

Abstract
It is widely accepted that the familial Parkinson's disease (PD)-linked gene product, parkin, functions as a ubiquitin ligase involved in protein turnover via the ubiquitin-proteasome system. Substrates ubiquitinated by parkin are hence thought to be destined for proteasomal degradation. Because we demonstrated previously that parkin interacts with and ubiquitinates synphilin-1, we initially expected synphilin-1 degradation to be enhanced in the presence of parkin. Contrary to our expectation, we found that synphilin-1 is normally ubiquitinated by parkin in a nonclassical, proteasomal-independent manner that involves lysine 63 (K63)-linked polyubiquitin chain formation. Parkin-mediated degradation of synphilin-1 occurs appreciably only at an unusually high parkin to synphilin-1 expression ratio or when primed for lysine 48 (K48)-linked ubiquitination. In addition we found that parkin-mediated ubiquitination of proteins within Lewy-body-like inclusions formed by the coexpression of synphilin-1, α-synuclein, and parkin occurs predominantly via K63 linkages and that the formation of these inclusions is enhanced by K63-linked ubiquitination. Our results suggest that parkin is a dual-function ubiquitin ligase and that K63-linked ubiquitination of synphilin-1 by parkin may be involved in the formation of Lewy body inclusions associated with PD.
Keywords: parkin, synphilin-1, ubiquitin, Parkinson's disease, Lewy body, dopamine
Introduction
Parkinson's disease (PD) is a prevalent neurodegenerative disorder that is characterized by the relatively selective loss of midbrain dopaminergic neurons and the presence of intracytoplasmic protein inclusions known as Lewy bodies (Siderowf and Stern, 2003). Although the etiology of PD remains poorly understood, several genetic factors are known to play a role in rare familial forms of PD (Dawson and Dawson, 2003; Greenamyre and Hastings, 2004). Of these, mutations in parkin are the most common cause of familial PD (Lucking et al., 2000). The recent association of parkin haploinsufficiency (West et al., 2002) and the S-nitrosylation of parkin in sporadic PD (Chung et al., 2004; Yao et al., 2004) also implicate a role for parkin in the more common form of PD.
We and others initially elucidated the function of parkin as a ubiquitin ligase and showed that pathological mutations of parkin impair its catalytic activity (Imai et al., 2000; Shimura et al., 2000; Zhang et al., 2000). We also speculated that a loss of parkin function would lead to the toxic accumulation of its substrates, eventually killing the cell (Dawson, 2000). Supporting this hypothesis are the observations that several substrates of parkin accumulate in brains of PD patients carrying parkin mutations (Imai et al., 2001; Shimura et al., 2001; Choi et al., 2003). Functional parkin may also be required for the formation of Lewy bodies because familial PD cases linked to parkin mutations are generally devoid of these signature inclusions (Chung et al., 2001a; Mata et al., 2004). Our identification of synphilin-1, an α-synuclein-interacting protein, as a parkin substrate, together with our demonstration that parkin mediates ubiquitination of proteins within Lewy-body-like inclusions formed by the coexpression of α-synuclein and synphilin-1, align with this speculation (Chung et al., 2001b). Furthermore, synphilin-1 is also a component of Lewy bodies in brains of sporadic PD patients (Wakabayashi et al., 2000), and a point mutation in synphilin-1 was found recently in a number of German patients with sporadic PD (Marx et al., 2003).
Synphilin-1 is a synaptic vesicle-enriched protein whose function is currently poorly understood. Although we have demonstrated previously that parkin interacts with, and ubiquitinates synphilin-1 (Chung et al., 2001b), recent studies indicate that synphilin-1 is also a substrate of two other really interesting new gene (RING) finger-containing ubiquitin E3 ligases, seven in absentia homologs (siah) and dorfin (Ito et al., 2003; Nagano et al., 2003; Liani et al., 2004). The functional similarity of parkin, siah, and dorfin suggests physiological redundancy and multiple pathways facilitating synphilin-1 degradation.
Here we show that the relative expression levels between parkin and synphilin-1 is an important determinant governing synphilin-1 degradation kinetics. Appreciable degradation of synphilin-1 by parkin occurs only at an unusually high parkin to synphilin-1 expression ratio or when the system is primed for lysine-48 (K48) ubiquitination. When synphilin-1 and parkin are coexpressed at equivalent levels, parkin ubiquitinates synphilin-1 via nonclassical K63-linked ubiquitin chains. Moreover, this proteasomal-independent mode of ubiquitination by parkin enhances the formation of Lewy-body-like inclusions formed by the coexpression of synphilin-1 and α-synuclein.
Materials and Methods
Plasmids, antibodies, and reagents. Plasmids expressing FLAG-tagged or myc-tagged parkin, myc-tagged synphilin-1, and myc-tagged siah-1 and siah-2 have been described previously (Zhang et al., 2000; Chung et al., 2001b; Liani et al., 2004). The plasmids pXJ40-hemagglutinin (HA)-Cbl and pXJ40-HA-human homology of Ariadne (HHART) were provided by Dr. G. R. Guy (Institute of Molecular and Cell Biology, Singapore); the plasmid containing HA-tagged E6-associated proteins (AP) was provided by Dr. P. Howley (Harvard Medical School, Boston, MA). The various forms of human ubiquitin cDNA, originally cloned into bacterial expression vectors, were generous gifts from Dr. C. Pickart (Johns Hopkins University, Baltimore, MD). These different ubiquitin cDNA species were amplified by PCR and subcloned into pRK5 vector in frame with an HA tag sequence to generate pRK5-HA-Ub wild type (WT), -Ub K48, and -Ub K48R, -Ub K33, -UbK63, and -Ub K0. Subsequent sequence analysis of these various ubiquitin constructs revealed no unintended mutations introduced by the PCR amplification. Monoclonal anti-myc and anti-HA were purchased from Roche Diagnostics (Welwyn Garden City, UK). Monoclonal anti-FLAG was purchased from Sigma (St. Louis, MO). Puromycin and clasto-lactacystin-β-lactone were purchased from Sigma and Affiniti Research (Exeter, UK), respectively.
Cell culture, immunoprecipitation, and Western blot analysis. Human embryonic kidney (HEK) 293 and SH-SY5Y cells were grown in DMEM containing 10% FBS in a 5% CO2 atmosphere. Cells were transiently transfected with various expression vectors using the LipofectAMINE PLUS reagent (Invitrogen, San Diego, CA) according to the manufacturer's instructions. Two days later, transfected HEK 293 cells were lysed in immunoprecipitation buffer containing 1% Triton X-100, 10 μg/ml aprotinin, and 1 mm PMSF in PBS. Immunoprecipitations from the transfected cell lysates were performed with anti-myc antibody and protein G PLUS/protein A-agarose (Oncogene Sciences, Uniondale, NY) and then washed six times in lysis buffer. Immunoprecipitates or total cell lysates were analyzed by Western blot analysis with ECL detection reagents (Amersham Biosciences, Arlington Heights, IL).
