

Abstract
Menin, a protein encoded by the MEN1 gene, suppresses cancers associated with multiple endocrine neoplasia type 1 (MEN1), but promotes the development of a subset of leukemia induced by mixed lineage leukemia (MLL)-derived fusion proteins (MLL-FPs). The crystal structure of menin indicates that it acts as a scaffold protein to bind the N-terminus of MLL via a central pocket. Small molecule menin-MLL inhibitors (MIs) bind the menin pocket to disrupt the menin/MLL interaction, resulting in suppression of MLL-FP-transformed acute myeoloid leukemia (AML). It is thought that MIs suppress the MLL-FP-induced leukemia by blocking the menin/MLL interaction and menin/MLL-induced HOX gene transcription. However, it is not clear whether MIs also affect other aspects of menin biology beyond disruption of the menin/MLL interaction. Here we show for the first time that MIs reduced menin protein levels and decreased the half-life of menin protein but have no effect on mRNA level in MLL-FP-expressing leukemia cells, and proteasome or E1 ligase inhibitor rescued the MI-induced menin degradation. Notably, the MI-induced reduction of H3K4m3 and HOXA9 expression was rescued with a proteasome inhibitor that blocks MI-induced menin protein degradation. Mechanistically, MIs promote the interaction of menin with Hsp70-associated ubiquitin ligase CHIP, resulting in increased menin ubiquitination, leading to increased menin degradation. Together, these findings uncover a novel mechanism whereby small molecule MIs increase menin degradation by triggering the Hsp70/CHIP-mediated ubiquitin-proteasome pathway that ultimately leads to the reduction in HOXA9 gene expression and leukemia suppression.
Keywords: Menin, menin-MLL inhibitor, ubiquitin-proteasome pathway, leukemia
Introduction
Approximately 10% of all leukemia and the majority of infant leukemia harbor the mixed-lineage leukemia gene (MLL)-related translocations, which involve fusion of the MLL gene with one of more than 90 different translocation partner genes, resulting in formation of MLL fusion proteins (MLL-FPs) which are critical for leukemogenesis [1,2]. MLL contains a well conserved SET domain that possess histone lysine 4 (H3K4) methyltransferase activity [3]. MLL-rearranged leukemia occurs across all age groups and develops as both acute myeloid leukemia (AML) and acute lymphoid leukemia (ALL) [4-6]. The 5-year survival rate of patients with MLL-rearranged leukemia is estimated to be only about 35% [7,8]. Chemotherapy is considered the standard of care, and while recently new drugs such as FLT3 inhibitor have been developed [9], more novel therapeutic strategies are still urgently needed [4,10].
Menin, a nuclear protein encoded by the multiple endocrine neoplasia 1 gene (MEN1), suppresses tumorigenesis in endocrine tumors but serves a tumor promoting role in MLL-rearranged leukemogenesis [11]. Structurally, menin directly interacts with partners, such as the N-terminal amino acid residues of MLL via menin’s central pocket, while also interacting with other partners such as LEDGF and c-Myb. Menin binds to the same N-terminal fragment of MLL retained in all MLL-FPs and wild-type MLL [12-15]. MLL is recruited to target genes such as HOXA cluster genes and MEIS1 loci to promote their expression [3,16]. When menin is removed from the promoter at targets gene loci, the recruitment of MLL and MLL-mediated H3K4 trimethylation (H3K4me3) decreases, indicating that menin is an essential cofactor for MLL function in these leukemia cells [12,13,15,17,18]. As the menin/MLL-FP interaction plays a critical role in controlling MLL-rearranged leukemias, small molecule inhibitors that block the menin-MLL interaction have been developed to investigate the impact of blocking menin/MLL interaction on MLL-FP leukemias [16,19-22].
Recently, two small molecular menin inhibitors (MIs), i.e., MI-463 and MI-503, which block the menin/MLL interaction, were synthesized and shown to be effective in suppressing HOXA9 transcription leading to potent inhibition of leukemia in mouse model xenografts of human MLL-FP-expressing cell lines or patient-derived leukemia cells, without impairing normal hematopoiesis [23]. Previous studies show that small molecule MIs inhibit protein-protein interaction of menin and its partners [20,23]. It is thought that MIs suppress the MLL-FP-induced leukemia by blocking the menin/MLL interaction, leading to failure of assembly of menin/MLL/MLL-FP complex and reduced H3K4me3 at targets like HOXA9, which are crucial for transformation and maintenance of MLL-FP-transformed leukemia. However, it is not clear whether MIs also affect other aspects of menin biology beyond disruption of the menin/MLL interaction.
Ubiquitin-mediated proteolysis is a major pathway for intracellular protein degradation. The ubiquitination process is carried out by three classes of enzymes, i.e., E1 (activating enzyme), E2 (conjugating enzyme) and E3 (ubiquitin ligase) [24]. The E1 enzyme starts the process by activating ubiquitin and then passes it to E2, and finally E3 adds ubiquitin onto a target or substrate protein, which can be recognized and degraded by the proteasome [24]. E3 ligases are substrate-specific and interact with both the target protein and the E2 enzyme. Cullin-RING ligases (CRLs) are the largest family of E3 ligases, consisting of RING proteins and a scaffold protein of Cullin-family, all of which are modified by NEDD8 [25]. Moreover, the C-terminus of Hsp70-interacting protein (CHIP) is a member of U-box-containing E3 ubiquitin ligases family, and it interacts with Hsc70 or heat-shock protein 70 (Hsp70), which binds misfolded proteins as a co-chaperone to trigger protein ubiquitination and substrate degradation [26]. Previous studies show that MEN1-related mutant menin, but not the wild type menin protein, is degraded through ubiquitin proteasome pathway in transfected cells [27]. Menin mutants lead to increased proteasome degradation by interacting with Hsp70 and the ubiquitin ligase CHIP, likely due to menin’s misfolded structure, while wild type menin is unaffected [27,28]. Further study shows that inhibition of Hsp70 or CHIP, mediated by small interfering RNA, restores the mutant menin protein level and menin’s ability to perform normal functions [28].
While it has been reported that MIs directly block menin/MLL protein interaction to suppress leukemia, and that mutant menin protein is subjected to proteasome-mediated degradation, little is known whether MIs have an impact on menin protein stability and degradation. Herein, we demonstrate the novel finding that MIs induce menin protein degradation in MLL-FP-expressing leukemia cells and inhibit HOX gene transcription.
Materials and methods
Cell culture
The human leukemia cell lines THP-1, MV4;11 and Jurkat cells were obtained from ATCC. All cell lines were cultured in RPMI-1640 medium with 2 mM L-glutamine (Gibco), supplemented with 10% fetal bovine serum and were maintained at 37°C in a humidified 5% CO2 atmosphere.
