

Abstract
Dishevelled-Associating Protein with a high frequency of LEucines (DAPLE) belongs to a group of unconventional activators of heterotrimeric G-proteins that are cytoplasmic factors rather than membrane proteins of the G-protein–coupled receptor superfamily. During neurulation, DAPLE localizes to apical junctions of neuroepithelial cells and promotes apical cell constriction via G-protein activation. While junctional localization of DAPLE is necessary for this function, the factors it associates with at apical junctions or how they contribute to DAPLE-mediated apical constriction are unknown. MPDZ is a multi-PDZ (PSD95/DLG1/ZO-1) domain scaffold present at apical cell junctions whose mutation in humans is linked to nonsyndromic congenital hydrocephalus (NSCH). DAPLE contains a PDZ-binding motif (PBM) and is also mutated in human NSCH, so we investigated the functional relationship between both proteins. DAPLE colocalized with MPDZ at apical cell junctions and bound directly to the PDZ3 domain of MPDZ via its PBM. Much like DAPLE, MPDZ is induced during neurulation in Xenopus and is required for apical constriction of neuroepithelial cells and subsequent neural plate bending. MPDZ depletion also blunted DAPLE-mediated apical constriction of cultured cells. These results show that DAPLE and MPDZ, two factors genetically linked to NSCH, function as cooperative partners at apical junctions and are required for proper tissue remodeling during early stages of neurodevelopment.
INTRODUCTION
Epithelial remodeling is crucial for the acquisition of organ and organismal three-dimensional shape, that is, for morphogenesis (Gilmour et al., 2017). It is well established that apical cell constriction is an important driver of epithelial remodeling in the context of embryo morphogenesis across evolutionarily distant organisms (Martin and Goldstein, 2014; Heer and Martin, 2017). For example, apical cell constriction is one of the key events in the formation of the ventral furrow in flies and in the formation of the neural tube in vertebrate model organisms like frogs or chicks (Gilmour et al., 2017; Heer and Martin, 2017; Nikolopoulou et al., 2017). A highly conserved effector machinery that directly acts on the cytoskeleton is responsible for generating the forces required for this process (Gilmour et al., 2017). Rho-dependent signaling pathways converge on promoting contractility of an actomyosin meshwork anchored at apical cell–cell junction complexes. It has been well documented in vertebrate cells that apical cell–cell junctions are key loci for the generation of forces that drive apical cell constriction. More specifically, contraction of cortical actomyosin associated with apical junctions causes the subsequent reduction of the diameter of the apical cell domain, analogous to a “purse-string” mechanism (Heer and Martin, 2017; Yano et al., 2017).
We have recently found a novel mechanism of heterotrimeric G-protein activation that promotes apical cell constriction of epithelial cells during embryonic development (Marivin et al., 2019). Although heterotrimeric G-proteins are primarily activated by membrane proteins of the G-protein–coupled receptor (GPCR) superfamily (Gilman, 1987), we found that the cytoplasmic factor Dishevelled-Associating Protein with a high frequency of LEucines (DAPLE, a.k.a. CCDC88C) triggers G-protein activation during apical cell constriction. DAPLE uses an evolutionarily conserved Gα-binding-and-activating (GBA) motif to activate G-proteins of the Gi subfamily (Aznar et al., 2015; de Opakua et al., 2017; DiGiacomo et al., 2018), which ultimately promotes Rho-dependent actomyosin contractility via a Gβγ-dependent RhoGEF (p114RhoGEF) (Marivin et al., 2019). Importantly, for DAPLE to activate this G-protein–dependent mechanism that drives apical cell constriction, it must be properly localized to apical cell junctions (Marivin et al., 2019). Although the association of DAPLE with apical junctions is mediated by its C-terminal PDZ-binding motif (PBM), the proteins it associates with in this subcellular location, or how they impact its ability to induce apical cell constriction, are unknown.
We were intrigued by multiple PDZ protein (MPDZ, a.k.a. Mupp-1) as a possible binding partner of DAPLE in the context of apical cell constriction. One reason for this is that MPDZ is a scaffold protein localized at apical junction complexes that contains 13 PDZ domains that could potentially serve as docking sites for DAPLE’s PBM (Adachi et al., 2009). What makes MPDZ stand out compared with other PDZ domain–containing proteins as possible partners for DAPLE is the genetic linkage to nonsyndromic congenital hydrocephalus (NSCH) in humans, a neurodevelopmental disorder that results in enlargement of the brain ventricles at birth. MPDZ and CCDC88C (encoding DAPLE) are two of the only four genes (along with L1CAM and AP1S2) whose mutation leads to NSCH (Van Camp et al., 1993; Ekici et al., 2010; Drielsma et al., 2012; Al-Dosari et al., 2013; Saugier-Veber et al., 2017; Shaheen et al., 2017; Ruggeri et al., 2018; Wallis et al., 2018), and recent evidence has confirmed that MPDZ and CCDC88C knockout mice display hydrocephalus (Feldner et al., 2017; Takagishi et al., 2017). Our results here reveal a direct physical interaction between MPDZ and DAPLE and that MPDZ is required for DAPLE-mediated apical cell constriction. Our data also suggest that both proteins might have a similar function during early stages of embryonic development, raising the intriguing question of whether the interplay between DAPLE and MPDZ is of significance for hydrocephalus.
RESULTS AND DISCUSSION
MPDZ colocalizes and coimmunoprecipitates with DAPLE
To begin characterizing a possible relationship between MPDZ and DAPLE, we used previously validated antibodies to investigate whether MPDZ and DAPLE colocalized in cells (Adachi et al., 2009; Aznar et al., 2015; Marivin et al., 2019). Consistent with previous reports (Adachi et al., 2009; Assemat et al., 2013), immunofluorescence staining of endogenous MPDZ in the epithelial cell line EpH4 showed enriched localization at cell–cell boundaries of the apical cell domain, where it partially colocalized with endogenous DAPLE (Figure 1A). To assess whether DAPLE and MPDZ can physically associate with each other in cells, we carried out coimmunoprecipitation (co-IP) experiments. For this, MYC-tagged DAPLE was coexpressed with FLAG-tagged MPDZ in HEK293T cells, and IPs were carried out with anti-FLAG antibodies. Cells expressing MYC-DAPLE in the absence of MPDZ-FLAG were used as a negative control. We found that DAPLE was efficiently immunoprecipitated in the presence of FLAG-MPDZ, whereas the co-IP of another cell–cell junction protein (β-catenin) or an unrelated transmembrane protein (integrin β1) was negligible (Figure 1B). Together, these results suggest that MPDZ and DAPLE bind each other at apical cell junctions.