Immunocytochemistry. Transfected SH-SY5Y cells were fixed after 48 h with 4% paraformaldehyde for 30 min and permeabilized with 0.2% Triton X-100 for 15 min at room temperature. The fixed cells were first labeled with either monoclonal anti-myc or polyclonal anti-HA primary antibody (Upstate Biotechnology, Lake Placid, NY). After washing, the cells were labeled with cyanine 3-conjugated anti-mouse and FITC-conjugated anti-rabbit secondary antibodies (Jackson ImmunoResearch, West Grove, PA) before analysis by confocal microscopy (LSM 410; Zeiss, Oberkochen, Germany).
Pulse-chase experiments. [35S]methionine pulse-chase experiments were performed as described previously (Zhang et al., 2000). Briefly, HEK 293 cells were transfected with myc-tagged synphilin-1 in the absence or presence of FLAG-tagged parkin. Twenty-four hours after transfection, cells were washed and incubated with methionine-free medium for 1 h. The cells were pulsed-chased with 100 μCi of [35S]methionine and harvested at the indicated times for immunoprecipitation with anti-myc antibody. The immunoprecipitates were resolved on a 8% SDS-PAGE gel, visualized, and quantitated with a PhosphorImager (Molecular Dynamics, Sunnyvale, CA). Pulse-chase analysis using the protein synthesis inhibitor puromycin or cycloheximide was performed as described by Ren et al. (2003) and Moore et al. (2003), respectively.
Results
Synphilin-1 degradation is not enhanced in the presence of parkin
Because parkin interacts with and polyubiquitinates synphilin-1 (Chung et al., 2001b), we initially expected an enhancement in synphilin-1 degradation in the presence of parkin. Accordingly, we performed pulse-chase experiments to analyze synphilin-1 turnover in the absence or presence of parkin. After a 6 h chase, ∼50% of de novo synthesized synphilin-1 remains in control cells (Fig. 1A). Surprisingly, in the presence of parkin, synphilin-1 degradation is not altered significantly (Fig. 1A). We obtained similar results when we repeated the experiments with an 18 h chase (data not shown). To confirm our pulse-chase experimental findings, we examined the turnover rate of synphilin-1 in the absence or presence of parkin using an alternate method as described previously by Ren et al. (2003). We expressed synphilin-1 in HEK 293 cells with or without parkin and treated the cells with the protein synthesis inhibitor puromycin (100 μm) for various durations. Without synthesis of new proteins, the levels of synphilin-1 in these cells should reflect their rates of degradation. Indeed, the level of synphilin-1 decreases with time, and its reduction is especially prominent at 24 h after puromycin treatment (Fig. 1B), an observation that is consistent with the report that synphilin-1 degradation occurs in part via the ubiquitin-proteasome pathway (Lee et al., 2002). The level of parkin is similarly affected by the puromycin treatment; however, we did not observe a significant further reduction in the steady-state levels of synphilin-1 at 24 h after puromycin treatment when it was coexpressed with parkin (Fig. 1B). This observation supports our pulse-chase experimental findings. Consistent with our previous report (Chung et al., 2001b), we also ascertained that synphilin-1 is polyubiquitinated by parkin under these conditions (Fig. 1C). Together, our results suggest that parkin-mediated ubiquitination of synphilin-1 does not enhance the degradation rate of synphilin-1.
Figure 1.

Effect of parkin on synphilin-1 turnover. A, Pulsechase analysis of the turnover rate of myc-tagged synphilin-1 in HEK 293 cells in the absence or presence of FLAG-tagged parkin. Data from three independent experiments are expressed as means ± SEM. B, Top panel, Anti-myc immunoblot of equal amount of total cell lysates prepared from HEK 293 cells transfected with myc-tagged synphilin-1 and treated with puromycin (100 μm) for different durations as indicated. Middle panel, Anti-myc and anti-FLAG immunoblots of equal amount of total cell lysates prepared from HEK 293 cells transfected with myc-tagged synphilin-1 without or with an equivalent amount of FLAG-tagged parkin and treated with puromycin (100 μm) for 24 h. The anti-myc immunoblots from three independent experiments were used to derive the relative densitometric units of synphilin-1, which is presented as a histogram in the bottom panel. Closed and opened bars represent units of synphilin-1 in the absence and presence of parkin respectively (*p > 0.1; **p < 0.05; Student's t test). C, Anti-HA and anti-myc immunoblots of synphilin immunoprecipitates (IPmyc) prepared from HEK 293 cells transfected with myc-tagged synphilin-1 in the presence of HA-tagged wild-type ubiquitin without or with FLAG-tagged parkin. Lysates prepared from these variously transfected cells were also subjected to anti-myc or anti-FLAG immunoblotting to show their expression (INPUT). These experiments were replicated at least three times.
Relative expression levels between parkin and synphilin-1 is an important determinant governing synphilin-1 degradation
Because parkin did not enhance synphilin-1 degradation, we reasoned that the stoichiometric ratio between parkin and synphilin-1 expression may be an important determinant governing synphilin-1 degradation. Alternatively, parkin-mediated ubiquitination of synphilin-1 under these conditions may be proteasomal independent. To ascertain whether a dose-dependent increase in parkin expression relative to synphilin-1 influences synphilin-1 degradation, we cotransfected myc-tagged synphilin-1 with an increasing amount of FLAG-tagged parkin in HEK 293 cells. At a parkin/synphilin-1 contransfection ratio of 1:1, the steady-state level of synphilin-1 compared with its steady-state level in the absence of parkin remained unaffected, an observation that is in agreement with our pulse-chase experiments; however, starting from a parkin/synphilin-1 cotransfection ratio of 2:1, we observed a progressive decrease in synphilin-1 steady-state levels that corresponded to the level of coexpressed parkin (Fig. 2A). The reduction in synphilin-1 steady-state levels is most obvious at the highest dose of coexpressed parkin, which represents a parkin/synphilin-1 cotransfection ratio of 4:1 (Fig. 2A). To rule out the possibility that the decrease in synphilin-1 at the elevated dose of cotransfected parkin is a nonspecific effect caused by parkin overexpression, we repeated the above experiment with myc-tagged α-synuclein in place of myc-tagged synphilin-1. Under the same conditions, the steady-state levels of α-synuclein remained essentially unchanged with an increasing dose of cotransfected parkin (Fig. 2A), an observation that is consistent with our previous description of the inability of parkin to interact with or ubiquitinate unmodified α-synuclein (Chung et al., 2001b). Thus, synphilin-1 steady-state levels are dependent on the ratio of parkin to synphilin-1.
