

Abstract
Nitric oxide (NO) is an important vasodilator with a well-established role in cardiovascular homeostasis. While mediator is synthesized from L-arginine by neuronal, endothelial, and inducible nitric oxide synthases (NOS1, NOS3 and NOS2 respectively), NOS3 is the most important isoform for NO formation in the cardiovascular system. NOS3 is a dimeric enzyme whose expression and activity are regulated at transcriptional, posttranscriptional, and posttranslational levels. The NOS3 gene, which encodes NOS3, exhibits a number of polymorphic sites including single nucleotide polymorphisms (SNPs), variable number of tandem repeats (VNTRs), microsatellites, and insertions/deletions. Some NOS3 polymorphisms show functional effects on NOS3 expression or activity, thereby affecting NO formation. Interestingly, many studies have evaluated the effects of functional NOS3 polymorphisms on disease susceptibility and drug responses. Moreover, some studies have investigated how NOS3 haplotypes may impact endogenous NO formation and disease susceptibility. In this article, we carried out a comprehensive review to provide a basic understanding of biochemical mechanisms involved in NOS3 regulation and how genetic variations in NOS3 may translate into relevant clinical and pharmacogenetic implications.
Keywords: Endothelial nitric oxide synthase, genetic polymorphisms, haplotypes, nitric oxide, NOS3 gene, pharmacogenetics
1. Introduction
The endogenous production of nitric oxide (NO), particularly in the cardiovascular system, is mainly dependent on the activity of the enzyme endothelial NO synthase (NOS3). Decreased NO formation promotes cardiovascular diseases, which are responsible for a large number of deaths worldwide (Pagidipati and Gaziano, 2013). It is now widely acknowledged that a healthy cardiovascular system depends on the integrity of the endothelium, a monolayer of cells lining the lumen of blood vessels (Bian et al., 2008), and dysfunctional endothelial cells trigger critical mechanisms involved in the pathogenesis of many cardiovascular diseases (Bian et al., 2008). Importantly, the endothelium regulates the vascular tone, structure, and function by controlling the release of multiple vasoactive substances including NO, a small gaseous and lipophilic molecule that acts as a ubiquitous mediator of a broad spectrum of biological processes (Cockcroft, 2005) (Villanueva and Giulivi, 2010).
NO is synthesized from L-arginine by three different synthases: neuronal (NOS1), endothelial (NOS3) and inducible (NOS2) synthases (Moncada and Higgs, 1993). While NOS2 typically produces high amounts of NO and is involved in host defense, inflammatory responses and airway epithelial NO formation, the constitutively expressed isoforms, NOS1 and NOS3, usually produce lower NO amounts that are important for physiological processes such as neuronal signaling, inhibition of the hemostatic system, vasodilation, and blood pressure control (Cortese-Krott and Kelm, 2014). This review is focused on NOS3.
Under physiological conditions, NOS3 is responsible for the production of most endothelium-derived NO, and for this reason it plays a pivotal role in cardiovascular homeostasis (Fish and Marsden, 2006). The relevance of this fact has been consistently shown in clinical and experimental studies (Albrecht et al., 2003). Indeed, NOS3 knockout mice are hypertensive and have increased susceptibility to stroke and other cardiovascular alterations (Li et al., 2002). Likewise, pharmacological inhibition of NOS3 induces similar alterations as compared to those found in NOS3 knockout mice, including increased blood pressure (Albrecht et al., 2003). Together, these observations highlight the protective role of a fully functional NOS3 against cardiovascular diseases.
Although NOS3 is constitutively expressed, numerous stimuli regulate NOS3 at the transcriptional, posttranscriptional and posttranslational levels (Rafikov et al., 2011). In this regard, variations in NOS3 gene have been described to influence NOS3 regulation, thereby affecting NO production (Cooke et al., 2007). Here, we briefly review the biochemical aspects underlying NOS3 structure, function, and regulation. Moreover, we discuss how polymorphisms in NOS3 gene may affect NOS3 regulation, endogenous NO production, and the impact of these polymorphisms on the susceptibility to diseases and responses to drugs.
2. An overview of vascular roles played by NO
NO has a well-established role in vascular homeostasis (Forstermann and Munzel, 2006), influencing vascular tone (Ignarro et al., 1987; Vallance et al., 1989; Lopez-Jaramillo et al., 1990; Imig and Roman, 1992), cellular proliferation (Albina et al., 1991; Yang et al., 1994), leukocyte adhesion (Kubes et al., 1991; Kubes et al., 1993), and platelet aggregation (Radomski et al., 1987; Benjamin et al., 1991). NO produced in endothelial cells diffuses across platelet or vascular smooth muscle cell membranes and binds to the heme moiety of soluble guanylate cyclase (sGC) forming a metal-nitrosyl adduct that is activated to catalyze the conversion of guanosine triphosphate into cyclic guanosine monophosphate (cGMP) (Craven and DeRubertis, 1978; Murad et al., 1978; Denninger and Marletta, 1999). While this process inhibits platelet reactivity (Walford and Loscalzo, 2003), increased cGMP concentrations activate protein kinase G-1 (PKG-1) in vascular smooth muscle cells, leading to multiple phosphorylation of cellular proteins and lower intracellular free calcium concentrations, thus promoting vascular relaxation (Surks et al., 1999). In more details, active PKG-1 phosphorylates large-conductance calcium-activated potassium channels, resulting in channel opening and allowing for loss of intracellular potassium, hyperpolarization of the plasma membrane, and reduced calcium influx through L-type calcium channels (White et al., 1993; Zhou et al., 1996; Alioua et al., 1998; Francis et al., 2010). In addition, L-type calcium channels may also be phosphorylated by PKG-1, thereby resulting in inhibition of channel function (Yang et al., 2007). PKG-1 also phosphorylates 1,4,5 inositoltriphosphate (IP3) receptor-associated cGMP kinase substrate (IRAG) and phospholamban in the sarcoplasmic reticulum (Schlossmann et al., 2000; Walford and Loscalzo, 2003). IRAG phosphorylation leads to inhibition of IP3-receptor activity, suppressing the release of calcium from intracellular stores, while phospholamban phosphorylation triggers its inhibitory effect on the sarcoplasmic reticulum ATPase (SERCA), promoting calcium sequestration in the sarcoplasmic reticulum (Schlossmann et al., 2000; Francis et al., 2010). The fall in calcium flux as result of these multiple phosphorylation causes a decrease in the formation of the calcium-calmodulin myosin light chain kinase complex, favoring smooth muscle relaxation (Horowitz et al., 1996) and vasodilation. Additionally, PKG-1 may activate myosin light chain phosphatase to decrease the level of myosin light chain phosphorylation, thereby also contributing to vasodilation (Surks et al., 1999) (Fig. 1).