FIGURE 1:
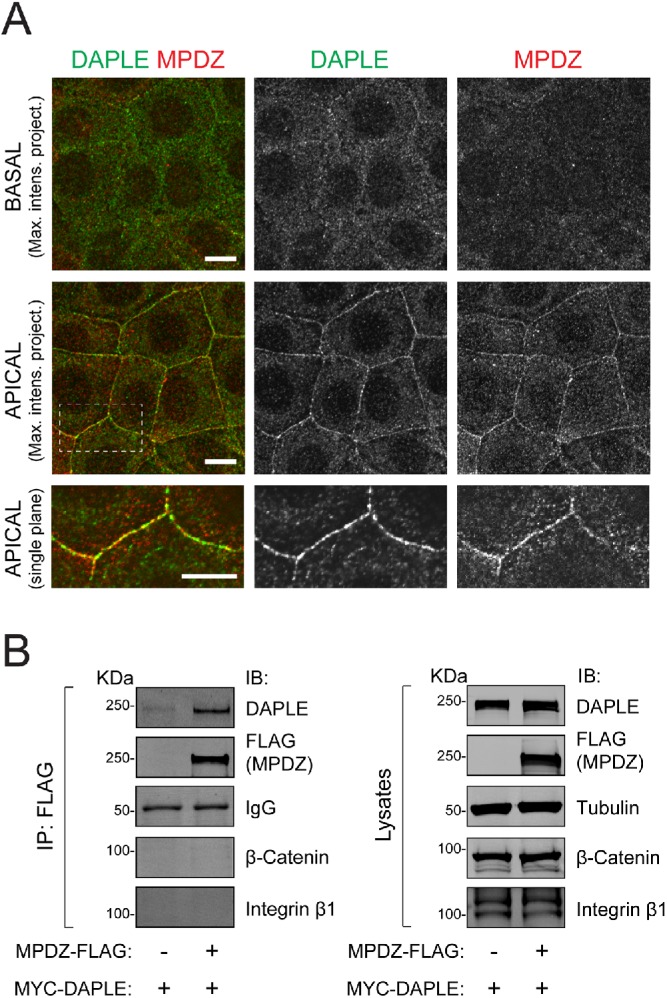
MPDZ colocalizes and coimmunoprecipitates with DAPLE. (A) Confocal fluorescence microscopy pictures of EpH4 cell monolayers stained for endogenous DAPLE (green) and endogenous MPDZ (red) as indicated. The two top rows correspond to maximum-intensity projections of basal and apical domains, respectively, of the cell monolayer viewed from the top. A single confocal plane of the apical area indicated by the dotted box is shown at the bottom. (B) Co-IP experiment showing that MYC-DAPLE, but not β-catenin or integrin β1, binds specifically to MPDZ-FLAG when coexpressed in HEK293T cells. Immunoblots (IB) of the FLAG IPs (left) and aliquots of the starting lysates (right) are shown. All results presented in this figure are representative of n ≥ 3 experiments. Scale bars: 5 µm.
DAPLE binds directly to the PDZ3 domain of MPDZ
Next, we set out to characterize the physical association between DAPLE and MPDZ. We carried out pull-down experiments using lysates of HEK293T cells expressing FLAG-MPDZ and purified DAPLE (aa 1650–2028) fused to glutathione S-transferase (GST). We also assessed the possible effect of deleting the C-terminal PBM of DAPLE (ΔPBM) (Figure 2A). The results revealed that GST-DAPLE wild type (WT), but not the ΔPBM mutant, binds to MPDZ (Figure 2B). Prompted by this result, we mapped which of the 13 PDZ domains of MPDZ (Figure 2A) mediates the interaction. For this, we made a series of MPDZ mutants that sequentially truncated the protein from the C-terminus, resulting in constructs that contained PDZ1-4 (aa 1–700), PDZ1-3 (aa 1–550), or PDZ1-2 (aa 1–370) domains. GST-DAPLE pull downs with lysates of HEK293T cells expressing these constructs showed that MPDZ proteins containing PDZ1-4 and PDZ1-3 bound similar to full-length MPDZ, whereas binding of the construct containing PDZ1-2 was undetectable (Figure 2C). This result suggests that PDZ3 is the binding site for DAPLE. To further substantiate this point, we followed a previously described strategy to disrupt PBM–PDZ interactions (Daniels et al., 1998; Joberty et al., 2000) based on mutating several residues in the carboxylate-binding loop within the PBM-binding pocket of PDZ3 (Q385A/G386A/L387A/G388A, PDZ3*). When this mutation was introduced in the backbone of full-length MPDZ, it completely disrupted binding to GST-DAPLE in pull-down experiments with cell lysates (Figure 2D). This indicates that PDZ3, but not any other PDZ domain of MPDZ, mediates the association of MPDZ with DAPLE. To rule out the possibility that the interaction between DAPLE and MPDZ is mediated by an intermediary protein, we tested whether their binding was direct by using purified proteins. Also, to mitigate concerns related to the presence of degradation products in the GST-DAPLE construct used in previous experiments (aa 1650–2028), we carried out these experiments with a shorter C-terminal fragment of DAPLE (aa 1908–2028) that yielded a better quality protein when purified as a GST-fused construct (i.e., with fewer degradation products). Purified His-MPDZ (aa 1–700, PDZ1-4) bound to purified GST-DAPLE, and this binding was disrupted when the PDZ3* mutation was introduced in His-MPDZ (Figure 2E). Taken together, these results demonstrate that DAPLE and MPDZ bind directly to each other and map the interaction to the PBM of DAPLE and the PDZ3 domain of MPDZ.
FIGURE 2:

DAPLE binds directly to the PDZ3 domain of MPDZ. (A) Bar diagram depicting DAPLE and MPDZ domains. (B) Protein–protein-binding experiment showing that full-length FLAG-MPDZ from HEK293T cell lysates binds to immobilized GST-DAPLE (aa1650–2028) WT but not to the ΔPBM mutant. Excess of GST-DAPLE ΔPBM compared with WT was used to ensure that lack of binding was not due to an insufficient amount of the immobilized ligand. (C) GST-DAPLE binds MPDZ full-length (FL), MPDZ-PDZ1-4 and MPDZ-PDZ1-3, but not MPDZ-PDZ1-2. The indicated FLAG-MPDZ constructs were expressed in HEK293T cells, and their corresponding lysates were used in pull downs with purified GST-DAPLE. (D) Mutation of the PDZ3 domain (PDZ3*) disrupts binding of MPDZ to GST-DAPLE. Full-length FLAG-MPDZ WT and PDZ3* mutant were expressed in HEK293T cells, and their corresponding lysates were used in pull downs with purified GST-DAPLE (aa1908–2028). (E) Purified MPDZ-PDZ1-4 binds directly to GST-DAPLE via its PDZ3 domain. Purified His-MPDZ-PDZ1-4 WT or PDZ3* mutant were incubated with purified GST-DAPLE (aa1908–2028) immobilized on glutathione–agarose resin. For all experiments, GST-fused proteins used as the immobilized ligands were visualized by Ponceau S staining, whereas soluble ligands were detected by immunoblotting (IB). All results presented are representative of n ≥ 3 experiments.
Loss of MPDZ causes apical cell constriction defects during Xenopus neurulation
Having established that MPDZ can bind directly to DAPLE, we set out to investigate whether it also shared cell biological functions. For this, we turned to Xenopus laevis as a model, as we have recently found that, in this system, loss of DAPLE impairs apical constriction of neuroepithelial cells during neurulation (Marivin et al., 2019). We also showed that this function of DAPLE requires an intact PBM motif (Marivin et al., 2019). Analysis of publicly available RNA-seq data (Peshkin et al., 2015) revealed that the time course of Xenopus (x)MPDZ mRNA expression during embryo development closely resembles that of Xenopus DAPLE (xDAPLE a.k.a. xDal), as both of them are virtually absent at fertilization and then become sharply induced during neurulation (Figure 3A). Interestingly, the close homologue of MPDZ named Pals-Associated Tight Junction protein (PATJ a.k.a. INADL) presents a completely different time course of expression, that is, mRNA is present at high levels at fertilization (maternally inherited) and then is cleared out at neurulation (Figure 3A). Thus, although PATJ might have redundant functions with MPDZ in mammalian cell lines (Adachi et al., 2009; Assemat et al., 2013), it is unlikely that this is the case during Xenopus neurulation, because only MPDZ appears to be expressed in this context. Based on this, we tested the hypothesis that depletion of xMPDZ alone might phenocopy the neurulation defects that occur upon loss of xDAPLE (Marivin et al., 2019). Unilateral microinjection of a morpholino (MO1) designed to block the translation of xMPDZ caused a delay in the closure of the neural tube, as determined by direct comparison with the uninjected control side at stage 17 (Figure 3B). A similar phenotype was also observed upon injection of a second morpholino (MO2) against a nonoverlapping sequence of xMPDZ that is predicted to interfere with RNA splicing at the exon5–intron5 boundary (Figure 3B). Next, we characterized the cytoarchitecture alterations in neuroepithelial cells associated with this phenotype. For this, embryos were unilaterally coinjected with xMPDZ MO1 and an mRNA-encoding membrane-bound red fluorescent protein (mRFP) as a lineage tracer (Figure 3C). xMPDZ MO1–targeted cells were identified by detection of RFP signal. We found that the apical area of neuroepithelial cells lining the outer limit of the neural plate in the MO-injected side was enlarged compared with the uninjected side at stage 15 (onset of neural plate bending) and at stage 16 (when the bending of the neural plate is more marked) (Figure 3C), which suggests a defect in apical cell constriction. Furthermore, these cells also failed to acquire a characteristic wedge shape that results from apical constriction during neural plate bending (Figure 3D). In contrast, MO-treated cells showed normal distribution of the cell junctional markers ZO-1 and β-catenin, which normally appear concentrated apically or along the cell–cell boundaries, respectively (Figure 3, D and E), suggesting that the observed apical constriction defects are not due to overt disruption of epithelial junction integrity. These cytoarchitectural features closely resemble those observed upon loss of xDAPLE in the same experimental system (Marivin et al., 2019). Thus, there is a strong correlation between the early development phenotypes associated with loss of xDAPLE and loss of xMPDZ.