Figure 2.

Dose-dependent effect of parkin on synphilin-1 steady-state levels. A, Anti-myc and anti-FLAG immunoblots of total cell lysates prepared from HEK 293 cells transfected with myc-tagged synphilin-1 (left panels) or myc-tagged α-synuclein (right panels) in the absence or presence of increasing relative amounts of FLAG-tagged parkin, as indicated. B, Anti-myc, anti-FLAG, and anti-HA immunoblots of total cell lysates prepared from HEK 293 cells transfected with myc-tagged synphilin-1 alone or with various FLAG-tagged or HA-tagged E3 ligases (as indicated) at an E3 ligase/synphilin-1 cDNA cotransfection ratio of 4:1. Equal loading of the different cell lysates was verified by anti-β-actin immunoblotting. These experiments were replicated at least three times.
To examine the specificity of parkin-mediated effects on the steady-state level of synphilin-1, we cotransfected synphilin-1 with other ubiquitin ligases in HEK 293 cells at an E3 ligase/synphilin-1 ratio of 4:1. At this transfection ratio, the expressed parkin reproducibly leads to a decline in the steady-state level of the coexpressed synphilin-1. In contrast, the RING finger-containing E3 ligase, Cbl, and the homologous to E6-AP C-terminus domain-containing E3 ligase, E6-AP, have no observable effects on the steady-state level of synphilin-1 (Fig. 2B). Interestingly, HHARI, an E3 ligase related to parkin (Ardley et al., 2001), reduces synphilin-1 levels, similar to parkin (Fig. 2B). Because dorfin, siah-1, and siah-2 also target synphilin-1 for ubiquitination and subsequent degradation (Ito et al., 2003; Nagano et al., 2003; Liani et al., 2004), it is not surprising that synphilin-1 levels may be regulated by E3 ligases other than parkin. Together, these results suggest that the stoichiometric ratio between parkin and synphilin-1 expression may affect the degradation kinetics of synphilin-1.
Degradation of synphilin-1 mediated by a high level of coexpressed parkin is dependent on the catalytic competencies of parkin and the proteasome
To further explore the observation that the relative expression between parkin and synphilin-1 governs the synphilin-1 turnover rate, we examined synphilin-1 degradation at different levels of parkin coexpression by means of the puromycin chase as described for Figure 1B. At a parkin/synphilin-1 cotransfection ratio of 1:1, we did not observe any parkin-mediated effects on the synphilin-1 turnover rate at 24 h after puromycin treatment; however, at a parkin/synphilin-1 cotransfection ratio of 4:1, parkin dramatically accelerated the degradation of synphilin-1 (Fig. 3A). We obtained similar results when we substituted puromycin for another protein synthesis inhibitor, cycloheximide (data not shown). Furthermore, the robust turnover rate of synphilin-1 mediated by parkin is clearly dependent on parkin catalytic competency, because parkin mutants containing the familial PD-associated mutations T240R and T415N, which have been shown previously to compromise its enzymatic activity (Shimura et al., 2000; Zhang et al., 2000), fail to accelerate the degradation of synphilin-1 under similar experimental conditions (Fig. 3B). To determine whether synphilin-1 is degraded by the proteasome in the presence of high levels of coexpressed parkin, we repeated the experiment with the addition of the potent proteasomal inhibitor lactacystin. The treatment of cells with 5 μm lactacystin effectively prevents parkin-mediated degradation of synphilin-1 via the proteasome (Fig. 3C). Together, our results suggest that synphilin-1 degradation by parkin requires a high parkin to synphilin-1 ratio and it is dependent on proteasomal function.
Figure 3.
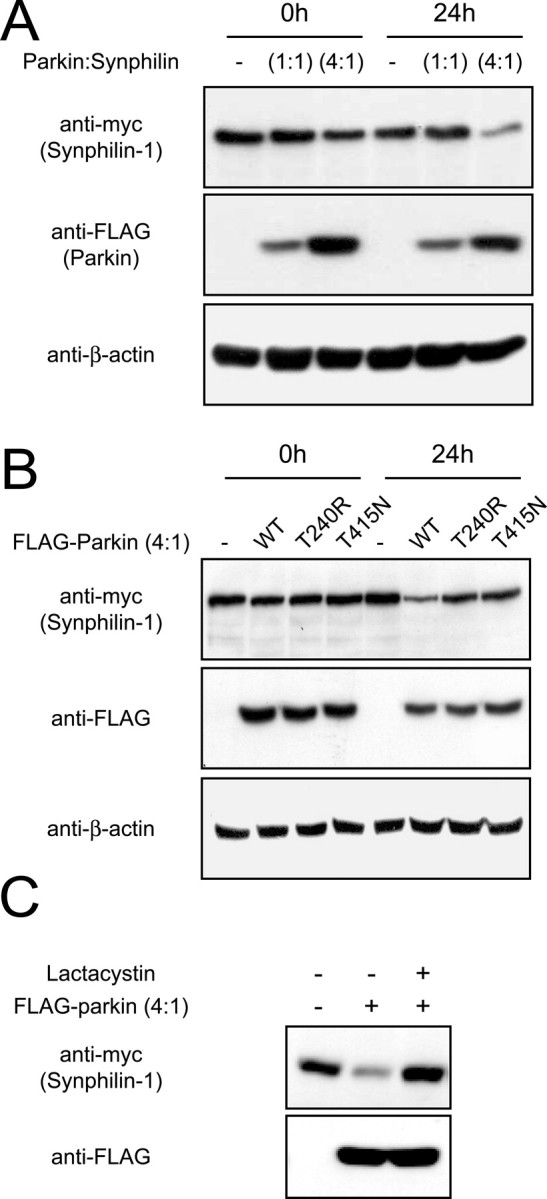
Degradation of synphilin-1 by parkin is dependent on their relative expression levels and on parkin catalytic competency. A, Anti-myc and anti-FLAG immunoblots of total cell lysates prepared from HEK 293 cells transfected with myc-tagged synphilin-1 without or with FLAG-tagged parkin at different cotransfection ratios (as indicated) and treated 24 h later with puromycin (100 μm) for 24 h. B, Anti-myc and anti-FLAG immunoblots of total cell lysates prepared from HEK 293 cells transfected with myc-tagged synphilin-1 without or with FLAG-tagged, wild-type parkin, or familial-PD parkin mutants, at a parkin/synphilin-1 cDNA cotransfection ratio of 4:1 and treated with puromycin (100 μm) for 24 h. In both cases, equal loading of the different cell lysates was verified by anti-β-actin immunoblotting. C, HEK 293 cells transfected with the indicated combinations of myc-tagged synphilin-1 and FLAG-tagged parkin were left untreated or treated with 5 μm clasto-lactacytstin β-lactone (lactacystin) for 16 h. Lysates prepared from these cells were immunoblotted with anti-myc or anti-FLAG as indicated. These experiments were duplicated with similar results.