Reagents and antibodies
Menin-MLL inhibitors MI-503 and MI-463 were synthesized by Wuxi Pharma, which was previously used [29]. MG-101 (#S7386) was obtained from Selleckchem. PYR-41 (#N2915), MG132 (#BML-PI102) and MLN4924 (#505477) were obtained from Sigma. The antibody for β-actin (#A5441) and ubiquitin (#051307) were purchased from Millipore-Sigma. The antibody for menin (#A300105) was purchased from Bethyl Laboratories. Antibodies for c-Myc (#ab32072), IgG (#ab46540), total H3 (#ab1791), and H3K4me3 (#ab8580) were purchased from Abcam. Anti-rabbit (#1706515) and anti-mouse (#1706516) secondary antibodies for Western blot were purchased from Bio-Rad. The antibodies for CHIP/STUB1 (#PA532046) and Hsp/Hsc70 (#ADI-SPA-820) were purchased from Thermo Fisher Scientific and Enzo Life Sciences, respectively. The anti-rabbit secondary antibody for IP (Clean-Blot™ IP Detection Reagent #21230) was purchased from Thermo Scientific. Anti-mouse secondary antibody for IP was Mouse TrueBlot® ULTRA: Anti-Mouse (#18881730), purchased from Rockland.
Western blot
THP-1, MV4;11 and Jurkat cells were plated in 6-well plates at a density of 5×105 cells/ml with fresh culture medium. The cells were treated as described for the reagents and indicated time. Cells were collected and then lysed with Radioimmunoprecipitation assay buffer (RIPA buffer) and protease inhibitors. Protein concentrations were determined using a BCA assay kit (Thermo Scientific). Quantified equivalent amounts of protein were subjected to polyacrylamide gel electrophoresis on Novex gels (Life Technologies), and then transferred to PVDF membranes (Life Technologies). Blocking was performed in TBST containing 5% non-fat dry milk. The proteins were visualized by detection with Amersham ECL Western blotting detection reagents (GE Healthcare). For quantitation of Western blot Menin band intensity, blots were scanned and the Menin and β-actin bands were obtained from Western blots with Image J of three independent experiments. The ratio of Menin/β-actin was calculated, averaged, normalized and presented as mean ± standard deviation, with statistics calculated based on an unpaired 2-tailed t-test.
Cycloheximide chase assay for the half-life of menin
THP-1 and MV4;11 cells were plated in 6-well plates at a density of 5×105 cells/ml with fresh culture medium, then treated with 10 μM cycloheximide for the indicated hours, and Western blotting was performed. The menin and β-actin protein levels were quantified with Image J scanning, then plotted as normalized menin/β-actin protein versus time, with 0 h treated sample as the 1 time-point, and a linear regression was performed by excel. We generally force the line through the origin. Divide 0.5 by the slope to determine the half-life.
RNA extraction and quantitative real time PCR (qRT-PCR)
Total RNA was extracted from 1×106 differently treated THP-1 and MV4;11 cells with Trizol and RNeasy extraction kit (Qiagen), and 1 μg was used as the template to synthesize cDNA. Real-time PCR (RT-PCR) was performed using a Quantitative SYBR-Green PCR Kit (Qiagen) and a 7500 Fast Real Time PCR System (Applied Biosystems). Transcript levels were normalized to actin transcript levels, with mean values ± SD reported for each group. All experiments were performed in at least triplicate.
Immunoprecipitation (IP)
THP-1 and MV4;11 cells (2×107) were plated on 10 cm plate, and treated with various indicated reagents for 6 hrs. Cells were collected and lysed in lysis buffer (50 mMTris-Cl, pH 7.4, 150 mMNa-Cl, 5% glycerol, 0.5% NP-40, 1 mM EDTA) with protease inhibitors. This lysate was briefly sonicated using the condition of 2 pulses of 5 s each, with 10 s rest on ice between each pulse. Cell lysates were incubated with either IgG or menin antibody at 4°C for 2 hrs with rotation. Then the cell lysates were bound to protein A agarose (#15918014, Invitrogen) at 4°C for 1 hour, and washed three times with the lysis buffer. Proteins were separated by SDS-PAGE and immunoblotted with the indicated antibodies.
Chromatin immunoprecipitation (ChIP) assay
THP-1 and MV4;11 cells (1×107) were plated on 10 cm plate, and treated for 10 hrs with three different conditions, DMSO, MI-503 and MI-503+MG132. After collection of cells, ChIP assays were performed according to the manufacturer’s instructions using a QuickChIP kit (Novus Biologicals). Briefly, cells were fixed with 1% formaldehyde for 10 minutes at 37°C, and glycine solution in the kit was used to stop the fixing process. The fixed cells were lysed according to the manufacturer protocol in a ChIP lysis buffer with protease inhibitors. Cellular DNA was sheared with sonication using condition of eleven pulses of 30 s each, with a 30 s rest between each pulse. The lysate was precleared and then incubated with either control IgG or a specific primary antibody (4 μg) at 4°C overnight, followed by collection with protein A/G agarose beads. The protein-DNA complexes were eluted from the beads. DNA was de-crosslinked and purified using a QIAquick PCR Purification Kit (Qiagen). DNA was amplified by real-time PCR using primer pairs specific to the HOXA9 promoter, a Quantitative SYBR-Green PCR Kit (Qiagen), and a 7500 Fast Real Time PCR System (Applied Biosystems). Reactions were done in triplicate, and results were normalized to input chromatin and reported as percent input +/- SD.
Primers and sequences of the primers
qRT-PCR primers: homo β-actin-For: 5’-GGTCATCACCATTGGCAATGA-3’; β-actin-Rev: 5’-GCACTGTGTTGGCGTACA-3’; homo MEN1-For: 5’-GTGGCCGACCTGTCTATCAT-3’; homo MEN1-Rev: 5’-GTGCCTGTGATGAAGCTGAA-3’; homo HOXA9-For: 5’-TACTACGTGGACTCGTTCCT-3’; homo HOXA9-Rev: 5’-CTTGGACTGGAAGCTGCA-3’.
ChIP assay primers for HOXA9: Amplicon 1-For: 5’-CCGCCTTTATTCCTCTCTCC-3’; Amplicon 1-Rev: 5’-AGTGCAACAGAGTGCCC-3’; Amplicon 2-For: 5’-TTTCGAGCCTTCCACGG-3’; Amplicon 2-Rev: 5’-AAGGTATTCCTGGGAGGGAG-3’.
Statistical analyses
Microsoft excel and GraphPad Prism were used for statistical analysis. The results were presented as mean values ± SD, with statistics calculated based on an unpaired 2-tailed t-test.