Figure 1.

Mechanisms involved in NO formation and vasodilation. Increases in intracellular Ca2+ in endothelial cells induced by stimuli such as shear stress, blood flow, and binding of agonists lead to formation of a Ca2+-calmodulin complex, which activates NOS3. Once activated, NOS3 forms NO and L-citrulline from L-arginine and molecular oxygen; tetrahydrobiopterin (BH4) and nicotinamide adenine dinucleotide phosphate (NADPH) play important roles in this process. Thereafter, NO diffuses across platelet or vascular smooth muscle cell membranes and activates soluble guanylate cyclase (sGC), which catalyzes the conversion of guanosine triphosphate into cyclic guanosine monophosphate (cGMP). In the platelets, this process leads to inhibition of platelet function. In the vascular smooth muscle cell, cGMP activates protein kinase G-1 (PKG-1), which leads to multiple phosphorylation of cellular proteins such as 1,4,5 inositoltriphosphate (IP3) receptor-associated cGMP kinase substrate (IRAG), phospholamban (P’Lamban), and myosin light chain phosphatase resulting in lower cellular calcium concentrations and vasodilation. In addition, PKG-1 phosphorylates large-conductance calcium-activated potassium channels and L-type calcium channels, reducing cellular calcium levels, thus promoting vascular relaxation.
Given that intracellular Ca2+ influx promotes proliferation of vascular smooth muscle cells (Baran, 1996), NO also exerts antiproliferative effects through cGMP-dependent inhibition of Ca2+ influx (Cornwell et al., 1994). Other antiproliferative mechanisms of NO include inhibition of arginase and ornithine decarboxylase activity, thus reducing the formation of polyamides required for DNA synthesis (Ignarro et al., 2001). Endogenous NO is important to regulate leukocyte adhesion to the vascular endothelium by inhibiting nuclear factor Kappa-B, which stimulates the vascular endothelial expression of chemokines and adhesion molecules (Chen et al., 1999a).
Endogenous NO may also indirectly affect both the vascular tonus and proliferation by regulating the redox environment of vascular cells (Walford and Loscalzo, 2003). NO exerts antioxidant effects on vascular cells by reacting with superoxide anion (Walford and Loscalzo, 2003) and by increasing the expression of the antioxidant enzyme superoxide dismutase, which catalyzes the dismutation of superoxide anion to hydrogen peroxide (Fukai et al., 2000). Hydrogen peroxide, in turn, may lead to activation of NOS3 via the oxidation of redox-sensitive protein kinases that promote NOS3 phosphorylation, an effect that increases the ability of vascular cells to attenuate oxidative stress (Thomas et al., 2002). In addition, antioxidant effects of NO are attributable to upregulation of heme-oxygenase-I and ferritin expression, which decrease superoxide anion concentrations in blood vessels (Balla et al., 1992; Durante et al., 1997; Maines, 1997; Recalcati et al., 1998).
NO interaction with superoxide anion results in peroxynitrite formation (Herce-Pagliai et al., 1998), and this highly reactive compound reacts with DNA (Virag et al., 2003), proteins (to form nitrotyrosine) (Herce-Pagliai et al., 1998), or lipids to cause oxidative stress (Salvemini and Cuzzocrea, 2002). The formation of peroxynitrite is usually limited by relatively high concentrations of superoxide dismutase, which outcompetes NO for superoxide anion (Koppenol, 1998). However, when NO levels increase, for example as a result of NOS2 upregulated expression and activity, the formation of peroxynitrite prevails and impaired vascular homeostasis results in endothelial dysfunction, a key feature of hypertension (Santhanam et al., 2007; Oliveira-Paula et al., 2014).
3. Structure and basic biochemistry of NOS3
In endothelial cells, NO is mostly synthetized by NOS3, which forms a dimer made up of two identical 134 kD monomers (List et al., 1997; Albrecht et al., 2003). The structure (Fig. 2) of this enzyme consists of a C-terminal reductase domain, which contains binding sites for nicotinamide adenine dinucleotide phosphate (NADPH), flavin mononucleotide (FMN), and flavin adenine dinucleotide (FAD), and an N-terminal oxidase domain, which contains binding sites for heme, zinc, tetrahydrobiopterin (BH4), and L-arginine (Bredt et al., 1991; Lamas et al., 1992; Alderton et al., 2001; Qian and Fulton, 2013). Importantly, the C-terminal is linked by a calmodulin-binding sequence to the N-terminal (Bredt et al., 1991; Alderton et al., 2001). Homodimers formation is essential for NOS3 activity and heme plays a fundamental role in this process (Panda et al., 2002). Indeed, heme is bound via a proximal cysteine zinc-thiolate cluster and this binding has been suggested as a key step in NOS3 dimerization (Raman et al., 1998; Albrecht et al., 2003). Moreover, the cofactor BH4 binding induces NOS3 to shift its heme iron to a high-spin state, stabilizing the active dimeric form of the enzyme (List et al., 1997; Rafikov et al., 2011) (Fig. 2).
Figure 2.

NOS3 structure. A. Monomeric structure containing a C-terminal reductase domain, which exhibits binding sites for nicotinamide adenine dinucleotide phosphate (NADPH), flavin mononucleotide (FMN), and flavin adenine dinucleotide (FAD), and an N-terminal oxidase domain, which exhibits binding sites for heme, zinc, tetrahydrobiopterin (BH4), and L-arginine. The C-terminal is linked by a calmodulin-binding sequence to the N-terminal. B. Dimerization of NOS3, an essential process for maximum enzyme activity.
NOS3 catalyzes flavin-mediated electron transfer from the C-terminally bound NADPH to the oxygenase domain of the other NOS3 monomer (Abu-Soud et al., 1994; Abu-Soud et al., 2000; Garcin et al., 2004; Fleming, 2010; Qian and Fulton, 2013). In the setting of increased intracellular Ca2+, the formation of a Ca2+-calmodulin complex disrupts NOS3 suppression from the NOS3-caveolin interaction, increasing the rate of electron transfer from NADPH via the reductase domain flavins to the oxygenase domain (Forstermann and Munzel, 2006; Maron and Michel, 2012; Piazza et al., 2012). Within the oxygenase domain, molecular oxygen is bound to heme and reduced and then incorporated into L-arginine to form NO and L-citrulline (List et al., 1997; Fleming and Busse, 1999; Verhaar et al., 2004).
