashi E, Terada T, Futai M, Sato R (1998) Structures of P-type transporting ATPases and chromosomal locations of their genes. Cell Struct Funct 23: 315–323. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ, Pellerin L (1999) Cellular mechanisms of brain energy metabolism and their relevance to functional brain imaging. Philos Trans R Soc Lond B Biol Sci 354: 1155–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolskee RF, McHendry-Rinde B, Horn R (1993) Panning transfected cells for electrophysiological studies. Biotechniques 15: 906–911. [PubMed] [Google Scholar]
- Mayrand RR, Fullerton DS, Ahmed K (1982) A simple method for the purification of rat brain Na+,K+-adenosine triphosphatase (ATPase). J Pharmacol Methods 7: 279–288. [DOI] [PubMed] [Google Scholar]
- Munzer JS, Daly SE, Jewell-Motz EA, Lingrel JB, Blostein R (1994) Tissue- and isoform-specific kinetic behavior of the Na,K-ATPase. J Biol Chem 269: 16668–16676. [PubMed] [Google Scholar]
- Pawson T, Nash P (2003) Assembly of cell regulatory systems through protein interaction domains. Science 300: 445–452. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ (1997) Glutamate uptake stimulates Na+,K+-ATPase activity in astrocytes via activation of a distinct subunit highly sensitive to ouabain. J Neurochem 69: 2132–2137. [DOI] [PubMed] [Google Scholar]
- Peng L, MartinVasallo P, Sweadner KJ (1997) Isoforms of Na,K-ATPase α and β subunits in the rat cerebellum and in granule cell cultures. J Neurosci 17: 3488–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopperle WM, Holmqvist MH, Zhou Y, Wang J, Wang Z, Griffith LC, Keselman I, Kusinitz F, Dagan D, Levitan IB (1998) Slob, a novel protein that interacts with the slowpoke calcium-dependent potassium channel. Neuron 20: 565–573. [DOI] [PubMed] [Google Scholar]
- See J, Zhang X, Eraydin N, Mun SB, Mamontov P, Golden JA, Grinspan JB (2004) Oligodendrocyte maturation is inhibited by bone morphogenetic protein. Mol Cell Neurosci 26: 481–492. [DOI] [PubMed] [Google Scholar]
- Skou JC (1998) Nobel Lecture. The identification of the sodium pump. Biosci Rep 18: 155–169. [DOI] [PubMed] [Google Scholar]
- Smith TW, Antman EM, Friedman PL, Blatt CM, Marsh JD (1984) Digitalis glycosides: mechanisms and manifestations of toxicity. III. Prog Cardiovasc Dis 27: 21–56. [DOI] [PubMed] [Google Scholar]
- Sweadner KJ, Rael E (2000) The FXYD gene family of small ion transport regulators or channels: cDNA sequence, protein signature sequence, and expression. Genomics 68: 41–56. [DOI] [PubMed] [Google Scholar]
- Wen H, Levitan IB (2002) Calmodulin is an auxiliary subunit of KCNQ2/3 potassium channels. J Neurosci 22: 7991–8001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox KS, Dichter MA (1994) Paired pulse depression in cultured hippocampal neurons is due to a presynaptic mechanism independent of GABAB autoreceptor activation. J Neurosci 14: 1775–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart MJ, Taylor GS, Dixon JE (2001) Phoxy lipids: revealing PX domains as phosphoinositide binding modules. Cell 105: 817–820. [DOI] [PubMed] [Google Scholar]
- Zeng H, Fei H, Levitan IB (2004) The slowpoke channel binding protein Slob from Drosophila melanogaster exhibits regulatable protein kinase activity. Neurosci Lett 365: 33–38. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Schopperie WM, Murrey H, Jaramillo A, Dagan D, Griffith LC, Levitan IB (1999) A dynamically regulated 14–3-3,slob, and slowpoke potassium channel complex in Drosophila presynaptic nerve terminals. Neuron 22: 809–818. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Fei H, Levitan IB (2003) An interaction domain in Slob necessary for its binding to the slowpoke calcium-dependent potassium channel. Neuropharmacology 45: 714–719. [DOI] [PubMed] [Google Scholar]