Results
Menin inhibitors reduce menin protein levels without altering mRNA levels
The menin inhibitor MI-503 blocks the menin/MLL interaction, thus reducing the transcription of target genes such as Hoxa9 in MLL-FP-transformed leukemia cells [23]. To determine whether MI-503 affects the expression of menin mRNA and protein, we treated MV4;11 and THP-1 cells, both human AML cell lines harboring MLL-AF4 and MLL-AF9, respectively [30], with MI-503 and then determined the impact on menin mRNA and protein levels. MI-503 treatment did not affect the MEN1 mRNA level of MV4;11 cells after various time (Figure 1A) nor at various MI-503 concentrations (Figure 1B). Similarly, the MI-503 treatment did not affect the MEN1 mRNA level in THP-1 leukemia cells (Figure 1A and 1C). In contrast, menin protein levels were decreased in both MV4;11 and THP-1 cell lines in a dose-dependent manner (Figure 1D, Lanes 2, 3, 5, 6). Similarly, treatment with a different menin inhibitor MI-463 [23] also led to a decrease in menin protein expression in MV4;11 cells (Figure 1E, Lanes 5 and 6). We also examined whether MI-503 affects menin protein levels in another human T cell leukemia cell line, Jurkat cells, and found that the MI-503 treatment did not reduce the menin protein level (Figure 1D, lanes 7-9). Consistently, MI-503 treatment reduced growth of MLL-FP-expressing MV4;11 cells and THP-1 cells, but not Jurkat cells, in a dose-dependent manner (Figure 1F). Together, these results demonstrated that the menin inhibitors reduce the level of menin protein, but not the level of menin mRNA.
Figure 1.
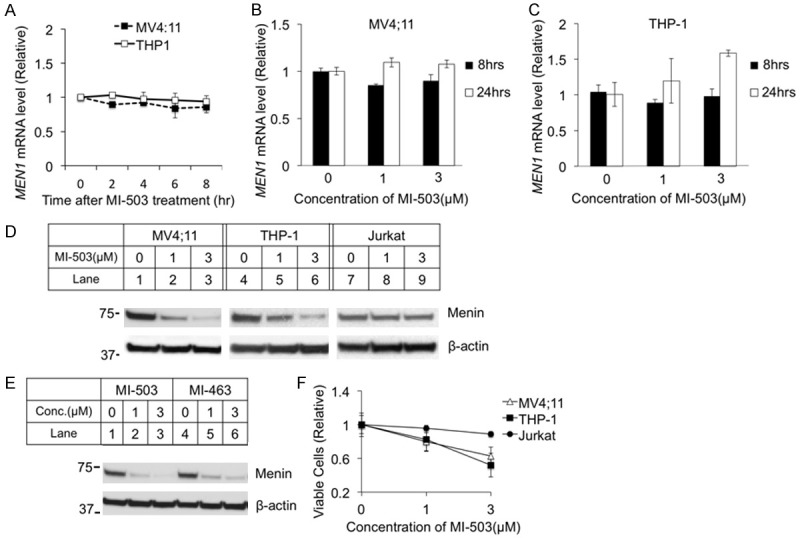
Menin inhibitor MI-503 reduced menin protein levels in MLL-FP transformed leukemia cell lines, but did not affect the menin mRNA level. MV4;11 and THP-1 cells were treated for 0-24 hours with analysis of mRNA levels at various time points. MV4;11, THP-1, and Jurkat cell lines were treated for 8 hours followed by analysis of protein levels by Western Blotting. Cells were counted by hand after treatment for 24 hours. A-C. MEN1 mRNA levels were detected by qRT-PCR. D. Western Blot of menin expression for MV4;11, THP-1, and Jurkat cell lines with treatment of 0, 1, or 3 μM MI-503. E. Western Blot of menin expression for MV4;11 with 8-hour treatment of 2 different menin inhibitors, MI-503 or MI-463, at varying concentrations. F. MV4;11, THP-1, and Jurkat cell lines were treated for 24 hours and cells were counted by hand.
Ubiquitin-activating enzyme and proteasome inhibitors rescue MI-induced menin degradation
Given the reduction of menin protein, but not mRNA levels following MI treatment, we next tested whether MIs induce menin protein degradation. As many proteins are degraded by ubiquitin-mediated proteasome degradation [24], we sought to determine the effect of proteasome inhibition and ubiquitin-activating enzyme inhibition on MI-induced reduction of menin protein. To this end, we examined whether the ubiquitin-activating enzyme (E1) inhibitor PYR-41 [31] affects MI-induced reduction of menin protein. We found that treatment of MV4;11 cells with PYR-41 abolished the MI-induced reduction of menin protein (Figure 2A, lanes 5-6). Likewise, PYR-41 also rescued MI-induced degradation at 50 μM in THP-1 cells (Figure 2A, lane 12).
Figure 2.
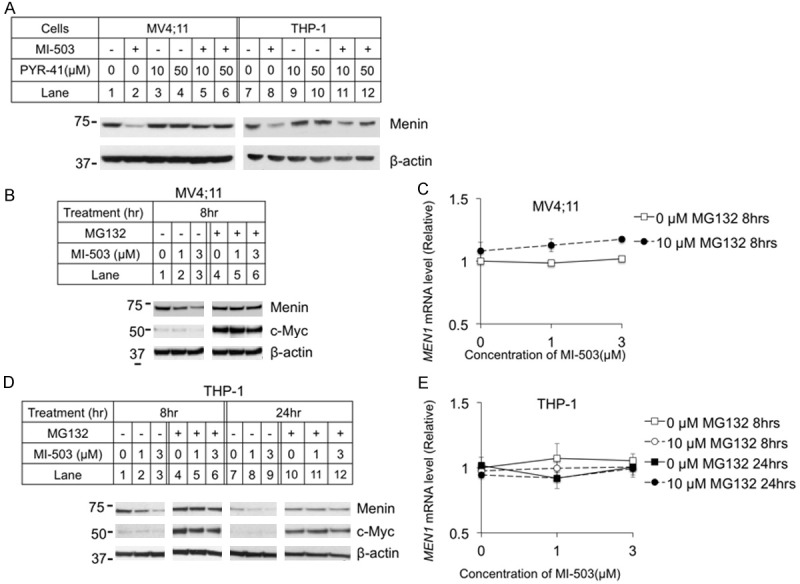
Ubiquitin pathway inhibitors blocked menin inhibitor-induced reduction of menin protein, but not mRNA. MV4;11 and THP-1 cell lines were treated for 8 hours with 3 μM MI-503 and E1 inhibitor PYR-41, followed by analysis of menin protein levels with Western blotting (A). MV4;11 and THP-1 cell lines were treated for 8 or 24 hours with 10 μM proteasome inhibitor MG132 and 0-3 μM MI-503 followed by analysis of protein levels by Western blotting, and mRNA levels by qRT-PCR. (B, D) Typical Western blot of menin and c-Myc expression in MV4;11 and THP-1 cell lines with indicated treatments. (C, E) MEN1 mRNA level was detected by qRT-PCR in MV4;11 and THP-1 cell lines.