The binding of multiple substrates and cofactors must be effectively controlled for NOS3 produce NO efficiently (Dudzinski et al., 2006). Disruption of this highly coordinated catalysis as a result of increased oxidative stress, for example, can impair NOS3 enzymatic activity (Shinozaki et al., 2000; Ulker et al., 2003; Li et al., 2014). Indeed, reactive oxygen species oxidize NOS3 cofactors such as BH4, leading to a shift from the dimeric to monomeric form of the enzyme (Kuzkaya et al., 2003; Landmesser et al., 2003; Maron and Michel, 2012). In the monomeric conformation, NOS3 is uncoupled and superoxide anion is synthesized instead of NO, potentially resulting in detrimental consequences to the cardiovascular system (Albrecht et al., 2003; Kuzkaya et al., 2003; Landmesser et al., 2003; Luo et al., 2014).
4. Clinical markers of endogenous NO formation
The search for reliable markers of endogenous NO formation has led to a few reasonably validated markers until now. Because NO has a very short half-life, there is no simple techniques available to assess NO formation in vivo, and therefore products of NO metabolism are usually measured because they may reflect NO production (Ellis et al., 1998; Metzger et al., 2006). NO is oxidized to nitrite and nitrate in vivo and in vitro, and therefore the measurement of plasma nitrates (which are stable products of NO oxidation) in plasma separated from blood collected after an overnight fast has been valued as index of endogenous production of NO. However, given the fact that many interfering factors may change the plasma levels of nitrate, including diet, clinical conditions, a variety of medications, smoking, and other environmental contributors (Ellis et al., 1998), the simple measurement of plasma nitrate (typically in the 20–80 μM range) concentrations is now taken as a very limited index of endogenous NO. In contrast, the measurement of plasma nitrite (typically in the 0.1–0.6 μM range) concentrations has been shown to reflect nitric oxide synthase activity, both under physiological or disease conditions (Kleinbongard et al., 2006). Moreover, because stimulation of sGC is the most important functions of NO, the measurement of cGMP concentrations in plasma samples has also been suggested as an index of NO production (Metzger et al., 2006).
While plasma nitrite concentration is probably the best marker of endogenous NO formation at this time, it is now clear that nitrate may recycle back to nitrite and to NO by mechanisms now described as the nitrate-nitrite-NO pathway, which is now regarded as a major source of NO independent of classic L-arginine NO synthases (Gladwin et al., 2005). This pathway complements NOS3-derived NO formation as nitrate is bioactivated after oral ingestion, when nitrate enters the circulation and is secreted into the oral cavity (by salivary glands), where it is converted into nitrite (Gladwin et al., 2005). Oral nitrite is swallowed and reduced to NO when it is exposed to the acidic conditions of the stomach. This pathway has been shown to play a critical role in the cardiovascular effects of exogenously nitrite (Montenegro et al., 2011; Pinheiro et al., 2012; Amaral et al., 2013). Therefore, plasma nitrite concentrations may clearly reflect a reservoir of NO.
5. NOS3 gene structure
The NOS3 gene (Fig. 3) is located in the 7q35–7q36 region of chromosome 7 in humans (Marsden et al., 1993). This gene is present as a single copy in the haploid human genome and contains 21–22 kb, 25 introns and 26 exons, which encode an mRNA of 4052 nucleotides (Marsden et al., 1993). Interestingly, the human coding NOS3 cDNA sequence showed about 90% of homology with murine, bovine, and porcine coding NOS3 cDNA sequence (Li et al., 2002), thus showing a high degree of similarity of NOS3 cDNA among different species.
Figure 3.
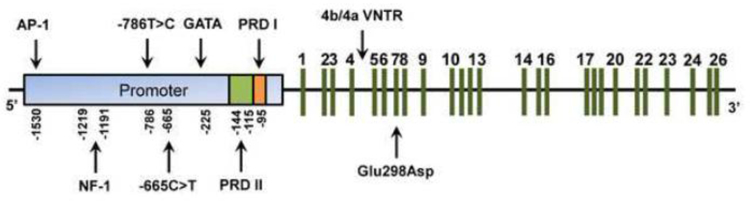
Schematic organization of NOS3 gene. This gene contains a promoter region displaying transcription factor binding sites such as GATA, NF-1 and AP-1, and the positive regulatory domains (PRD) I and II. NOS3 gene also contains 25 introns and 26 exons (represented by green lines) and exhibits functional polymorphisms such as the g.-786T>C and g.-665C>T in the promoter region, the 4b/4a VNTR in intron 4, and the Glu298Asp in exon 7.
Characterization of 5’flanking genomic region showed that human NOS3 promoter is “TATA-less” and displays proximal elements such as Sp1 and GATA motifs compatible with a constitutively expressed gene (Zhang et al., 1995; Karantzoulis-Fegaras et al., 1999). Moreover, NOS3 promoter has homologies to several binding sites for transcription factors, including NF-1, AP-1, NF-κB, shear-stress response elements and sterol regulatory elements (Li et al., 2002). Indeed, NOS3 promoter, as well as posttranscriptional and posttranslational modifications play an important role in NOS3 regulation.
6. Regulation of NOS3 expression and activity
NOS3 was originally identified and isolated from bovine aortic endothelial cells (Forstermann et al., 1991; Pollock et al., 1991). Further studies revealed that several nonendothelial cell types also express NOS3, such as cardiomyocytes (Balligand et al., 1993), platelets (Sase and Michel, 1995) and neurons (Dinerman et al., 1994). Consistent with transiently and spatially precise NO-dependent signaling, NOS3 expression and activity are carefully controlled by multiple interconnected mechanisms of regulation (Dudzinski et al., 2006). These mechanisms are effective at transcriptional, posttranscriptional, and posttranslational levels and will be discussed below.
6.1. Transcriptional regulation
The most important level of NOS3 regulation corresponds to NOS3 transcription, when NOS3 promoter plays a fundamental role. Indeed, a detailed analysis of human NOS3 promoter revealed two regulatory regions involved in basal NOS3 transcription (Karantzoulis-Fegaras et al., 1999). A positive regulatory domain I (PRD I; −104 to −95 relative to transcription initiation; Fig. 3) was mapped to a 10-bp cis-region corresponding to a high-affinity Sp1 transcription factor recognition site and was found to bind three nucleoproteins identified as Sp1 and two variants of Sp3 (Karantzoulis-Fegaras et al., 1999). The other regulatory region, a positive regulatory domain II (PRD II; −144 to −115; Fig. 3), includes a 30-bp region of the promoter and forms nucleoprotein complexes with the transcription factors Ets-1, Elf-1, YY1, Sp1, and MYC-associated zinc finger protein (MAZ) (Karantzoulis-Fegaras et al., 1999). Particularly, Sp1, Sp3, Ets-1, Elf-1 and YY1 positively regulate human NOS3 promoter activity, while MAZ seems to inhibit the NOS3 promoter (Karantzoulis-Fegaras et al., 1999).