Parkin is an inefficient “degradation-associated” E3 ligase for synphilin-1
Because the degradation of synphilin-1 is promoted by parkin only when the latter is coexpressed at high levels, our results suggest that parkin may not be an efficient degradation-associated ubiquitin ligase for synphilin-1. Indeed, when we compared the respective efficiencies of parkin, siah-1, and siah-2 in promoting the degradation of synphilin-1, it was apparent that both siah-1 and siah-2 enhance synphilin-1 degradation much more efficiently (Fig. 4A). As described previously (Nagano et al., 2003; Liani et al., 2004), at a cotransfection ratio of 1:1, we could barely detect synphilin-1 when it was coexpressed with siah-1 or siah-2 (Fig. 4A).
Figure 4.
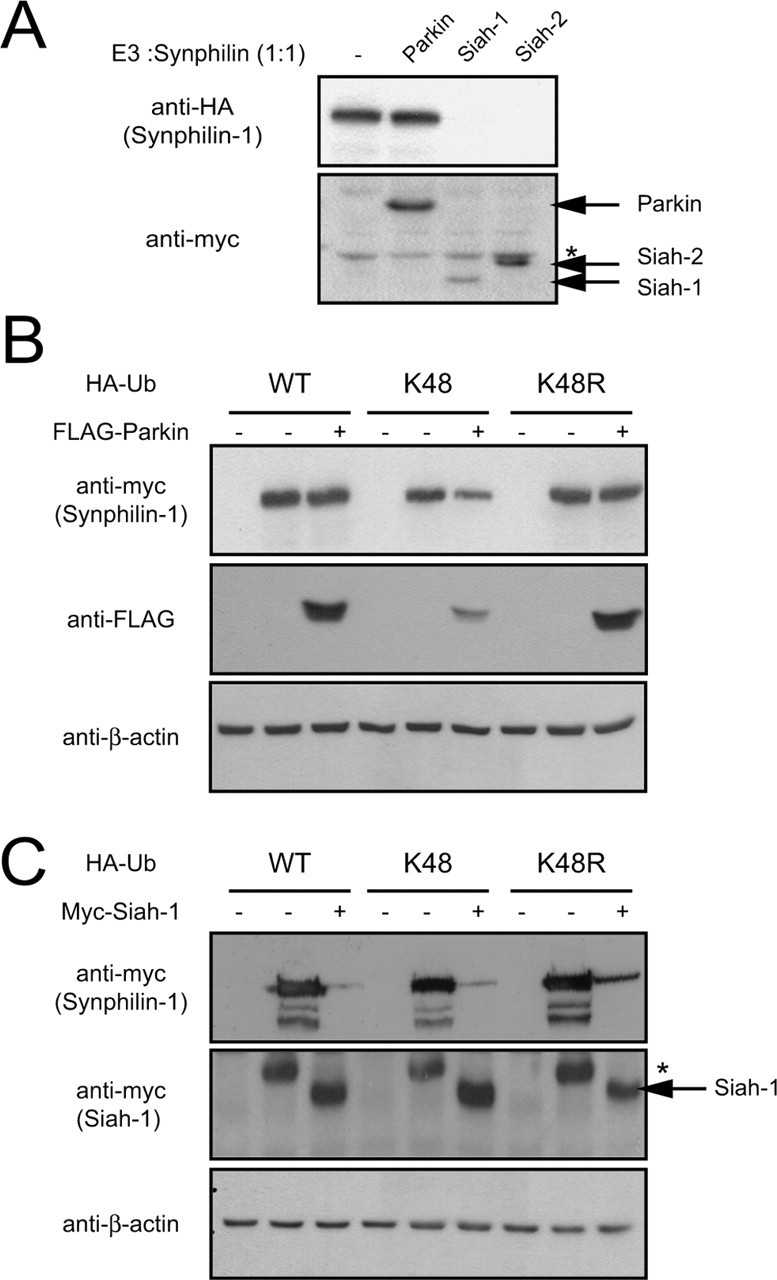
Parkin is not an efficient degradation-associated E3 ligase. A, Anti-HA and anti-myc immunoblots of total cell lysates prepared from HEK 293 cells transfected with HA-tagged synphilin-1 alone or with myc-tagged parkin, siah-1, or siah-2 at 1:1 cotransfection ratio. Asterisk indicates nonspecific band. B, Anti-myc and anti-FLAG immunoblots of total cell lysates prepared from HEK 293 cells cotransfected with the indicated combinations of myc-tagged synphilin-1, FLAG-tagged parkin, and various HA-tagged ubiquitin species. C, Anti-myc immunoblots of total cell lysates prepared from HEK 293 cells cotransfected with the indicated combinations of myc-tagged synphilin-1, myc-tagged siah-1, and various HA-tagged ubiquitin species. Asterisk indicates nonspecific band. For both B and C, equal loading of the different cell lysates was verified by anti-β-actin immunoblotting. These experiments were duplicated with similar results.
We reasoned that the poor enhancement of synphilin-1 degradation in the presence of parkin may be related to the nature of synphilin-1 ubiquitination mediated by parkin under normal conditions. A critical determinant of whether a polyubiquitinated protein undergoes proteolysis is the nature of the ubiquitin chain linkage (Chau et al., 1989; Pickart, 2001). The ubiquitin molecule contains a total of seven lysine residues at positions 6, 11, 27, 29, 33, 48 (K48), and 63. Potentially, these lysine residues could mediate ubiquitin chain elongation. Most of the cellular proteins undergo proteasomal-dependent ubiquitination, which is usually signaled by ubiquitin chains linked through isopeptide bonds between the terminal glycine (G76) and the lysine at position 48.
To test whether parkin could mediate proteasomal-dependent degradation of synphilin-1 under conditions that promote G76-K48 ubiquitin chain formation, we cotransfected HEK 293 cells with myc-tagged synphilin-1 and FLAG-tagged parkin at a cotransfection ratio of 1:1 in the presence of a HA-tagged ubiquitin mutant, designated K48, that would enhance the G76-K48-linked polyubiquitination of proteins. The K48 ubiquitin mutant contains arginine substitutions on all of its lysine residues except the one at position 48. We also included HA-tagged wild-type ubiquitin and K48R ubiquitin mutant as controls. The K48R mutant ubiquitin, containing a single lysine to arginine mutation at position 48, is expected to disrupt G76-K48 ubiquitin chain assembly.