Further results indicated that MG132, a pan-active proteasome inhibitor, blocked MI-induced menin protein reduction in MV4;11 cells [32] (Figure 2B, lane 5 and 6 vs 2 and 3, respectively). We examined c-Myc protein as a positive control of MG132. The MEN1 mRNA level in MV4;11 cells were only slightly increased with the MI-503 treatment, in both control and MG132-treated cells (Figure 2C). Similarly, MG132 treatment blocked MI-induced reduction of menin protein in THP-1 cells, after both 8 hours (Figure 2D, lanes 1-6) and 24 hours (Figure 2D, lanes 7-12) of treatment. However, MI-503 and MG132 did not significantly affect the MEN1 mRNA levels in the THP-1 cells (Figure 2E).
Collectively, these results demonstrate that both E1 and proteasome inhibitor rescued MI-induced reductions of menin protein, but did affect menin mRNA levels, suggesting that MIs alter menin protein stability and degradation in the leukemia cells.
MI decreases the half-life of menin protein
To further determine the effect of MI on half-life of menin protein, we treated MV4;11 cells for various periods of time (0-6 hours), in the presence or absence of cycloheximide (CHX), which inhibits protein translation and thus de novo protein synthesis [33]. Over the times examined, menin protein levels collected from cells not treated with MIs were comparable (Figure 3A, top 2 panels). Treatment with MI proportionally reduced the steady level of menin protein in cells without CHX treatment (Figure 3A, middle two panels, lanes 1-6). Treating these cells with CHX, which blocks new menin protein synthesis, further reduced the menin protein level (Figure 3A, middle two panels, lanes 7-12). Furthermore, to test whether MG132 blocks the MI-induced reduction, MV4;11 cells treated at the above conditions were also co-treated with MG132, which blocked the MI-induced reduction of the menin protein levels (Figure 3A, bottom two panels). Quantitation of the protein levels show that MI treatment reduced the half-life (T1/2) of menin protein to ~3 hrs, while only CHX treated menin protein was quite stable, and this reduction was almost completely rescued by MG132 treatment (Figure 3E).
Figure 3.
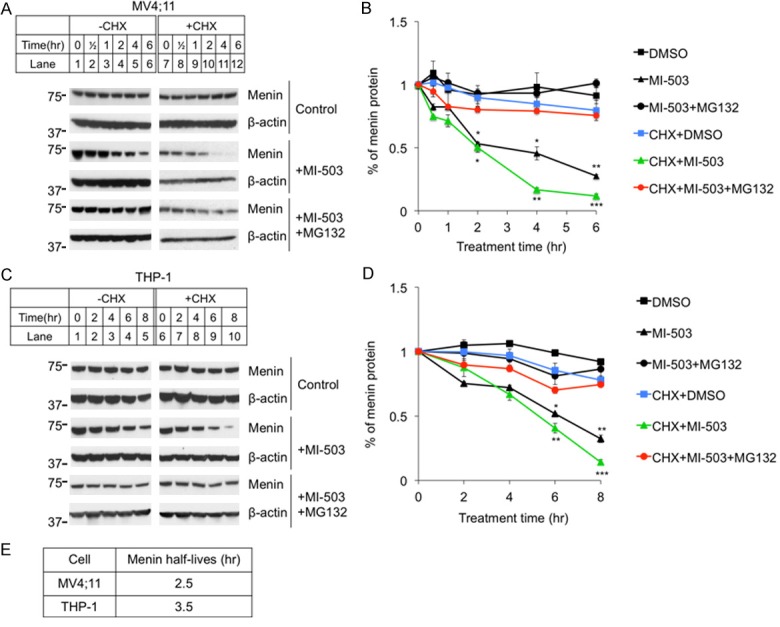
Protein translation inhibitor Cycloheximide (CHX) enhanced MI-503 induced reduction of menin protein levels, but the reduction was blocked by MG132 treatment. MV4;11 and THP-1 cell lines were treated for up to 8 hours followed by analysis of protein levels by Western blotting, 10 μM CHX, 3 μM MI-503, 10 μM MG132. (A, C) Typical Western blot of menin expression in MV4;11 and THP-1 cell lines. (B, D) Image J Western Blot quantification of menin protein levels and analyzed by t-test, compared with vehicle (n=3). *P<0.05, **P<0.01, ***P<0.001. (E) Calculated menin half-life equations in MI treated MV4;11 and THP-1 cells from (B) and (D).
We also extended these experiments to THP-1 cells. Similarly, we found that MI treatment substantially reduced menin protein level in a time-dependent manner (Figure 3C, middle two panels, lanes 1-10). Notably, co-treatment with MG132 blocked MI-induced reduction of menin protein (Figure 3C, bottom two panels). Quantitation of menin protein levels showed that MI-503 treatment substantially reduced the T1/2 of menin protein to 3.5 hrs, and this reduction was rescued by MG132 (Figure 3D, 3E). Together, these studies demonstrate that MI reduces the T1/2 of menin substantially, to 2-3 hrs, and this reduction is blocked by a proteasome inhibitor, thus demonstrating that MI likely destabilizes menin protein, triggering the ubiquitin-mediated menin degradation.
Neddylation inhibitor MLN4924 only partially rescues MI-induced menin degradation
To elucidate the pathway underlying MG132 rescue of MI-induced menin degradation, we first examined potential involvement of the family of Cullin-RING ligases (CRLs), as this family has the largest number of E3 ubiquitin ligases [25]. We used the NEDD8 Activating Enzyme (NAE) inhibitor MLN4924 to inhibit CRLs by blocking Cullin neddylation, as neddylation of Cullins is crucial for activating CRLs [34]. To this end, we treated ML4;11 cells and THP-1 cells with MI-503 and/or MLN4924 and found that the MI-induced reduction of menin protein in the cells was only moderately rescued by treatment with MLN4924 (Figure 4A, Lanes 4 and 8, vs Lanes 2 and 6, respectively). Notably, treating either MV4;11 cells or THP-1 cells with the proteasome inhibitor MG132 almost completely rescued the MI-induced reduction of menin protein (Figure 4B, Lanes 4 and 8 vs Lanes 3 and 7, respectively). We quantified the change of menin protein, and the results showed that menin protein was significantly reduced by MI-503 treatment (Figure 4C, 4D, >50%) and this protein reduction was rescued some by both MLN4924 and MG132 (Figure 4C, 4D). However, treatment of the cells with either MLN4924 or MG132 did not substantially increase the menin protein levels alone, suggesting that normal menin protein without MI treatment is quite stable without noticeable proteasome-mediated degradation in the experimental timeframe.
Figure 4.