In addition to these regulatory regions, a 269-nucleotide sequence located at positions −4907 to −4638 upstream of the transcription start site acts as an enhancer of NOS3 expression in endothelial cells (Laumonnier et al., 2000). Indeed, this sequence increases the activity of NOS3 promoter, independent of the distance from the promoter and the orientation of the enhancer element, thereby fulfilling the criteria of a classic enhancer element (Laumonnier et al., 2000). Characterization of this element identified multiple protein complexes that are important for the enhancer function, such as Ets-related factors, AP-2, and Sp1-related factor (Laumonnier et al., 2000). In fact, experiments reveled that Erg (an Ets factor) binds to cognate sites in the enhancer region and transactivates the promoter (Laumonnier et al., 2000).
DNA methylation at CpG dinucleotides has been involved in several cellular processes, including transcriptional regulation (Jaenisch and Bird, 2003; Postberg et al., 2015). Methylation of NOS3 promoter was associated with dramatic impairment of promoter activity in mammalian cells, suggesting that DNA methylation plays an important role in endothelial cell-specific expression of the human NOS3 gene (Chan et al., 2004). Decreased promoter activity is associated with reduced ability of Sp1, Sp3 and Ets-1 to transactivate the NOS3 promoter (Chan et al., 2004). In addition to interfering with the binding of transcription factors, DNA methylation of promoters is often accompanied by histone modifications such that chromatin is effectively inaccessible to transcription factors (Schubeler et al., 2000). Interestingly, this represents an additional repressive mechanism of NOS3 promoter (Fish and Marsden, 2006).
Specific compounds and conditions may also regulate NOS3 transcription. For example, transforming growth factor β1 (TGF-β1), a homodimeric peptide that plays an important role in the pathogenesis of atherosclerosis, hypertensive vessel remodeling, and angiogenesis, increases NOS3 promoter activity (Inoue et al., 1995). Another important example is vascular shear stress, which induces NOS3 promoter activation due to binding of NF-κB subunits p50 and p65 to a shear-responsive element (GAGACC) −990 to −984 bp upstream from the NOS3 transcription start site, leading to increased NOS3 expression (Silacci et al., 2000; Davis et al., 2004). Moreover, NOS3 transcription shear stress-mediated is regulated by Kruppel-like factor (KLF2) 2 and Kruppel-like factor (KLF4), which are key regulators of endothelial function (Dekker et al., 2002; Villarreal et al., 2010; Mun and Boo, 2012). Finally, several others compounds and conditions such as estrogen, hydrogen peroxide, protein kinase C, hypoxia, and cyclic strain contribute to the regulation of NOS3 transcription (Searles, 2006).
6.2. Posttranscriptional regulation
Posttranscriptional NOS3 regulation includes modifications of the primary transcript, mRNA stability, subcellular localization, and nucleocytoplasmatic transport (Searles, 2006). Importantly, cis-acting RNA elements located in 5’- and 3’- mRNA untranslated regions (5’-UTRs and 3’-UTRs) are critical mediators of these modifications (Pesole et al., 2001). They result of the combination of the primary and secondary structures of cis-acting RNA elements and their recognition by trans-acting RNA binding proteins (Liebhaber, 1997).
Studies involving cis elements in posttranscriptional regulation of NOS3 focused on the 3’-UTR (Sanchez de Miguel et al., 1999; Searles et al., 1999; Lai et al., 2003; Chaudhury et al., 2010). Characterization of the NOS3 3’-UTR showed that some sequences at the origin of the 3’-UTR are critical for the binding of certain cytosolic proteins and modify its configuration increasing its susceptibility to RNase activity (Searles et al., 1999; Fleming and Busse, 2003; Chaudhury et al., 2010). Interestingly, a mutant 3’-UTR lacking a 43-nucleotide sequence increased the half-life of NOS3 mRNA, indicating that this sequence plays a crucial role in destabilizing NOS3 mRNA (Searles et al., 1999). In addition, a 25-nucleotide UC-rich sequence and another 158-nucleotide CU-rich sequence, both located in NOS3 3’-UTR, are also important in regulating NOS3 mRNA stability (Sanchez de Miguel et al., 1999; Lai et al., 2003). Together, these studies indicate that cis-regulatory elements in the NOS3 3’-UTR are highly relevant to posttranscriptional regulation of NOS3.
Another mechanism for the posttranscriptional regulation of NOS3 involves a cis-natural antisense transcript to NOS3 called sONE (also known as ATG9B, NOS3AS, and APG9L2) (Robb et al., 2004). The transcripts for NOS3 and sONE genes are complementary for a total of 662 nucleotides, including significant exon/exon overlap (Robb et al., 2004). Under basal conditions, sONE transcripts are poorly expressed in endothelial cells due to posttranscriptional regulation, while NOS3 transcripts are highly abundant (Robb et al., 2004). Exposure of endothelial cells to hypoxia, a condition known to downregulate NOS3 mRNA and protein expression, substantially increased the steady-state levels of sONE mRNA (Fish et al., 2007). This dowregulation of NOS3 mRNA was related, at least in part, to the destabilization of NOS3 mRNA and increased sONE levels (Fish et al., 2007). Together, these findings indicate that sONE is a posttranscriptional inhibitor of NOS3 mRNA and protein expression, particularly under hypoxic conditions.
In addition to antisense RNAs, micro (mi)RNA-mediated processes were also shown to modulate NOS3 at posttranscriptional level (Suarez et al., 2007; Sun et al., 2012; Yan et al., 2013). miRNA are approximately 22-nucleotide endogenous small RNAs that negatively regulate gene expression by targeting the 3´-UTR of specific mRNAs and promote mRNA degradation or translational repression (Bartel, 2009; Sun and Lai, 2013). Indeed, studies have demonstrated that miRNAs inhibit NOS3 expression at posttranscriptional level (Sun et al., 2012; Yan et al., 2013). Moreover, genetic knockdown of Dicer (an enzyme necessary for miRNA maturation) increases NOS3 expression (Suarez et al., 2007), thereby supporting a critical role of miRNAs for posttranscriptional regulation of NOS3.