FIGURE 3:

Loss of MPDZ causes apical cell constriction defects during Xenopus neurulation. (A) Quantification of DAPLE, MPDZ, and PATJ mRNA abundance in whole X. laevis embryos at different stages by RNAseq (extracted from Peshkin et al., 2015). xMPDZ and xDAPLE mRNAs are induced at neurulation, whereas PATJ mRNA is abundant at fertilization and is cleared out at neurulation. (B) Quantification of neural plate bending defects in embryos unilaterally injected with xMPDZ MOs. n = 50–100 embryos/condition analyzed at stage 17; ***, p < 0.001 using the χ2 test. Images of a representative embryo phenotypes are shown on the left. (C) Whole-mount F-actin staining (green) of Xenopus embryos unilaterally coinjected with xMPDZ MO1 and a lineage tracer (mRFP, magenta) showing enlarged apical surface of DAPLE-depleted neuroepithelial cells compared with uninjected control sides at stage 15 and stage 16. Red dashed boxes indicate the areas enlarged on the adjacent right panels. (D) Transverse view of the anterior neural plate of a stage 16 embryo stained with β-catenin (magenta) after unilateral coinjection with xMPDZ MO1 and a lineage tracer (GFP-CAAX, green). (E) Transverse view of the anterior neural plate of a stage 15 embryo stained with ZO-1 (magenta) after unilateral coinjection with xMPDZ MO1 and a lineage tracer (GFP-CAAX, green). All images presented in this figure are representative results of n ≥ 3 experiments. Scale bars: 250 µm (B); 25 µm (all other panels).
Dominant-negative disruption of DAPLE-MPDZ binding impairs DAPLE localization at apical cell junctions
We have previously found that deletion of DAPLE’s PBM disrupts its localization at cell–cell junctions and DAPLE-mediated apical cell constriction (Marivin et al., 2019). Because MPDZ can bind directly to DAPLE’s PBM (Figure 2), we reasoned that it might be required for DAPLE localization and function at apical cell junctions. To begin testing this idea, we investigated whether transient overexpression of a truncated, N-terminally tagged MPDZ construct lacking PDZ domains 5 through 13 (MYC-MPDZ-PDZ1-4) would disrupt DAPLE localization at cell–cell junctions in EpH4 cells. Similar N-terminally tagged MPDZ constructs have been shown to localize diffusely in the cytosol and exert dominant-negative effects in similar contexts (Adachi et al., 2009). As expected, MYC-MPDZ-PDZ1-4 localized diffusely in the cytosol rather than at cell–cell boundaries (Figure 4A). Quantification of fluorescence intensity revealed that the signal for DAPLE at the boundaries between cells overexpressing MYC-MPDZ-PDZ1-4 was reduced ∼40% compared with the signal at cell–cell boundaries of cells not expressing MYC-MPDZ-PDZ1-4 in the same field (Figure 4, A and B). On the other hand, the construct bearing the DAPLE binding–deficient PDZ3* mutation failed to reproduce this effect (Figure 4, A and B). The incomplete loss of DAPLE localization at cell junctions could be attributed to insufficient expression of the dominant-negative WT construct and/or the existence of MPDZ-independent mechanisms for targeting DAPLE to cell junctions. Nevertheless, these results suggest that MPDZ binding might be required for the optimal localization and function of DAPLE at apical cell junctions.
FIGURE 4:

MPDZ is required for DAPLE-induced apical cell constriction. (A) Representative fluorescence microscope pictures of EpH4 cells sparsely expressing MYC-MPDZ-PDZ1-4 and costained for MYC (magenta) and DAPLE (green). (B) Quantification of DAPLE at cell–cell boundaries of EpH4 cells expressing MYC-MPDZ-PDZ1-4 (FMYC-MYC) relative to the signal in control cells (FControl). Results are presented as box-and-whisker plots of n = 3 independent experiments per condition. The “+” indicates the average. ***, p < 0.001 using the Mann-Whitney test. (C) Diagram depicting the assay used to quantify the apical cell constriction induced by expression of DAPLE. The “relative apical area” of MYC-DAPLE-transfected cells is calculated by dividing the area of the DAPLE-expressing cell by the average of the area of the adjacent cells. (D, E) Validation of shRNA-mediated depletion of MPDZ and PATJ in EpH4 cells. Cell lines stably expressing the indicated shRNAs were generated by lentiviral transduction followed by selection, and the reduction in MPDZ and PATJ protein expression was confirmed by immunoblotting (“IB” in D) and immunofluorescence staining (E). (F) Quantification of the relative apical area of DAPLE-transfected cells compared with neighboring, untransfected cells shows that depletion of MPDZ or MPDZ and PATJ impairs DAPLE-induced apical cell constriction. Representative fluorescence microscopy pictures of the indicated EpH4 cell lines sparsely expressing MYC-DAPLE and costained for MYC (magenta) and ZO-1 (green) are shown on top, and a graph with the quantification data across independent experiments is shown on the bottom. Results are presented as box-and-whisker plots of n = 4 independent experiments per condition. The “+” indicates the average. *, p < 0.05 using the Mann-Whitney test. (G) Whole-mount staining (MYC-xDAPLE, green; mRFP, magenta) of Xenopus embryos bilaterally injected with xDAPLE mRNA and unilaterally coinjected with xMPDZ MO1 and a lineage tracer (mRFP). (H) Confocal fluorescence microscopy pictures of monolayers of EpH4 cell lines depleted of MPDZ or MPDZ/PATJ show no difference in endogenous DAPLE staining compared with control cells. Maximum-intensity projection views of the apical domain of the cell monolayers are shown. All images presented in this figure are representative results of n ≥ 3 experiments. Scale bars: 25 µm (G); 5 µm (all others).