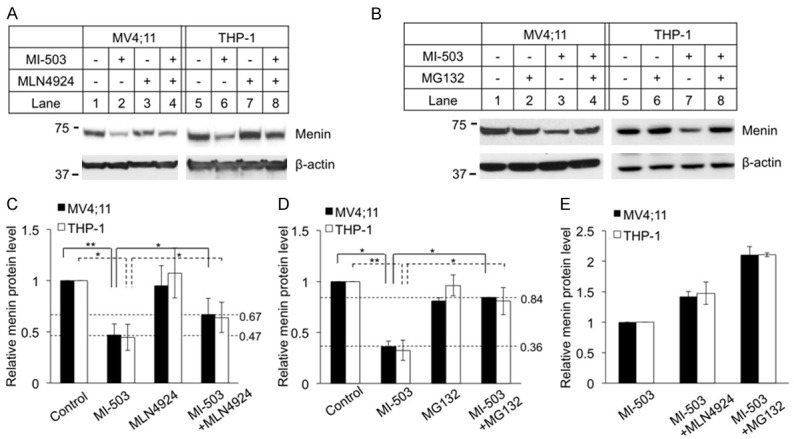
Proteasome inhibitor MG132 is more potent to block MI-503 induced menin degradation than neddylation inhibitor MLN4924. MV4;11 and THP-1 cells treated for 8 hours followed by analysis of protein levels by Western blotting, 3 μM MI-503, 5 μM MLN4924, 10 μM MG132. Experiments were performed at least three times independently. A, B. Typical Western blot of menin expression in MV4;11 and THP-1 cell lines with indicated treatments. C, D. Image J Western blot quantification of menin protein level and analyzed by t-test (n=3). *P<0.05, **P<0.01. E. Image J quantified the percentage of menin rescued by either MLN4924 or MG132 in MI-503 treated MV4;11 and THP-1 cells.
To further quantify the ability of MLN4924 and MG132 to rescue MI-induced menin degradation, the menin protein level in MV4;11 and THP-1 cell lines were normalized to the menin protein level of cell treated with MI-503 alone (Figure 4E). The results showed that menin protein level was increased nearly 1.5-fold after treatment with MLN4924, but nearly 2-fold after treatment with MG132 (Figure 4E). Moreover, the menin protein in the MI-503+MLN4924 treated cells was rescued to ~60% of the normal control level (Figure 4C). Notably, the menin protein levels in the MI503+MG132 treated MV4;11 and THP-1 cells were increased to 80% of control level (Figure 4D). Taken together, these results indicate that MLN4924 only partially rescues MI-induced and CRL-mediated menin protein degradation, and therefore there are likely other proteasome-mediated degradation pathways mediating MI-induced menin degradation.
MI promotes the ubiquitination of menin by increasing the interaction with Hsp70 and ubiquitin ligase carboxyl terminus of CHIP
It has been reported that menin mutants associated with MEN1 syndrome result in aberrant folding allowing them to be recognized by chaperone Hsp70, which interacts with the Hsp70 co-chaperone, CHIP [27]. Subsequently, the complex (Hsp70/CHIP/menin mutants) ubiquitinates and degrades mutant menin, but natively folded wild type menin protein is not degraded by this pathway [27]. Our previous crystal structural studies showed that menin acts as a scaffold protein interacting via a central deep pocket with various partners including MLL, JunD and LEDGF [12], with MIs being able to block menin/MLL interaction [12,20]. We thus sought to determine whether MI blocks wild type menin interaction, and thus destabilizes menin leading menin to interact with Hsp70/Hsc70 or CHIP in leukemia cells. To this end, we treated MV4;11 cells with MI-503 and proteasome inhibitor MG132 to enrich endogenous protein, and investigated whether wild type menin, when deprived of binding partners by MI503, can bind Hsp70/Hsc70 and CHIP. Immunoprecipitation showed that the menin antibody pulled down CHIP and Hsp70/Hsc70 proteins selectively from the cells treated with MI503, but not from control cells (Figure 5A, lane 6 vs 5, top 2nd and 3rd panels). Consistently, the pulled down menin was also more ubiquitinated from the MI-503 treated cells than the control cells (Figure 5A, lane 6 vs 5, bottom panel). Likewise, in THP-1 cells, co-IP also showed that the menin antibody pulled down CHIP, Hsp70/Hsc70, and ubiquitinylated menin from the MI503-treated cells, but not from the control cells (Figure 5B, lane 6 vs 5). In aggregate, these results suggest that MI-503 increases menin degradation via formation of menin, CHIP, and Hsp70/Hsc70 complex, leading to menin ubiquitination and subsequence proteasome-mediated degradation.
Figure 5.
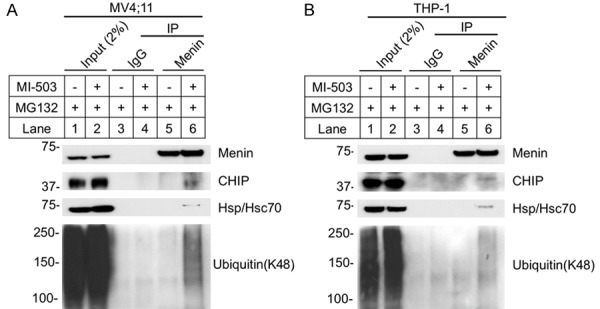
MI-503 increased Menin interacted with CHIP, Hsp70 and Ubiquitin. A, B. MV4;11 and THP-1 cells treated with 10 μM MG132 +/- 3 μM MI-503 for 6 hours, followed by IP and Western blot using the indicated antibodies.
MI-503 reduced H3K4 trimethylation at the HOXA9 locus to repress its expression
Whereas prior work proposed that MIs block the menin/MLL interaction, reduce H3K4m3 and thus reduce HOX gene transcription [11,23], our new findings also indicate that MIs destabilize menin protein and trigger ubiquitin-mediated degradation. It is therefore intriguing to determine the potential role of MI-mediated degradation in menin-regulated HOX gene transcription. To this end, MV4;11 and THP-1 cells were treated with either vehicle, MI-503 and/or MG132. The resulting cells were subjected to qRT-PCR and chromatin immunoprecipitation (ChIP) assay to determine the impact of the treatments on HOX gene mRNA levels, menin binding to the HOXA9 locus, and H3K4me3 at the HOXA9 locus. QRT-PCR results showed that HOXA9 mRNA level was reduced by MI-503 treatment (Figure 6A), as expected. ChIP assay revealed that menin and the menin-dependent active histone mark H3K4me3 [3,23], catalyzed by MLL, were detected at the HOXA9 promoter (Figure 6B-F).
Figure 6.