Finally, it should be mentioned that a variety of compounds and conditions, such as lipopolysaccharide, cell growth, shear stress, thrombin, and hypercholesterolemia and statins may have relevant effects on posttranscriptional NOS3 regulation (Searles, 2006). Statins are usually prescribed because they inhibit cholesterol synthesis in the liver after blocking the conversion of 3-hydroxy-3-methylglutaryl coenzyme A to mevalonate. However, statins also exert other pleiotropic, cholesterol-independent effects including increased NOS3 expression (Laufs and Liao, 1998; Liao and Laufs, 2005). These effects are attributed to increased NOS3 mRNA stability mediated by interference with the small GTP binding protein Rho (Laufs and Liao, 1998; Liao and Laufs, 2005; Mason and Cockcroft, 2006). Satins inhibit the geranyl-geranylation and consequent translocation of Rho from the cytosol to the membrane, a fundamental step for Rho activation, resulting in an induction of NOS3 mRNA levels and enhanced NO production (Laufs and Liao, 1998; Lefer et al., 2001). Indeed, these pleiotropic effects of statins have clinical and pharmacogenetics implications (Lacchini et al., 2010), as will be discussed in the last part of this review.
6.3. Posttranslational regulation
Important posttranslational modifications that affect NOS3 activity include fatty acid acylation, acetylation, protein-protein interactions, substrate and co-factor availability, and degree of phosphorylation (Qian and Fulton, 2013). In resting endothelial cells, most NOS3 is attached to caveolae, a pocket-like invagination of the membrane enriched in cholesterol and sphingolipids (Lisanti et al., 1994). This is mediated by protein fatty acid acylation and is represented mainly by a co-translational N-myristoylation on glycine-2 and a posttranslational palmitoylation on cysteines-15 and 26 within the oxygenase domain of the enzyme (Liu and Sessa, 1994; Liu et al., 1995; Shaul et al., 1996). Many studies showed that both myristoylation and palmitoylation are important for membrane localization and maximum NOS3 activity (Robinson and Michel, 1995; Sakoda et al., 1995). Moreover, NOS3 activity is influenced by acetylation of NOS3 protein (Jung et al., 2010). Deacetlyation of NOS3, acetylated at lysine 609, is mediated by histone deacetylase 3, resulting in reduced NO production by decreased calmodulin association (Jung et al., 2010).
The localization of NOS3 within the endothelial cell caveolae inactivates the enzyme as a result of strong and direct interaction of NOS3 with caveolin-1 (Ju et al., 1997). This protein-protein interaction inhibits NOS3 activity mainly by interfering with calmodulin binding site (Ju et al., 1997). Conversely, binding of calcium-activated calmodulin to NOS3 displaces caveolin-1 and favors NOS3 activation (Qian and Fulton, 2013). Calmodulin interacts with a cognate binding site on NOS3 located between the oxygenase and reductase domains (Qian and Fulton, 2013). This binding shifts an adjacent autoinhibitory loop and allows NADPH-dependent electron flux to the heme moiety (Fulton et al., 2001). However, in the absence of bound calmodulin, electron transfer is blocked and NOS3 catalytic activity is suppressed (Qian and Fulton, 2013). Besides caveolin-1, NOS3 may interact with heat-shock protein 90 (Hsp90), a protein involved in a multicomponent chaperone system that is responsible for the folding of several proteins (Caplan, 1999). Indeed, Hsp90 is involved in the folding of NOS3 and may modulate the enzyme allosterically by inducing a conformational change or by stabilizing the dimeric form, thereby activating NOS3 (Garcia-Cardena et al., 1998).
NOS3 activity also depends on substrate and co-factor availability (Fleming, 2010). L-arginine is the substrate for NOS3 and its catalytic activity requires NADPH and the co-factor BH4 (Forstermann and Munzel, 2006). Many studies have demonstrated that suboptimal concentrations of L-arginine or BH4 reduce the generation of NO and promote NOS3 uncoupling, leading to NOS3-mediated reduction of oxygen and formation of superoxide anion instead of NO (Qian and Fulton, 2013). As previously discussed, an important consequence of NOS3 uncoupling is endothelial dysfunction, which results in deleterious cardiovascular events (Albrecht et al., 2003; Luo et al., 2014).
Multiple phosphorylation sites at tyrosine, serine, and threonine residues dynamically regulate NOS3 activity (Michel et al., 1993; Corson et al., 1996; Garcia-Cardena et al., 1996). Despite numerous phosphorylation sites in human NOS3, the most functionally relevant sites are Ser1177 and Thr495 (corresponding to Ser1179 and Thr497 in bovine NOS3, respectively) (Fleming and Busse, 2003). Indeed, NOS3 phosphorylation at Ser1177 leads to NOS3 activation at basal levels and in response to agonists (Fleming and Busse, 2003). This process is mediated by protein kinases, such as Akt (Dimmeler et al., 1999), AMP-activated protein kinase (AMPK) (Chen et al., 1999b) and calmodulin-dependent kinase II (CaMKII) (Fleming et al., 2001). In addition, NOS3 can be activated by phosphorylation at Ser633 mediated by AMPK (Chen et al., 2009). In contrast, NOS3 phosphorylation at Thr495 decreases enzyme activity by impairing the binding of calcium-activated calmodulin (Fleming et al., 2001). However, phosphorylation at Thr495 is important for the coupling of L-arginine metabolism to NO synthesis, whereas under Thr495 dephosphorylation NOS3 may generate both NO and superoxide anion (Lin et al., 2003). Together, these findings highlight the important role of phosphorylation in regulating NOS3 activity.
7. Genetic polymorphisms in the NOS3 gene
NOS3 regulation at the transcriptional, posttranscriptional, and posttranslational levels may be influenced by genetic polymorphisms in the NOS3 gene. Since its characterization in the mid-1990s, many polymorphic sites have been described in human NOS3 gene, including single nucleotide polymorphisms (SNPs), variable number of tandem repeats (VNTR), microsatellites and insertions/deletions (Cooke et al., 2007). Currently, more than 1700 genetic variations in human NOS3 gene are reported in the SNP database. More importantly, several studies have shown functional consequences and clinical implications of NOS3 polymorphisms (Wattanapitayakul et al., 2001).
7.1. Functionality of NOS3 polymorphisms
Some NOS3 polymorphisms are considered functional because they affect NOS3 expression or activity. Among these polymorphisms, the SNPs rs2070744 and rs1799983, and a VNTR in intron 4 have been widely studied (Lacchini et al., 2010). In addition, the functionality of the SNP rs3918226, which is associated with cardiovascular diseases, has recently been shown (Salvi et al., 2013). The functional mechanisms of these polymorphisms are illustrated in Fig. 4 and summarized in Table 1. Here we will briefly discuss the most important.
Figure 4.