Anti-myc immunoblotting of the transfected cell lysates reveals that in the absence of parkin, the steady-state level of synphilin-1 remains essentially unchanged when it is coexpressed with wild-type ubiquitin or K48R ubiquitin but decreases moderately when it is coexpressed with the K48 ubiquitin mutant (Fig. 4B). When parkin was introduced into the experiment at the parkin/synphilin-1 cotransfection ratio of 1:1, we did not observe any significant difference in the steady-state level of synphilin-1 in the presence of wild-type ubiquitin with or without exogenously introduced parkin (Fig. 4B). This is consistent with our previous results described in this study. In contrast, we observe a significant reduction in the steady-state level of synphilin-1 in the presence of parkin when wild-type ubiquitin was replaced with K48 ubiquitin, an effect that was abolished completely when the exogenously introduced K48 ubiquitin was substituted with K48R ubiquitin (Fig. 4B). It is interesting to note that the effects of parkin, or the lack of it, on the steady-state level of synphilin-1 in the presence of wild-type ubiquitin or the K48R mutant mirror each other. This similarity suggests that parkin-mediated ubiquitination of synphilin-1 takes place predominantly via non-K48-linked ubiquitin chains. In support of this speculation, we found an opposite picture when we repeated the experiment with siah-1 in place of parkin. Siah-1, which robustly mediates degradation of synphilin-1, expectedly does so in the presence of either wild-type ubiquitin or K48 ubiquitin, but the degradation of synphilin-1 mediated by siah-1 is compromised in the presence of the K48R mutant (Fig. 4C). Together, our results suggest that parkin ubiquitination of synphilin-1 proceeds predominantly via non-K48-linked chains and that parkin is capable of mediating proteasomal-independent ubiquitination.
Parkin-mediated autoubiquitination and synphilin-1 ubiquitination occur via K63-linked chains
Although ubiquitin chain assembly typically involves K48-G76 linkage, it is now clear that polyubiquitination could also occur through alternative lysine residues on ubiquitin. Several groups have shown that K63-linked, proteasomal-independent polyubiquitination takes place in eukaryotic cells (Arnason and Ellison, 1994; Spence et al., 1995; Galan and Haguenauer-Tsapis, 1997; Fisk and Yaffe, 1999; Wang et al., 2001). To investigate whether parkin could mediate non-G76-K48-linked but possibly G76-K63-linked polyubiquitination, we examined parkin self-ubiquitination properties in HEK 293 cells in the presence of wild-type ubiquitin, ubiquitin K48R, or ubiquitin K63 mutants. Parkin self-ubiquitination has been described in various studies and is a straightforward assay to analyze parkin catalytic activity. Anti-HA immunoblotting of parkin immunoprecipitated from lysates containing parkin coexpressed with wild-type ubiquitin reveals a ladder of anti-HA immunoreactivity that is consistent with parkin polyubiquitination (Fig. 5A). Similarly, we observed robust anti-HA immunoreactivity of parkin immunoprecipitated from lysates containing parkin coexpressed with either the K48R or the K63 ubiquitin mutants. In all three cases, as revealed by anti-FLAG immunoblotting, the amount of high molecular parkin species representing ubiquitinated forms of parkin also appears more prominent relative to control parkin immunoprecipitates. Our results thus suggest that parkin may promote its self-ubiquitination via K63-linked chains.
Figure 5.
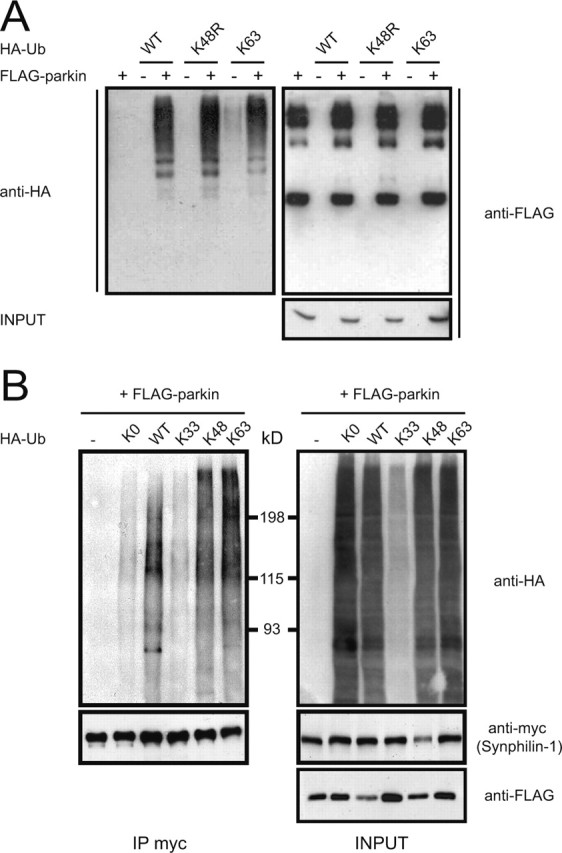
Parkin mediates K63-linked polyubiquitination of synphilin-1. A, A portion of lysates prepared from HEK 293 cells transfected with FLAG-tagged parkin alone or with various HA-tagged ubiquitin species were subjected to anti-FLAG immunoprecipitation followed by anti-HA immunoblotting (left panel). The blot was stripped and reprobed with anti-FLAG to show the amounts of immunoprecipitated parkin (top right panel). Lysates prepared from these variously transfected cells (INPUT) were also subjected to anti-FLAG immunoblotting to show FLAG-parkin expression levels (bottom right panel). B, A portion of lysates prepared from HEK 293 cells transfected with the indicated combinations of myc-tagged synphilin-1, FLAG-tagged parkin, and various HA-tagged ubiquitin species were subjected to anti-myc immunoprecipitation followed by anti-HA immunoblotting (top left panel). The blot was stripped and reprobed with anti-myc (bottom left panel) to illustrate that relatively equal amounts of synphilin were immunoprecipitated. The remaining portion of the transfected cell lysates was immunoblotted with anti-HA (top right panel) and anti-myc (bottom right panel) to show their respective expression levels. These experiments were replicated at least three times.