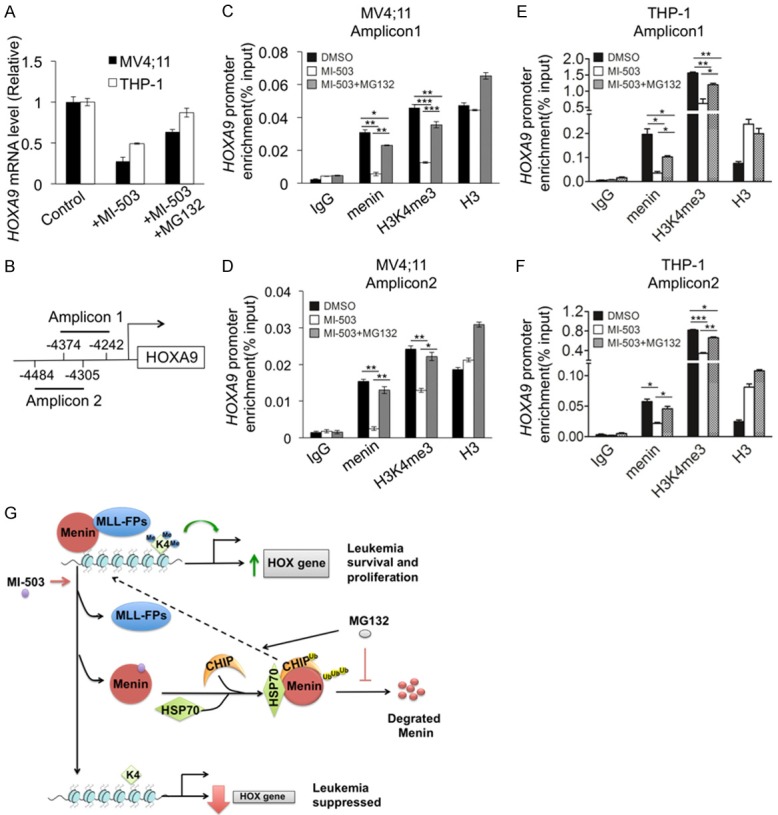
Menin inhibitor repressed HOXA9 gene expression by reducing H3K4me3 in the HOXA9 locus, which was rescued by MG132. MV4;11 and THP-1 cells treated for 10 h, with either control DMSO or 3 μM MI-503 +/- 10 μM MG132. A. HOXA9 mRNA level was detected by qRT-PCR. B. Schematic diagram to show two amplicons in HOXA9 promoter. C-F. ChIP assays were performed to look for menin, total H3, and H3K4m3 at amplicon 1 and 2 of the HOXA9 promoter (n=3). *P<0.05, **P<0.01, ***P<0.001. G. A molecular model for menin inhibitor-induced menin degradation and repression of HOX gene transcription.
Consistently, treating the cells with MI-503 reduced menin binding with H3K4me3 at the HOXA9 promoter, with two distinct amplicons (Figure 6C, 6D). Notably, co-treatment of MV4;11 cells with MI-503 and MG132 rescued the MI-503-induced reduction of menin binding at the HOXA9 promoter as well as H3K4me3 at the promoter (Figure 6C, 6D). Likewise, treatment of THP-1 cells with MI-503 also reduced menin binding and H3K4me3 at the HOXA9 promoter (Figure 6E, 6F), and importantly, co-treatment with MG132 also rescued the MI503-induced reduction of menin binding and H3K4me3 at the HOXA9 promoter (Figure 6E, 6F). Taken together, these results indicate that MI-induced menin protein degradation is more crucial than disruption of menin/MLL interaction in repressing HOXA9 transcription. This likely results from MI-503 losing the majority of its ability to repress HOXA9 expression (Figure 6A), menin binding and H3K4m3 (Figure 6B-F) when MG132 was added to the cells to maintain menin protein levels, even though MI-503 should theoretically inhibit the menin/MLL interaction in the presence of MG132.
Discussion
Menin acts as a scaffold protein to bind MLL and other partner proteins, and recruits them to activate target gene transcription and block myeloid differentiation. Menin-MLL inhibitors show benefit in the treatment of murine models of MLL-FP-expressing leukemia in vitro and in vivo [20,22,23], representing a promising therapeutic approach for MLL- FP leukemia. The U.S. Food and Drug Administration (FDA) has currently cleared the company’s investigational new drug application for Menin-MLL inhibitor KO-539. However, little is known regarding the effect of menin/MLL inhibitors on other aspects of menin biology beyond disruption of the menin/MLL interaction. Herein we demonstrated that menin-MLL inhibitors induce menin protein degradation in MLL-FP leukemia cells via the ubiquitin-proteasome pathway and repress HOXA9 gene expression by decreasing H3K4me3 at the HOXA9 promoter, which is in addition to disruption of the interaction between menin and the MLL proteins as previously reported [20,23]. These findings are significant for the following reasons: first, we demonstrate that MIs reduce menin protein level, but not mRNA level in MLL-FPs leukemia cell lines. Second, we show that the MI-induced reduction of menin protein is through the ubiquitin pathway, mainly through CHIP/Hsp70 and partly through Cullin-RING E3 ligases (CRLs), as the NEDD8 activating enzyme inhibitor MLN4924 only partially rescued menin degradation. Third, the major impact of MIs on reducing HOXA9 expression was rescued by the proteasome inhibitor MG132, suggesting that the major impact of MIs is through reducing the total menin protein level. Furthermore, this suggests that the impact of MI on degrading menin is perhaps more potent than its reversible inhibition of the menin/MLL interaction at the HOX gene promoters.
E3 ubiquitin ligases bind different substrate proteins, polyunbiquitinate these proteins, and then transport them to proteasomes for degradation [35,36]. There are over 600 E3 ligases in humans, and they are classified into four families: HECT, RING-finger, U-box and PHD-finger [35]. We used NEDD8 activating enzyme inhibitor MLN4924 to block cullin neddylation and inactivate Cullin-RING E3 ligases (CRLs) [25]. Our results suggest that menin degradation induced by MI-503 is only partially rescued by MLN4924, but almost completely rescued by the pan-active proteasome inhibitor MG132. These findings suggest that other ubiquitin/proteasome pathways may also be involved in MI-induced menin degradation. Consistently, we found that MI increases the interaction of menin with both the molecular chaperone Hsp70 and the Hsp70-associated ubiquitin E3 ligase CHIP, and promotes menin ubiquitination in MLL-FPs leukemia cells. These results illustrate that CHIP/Hsp70 plays a crucial role in triggering wild type menin degradation, while the CRL pathway may also be partially involved in menin degradation.
MEN1-related mutant menin, but not wild type menin, has previously been reported to be degraded via the CHIP/Hsp70 complex in transfected HEK 293 cells [28]. It is likely that menin binds numerous partner proteins, like MLL-FP, c-Myb and LEDGF [13,15,37], and that by disrupting menin interaction with these partners, MIs destabilize menin protein (without the ability of menin to bind to stabilizing partners), triggering its association with chaperone Hsp70/CHIP, leading to increased ubiquitination and degradation (Figure 6G).
We also noted that MIs did not reduce menin protein levels in Jurkat cells. Further studies are needed to determine whether the CHIP pathway is not active in Jurkat cells, or whether there are other partners beyond the MI-blockable partners that remain functional to stabilize menin in the presence of MIs in a cell type specific manner. Furthermore, it would also be interesting to see if MIs destabilize menin in other cell types, especially prostate cancer where menin/MLL has also been found to be important for tumorigenesis [38].