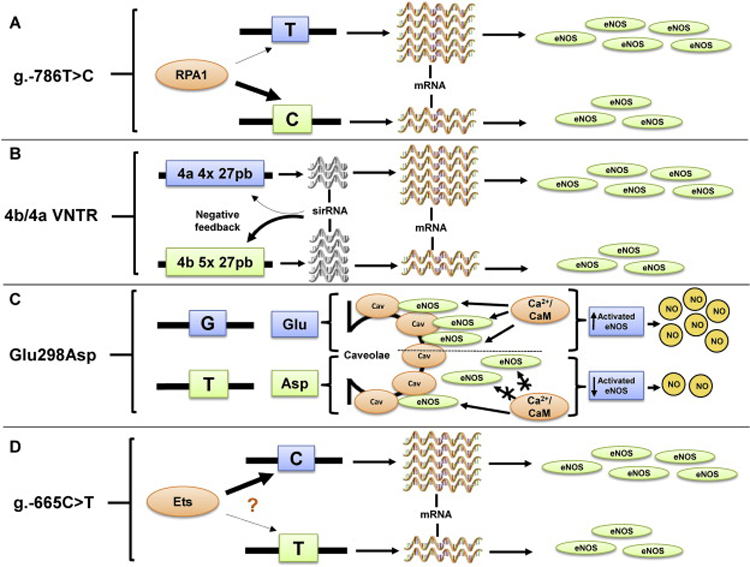
Functional mechanisms of NOS3 polymorphisms. A. The functional consequence of the g.-786T>C polymorphism is related to replication protein A1 (RPA1), which binds to NOS3 promoter with more affinity when the C allele is present, resulting in reduced NOS3 expression. B. The functionality of 4b/4a VNTR is associated with sirRNA formation. Endothelial cells containing the 4b allele (five copies of 27 pb) show increased levels of sirRNA, leading to lower NOS3 expression compared with cells containing the 4a allele (four copies of 27 pb). C. The Glu298Asp polymorphism corresponds to a guanine (G) to thymine (T) change in position 894 of the NOS3 gene, resulting in a glutamine (Glu) to aspartate (Asp) substitution in position 298 of NOS3. This substitution leads to reduced NOS3 binding to caveolin-1 and decreased NOS3 availability in the caveolar fraction in the endothelial cell, resulting in lower NOS3 available for calcium-activated calmodulin activation, thus reducing NOS3 activity and NO production. D. The functional aspects of g.-665C>T polymorphism are probably involved with transcription factor binding site for the Ets family domain. It is possible that these transcription factors, which positively regulate NOS3 promoter activity, bind to NOS3 promoter with less affinity in the presence of the T allele, leading to decreased NOS3 expression.
Table 1 -.
Functional findings of commonly studied NOS3 polymorphisms.
| Rs number | Common name | Location | Functional findings | Reference |
|---|---|---|---|---|
| rs2070744 | g.-786T>C | Promoter | C allele results in lower NOS3 transcriptional activity compared with T allele. | (Nakayama et al., 1999; Wang et al., 2002) |
| - | 4b/4a VNTR | Intron 4 | 4a allele leads to decreased levels of NOS3 mRNA in relation to 4b allele. | (Zhang et al., 2008a; Zhang et al., 2008b) |
| rs1799983 | Glu298Asp | Exon 7 | Variant allele results in lower NOS3 activity than ancestral allele. | (Joshi et al., 2007) |
| rs3918226 | g.-665C>T | Promoter | T allele is associated with reduced NOS3 transcriptional activity compared with C allele. | (Salvi et al., 2013) |
The SNP rs2070744, commonly known as g.-786T>C (Fig. 3), affects NOS3 at transcriptional level. Studies with luciferase reporter gene assays demonstrated that the thymine replacement by cytosine at the position −786 of NOS3 promoter dramatically reduces NOS3 transcriptional activity (Nakayama et al., 1999; Wang et al., 2002). This effect is probably related to a gene repressor protein called replication protein A1 (RPA1), which binds to NOS3 promoter with more affinity when the C allele is present (Miyamoto et al., 2000) (Fig. 4A). Indeed, inhibition of RPA1 expression using antisense oligonucleotide restored transcriptional activity in the NOS3 promoter with C allele, while overexpression of RPA1 showed the opposite effect (Miyamoto et al., 2000). Interestingly, these in vitro studies are in line with in vivo findings, which revealed a tendency for lower circulating NO-related markers levels in individuals carrying the C allele (Nagassaki et al., 2006) compared with T allele carriers, corroborating the functional role of this SNP (Miyamoto et al., 2000).
A VNTR characterized by 27 bp repeat in intron 4 of NOS3 gene regulates NOS3 posttranscriptionally by affecting the formation of small interference RNA (sirRNA) (Zhang et al., 2008a; Zhang et al., 2008b). This polymorphism is commonly known as 4b/4a VNTR (Fig. 3) and the most common alleles are those with five (variant 4b) or four copies (variant 4a) of the 27 bp fragment (Cooke et al., 2007), even though other rarer alleles were described (Tanus-Santos et al., 2001). In vitro studies indicate that endothelial cells containing five copies show higher quantities of sirRNA, thereby leading to lower levels of NOS3 mRNA compared with cells containing four copies (Zhang et al., 2008a; Zhang et al., 2008b) (Fig. 4B).
The SNP rs1799983 is located in exon 7 and corresponds to a guanine to thymine change in position 894 of the NOS3 gene, resulting in a glutamine to aspartate substitution at position 298 of the protein, and hence this SNP is known as Glu298Asp (Marsden et al., 1993) (Fig. 3). The variant Asp allele for Glu298Asp polymorphism reduces NOS3 binding to caveolin-1 and results in decreased NOS3 availability in its caveolar fraction in the endothelial cells (Joshi et al., 2007). When calcium-activated calmodulin dissociates NOS3 from its binding to caveolin-1, the reduced binding of NOS3 to the caveolar fraction results in lower amounts of NOS3 available for activation and reduced NOS3 activity and NO production (Qian and Fulton, 2013) (Fig. 4C). In fact, decreased NOS3 activity was observed in endothelial cells carrying the variant allele for the Glu298Asp polymorphism (Joshi et al., 2007). In support of these in vitro findings, reduced platelet NO formation was found in subjects carrying the variant allele (Tanus-Santos et al., 2002; Godfrey et al., 2007).
Although a previous study showing that the SNP rs3918226 does not affect plasma nitrite levels (a relevant index of endogenous NO production) (Luizon et al., 2012), Salvi and cols recently showed that this polymorphism affects NOS3 expression (Salvi et al., 2013). Using a luciferase assay, the authors observed that the cytosine to thymine conversion at the position −665 of NOS3 promoter (g.-665C>T; Fig. 3) reduces NOS3 promoter activity. Taking into account that this SNP is located next to a potential transcription factor binding site for the Ets family domain, it is possible that g.-665C>T polymorphism modulates NOS3 transcription by affecting transcription factor-binding affinity (Salvi et al., 2012). As previously discussed, the Ets family domain positively regulates NOS3 promoter activity (Karantzoulis-Fegaras et al., 1999) and therefore it is reasonable to suspect that these transcription factors bind to NOS3 promoter with lower affinity in the presence of the variant allele, resulting in less NOS3 transcription compared with the ancestral allele (Fig. 4D). However, this suggestion still requires further testing.