MPDZ is required for DAPLE-induced apical cell constriction
To determine more directly whether MPDZ affects DAPLE-mediated apical cell constriction, we used a previously established assay in cultured epithelial cells. Essentially, we have previously shown that ectopic expression of DAPLE in epithelial cell lines mimics the natural induction of DAPLE expression in neuroepithelial cells during neurulation and demonstrated its consequence of promoting apical cell constriction (Marivin et al., 2019). In this assay, DAPLE is sparsely expressed in a cell monolayer, and apical constriction is quantified by calculating the ratio between the apical area of the cells expressing DAPLE and its neighboring cells (Figure 4C). We generated a stable EpH4 cell line with knocked-down expression of MPDZ (Figure 4, D and E) by using a previously validated short hairpin RNA (shRNA) sequence (Kruse et al., 2014) and assessed the ability of DAPLE to induce apical constriction in these cells. We found that, compared with cells stably expressing a control shRNA, MPDZ-depleted cells failed to undergo apical cell constriction upon expression of DAPLE (Figure 4F). To our surprise, DAPLE still localized predominantly to cell–cell junctions in the absence of MPDZ (Figure 4F). This is surprising, because deletion of DAPLE’s PBM, which binds directly to MPDZ (Figure 2), disrupts both apical cell constriction and localization at cell–cell junctions (Marivin et al., 2019) and because MYC-MPDZ-PDZ1-4 WT, but not the DAPLE binding–deficient PDZ3*, disrupts DAPLE localization (Figure 4, A and B). Similarly, we found that the apical junction localization of MYC-xDAPLE ectopically expressed in the neural plate of Xenopus embryos (available antibodies cannot detect endogenous xDAPLE) was not disrupted in MPDZ morphants despite clear defects in apical constriction (Figure 4G). Taken together, these results indicate that MPDZ is required for apical cell constriction induced by DAPLE, while it might not be essential for its localization at apical cell junctions.
MPDZ depletion does not impair DAPLE localization at apical cell junctions
To rule out that the apparent localization of ectopically expressed DAPLE in cell–cell junctions observed in MPDZ-depleted cells and embryos was due to overexpression, we investigated the localization of endogenous DAPLE. We found that endogenous DAPLE localization is not significantly altered in MDPZ-depleted EpH4 cells compared with control cells (Figure 4H). One possible explanation for this result is that DAPLE still localizes at apical junctions due to a compensatory role of PATJ, as it has been previously reported that MPDZ and PATJ might have redundant functions in cell lines (Adachi et al., 2009; Assemat et al., 2013). To address this issue, we generated a new stable EpH4 cell line depleted of both MPDZ and PATJ (Figure 4, D and E) by using a previously validated PATJ shRNA sequence (Adachi et al., 2009) in MPDZ-depleted cells. Not surprisingly, EpH4 depleted of both MPDZ and PATJ failed to undergo apical cell constriction upon expression of DAPLE (Figure 4F), much like in the cell line depleted of only MPDZ. The localization of ectopically expressed (Figure 4F) or endogenous (Figure 4H) DAPLE was also unchanged in these cells, where it remained concentrated at apical cell junctions.
There are several, non–mutually exclusive possible explanations for the unaltered localization of DAPLE upon loss of MPDZ (and PATJ). While dominant-negative experiments (Figure 4, A and B) suggest that DAPLE localization at apical junctions might rely on MPDZ binding, the incomplete loss of DAPLE from junctions also suggested the existence of other redundant mechanisms to target DAPLE to junctions. Such mechanisms could compensate for the loss of MPDZ through adaptation, especially considering the length of the process to generate MPDZ-depleted stable cell lines compared with the transient transfection experiments with dominant-negative MPDZ (Figure 4, A and B). The incomplete depletion of MPDZ in EpH4 cells (Figure 4D) might also contribute to masking of any possible effect on the localization of DAPLE. Regardless of the possible explanation for the unchanged localization of DAPLE, what is clear is that MPDZ-depleted cells still fail to undergo DAPLE-mediated apical constriction (Figure 4F). To reconcile our results, we propose that localization of DAPLE at apical junctions is necessary but not sufficient to promote apical cell constriction. While other PDZ domain–containing proteins are likely to contribute to the proper targeting of DAPLE to apical junctions (as DAPLE ΔPBM does not localize to junctions; Marivin et al., 2019), our findings suggest that association with MPDZ is specifically required to form a complex competent to transmit the signal triggered by DAPLE to promote apical constriction (Marivin et al., 2019).
Conclusions and future perspectives
The main finding of this brief report is the identification of a physical and functional interaction between DAPLE and MPDZ, which work cooperatively to control apical cell constriction and influence the proper course of an early stage of development such as neurulation in Xenopus embryos. This is significant for two reasons. One is that it identifies MPDZ as a factor that contributes to the function of DAPLE at apical cell junctions, potentially providing spatial specificity to the unconventional mechanism of heterotrimeric G-protein signaling triggered by DAPLE. Although additional investigation will be required to elucidate the mechanism by which MPDZ impacts DAPLE function, it is possible that MPDZ’s role is to act as a scaffold that brings together DAPLE and other proteins involved in apical cell constriction to facilitate the process. On the other hand, MPDZ might not be essential for the localization of DAPLE at apical junctions, suggesting that it is the nature of the specific molecular complex at apical junctions rather than just subcellular localization that determines its function of promoting apical constriction. A question left open by this work is the identity of other PDZ domain–containing proteins that contribute to the targeting of DAPLE to apical cell junctions. Ongoing work in our laboratory is pursuing this avenue of research.
The second significant observation derived from our findings relates to a possible function of the MPDZ/DAPLE complex in the etiology of NSCH. The mainstream thinking on the etiology of hydrocephalus is that it is caused by defects in the multiciliated ependymal cells that line the brain ventricles and clear excess cerebrospinal fluid (Vogel et al., 2012), although there are other possible causes, like cytoarchitectural defects at early stages of development (Ma et al., 2007; Rodriguez et al., 2012; Rodriguez and Guerra, 2017; Guerra et al., 2015). Our findings reveal that both MPDZ and DAPLE, possibly in cooperation, affect very early stages of neurodevelopment such as neurulation. It will be of interest for future investigations to assess whether alterations in early development upon loss of MPDZ/DAPLE contribute to NSCH, or whether dysregulation of the same complex at later stages of development also participates in the onset of hydrocephaly.
MATERIALS AND METHODS
Plasmids and morpholinos
The full-length cDNA of human MPDZ was obtained from Harvard repository plasmid database clone HsCD00399210 (encoding the protein sequence of Uniprot O75970-3) and cloned into the plasmids p3XFLAG-CMV10 at NotI/EcoRI sites or p3XFLAG-CMV14 NotI/EcoRI sites to generate N-terminally (FLAG-MPDZ) or C-terminally (MPDZ-FLAG) FLAG-tagged constructs, respectively. The plasmids for expression of human MYC-DAPLE (pCS2-6XMYC-hDAPLE) and GST-DAPLE (aa 1650–2028, pGEX-4T-1-DAPLE-CT) have been described previously (Aznar et al., 2015; Marivin et al., 2019). Plasmids for bacterial production of His-MPDZ-PDZ1-4 (aa 1–700, pLIC-His-MPDZ-PDZ1-4) or GST-DAPLE (aa 1908–2028, pLIC-GST-DAPLE-1908-2028) were generated by ligation-independent cloning (LIC) (Stols et al., 2002) of PCR-amplified fragments into pLIC-His (a.k.a. pMCSG7) (Stols et al., 2002) or pLIC-GST (kindly provided by J. Sondek, University of North Carolina–Chapel Hill) (Cabrita et al., 2006), respectively. The plasmid used for the expression of full-length MYC-xDAPLE was kindly provided by T. Michiue (University of Tokyo) (Kobayashi et al., 2005). Plasmids for the expression of GFP-CAAX (pCS2-eGFP-CAAX) and membrane(m)RFP (pCS2-mRFP) in Xenopus were a gift from S. Sokol (Icahn School of Medicine at Mount Sinai) (Ossipova et al., 2015). Site-directed mutagenesis in DAPLE and MPDZ was carried out using QuikChange II (Agilent). DAPLE ΔPBM and MPDZ truncation mutants were generated by introducing stop codons, and the multisite PDZ3* mutant was generated in two mutagenesis steps. Plasmids for the expression of shRNAs were generated by annealing complementary primers encoding the hairpin and designed to have overhanging sequences complementary to the AgeI/EcoRI sites of pLKO.1 plasmids. shRNA hairpins for a previously validated sequence targeting mouse MPDZ (GCCTTCAGGAATCTTTGTAAA, MPDZ shRNA) (Kruse et al., 2014) and for a nontargeting sequence (TGGAGTACAACTACAACAGCCA, control shRNA) were cloned into pLKO.1-puro (Addgene 8453), whereas the shRNA hairpin for a previously validated sequence targeting mouse PATJ (GAGACGAGCTGCTAGAGAT, PATJ shRNA) (Adachi et al., 2009) was cloned into pLKO.1-blast (Addgene 26655).