Next we sought to determine whether parkin uses K63-linked ubiquitin chain in the ubiquitination of synphilin-1. We cotransfected HEK 293 cells with FLAG-tagged parkin, myc-tagged synphilin-1, and different forms of HA-tagged ubiquitin mutants carrying a single lysine residue at positions 33 (K33), 48 (K48), or 63 (K63), or carrying zero lysines (K0) (Fig. 5B). Two days later, we immunoprecipitated the transfected cells with an antibody against myc and probed the immunoprecipitates with an antibody against HA to monitor ubiquitination. Immunoprecipitated myc-synphilin-1 shows significant anti-HA immunoreactivity in lanes containing WT-, K48-, and K63-ubiquitin consistent with parkin-mediated ubiquitination (Fig. 5B). Similar to our results shown in Figure 1C, a ladder of anti-HA immunoreactivity is observed in all three cases indicating polyubiquitination of synphilin-1 (Fig. 5B). The robust anti-HA immunoreactivity associated with K63-linked polyubiquitination of synphilin-1 indicates that parkin can mediate this form of ubiquitin chain formation on synphilin-1 (Fig. 5B); however, despite being robustly ubiquitinated by parkin, the level of synphilin-1 in the presence of K63 ubiquitin mutant remains unaffected relative to those in control transfections (Fig. 5B). These results are consistent with several reported observations that K63-linked polyubiquitination of proteins does not affect their degradation kinetics (Arnason and Ellison, 1994; Spence et al., 1995; Galan and Haguenauer-Tsapis, 1997; Fisk and Yaffe, 1999). In contrast, negligible anti-HA immunoreactivity is seen in synphilin-1 immunoprecipitates in lanes containing K0, K33, or no ubiquitin (Fig. 5B). These result are not surprising because K0 is a lysine-less ubiquitin mutant that is presumably incapable of polyubiquitin chain formation; thus far, there are no reports implicating K33 in polyubiquitination. The anti-myc immunoblot of the whole-cell extracts that we observed here mirrors our previous observations in Figure 4 in that reduction of synphilin-1 levels by parkin is observed only in the presence of K48 ubiquitin. Together, our results suggest that K63-linked ubiquitination of synphilin-1 by parkin may be the predominant form of polyubiquitinated chain assembly. This phenomenon provides an explanation for our inability to observe a significant alteration in synphilin-1 levels when it is coexpressed with parkin alone or in the presence of ectopically expressed wild-type ubiquitin.
Parkin-mediated ubiquitination of proteins within cytoplasmic inclusions formed by coexpression of α-synuclein and synphilin-1 occurs via ubiquitin K63 linkages
Because K63-linked ubiquitination of proteins is not associated with proteasomal degradation, it is conceivable that K63-linked ubiquitination of proteins could promote the half-life of the proteins and lead to their accumulation and subsequent aggregation within the cell. In this light, parkin-mediated K63-linked ubiquitination of proteins may be a mechanism by which Lewy bodies are formed within the cell. We showed previously that coexpression of α-synuclein, synphilin-1, and parkin in cultured cells results in the formation of Lewy-body-like inclusions and that parkin is involved in the ubiquitination of these inclusions (Chung et al., 2001b). To ascertain whether the ubiquitin-positive Lewy-body-like inclusions consist of K48- or K63-linked ubiquitin, we examined the role of K48 versus K63 ubiquitination on the inclusions in SH-SY5Y neuroblastoma cells by cotransfecting α-synuclein, myc-tagged synphilin-1, and FLAG-tagged parkin in the presence of either HA-tagged K48 or K63 ubiquitin. Two days later, we visualized the transfected cells using immunocytochemical methods with anti-myc and anti-HA. In both transfections, immunofluorescence of the cells with anti-myc revealed the formation of cytosolic inclusions (Fig. 6A). With anti-HA immunofluorescence, however, we observed that most of the anti-HA-positive inclusions occur in cells expressing HA-tagged K63 ubiquitin. These anti-HA-positive inclusions tend to colocalize with the anti-myc-positive inclusions, indicating that most of the K63-linked ubiquitinated proteins are contained within the inclusions (Fig. 6A). Indeed, quantitative analyses performed on these inclusions showed a threefold increase in the number of K63-linked ubiquitinated inclusions over that of K48-linked ubiquitinated inclusions (Fig. 6C). Together, these results suggest that parkin-mediated ubiquitination of proteins contained within the Lewy-body-like inclusions occurs predominantly via K63 of ubiquitin. In addition, we observed that the number of protein inclusions formed in the presence of K63 ubiquitin more than doubled those formed in the presence of K48 ubiquitin (Fig. 6C), indicating that the formation of α-synuclein/synphilin-1 inclusions is enhanced under conditions that promote cellular K63-linked ubiquitination. Our results therefore suggest that K63-linked ubiquitination of synphilin-1 by parkin may be involved in Lewy body pathology associated with PD.
Figure 6.

Parkin mediates predominantly K-63-linked ubiquitination of cytosolic inclusions formed by coexpression of synphilin-1 and α-synuclein. A, Coexpression of α-synuclein, myc-tagged synphilin-1, FLAG-tagged parkin, and either HA-tagged K48 ubiquitin or K63 ubiquitin results in the formation of myc-synphilin-1-positive inclusions (red). Positive anti-HA staining of the inclusion bodies is observed only in transfections containing HA-tagged K63 ubiquitin (green). The merged pictures show colocalization of the anti-myc and anti-HA staining (yellow). B, Coexpression of mutant huntingtin and either HA-tagged K-48 ubiquitin or HA-tagged K-63 ubiquitin results in the formation of huntingtin-positive inclusions (red). Positive anti-HA staining of the inclusion bodies is observed only in transfections containing HA-tagged K-48 ubiquitin (green). The merged pictures show colocalization of the anti-huntingtin and anti-HA staining (yellow). C, Histograms showing quantitative data of inclusion bodies in SH-SY5Y cells cotransfected with the various construct as described in A and B. These experiments were replicated at least three times.
To investigate whether K63-linked ubiquitination of proteins found within cytoplasmic inclusions formed by α-synuclein and synphilin-1 coexpression is a property unique to parkin activity, we examined the ubiquitination profile of huntingtin-mediated inclusions in the presence of either K48 or K63 ubiquitin. Ectopic expression of a mutant huntingtin containing 99 polyglutamine repeats generates protein inclusions equally well in cells coexpressing either HA-tagged K48 or K63 ubiquitin (Fig. 6B). The number of huntingtin-positive inclusions formed in each case is comparable with that generated by the coexpression of α-synuclein and synphilin-1 in the presence of K63 ubiquitin (Fig. 6C); however, unlike parkin-mediated ubiquitination of α-synuclein/synphilin-1-mediated inclusions, ubiquitination of proteins contained within the huntingtin-positive inclusions occurs mainly via K48 of ubiquitin (Fig. 6C). Additionally, although the percentage ratios between K48-ubiquitinated huntingtin-positive inclusions/total inclusions and K48-ubiquitinated synphilin-1-positive/total inclusions compare well with each other, the percentage ratio of K63-ubiquitinated huntingtin-positive inclusions/total inclusions is negligible compared with that of K63-ubiquitinated synphilin-1-positive inclusions/total inclusions (Fig. 6C). Presumably, the differences in the ubiquitination profiles relate to distinct mechanisms of protein inclusion formation in the two experimental paradigms. Nonetheless, these results suggest that K63-linked ubiquitination of cytoplasmic inclusions formed by coexpression of α-synuclein and synphilin-1 is a unique property of parkin-mediated ubiquitination.