7.2. Haplotype analysis approach
Despite the relevance of functional effects produced by NOS3 polymorphisms individually, the haplotype analysis approach may provide improved genetic information (Crawford and Nickerson, 2005). Haplotypes are a combination of alleles at different markers within the single chromosome that are inherited as a unit (Crawford and Nickerson, 2005). The main difference between haplotypes and individual genotypes is that the alleles are attributed to a chromosome. Each individual has two haplotypes for a given fragment of the genome, representing the maternal and paternal chromosomes (Crawford and Nickerson, 2005). Taking into account that the determination of individual haplotypes is time-consuming and demands isolation of individual chromosomes for sequencing or genotyping of family members, statistical methods have been developed to provide estimation of haplotype frequencies from unphased genotype data (Cox et al., 1998; Stephens et al., 2001). Using this approach, studies have shown the effects of NOS3 haplotypes on NO-related markers in vivo.
The effects of haplotypes involving the functional NOS3 polymorphisms g.-786T>C, 4b/4a VNTR and Glu298Asp on plasma nitrite/nitrate (NOx) were assessed in healthy white males (Metzger et al., 2005). Interestingly, although genotypes for the three polymorphisms did not affect plasma NOx levels, the haplotype including the C, 4b, and Glu alleles (C-4b-Glu haplotype) was associated with decreased NOx levels. Consistently, these findings were later confirmed in further populations of white (Metzger et al., 2007) and black (Metzger et al., 2011) healthy subjects, suggesting that the C-4b-Glu haplotype plays an important role in NO bioavailability. Indeed, a previous in vitro functional study observed that the g.-786T>C and 4b/4a VNTR polymorphisms exert an haplotype-dependent effect on NOS3 transcriptional activity (Wang et al., 2002), supporting the idea that NOS3 haplotype analysis approach may offer improved genetic information as compared to the analysis of each polymorphism.
7.3. Interethnic distribution of NOS3 polymorphisms
There are interethnic differences in NO-mediated vasodilation (Cardillo et al., 1998; Jones et al., 1999), which suggest that relevant differences in the distribution of NOS3 variants among different ethnic groups. Such differences were consistently shown is different populations (Tanus-Santos et al., 2001; Marroni et al., 2005; Luizon et al., 2009; Thomas et al., 2013). The variant alleles for the g.-786T>C and Glu298Asp polymorphisms were more commonly found in Caucasians than in African-Americans, whereas the 4a allele for the 4b/4a VNTR was more commonly found in African-Americans than in Caucasians (Tanus-Santos et al., 2001; Thomas et al., 2013). In parallel with genotypes, major interethnic differences exist with regards to haplotype distribution. For example, the T-4a-Glu haplotype is more commonly found in blacks, whereas the C-4b-Asp haplotype is more commonly found in whites (Tanus-Santos et al., 2001), and those differences may underlie differences in the susceptibility to a variety of diseases involving alterations in NO formation (Sandrim et al., 2007).
Interestingly, such interethnic differences are also found in more admixed populations, as shown in a study examining the distribution of the g.-786T>C, Glu298Asp, and 4b/4a VNTR polymorphisms in black and white Brazilians (Marroni et al., 2005). Although the Brazilian population was formed after extensive interethnic crosses between subjects from different continents including Europeans, Africans, and autochthonous Amerindians (Parra et al., 2003; Ferreira et al., 2006), the interethnic distribution of NO3 genotypes and haplotypes in Brazilians was very similar to that found in the American population (Tanus-Santos et al., 2001; Thomas et al., 2013). These findings strongly indicate that the interethnic differences in the distribution of NOS3 polymorphisms do not depend on geographic origin, at least when two populations from North America and South America are compared.
In addition to these findings, a recent study observed important differences in the distribution of NOS3 polymorphisms in Amerindians compared with white and black subjects from Brazil (Luizon et al., 2009). In this study, the variant alleles for the g.-786T>C, Glu298Asp, and 4b/4a VNTR polymorphisms were much less frequent in Amerindians than in blacks or whites, and the T-4b-Glu haplotype was more frequent in Amerindians than in blacks or whites. The data shown in that study support the idea of reduced genetic diversity in Amerindians in relation to black and white subjects, and may be relevant for studies involving NOS3 polymorphisms in admixed populations such as the American and the Brazilian populations (Luizon et al., 2009).
7.4. Clinical relevance of NOS3 polymorphisms
Given the major role played by NO in the cardiovascular system and the effects of NOS3 polymorphisms on NOS3 expression and activity, as well as on circulating levels of markers of endogenous NO formation, a large number of studies have evaluated the influence of these polymorphisms in cardiovascular diseases. However, discrepant results have been described (Cooke et al., 2007; Pereira et al., 2007). A possible explanation for these conflicting findings may be the analysis of genetic markers individually instead of the haplotype analysis approach (Crawford and Nickerson, 2005; Tanus-Santos and Casella-Filho, 2007). Indeed, the analysis of genetic polymorphisms one by one may not have enough power to detect small effects, and therefore, the analysis of polymorphisms combined within haplotypes is imperative (Tanus-Santos and Casella-Filho, 2007). Another explanation for the controversial clinical effects of NOS3 polymorphisms may be that population stratification may dilute the power of case-control studies that are commonly designed to identify genetic risk factors for a disease (Cardon and Bell, 2001).
Examples of highly prevalent diseases that include impaired endogenous NO formation as relevant pathogenetic mechanism are chronic hypertension and hypertensive disorders of pregnancy, erectile dysfunction, migraine, and metabolic disorders (Rajfer et al., 1992; Pieper, 1999; Thomas et al., 2001; Sandrim et al., 2008b; Olesen, 2010; Sansbury and Hill, 2014). Therefore, many studies have focused in the possibility of functional NOS3 polymorphisms affecting the susceptibility to these diseases. The main findings of these studies are summarized in Table 2 and will be briefly discussed below.
Table 2 -.