The following MOs were purchased from Gene Tools: xMPDZ MO1 (translation start site [TSS]-15/+11nt: 5′-GTTTCCAACATGGTTTTCAGTAGTG-3′), xMPDZ MO2 (targeting exon5–intron5 boundary 5′-TGAGGATTTCAGTACTTACAGGACT-3′), and control MO (5′-CCTCTTACCTCAGTTACAATTTATA-3′).
Cell culture, transfections, and generation of stable cell lines
HEK293T (ATCC CRL3216) and EpH4 (from X. Varelas, Boston University) were grown at 37°C, 5% CO2 in DMEM supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 µg/ml streptomycin, and 1% l-glutamine. HEK293T cells were transfected using the calcium phosphate method. EpH4 cells were seeded on glass coverslips placed in 24-well plates at a density of 250,000 cells per well. Next day, cells were transfected with 1 µg of plasmid DNA encoding MYC-DAPLE using Lipofectamine LTX (Life Technologies 15338100) following the manufacturer’s instructions. At 30 h after transfection, confluent cell monolayers were processed for immunofluorescence staining as described in Immunofluorescence analysis of cultured cells.
EpH4 stable cell lines were generated by lentiviral transduction followed by selection with the appropriate antibiotics. Briefly, lentivirus packaging was performed in HEK293T cells by cotransfection of the lentiviral plasmid of interest with the packaging plasmid psPAX2 and a VSV-G–encoding plasmid at a 1:1:0.5 ratio. Approximately 6 h after transfection, the media was changed, and ∼42 h later, lentivirus-containing media were collected, centrifuged at 500 × g for 3 min, and filtered through a 0.45-µm filter. EpH4 cells were incubated with supernatants of pLKO.1-puro-packaged MPDZ shRNA or control shRNA lentivirus (mixed 1:1 with fresh media) for 2 d, followed by selection with 5 µg/ml puromycin. All surviving clones were pooled and maintained in the presence of 5 µg/ml puromycin. MPDZ knockdown cells were used as the starting point to knock down PATJ. Essentially, they were incubated with supernatants of pLKO.1-blast-packaged PATJ shRNA lentivirus as described above and subjected to double selection with 4 µg/ml blasticidin and 5 µg/ml puromycin. All surviving clones were pooled and maintained in the presence of both antibiotics.
Immunofluorescence analysis of cultured cells
Cells grown on glass coverslips were fixed in 100% methanol at −20°C for 10 min, washed three times with phosphate-buffered saline (PBS), and incubated in blocking solution (10% [vol/vol] normal goat serum, 0.1% [vol/vol] Triton X-100 in PBS) for 1 h. For endogenous DAPLE immunostaining (Figures 1A and 4H), the blocking solution described above was supplemented with 2% bovine serum albumin (BSA). Cells were incubated overnight at 4°C in blocking solution with the following primary antibodies: rabbit polyclonal MYC (Sigma C3956, 1:300), rabbit polyclonal DAPLE (Millipore ABS515, 1:100), rabbit polyclonal PATJ (provided by A. Le Bivic, Aix-Marseille University, 1:400); mouse monoclonal MYC (Cell Signaling Technology no. 2276 [9B11], 1:200), mouse monoclonal MPDZ (BD Transduction Laboratories no. 611558, 1:100); and rat monoclonal ZO1 (Developmental Studies Hybridoma Bank [DSHB] R26.4c, 1:100). Coverslips were washed three times in PBS and incubated in secondary antibodies for 1 h at room temperature in blocking solution. The secondary antibodies used were goat anti-mouse Alexa Fluor 488 (Invitrogen A11017, 1:400) and goat anti-rabbit Alexa Fluor 594 (Invitrogen A11072, 1:400) and goat anti-rat Alexa Fluor 647 (Invitrogen A21247, 1:400) diluted in blocking solution. Coverslips were mounted in ProLong Diamond Antifade (Invitrogen P36965) and imaged by fluorescence microscopy.
For experiments to determine the localization of endogenous DAPLE (Figures 1A and 4, A, B, and H), a Zeiss LSM 700 microscope was used. Stacks of confocal pictures of 0.39-µm thickness along the z-axis were taken with a 63× oil-immersion objective (numerical aperture [NA] = 1.4; working distance = 0.19 mm) using ZEN software. Maximum-intensity projections (as indicated in figure legends) were generated with ImageJ, and images were adjusted for brightness/contrast and assembled for presentation using Photoshop and Illustrator software (Adobe). The quantification of endogenous DAPLE localization upon overexpression of dominant-negative MPDZ (Figure 4, A and B) was carried out using ImageJ (National Institutes of Heath [NIH]). Lines of 30-pixel width were drawn orthogonal and centered on a cell–cell boundary. The peak value of the fluorescence intensity signal of DAPLE at the cell–cell boundaries of adjacent cells expressing MYC-MPDZ-PDZ1-4 (FMYC-MYC) was extracted from the ImageJ plot profile. This value was normalized by dividing it by the average of the peak-intensity value of DAPLE signal in 4−6 cell–cell boundaries of cells not expressing MYC-MPDZ-PDZ1-4 in the same image field (FControl). Results are presented as box-and-whisker plots (min to max) of 19−25 images analyzed from three independent experiments. Statistical p values were calculated with the Mann-Whitney test in Prism (GraphPad). For each experiment, the expression of MYC-MPDZ-PDZ1-4 constructs was within the same order of magnitude as the average fluorescence intensity of WT. The average fluorescence intensity of MYC-MPDZ-PDZ1-4 PDZ3* mutant was not significantly different from that of WT (p > 0.05, Mann-Whitney test).
For experiments to assess apical constriction of cells (Figure 4F), a Zeiss Axio Observer Z1 fluorescence microscope equipped with a digital camera (C10600/ORCA-R2 Hamamatsu Photonics) was used. Images were taken with a 63× oil-immersion objective (NA = 1.4; working distance = 0.19 mm) using ZEN software. Cells were costained for MYC (MYC-DAPLE) and ZO-1 as described above. Random fields containing MYC-positive cells were selected, and images of the apical cell domain were acquired by bringing into focus the ZO-1 signals. Image analysis was done with ImageJ, and the apical areas of MYC-DAPLE-expressing and neighboring cells (defined by shared cell–cell apical boundaries) were determined by delineating the cell boundaries manually or by using the “magic wand” plug-in when possible. The relative apical area was calculated by dividing the apical area of the MYC-positive cell by the average of the apical area of its neighboring cells (as represented in Figure 4C). MYC-positive cells in direct contact with another MYC-positive cell or overexpressing high levels of MYC-DAPLE in aggregate-like structures were excluded from the analysis. Results are presented as box-and-whisker plots (min to max) of the median relative apical area of individual experiments (18–25 cells analyzed per experiment). Statistical p values were calculated with the Mann-Whitney test in Prism (GraphPad).