It is also noteworthy that even in the presence of a MI, when menin degradation was blocked by MG132 (Figure 4B), the menin binding, H3K4m3 at the HOXA9 promoter, and HOXA9 expression persisted (Figure 6A-F). These results suggest that the major impact of MIs on reducing HOXA9 expression and suppressing MLL-FP AML is through reducing the total menin level, rather than reversibly inhibiting the menin/MLL interaction at the promoter. Along these lines, when the total menin protein level is reduced, menin is unable to assist at the HOXA9 gene promoter to sustain the menin/MLL interaction and H3K4 trimethylation necessary for HOXA9 gene transcription.
Together, our findings uncover a new mechanism whereby menin inhibitors that disrupt the menin-MLL interaction trigger the association of menin with Hsp70/CHIP, inducing the ubiquitin/proteasome pathway to degrade menin protein (Figure 6G). Menin degradation leads to reduced menin binding to the HOXA9 promoter, and decreased H3K4me3 marker at the HOXA9 locus irreversibly repressing its transcription and leukemia cell growth (Figure 6G).
Acknowledgements
This work was supported by Natural Science Foundation of China Grants (No. 81773238, Fuxiang Zhou; No. 81401913 Yuan Wu), a Harrington Takeda Rare Disease Award (Xianxin Hua), and a Foster Foundation for Initiative Research in Zhongnan Hospital of Wuhan University (Fuxiang Zhou).
Disclosure of conflict of interest
None.
References
- 1.Meyer C, Burmeister T, Groger D, Tsaur G, Fechina L, Renneville A, Sutton R, Venn NC, Emerenciano M, Pombo-de-Oliveira MS, Barbieri Blunck C, Almeida Lopes B, Zuna J, Trka J, Ballerini P, Lapillonne H, De Braekeleer M, Cazzaniga G, Corral Abascal L, van der Velden VHJ, Delabesse E, Park TS, Oh SH, Silva MLM, Lund-Aho T, Juvonen V, Moore AS, Heidenreich O, Vormoor J, Zerkalenkova E, Olshanskaya Y, Bueno C, Menendez P, Teigler-Schlegel A, Zur Stadt U, Lentes J, Gohring G, Kustanovich A, Aleinikova O, Schafer BW, Kubetzko S, Madsen HO, Gruhn B, Duarte X, Gameiro P, Lippert E, Bidet A, Cayuela JM, Clappier E, Alonso CN, Zwaan CM, van den Heuvel-Eibrink MM, Izraeli S, Trakhtenbrot L, Archer P, Hancock J, Moricke A, Alten J, Schrappe M, Stanulla M, Strehl S, Attarbaschi A, Dworzak M, Haas OA, Panzer-Grumayer R, Sedek L, Szczepanski T, Caye A, Suarez L, Cave H, Marschalek R. The MLL recombinome of acute leukemias in 2017. Leukemia. 2018;32:273–284. doi: 10.1038/leu.2017.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muntean AG, Hess JL. The pathogenesis of mixed-lineage leukemia. Annu Rev Pathol. 2012;7:283–301. doi: 10.1146/annurev-pathol-011811-132434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen YX, Yan J, Keeshan K, Tubbs AT, Wang H, Silva A, Brown EJ, Hess JL, Pear WS, Hua X. The tumor suppressor menin regulates hematopoiesis and myeloid transformation by influencing Hox gene expression. Proc Natl Acad Sci U S A. 2006;103:1018–1023. doi: 10.1073/pnas.0510347103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andersen MK, Christiansen DH, Jensen BA, Ernst P, Hauge G, Pedersen-Bjergaard J. Therapy-related acute lymphoblastic leukaemia with MLL rearrangements following DNA topoisomerase II inhibitors, an increasing problem: Report on two new cases and review of the literature since 1992. Br J Haematol. 2001;114:539–543. doi: 10.1046/j.1365-2141.2001.03000.x. [DOI] [PubMed] [Google Scholar]
- 5.Super HJ, McCabe NR, Thirman MJ, Larson RA, Le Beau MM, Pedersen-Bjergaard J, Philip P, Diaz MO, Rowley JD. Rearrangements of the MLL gene in therapy-related acute myeloid leukemia in patients previously treated with agents targeting DNA-topoisomerase II. Blood. 1993;82:3705–3711. [PubMed] [Google Scholar]
- 6.Winters AC, Bernt KM. MLL-rearranged leukemias-an update on science and clinical approaches. Front Pediatr. 2017;5:4. doi: 10.3389/fped.2017.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hilden JM, Dinndorf PA, Meerbaum SO, Sather H, Villaluna D, Heerema NA, McGlennen R, Smith FO, Woods WG, Salzer WL, Johnstone HS, Dreyer Z, Reaman GH, Children’s Oncology G. Analysis of prognostic factors of acute lymphoblastic leukemia in infants: report on CCG 1953 from the Children’s Oncology Group. Blood. 2006;108:441–451. doi: 10.1182/blood-2005-07-3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tomizawa D, Koh K, Sato T, Kinukawa N, Morimoto A, Isoyama K, Kosaka Y, Oda T, Oda M, Hayashi Y, Eguchi M, Horibe K, Nakahata T, Mizutani S, Ishii E. Outcome of risk-based therapy for infant acute lymphoblastic leukemia with or without an MLL gene rearrangement, with emphasis on late effects: a final report of two consecutive studies, MLL96 and MLL98, of the Japan Infant Leukemia Study Group. Leukemia. 2007;21:2258–2263. doi: 10.1038/sj.leu.2404903. [DOI] [PubMed] [Google Scholar]
- 9.Armstrong SA, Kung AL, Mabon ME, Silverman LB, Stam RW, Den Boer ML, Pieters R, Kersey JH, Sallan SE, Fletcher JA, Golub TR, Griffin JD, Korsmeyer SJ. Inhibition of FLT3 in MLL. Validation of a therapeutic target identified by gene expression based classification. Cancer Cell. 2003;3:173–183. doi: 10.1016/s1535-6108(03)00003-5. [DOI] [PubMed] [Google Scholar]
- 10.Kuhn MW, Armstrong SA. Designed to kill: novel menin-MLL inhibitors target MLL-rearranged leukemia. Cancer Cell. 2015;27:431–433. doi: 10.1016/j.ccell.2015.03.012. [DOI] [PubMed] [Google Scholar]