Discussion
The major finding of this report is that parkin mediates both classical K48-linked ubiquitination and nonclassical K63-linked ubiquitination. K48-linked ubiquitination of synphilin-1 leads to its proteasomal degradation, whereas K63-linked ubiquitination of synphilin-1 fails to enhance its proteasomal degradation. Moreover, parkin-mediated K63-linked ubiquitination of synphilin-1 promotes the formation of Lewy-body-like inclusions when parkin, α-synuclein, and synphilin-1 are coexpressed.
The covalent attachment of ubiquitin to proteins via K48-linked ubiquitin chains and their subsequent degradation by the 26S proteasome represents the most commonly ascribed role for the protein ubiquitination system (Pickart, 2001). We showed that the relative expression level between parkin and its substrate synphilin-1 is an important determinant governing synphilin-1 degradation. We observed appreciable degradation of synphilin-1 by parkin only at an unusually high parkin to synphilin-1 expression ratio or when the system was primed for K48-linked ubiquitination. In contrast, synphilin-1 appeared to be predominantly ubiquitinated via K63-linked ubiquitin chains by parkin when they were coexpressed at equivalent levels in cells. Thus, parkin-mediated ubiquitination of synphilin-1, which appears to proceed predominantly via non K63-ubiquitin chains, is a likely explanation for the failure to observe appreciable degradation of synphilin-1 by parkin under normal experimental conditions. Consistent with the notion that parkin ubiquitinates synphilin-1 via non-K48-linked ubiquitin chains is the lack of differences in the steady-state levels of synphilin-1 in parkindeficient mice compared with their wild-type littermates (Goldberg et al., 2003; Von Coelln et al., 2004).
Our results suggest that parkin may act as a dual-function ubiquitin protein ligase capable of mediating both proteasomal-linked and proteasomal-independent ubiquitination under different cellular conditions. In this regard, the dual catalytic function of parkin is reminiscent of recent findings with ubiquitin C-terminal hydrolase-L1 (UCHL1), a ubiquitin hydrolase that appears to also possess a second ubiquitin ligase activity (Liu et al., 2002). Similar to the two activities of parkin reported here, the dual catalytic activities of UCHL1 apparently oppose each other. When UCHL1 functions as a ligase it promotes K63-linked ubiquitination of α-synuclein, which results in α-synuclein accumulation within the cell (Liu et al., 2002). The association of K63-linked ubiquitination activity with two familial PD-linked gene products indicates an important functional relationship between this unique mode of ubiquitin tagging and PD pathogenesis, especially in light of our current findings that parkin-mediated K63-linked ubiquitination enhances Lewy-body-like protein inclusion formation. Moreover, these results are consistent with the suggestion that functional parkin is required for Lewy body formation and the findings that PD patients with homologous mutations in parkin lack the Lewy body hallmark (Takahashi et al., 1994; Shimura et al., 1999). Although more work undoubtedly needs to be done to clarify the relevance of parkin-mediated K63 ubiquitination to PD pathology, it is tempting for us to speculate that K63 ubiquitination may represent a general mechanism that underlies protein inclusion formation associated with neurodegenerative disorders other than PD. Although our data on the ubiquitination of huntingtin-mediated inclusions would suggest otherwise, we cannot yet rule out potential changes in their ubiquitination profile in the presence of a huntingtin-specific ubiquitin ligase. Mechanistically, the involvement of a “nondegradative” mode of ubiquitination in protein inclusion formation is an attractive explanation of how proteins within inclusion bodies escape proteasomal degradation despite being heavily ubiquitinated.
In summary, our results suggest that parkin could ubiquitinate proteins via two distinct ubiquitin chain topologies. K48-linked ubiquitination leads to proteasomal-dependent degradation, whereas K63-linked ubiquitination is uncoupled from the proteasome. Furthermore, the proteasomal-independent K63-linked ubiquitination activity of parkin may influence the dynamics of Lewy body formation in PD. We believe that the novel parkin function uncovered here will help shape our understanding of the role of parkin-mediated ubiquitination in normal and abnormal CNS biology.
Footnotes
This work was supported by grants from the National Institutes of Health-National Institute of Neurological Disorders and Stroke (NIH-NINDS) Morris K. Udall Parkinson's Disease Research Center (NS38377), NS48206, The NIH-NINDS Huntington's Disease Center (NS16375), the Edward D. and Anna Mitchell Family Foundation, the National Medical Research Council (0776/2003), Singapore (L.K.L.), and AstraZeneca (London, UK) (Y.T.). T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases.
Correspondence should be addressed to Dr. Ted M. Dawson, Institute for Cell Engineering, Departments of Neurology and Neuroscience, Johns Hopkins University School of Medicine, 733 Broadway, Suite 731, Baltimore, MD 21205. E-mail: tdawson@jhmi.edu.
Y. Tanaka's present address: Okayama University Graduate School of Medicine and Dentistry, 2-5-1 Shikata-cho, Okayama City, Okayama 700-8558, Japan.