Immunoprecipitation
Approximately 3 million HEK293T cells were seeded on 10-cm dishes and transfected the following day using calcium phosphate with plasmids encoding MYC-DAPLE (8 µg) and MPDZ-FLAG (8 µg) or MYC-DAPLE (8 µg) and an empty plasmid (8 µg). At 24 h after transfection, confluent cell monolayers were washed once with PBS and lysed on ice by adding 300 µl of RIPA buffer (pH 7.4, 20 mM Tris-Cl, 150 mM NaCl, 1% [vol/vol] NP-40, 0.5% [wt/vol] sodium deoxycholate, 0.1% [wt/vol] SDS, 10 mM β-glycerophosphate, 0.5 mM Na3VO4, 1 mM dithiothreitol [DTT], and a protease inhibitor cocktail [Sigma S8830]). These lysates were rapidly passed through a 30G insulin syringe (3–4 strokes), and immediately diluted with 1.2 ml of a buffer containing TX-100 as the only detergent (“binding buffer” pH 6.8, 10 mM PIPES, 300 mM sucrose, 50 mM NaCl, 3 mM MgCl2, 0.5% [vol/vol] Triton X-100, 10 mM β-glycerophosphate, 0.5 mM Na3VO4, 1 mM DTT, and a protease inhibitor cocktail [Sigma S8830]). Lysates were syringed 5–10 times in ice and cleared twice (14,000 × g for 10 min, then 21,000 × g for 3 min). Cleared lysates were incubated with 2 μg of FLAG antibodies (Sigma F1804) for 2.5 h at 4°C with constant tumbling. Thirty-five microliters of protein G agarose beads preblocked with 5% (wt/vol) BSA (2 h at room temperature) was added to the tubes and incubated for 90 min at 4°C. Beads were washed three times in binding buffer, and resin-bound proteins were eluted by adding Laemmli sample buffer and boiling for 5 min. Proteins were separated by SDS–PAGE and immunoblotted with antibodies as indicated in Immunoblotting.
Immunoblotting
Proteins were separated by SDS–PAGE and transferred to polyvinylidene fluoride (PVDF) membranes, which were sequentially incubated with primary and secondary antibodies. The primary antibodies were used at the following dilutions: MYC (Cell Signaling Technology no. 2276 [9B11], 1:1000), MPDZ (BD Transduction Laboratories no. 611558, 1:1000), β-catenin (BD Transduction Laboratories no. 610153, 1:1000), integrin β1 (Cell Signaling Technology no. 9699, 1:1000), PATJ (provided by A. Le Bivic, Aix-Marseille University, 1:400), His (Sigma H1029, 1:1,2500), FLAG (Sigma F1804, 1:2,000), α-tubulin (Sigma T6074, 1:2500). The secondary antibodies were goat anti-rabbit Alexa Fluor 680 (Invitrogen A21077, 1:10,000) and goat anti-mouse IRDye 800 (Li-Cor Biosciences 926-32210, 1:10,000). Infrared imaging of immunoblots was performed using an Odyssey Infrared Imaging System (Li-Cor Biosciences). Images were adjusted for brightness/contrast and assembled for presentation using ImageJ (NIH), Photoshop (Adobe), and Illustrator software (Adobe).
Protein expression and purification
His-tagged and GST-tagged proteins were expressed in BL21(DE3) Escherichia coli (Life Technologies) transformed with the corresponding plasmids by overnight induction at 23°C with 1 mM isopropyl-β-d-1-thio-galactopyranoside. Protein purification was carried out following previously described protocols (Garcia-Marcos et al., 2009, 2010). Briefly, bacteria pelleted from 1 l of culture were resuspended in 25 ml of buffer (50 mM NaH2PO4, pH 7.4, 300 mM NaCl, 10 mM imidazole, 1% [vol/vol] Triton X-100 supplemented with protease inhibitor cocktail [leupeptin 1 µM, pepstatin 2.5 µM, aprotinin 0.2 µM, phenylmethylsulfonyl fluoride 1 mM]). After sonication (four cycles, with pulses lasting 20 s/cycle, and with a 1-min interval between cycles to prevent heating), lysates were centrifuged at 12,000 × g for 20 min at 4°C. The soluble fraction (supernatant) of the lysate was used for affinity purification on HisPur Cobalt or Glutathione Agarose resins (Pierce) and eluted with lysis buffer supplemented with 250 mM imidazole or with 50 mM Tris-HCl (pH 8), 100 mM NaCl, and 30 mM reduced glutathione, respectively. Proteins were dialyzed overnight at 4°C against PBS. All protein samples were aliquoted and stored at −80°C.
In vitro protein-binding assays with GST-fused proteins
GST-fused DAPLE proteins (30 µg of total protein) or a matching amount of GST were immobilized on glutathione-agarose beads in PBS for 90 min at room temperature. Beads were washed twice with PBS, resuspended in 300 µl of binding buffer (50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 0.4% [vol/vol] NP-40, 10 mM MgCl2, 5 mM EDTA, 2 mM DTT), and incubated 4 h at 4°C with constant tumbling in the presence of purified His-MPDZ-PDZ1-4 (3.5 µg) or lysates of HEK293T cells expressing FLAG-MPDZ. For the preparation of lysates used as the source of the soluble ligand, HEK293T cells expressing FLAG-MPDZ were washed with PBS, scraped in PBS, and pelleted before resuspension in cold lysis buffer (20 mM HEPES, pH 7.2, 5 mM Mg(CH3COO)2, 125 mM K(CH3COO), 0.4% [vol/vol] Triton X-100, 1 mM DTT, 10 mM β-glycerophosphate, and 0.5 mM Na3VO4 supplemented with a protease inhibitor cocktail [Sigma S8830]) and clearing (14,000 × g, 10 min). Approximately one-third of a p10 dish transfected with 10 µg of DNA by the calcium phosphate method was used for each pull-down condition. Beads were washed four times with 1 ml of wash buffer (4.3 mM Na2HPO4, 1.4 mM KH2PO4, pH 7.4, 137 mM NaCl, 2.7 mM KCl, 0.1% [vol/vol] Tween-20, 10 mM MgCl2, 5 mM EDTA, 1 mM DTT), and resin-bound proteins were eluted with Laemmli sample buffer by incubation at 37°C for 10 min. Proteins were separated by SDS–PAGE, transferred to PVDF membranes, and stained with Ponceau S or immunoblotted as indicated in Immunoblotting.
Xenopus embryo culture, microinjections, and morphological analyses
Frog studies were carried out with WT Xenopus laevis animals (Nasco) according to the Boston University Institutional Animal Care and Use Committee’s (IACUC) approved protocol AN14092, in compliance with the Guide for the Care and Use of Laboratory Animals. Egg laying was induced by dorsal lymph injection of 600 U of human chorionic gonadotrophin (Merck 028938). In vitro fertilization and embryo culture were carried out in 0.1× Marc’s Modified Ringer’s medium as previously described (Newport and Kirschner, 1982). Dejellied embryos were used in all of the experiments described below.
mRNAs for injections in frog embryos were prepared using the SP6 mMessage mMachine Kit (Ambion cat no. AM1340). MOs and mRNAs were injected dorsally at the 2- to 4-cell stage to target the presumptive neuroectoderm in a single (unilateral injections) or both blastomeres (bilateral injections), as indicated in the figure legends, and cultured at 16°C. Morphological analyses to score neural plate bending defects were performed on embryos manually devitellinized with blunt-ended fine forceps with a Leica MZ6 dissection microscope. The total number of embryos analyzed per condition is indicated in the figures, and they were from at least four independent clutches from different females obtained on at least two different days. All the pictures of whole Xenopus embryos were taken with a Canon XSi camera connected to a Leica MZ6 dissection microscope.