- 11.Matkar S, Thiel A, Hua X. Menin: a scaffold protein that controls gene expression and cell signaling. Trends Biochem Sci. 2013;38:394–402. doi: 10.1016/j.tibs.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang J, Gurung B, Wan B, Matkar S, Veniaminova NA, Wan K, Merchant JL, Hua X, Lei M. The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature. 2012;482:542–546. doi: 10.1038/nature10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin S, Zhao H, Yi Y, Nakata Y, Kalota A, Gewirtz AM. c-Myb binds MLL through menin in human leukemia cells and is an important driver of MLL-associated leukemogenesis. J Clin Invest. 2010;120:593–606. doi: 10.1172/JCI38030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thiel AT, Huang J, Lei M, Hua X. Menin as a hub controlling mixed lineage leukemia. Bioessays. 2012;34:771–780. doi: 10.1002/bies.201200007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell. 2008;14:36–46. doi: 10.1016/j.ccr.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123:207–218. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 17.Thiel AT, Blessington P, Zou T, Feather D, Wu X, Yan J, Zhang H, Liu Z, Ernst P, Koretzky GA, Hua X. MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell. 2010;17:148–159. doi: 10.1016/j.ccr.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thiel AT, Feng Z, Pant DK, Chodosh LA, Hua X. The trithorax protein partner menin acts in tandem with EZH2 to suppress C/EBPalpha and differentiation in MLL-AF9 leukemia. Haematologica. 2013;98:918–927. doi: 10.3324/haematol.2012.074195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caslini C, Yang Z, El-Osta M, Milne TA, Slany RK, Hess JL. Interaction of MLL amino terminal sequences with menin is required for transformation. Cancer Res. 2007;67:7275–7283. doi: 10.1158/0008-5472.CAN-06-2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grembecka J, He S, Shi A, Purohit T, Muntean AG, Sorenson RJ, Showalter HD, Murai MJ, Belcher AM, Hartley T, Hess JL, Cierpicki T. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol. 2012;8:277–284. doi: 10.1038/nchembio.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manka J, Daniels RN, Dawson E, Daniels JS, Southall N, Jadhav A, Zheng W, Austin C, Grembecka J, Cierpicki T, Lindsley CW, Stauffer SR. Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD): 2010. Inhibitors of the menin-mixed lineage leukemia (MLL) Interaction. [PubMed] [Google Scholar]
- 22.Shi A, Murai MJ, He S, Lund G, Hartley T, Purohit T, Reddy G, Chruszcz M, Grembecka J, Cierpicki T. Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood. 2012;120:4461–4469. doi: 10.1182/blood-2012-05-429274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borkin D, He S, Miao H, Kempinska K, Pollock J, Chase J, Purohit T, Malik B, Zhao T, Wang J, Wen B, Zong H, Jones M, Danet-Desnoyers G, Guzman ML, Talpaz M, Bixby DL, Sun D, Hess JL, Muntean AG, Maillard I, Cierpicki T, Grembecka J. Pharmacologic inhibition of the Menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell. 2015;27:589–602. doi: 10.1016/j.ccell.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell. 1994;79:13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- 25.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 26.McDonough H, Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones. 2003;8:303–308. doi: 10.1379/1466-1268(2003)008<0303:calbtc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yaguchi H, Ohkura N, Takahashi M, Nagamura Y, Kitabayashi I, Tsukada T. Menin missense mutants associated with multiple endocrine neoplasia type 1 are rapidly degraded via the ubiquitin-proteasome pathway. Mol Cell Biol. 2004;24:6569–6580. doi: 10.1128/MCB.24.15.6569-6580.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Canaff L, Vanbellinghen JF, Kanazawa I, Kwak H, Garfield N, Vautour L, Hendy GN. Menin missense mutants encoded by the MEN1 gene that are targeted to the proteasome: restoration of expression and activity by CHIP siRNA. J Clin Endocrinol Metab. 2012;97:E282–291. doi: 10.1210/jc.2011-0241. [DOI] [PubMed] [Google Scholar]
- 29.Katona BW, Glynn RA, Paulosky KE, Feng Z, Davis CI, Ma J, Berry CT, Szigety KM, Matkar S, Liu Y, Wang H, Wu Y, He X, Freedman BD, Brady DC, Hua X. Combined menin and EGFR inhibitors synergize to suppress colorectal cancer via EGFR-independent and calcium-mediated repression of SKP2 transcription. Cancer Res. 2019;79:2195–2207. doi: 10.1158/0008-5472.CAN-18-2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drexler HG, Quentmeier H, MacLeod RA. Malignant hematopoietic cell lines: in vitro models for the study of MLL gene alterations. Leukemia. 2004;18:227–232. doi: 10.1038/sj.leu.2403236. [DOI] [PubMed] [Google Scholar]
- 31.Yang Y, Kitagaki J, Dai RM, Tsai YC, Lorick KL, Ludwig RL, Pierre SA, Jensen JP, Davydov IV, Oberoi P, Li CC, Kenten JH, Beutler JA, Vousden KH, Weissman AM. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res. 2007;67:9472–9481. doi: 10.1158/0008-5472.CAN-07-0568. [DOI] [PubMed] [Google Scholar]
- 32.Tsubuki S, Saito Y, Tomioka M, Ito H, Kawashima S. Differential inhibition of calpain and proteasome activities by peptidyl aldehydes of di-leucine and tri-leucine. J Biochem. 1996;119:572–576. doi: 10.1093/oxfordjournals.jbchem.a021280. [DOI] [PubMed] [Google Scholar]
- 33.Ennis HL, Lubin M. Cycloheximide: aspects of inhibition of protein synthesis in mammalian cells. Science. 1964;146:1474–1476. doi: 10.1126/science.146.3650.1474. [DOI] [PubMed] [Google Scholar]
- 34.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A, Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, McDonald A, Mizutani H, Narayanan U, Olhava EJ, Peluso S, Rezaei M, Sintchak MD, Talreja T, Thomas MP, Traore T, Vyskocil S, Weatherhead GS, Yu J, Zhang J, Dick LR, Claiborne CF, Rolfe M, Bolen JB, Langston SP. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 35.Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6:369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 36.Behrends C, Harper JW. Constructing and decoding unconventional ubiquitin chains. Nat Struct Mol Biol. 2011;18:520–528. doi: 10.1038/nsmb.2066. [DOI] [PubMed] [Google Scholar]
- 37.Milne TA, Hughes CM, Lloyd R, Yang Z, Rozenblatt-Rosen O, Dou Y, Schnepp RW, Krankel C, Livolsi VA, Gibbs D, Hua X, Roeder RG, Meyerson M, Hess JL. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci U S A. 2005;102:749–754. doi: 10.1073/pnas.0408836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malik R, Khan AP, Asangani IA, Cieslik M, Prensner JR, Wang X, Iyer MK, Jiang X, Borkin D, Escara-Wilke J, Stender R, Wu YM, Niknafs YS, Jing X, Qiao Y, Palanisamy N, Kunju LP, Krishnamurthy PM, Yocum AK, Mellacheruvu D, Nesvizhskii AI, Cao X, Dhanasekaran SM, Feng FY, Grembecka J, Cierpicki T, Chinnaiyan AM. Targeting the MLL complex in castration-resistant prostate cancer. Nat Med. 2015;21:344–352. doi: 10.1038/nm.3830. [DOI] [PMC free article] [PubMed] [Google Scholar]