Copyright © 2005 Society for Neuroscience 0270-6474/05/252002-08$15.00/0
References
- Ardley HC, Tan NG, Rose SA, Markham AF, Robinson PA (2001) Features of the parkin/ariadne-like ubiquitin ligase, HHARI, that regulate its interaction with the ubiquitin-conjugating enzyme, Ubch7. J Biol Chem 276: 19640-19647. [DOI] [PubMed] [Google Scholar]
- Arnason T, Ellison MJ (1994) Stress resistance in Saccharomyces cerevisiae is strongly correlated with assembly of a novel type of multiubiquitin chain. Mol Cell Biol 14: 7876-7883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, Varshavsky A (1989) A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science 243: 1576-1583. [DOI] [PubMed] [Google Scholar]
- Choi P, Snyder H, Petrucelli L, Theisler C, Chong M, Zhang Y, Lim K, Chung KK, Kehoe K, D'Adamio L, Lee JM, Cochran E, Bowser R, Dawson TM, Wolozin B (2003) SEPT5_v2 is a parkin-binding protein. Brain Res Mol Brain Res 117: 179-189. [DOI] [PubMed] [Google Scholar]
- Chung KK, Dawson VL, Dawson TM (2001a) The role of the ubiquitin-proteasomal pathway in Parkinson's disease and other neurodegenerative disorders. Trends Neurosci 24: S7-14. [DOI] [PubMed] [Google Scholar]
- Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL, Dawson TM (2001b) Parkin ubiquitinates the alpha-synucleininteracting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat Med 7: 1144-1150. [DOI] [PubMed] [Google Scholar]
- Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM (2004) S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science 304: 1328-1331. [DOI] [PubMed] [Google Scholar]
- Dawson TM (2000) New animal models for Parkinson's disease. Cell 101: 115-118. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL (2003) Molecular pathways of neurodegeneration in Parkinson's disease. Science 302: 819-822. [DOI] [PubMed] [Google Scholar]
- Fisk HA, Yaffe MP (1999) A role for ubiquitination in mitochondrial inheritance in Saccharomyces cerevisiae J Cell Biol 145: 1199-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan JM, Haguenauer-Tsapis R (1997) Ubiquitin lys63 is involved in ubiquitination of a yeast plasma membrane protein. EMBO J 16: 5847-5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J (2003) Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem 278: 43628-43635. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, Hastings TG (2004) Parkinson's-divergent causes, convergent mechanisms. Science 304: 1120-1122. [DOI] [PubMed] [Google Scholar]
- Imai Y, Soda M, Takahashi R (2000) Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J Biol Chem 275: 35661-35664. [DOI] [PubMed] [Google Scholar]
- Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R (2001) An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell 105: 891-902. [DOI] [PubMed] [Google Scholar]
- Ito T, Niwa J, Hishikawa N, Ishigaki S, Doyu M, Sobue G (2003) Dorfin localizes to Lewy bodies and ubiquitylates synphilin-1. J Biol Chem 278: 29106-29114. [DOI] [PubMed] [Google Scholar]
- Lee G, Junn E, Tanaka M, Kim YM, Mouradian MM (2002) Synphilin-1 degradation by the ubiquitin-proteasome pathway and effects on cell survival. J Neurochem 83: 346-352. [DOI] [PubMed] [Google Scholar]
- Liani E, Eyal A, Avraham E, Shemer R, Szargel R, Berg D, Bornemann A, Riess O, Ross CA, Rott R, Engelender S (2004) Ubiquitylation of synphilin-1 and alpha-synuclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson's disease. Proc Natl Acad Sci USA 101: 5500-5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Fallon L, Lashuel HA, Liu Z, Lansbury Jr PT (2002) The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson's disease susceptibility. Cell 111: 209-218. [DOI] [PubMed] [Google Scholar]
- Lucking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW, Agid Y, Brice A (2000) Association between early-onset Parkinson's disease and mutations in the parkin gene. French Parkinson's Disease Genetics Study Group. N Engl J Med 342: 1560-1567. [DOI] [PubMed] [Google Scholar]
- Marx FP, Holzmann C, Strauss KM, Li L, Eberhardt O, Gerhardt E, Cookson MR, Hernandez D, Farrer MJ, Kachergus J, Engelender S, Ross CA, Berger K, Schols L, Schulz JB, Riess O, Kruger R (2003) Identification and functional characterization of a novel R621C mutation in the synphilin-1 gene in Parkinson's disease. Hum Mol Genet 12: 1223-1231. [DOI] [PubMed] [Google Scholar]
- Mata IF, Lockhart PJ, Farrer MJ (2004) Parkin genetics: one model for Parkinson's disease. Hum Mol Genet 13: R127-133. [DOI] [PubMed] [Google Scholar]
- Moore DJ, Zhang L, Dawson TM, Dawson VL (2003) A missense mutation (L166P) in DJ-1, linked to familial Parkinson's disease, confers reduced protein stability and impairs homo-oligomerization. J Neurochem 87: 1558-1567. [DOI] [PubMed] [Google Scholar]
- Nagano Y, Yamashita H, Takahashi T, Kishida S, Nakamura T, Iseki E, Hattori N, Mizuno Y, Kikuchi A, Matsumoto M (2003) Siah-1 facilitates ubiquitination and degradation of synphilin-1. J Biol Chem [DOI] [PubMed]
- Pickart CM (2001) Mechanisms underlying ubiquitination. Annu Rev Biochem 70: 503-533. [DOI] [PubMed] [Google Scholar]
- Ren Y, Zhao J, Feng J (2003) Parkin binds to α/β tubulin and increases their ubiquitination and degradation. J Neurosci 23: 3316-3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Yoshikawa M, Kitada T, Matsumine H, Asakawa S, Minoshima S, Yamamura Y, Shimizu N, Mizuno Y (1999) Immunohistochemical and subcellular localization of Parkin protein: absence of protein in autosomal recessive juvenile parkinsonism patients. Ann Neurol 45: 668-672. [DOI] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T (2000) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 25: 302-305. [DOI] [PubMed] [Google Scholar]
- Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ (2001) Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson's disease. Science 293: 263-269. [DOI] [PubMed] [Google Scholar]
- Siderowf A, Stern M (2003) Update on Parkinson disease. Ann Intern Med 138: 651-658. [DOI] [PubMed] [Google Scholar]
- Spence J, Sadis S, Haas AL, Finley D (1995) A ubiquitin mutant with specific defects in DNA repair and multiubiquitination. Mol Cell Biol 15: 1265-1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Ohama E, Suzuki S, Horikawa Y, Ishikawa A, Morita T, Tsuji S, Ikuta F (1994) Familial juvenile parkinsonism: clinical and pathologic study in a family. Neurology 44: 437-441. [DOI] [PubMed] [Google Scholar]
- Von Coelln R, Thomas B, Savitt JM, Lim KL, Sasaki M, Hess EJ, Dawson VL, Dawson TM (2004) Loss of locus coeruleus neurons and reduced startle in parkin null mice. Proc Natl Acad Sci USA 101: 10744-10749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi K, Engelender S, Yoshimoto M, Tsuji S, Ross CA, Takahashi H (2000) Synphilin-1 is present in Lewy bodies in Parkinson's disease. Ann Neurol 47: 521-523. [PubMed] [Google Scholar]
- Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ (2001) TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412: 346-351. [DOI] [PubMed] [Google Scholar]
- West A, Periquet M, Lincoln S, Lucking CB, Nicholl D, Bonifati V, Rawal N, Gasser T, Lohmann E, Deleuze JF, Maraganore D, Levey A, Wood N, Durr A, Hardy J, Brice A, Farrer M (2002) Complex relationship between Parkin mutations and Parkinson disease. Am J Med Genet 114: 584-591. [DOI] [PubMed] [Google Scholar]
- Yao D, Gu Z, Nakamura T, Shi ZQ, Ma Y, Gaston B, Palmer LA, Rockenstein EM, Zhang Z, Masliah E, Uehara T, Lipton SA (2004) Nitrosative stress linked to sporadic Parkinson's disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci USA 101: 10810-10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM (2000) Parkin functions as an E2-dependent ubiquitin-protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci USA 97: 13354-13359. [DOI] [PMC free article] [PubMed] [Google Scholar]