Fluorescence microscopy analysis of Xenopus embryos
For whole-mount F-actin staining (Figure 3C), embryos were fixed in MEMFA for 2 h and washed three times in PBT (PBS supplemented with 0.1% [vol/vol] Tx-100) before dissecting the neural plates to be used in subsequent steps. Solutions for staining F-actin in neural plates were prepared by evaporating the methanol in Alexa Fluor 488–conjugated phalloidin (Cytoskeleton, PHDG1-A) stocks at 37°C followed by reconstitution in PBT at a final concentration of 0.28 µM Alexa Fluor 488–conjugated phalloidin. Neural plates were incubated overnight at 4°C in staining solution, washed three times for 20 min with PBT and imaged in PBS in homemade chambers built as previously described (Wallingford, 2010) with a Zeiss LSM 700 laser-scanning inverted confocal microscope controlled by the manufacturer’s software. Pictures of 2-µm thickness were taken with a 40× objective (NA = 1.3; working distance = 0.21 mm) with a digital 0.5× zoom. Images are presented as maximum-intensity projections (generated with ImageJ) of views from the top or transversal optical sections (as indicated in the figure legends). Images were adjusted for brightness/contrast and assembled for presentation using Photoshop and Illustrator software (Adobe).
For whole-mount immunostaining of β-catenin and ZO-1 (Figure 3, D and E), embryos were fixed in MEMFA as described above. For whole-mount immunostaining of MYC-xDAPLE (Figure 4G), embryos were fixed in 2% (vol/vol) trichloroacetic acid solution for 30 min at room temperature and washed three times with 0.3% (vol/vol) Triton X-100 in PBS for 30 min (Ossipova et al., 2015). Neural plates were dissected and incubated in blocking solution (PBS supplemented with 0.1% [vol/vol] Triton X-100 and 10% [vol/vol] normal goat serum). Neural plate explants were incubated with the following primary antibodies overnight in blocking solution: ZO-1 (rabbit polyclonal, Zymed 61-7300, 1:100); β-catenin (rabbit polyclonal, Abcam ab16051, 1:400); MYC (mouse monoclonal, DSHB 9E10); GFP (mouse monoclonal, Santa Cruz Biotechnology, sc-9996, 1:100); and RFP (rabbit polyclonal, Abcam ab62341,1:200). Neural plate explants were washed five times for 1 h in PBT and incubated overnight with secondary antibodies, goat anti-mouse Alexa Fluor 488 (Invitrogen A11017 1:400) and goat anti-rabbit Alexa Fluor 594 (Invitrogen A11072, 1:400), diluted in blocking solution. Explants were washed five times for 1 h in PBT, cleared in Murray’s clear (benzyl benzoate: benzyl alcohol, 2:1) before being imaged in homemade chambers with a Zeiss LSM 700 laser-scanning inverted confocal microscope controlled by the manufacturer’s software. Stacks of confocal pictures of 1-µm thickness along the z-axis were taken with a 40× objective (NA = 1.3; working distance = 0.21 mm) using the ZEN software. Images are presented as transversal optical sections (cross-section) or maximum-intensity projections (generated with Image J) of the superficial layer (top view), as indicated in the figure legends. All images were adjusted for brightness/contrast and assembled for presentation using Photoshop and Illustrator software (Adobe).
Statistical analysis
Each experiment was performed at least three times. For experiments displaying pooled data, results are expressed as frequency distributions or as box-and-whisker plots (as indicated in the figure legends). For other experiments, one representative result is presented. Statistical significance between various conditions was assessed by determining p values using Student’s t test, chi-square (χ2), or Mann-Whitney test (as indicated in the figure legends) in Prism (GraphPad).
Acknowledgments
This work was supported by NIH grant R01GM108733 (to M.G.-M.). We thank Isha Walawalkar, Anthony Cheung, and Covadonga Lopez-Ramos for their technical help. We are indebted to I. Dominguez (Boston University) for providing advice and access to equipment for frog studies. We thank V. Trinkaus-Randall (Boston University) for providing access to the Confocal Microscopy Facility at Boston University School of Medicine. We thank S. Sokol (Icahn School of Medicine at Mount Sinai) for plasmids, P. Ghosh (University of California, San Diego) for the full-length human DAPLE cDNA, T. Michiue (University of Tokyo) for the full-length Xenopus DAPLE cDNA, A. Le Bivic (Aix-Marseille University) for the rabbit PATJ antibody, J. Sondek (University of North Carolina–Chapel Hill) for plasmids, and X. Varelas (Boston University) for sharing reagents and for helpful discussions.
Abbreviations used:
- BSA
bovine serum albumin
- co-IP
coimmunoprecipitation
- DAPLE
Dishevelled-Associating Protein with a high frequency of LEucines
- DTT
dithiothreitol
- GBA
Gα-binding-and-activating
- GPCR
G-protein–coupled receptor
- GST
glutathione S-transferase
- LIC
ligation-independent cloning
- MO
morpholino
- MPDZ
multiple PDZ protein
- mRFP
membrane-bound red fluorescent protein
- NA
numerical aperture
- NSCH
nonsyndromic congenital hydrocephalus
- PATJ
Pals-Associated Tight Junction protein
- PBM
PDZ-binding motif
- PBS
phosphate-buffered saline
- PDZ
Psd-95 (postsynaptic density protein)
- PVDF
polyvinylidene fluoride
- shRNA
short hairpin RNA
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E19-02-0091) on July 3, 2019.
REFERENCES
- Adachi M, Hamazaki Y, Kobayashi Y, Itoh M, Tsukita S, Furuse M. (2009). Similar and distinct properties of MUPP1 and Patj, two homologous PDZ domain-containing tight-junction proteins. Mol Cell Biol , 2372–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Dosari MS, Al-Owain M, Tulbah M, Kurdi W, Adly N, Al-Hemidan A, Masoodi TA, Albash B, Alkuraya FS. (2013). Mutation in MPDZ causes severe congenital hydrocephalus. J Med Genet , 54–58. [DOI] [PubMed] [Google Scholar]
- Assemat E, Crost E, Ponserre M, Wijnholds J, Le Bivic A, Massey-Harroche D. (2013). The multi-PDZ domain protein-1 (MUPP-1) expression regulates cellular levels of the PALS-1/PATJ polarity complex. Exp Cell Res , 2514–2525. [DOI] [PubMed] [Google Scholar]
- Aznar N, Midde KK, Dunkel Y, Lopez-Sanchez I, Pavlova Y, Marivin A, Barbazan J, Murray F, Nitsche U, Janssen KP, et al. (2015). Daple is a novel non-receptor GEF required for trimeric G protein activation in Wnt signaling. eLife , e07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrita LD, Dai W, Bottomley SP. (2006). A family of E. coli expression vectors for laboratory scale and high throughput soluble protein production. BMC Biotechnol , 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels DL, Cohen AR, Anderson JM, Brunger AT. (1998). Crystal structure of the hCASK PDZ domain reveals the structural basis of class II PDZ domain target recognition. Nat Struct Biol , 317–325. [DOI] [PubMed] [Google Scholar]
- de Opakua AI, Parag-Sharma K, DiGiacomo V, Merino N, Leyme A, Marivin A, Villate M, Nguyen LT, de la Cruz-Morcillo MA, Blanco-Canosa JB, et al. (2017). Molecular mechanism of Gαi activation by non-GPCR proteins with a Gα-binding and activating motif. Nat Commun , 15163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiacomo V, Marivin A, Garcia-Marcos M. (2018). When heterotrimeric G proteins are not activated by G protein-coupled receptors: structural insights and evolutionary conservation. Biochemistry , 255–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drielsma A, Jalas C, Simonis N, Desir J, Simanovsky N, Pirson I, Elpeleg O, Abramowicz M, Edvardson S. (2012). Two novel CCDC88C mutations confirm the role of DAPLE in autosomal recessive congenital hydrocephalus. J Med Genet , 708–712. [DOI] [PubMed] [Google Scholar]
- Ekici AB, Hilfinger D, Jatzwauk M, Thiel CT, Wenzel D, Lorenz I, Boltshauser E, Goecke TW, Staatz G, Morris-Rosendahl DJ, et al. (2010). Disturbed Wnt signalling due to a mutation in CCDC88C causes an autosomal recessive non-syndromic hydrocephalus with medial diverticulum. Mol Syndromol , 99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldner A, Adam MG, Tetzlaff F, Moll I, Komljenovic D, Sahm F, Bauerle T, Ishikawa H, Schroten H, Korff T, et al. (2017). Loss of Mpdz impairs ependymal cell integrity leading to perinatal-onset hydrocephalus in mice. EMBO Mol Med , 890–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Marcos M, Ghosh P, Ear J, Farquhar MG. (2010). A structural determinant that renders Gαi sensitive to activation by GIV/girdin is required to promote cell migration. J Biol Chem , 12765–12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Marcos M, Ghosh P, Farquhar MG. (2009). GIV is a nonreceptor GEF for Gαi with a unique motif that regulates Akt signaling. Proc Natl Acad Sci USA , 3178–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman AG. (1987). G proteins: transducers of receptor-generated signals. Annu Rev Biochem , 615–649. [DOI] [PubMed] [Google Scholar]
- Gilmour D, Rembold M, Leptin M. (2017). From morphogen to morphogenesis and back. Nature , 311–320. [DOI] [PubMed] [Google Scholar]
- Guerra MM, Henzi R, Ortloff A, Lichtin N, Vio K, Jimenez AJ, Dominguez-Pinos MD, Gonzalez C, Jara MC, Hinostroza F, et al. (2015). Cell junction pathology of neural stem cells is associated with ventricular zone disruption, hydrocephalus, and abnormal neurogenesis. J Neuropathol Exp Neurol , 653–671. [DOI] [PubMed] [Google Scholar]
- Heer NC, Martin AC. (2017). Tension, contraction and tissue morphogenesis. Development , 4249–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joberty G, Petersen C, Gao L, Macara IG. (2000). The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat Cell Biol , 531–539. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Michiue T, Yukita A, Danno H, Sakurai K, Fukui A, Kikuchi A, Asashima M. (2005). Novel Daple-like protein positively regulates both the Wnt/beta-catenin pathway and the Wnt/JNK pathway in Xenopus. Mech Dev , 1138–1153. [DOI] [PubMed] [Google Scholar]
- Kruse LC, Walter NA, Buck KJ. (2014). Mpdz expression in the caudolateral substantia nigra pars reticulata is crucially involved in alcohol withdrawal. Genes Brain Behav , 769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Bao J, Adelstein RS. (2007). Loss of cell adhesion causes hydrocephalus in nonmuscle myosin II-B-ablated and mutated mice. Mol Biol Cell , 2305–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marivin A, Morozova V, Walawalkar I, Leyme A, Kretov D, Cifuentes D, Dominguez I, Garcia-Marcos M. (2019). GPCR-independent activation of G proteins promotes apical cell constriction in vivo. J Cell Biol , 1743–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AC, Goldstein B. (2014). Apical constriction: themes and variations on a cellular mechanism driving morphogenesis. Development , 1987–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newport J, Kirschner M. (1982). A major developmental transition in early Xenopus embryos: I. characterization and timing of cellular changes at the midblastula stage. Cell , 675–686. [DOI] [PubMed] [Google Scholar]
- Nikolopoulou E, Galea GL, Rolo A, Greene ND, Copp AJ. (2017). Neural tube closure: cellular, molecular and biomechanical mechanisms. Development , 552–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossipova O, Kim K, Sokol SY. (2015). Planar polarization of Vangl2 in the vertebrate neural plate is controlled by Wnt and myosin II signaling. Biol Open , 722–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peshkin L, Wuhr M, Pearl E, Haas W, Freeman RM, Jr, Gerhart JC, Klein AM, Horb M, Gygi SP, Kirschner MW. (2015). On the relationship of protein and mRNA dynamics in vertebrate embryonic development. Dev Cell , 383–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez EM, Guerra MM. (2017). Neural stem cells and fetal-onset hydrocephalus. Pediatr Neurosurg , 446–461. [DOI] [PubMed] [Google Scholar]
- Rodriguez EM, Guerra MM, Vio K, Gonzalez C, Ortloff A, Batiz LF, Rodriguez S, Jara MC, Munoz RI, Ortega E, et al. (2012). A cell junction pathology of neural stem cells leads to abnormal neurogenesis and hydrocephalus. Biol Res , 231–242. [DOI] [PubMed] [Google Scholar]
- Ruggeri G, Timms AE, Cheng C, Weiss A, Kollros P, Chapman T, Tully H, Mirzaa GM. (2018). Bi-allelic mutations of CCDC88C are a rare cause of severe congenital hydrocephalus. Am J Med Genet A , 676–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saugier-Veber P, Marguet F, Lecoquierre F, Adle-Biassette H, Guimiot F, Cipriani S, Patrier S, Brasseur-Daudruy M, Goldenberg A, Layet V, et al. (2017). Hydrocephalus due to multiple ependymal malformations is caused by mutations in the MPDZ gene. Acta Neuropathol Commun , 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen R, Sebai MA, Patel N, Ewida N, Kurdi W, Altweijri I, Sogaty S, Almardawi E, Seidahmed MZ, Alnemri A, et al. (2017). The genetic landscape of familial congenital hydrocephalus. Ann Neurol , 890–897. [DOI] [PubMed] [Google Scholar]
- Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. (2002). A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr Purif , 8–15. [DOI] [PubMed] [Google Scholar]
- Takagishi M, Sawada M, Ohata S, Asai N, Enomoto A, Takahashi K, Weng L, Ushida K, Ara H, Matsui S, et al. (2017). Daple coordinates planar polarized microtubule dynamics in ependymal cells and contributes to hydrocephalus. Cell Rep , 960–972. [DOI] [PubMed] [Google Scholar]
- Van Camp G, Vits L, Coucke P, Lyonnet S, Schrander-Stumpel C, Darby J, Holden J, Munnich A, Willems PJ. (1993). A duplication in the L1CAM gene associated with X-linked hydrocephalus. Nat Genet , 421–425. [DOI] [PubMed] [Google Scholar]
- Vogel P, Read RW, Hansen GM, Payne BJ, Small D, Sands AT, Zambrowicz BP. (2012). Congenital hydrocephalus in genetically engineered mice. Vet Pathol , 166–181. [DOI] [PubMed] [Google Scholar]
- Wallingford JB. (2010). Low-magnification live imaging of Xenopus embryos for cell and developmental biology. Cold Spring Harb Protoc , pdb prot5425. [DOI] [PubMed] [Google Scholar]
- Wallis M, Baumer A, Smaili W, Jaouad IC, Sefiani A, Jacobson E, Bowyer L, Mowat D, Rauch A. (2018). Surprisingly good outcome in antenatal diagnosis of severe hydrocephalus related to CCDC88C deficiency. Eur J Med Genet , 189–196. [DOI] [PubMed] [Google Scholar]
- Yano T, Kanoh H, Tamura A, Tsukita S. (2017). Apical cytoskeletons and junctional complexes as a combined system in epithelial cell sheets. Ann NY Acad Sci , 32–43. [DOI] [PubMed] [Google Scholar]