Clinical implications of NOS3 polymorphisms.
| Disease | Genetic marker | Findings | Reference |
|---|---|---|---|
| Hypertension | g.-786T>C | C allele associated with higher risk of hypertension in different populations | (Hyndman et al., 2002; Nejatizadeh et al., 2008; Niu and Qi, 2011) |
| Lack of association in different populations | (Tsujita et al., 2001; Sandrim et al., 2006a; Sandrim et al., 2006c; Pereira et al., 2007) | ||
| 4b/4a VNTR | 4a allele associated with higher risk of hypertension in Asians | (Uwabo et al., 1998; Niu and Qi, 2011) | |
| Lack of association in different populations | (Miyamoto et al., 1998; Benjafield and Morris, 2000; Sandrim et al., 2006a; Sandrim et al., 2006c) | ||
| Glu298Asp | Asp allele associated with higher risk of hypertension in different populations | (Jachymova et al., 2001; Pereira et al., 2007; Niu and Qi, 2011; Yang et al., 2013; Liu et al., 2015) | |
| Lack of association in different populations | (Kato et al., 1999; Schneider et al., 2000; Sandrim et al., 2006a; Sandrim et al., 2006c) | ||
| g.-665C>T | T allele associated with higher risk of hypertension in different populations | (Salvi et al., 2012; Salvi et al., 2013; Levinsson et al., 2014; de Miranda et al., 2015) | |
| Lack of association in Caucasian women | (Conen et al., 2008) | ||
| Haplotypes | Haplotypes including g.-786T>C, 4b/4a VNTR and Glu298Asp polymorphisms affect the susceptibility to hypertension. | (Sandrim et al., 2006a; Sandrim et al., 2006b; Sandrim et al., 2006c; Vasconcellos et al., 2010) | |
| Hypertensive disorders of pregnancy | g.-786T>C | C allele associated with higher risk of preeclampsia in different populations | (Seremak-Mrozikiewicz et al., 2011; Dai et al., 2013) |
| Lack of association in different populations | (Serrano et al., 2004; Sandrim et al., 2010b) | ||
| 4b/4a VNTR | 4a allele associated with higher risk of preeclampsia in different populations | (Tempfer et al., 2001; Dai et al., 2013) | |
| Lack of association in different populations | (Serrano et al., 2004; Sandrim et al., 2010b) | ||
| Glu298Asp | Asp allele associated with higher risk of preeclampsia in Colombians | (Serrano et al., 2004) | |
| Lack of association in different populations | (Sandrim et al., 2010b; Dai et al., 2013) | ||
| Haplotypes | Haplotypes including g.-786T>C, 4b/4a VNTR and Glu298Asp polymorphisms and the tagSNPs rs743506 and rs7830 affect the susceptibility to hypertensive disorders of pregnancy | (Serrano et al., 2004; Sandrim et al., 2010b; Muniz et al., 2012) | |
| Obesity and associated disorders | g.-786T>C | CC genotype associated with metabolic syndrome in children and adolescents | (Miranda et al., 2013) |
| 4b/4a VNTR | 4a4a genotype associated with obesity in children and adolescents | (Souza-Costa et al., 2011) | |
| Glu298Asp | Asp allele associated a larger mean body mass index, waist circumference and sum of skinfolds | (Podolsky et al., 2007) | |
| Haplotypes | CTGC haplotype, including the NOS3 tagSNPs rs3918226, rs3918188, rs743506 and rs7830 associated with obesity in children and adolescents | (de Miranda et al., 2015) | |
| C-4b-Glu haplotype including the g.-786T>C, 4b/4a VNTR and Glu298Asp polymorphisms affect the susceptibility to metabolic syndrome and hypertension in obese children and adolescents | (Souza-Costa et al., 2011; Miranda et al., 2013) | ||
| Diabetes mellitus and its complications | g.-786T>C | C allele associated with diabetic nephropathy in Egyptians | (Shoukry et al., 2012) |
| CC genotype associated with diabetic retinopathy in Caucasians | (Taverna et al., 2005) | ||
| 4b/4a VNTR | 4a allele associated with higher risk of diabetes mellitus in different populations | (Mehrab-Mohseni et al., 2011; Jia et al., 2013) (Galanakis et al., 2008) | |
| Lack of association with diabetic retinopathy in different populations | (Ma et al., 2014) | ||
| Glu298Asp | Asp allele associated with higher risk of diabetes mellitus in different populations | (Monti et al., 2003; Jia et al., 2013) | |
| Asp allele associated with diabetic nephropathy in different populations | (Shoukry et al., 2012; Kuricova et al., 2013) | ||
| Haplotypes | Haplotypes including the g.-786T>C, 4b/4a VNTR and Glu298Asp polymorphisms are not associated with diabetic retinopathy but the C-4b-Glu haplotype protects against type 2 diabetes mellitus | (de Syllos et al., 2006) | |
| Migraine | g.-786T>C | C allele associated with higher risk of migraine in Caucasians | (Eroz et al., 2014) |
| Glu298Asp | ‘AspAsp’ genotype associated with higher risk of migraine in Caucasians | (Borroni et al., 2006; Eroz et al., 2014) | |
| Lack of association with migraine in Caucasians | (Toriello et al., 2008) | ||
| Haplotypes | Lack of association with migraine in different populations Haplotypes including the g.-786T>C, g.-665C>T, 4b/4a VNTR and Glu298Asp polymorphisms and the tagSNP rs743506 associated with aura in patients with migraine | (Toriello et al., 2008; Goncalves et al., 2011) (Goncalves et al., 2011) | |
| Erectile dysfunction | g.-786T>C | CC genotype associated with higher risk of erectile dysfunction in different populations | (Sinici et al., 2010; Safarinejad et al., 2011) |
| 4b/4a VNTR Glu298Asp | Lack of association in different populations Asp allele associated with higher risk of erectile dysfunction in different populations | (Erol et al., 2009; Wang et al., 2010) (Rosas-Vargas et al., 2004; Erol et al., 2009; Lee et al., 2009; Wang et al., 2010; Safarinejad et al., 2011; Hermans et al., 2012; Lee et al., 2012) |
7.4.1. Hypertension
Hypertension is a multifactorial disease affecting approximately 1 billion subjects, and is a major risk factor for coronary heart disease and cerebrovascular accidents (Chobanian et al., 2003). There is strong evidence that abnormalities in NOS3 regulation may result in NO deficiency and cause hypertension (Thomas et al., 2001). Therefore, several studies have examined whether NOS3 polymorphisms are associated with hypertension. Increased risk of hypertension associated with the variant alleles for the g.-786T>C, g.-665C>T, 4b/4a VNTR and Glu298Asp polymorphisms have been observed in some studies (Uwabo et al., 1998; Jachymova et al., 2001; Hyndman et al., 2002; Pereira et al., 2007; Nejatizadeh et al., 2008; Niu and Qi, 2011; Salvi et al., 2013; Yang et al., 2013; Levinsson et al., 2014; de Miranda et al., 2015; Liu et al., 2015). However, lack of association of these polymorphisms with hypertension was reported in many other studies (Miyamoto et al., 1998; Kato et al., 1999; Benjafield and Morris, 2000; Schneider et al., 2000; Tsujita et al., 2001; Sandrim et al., 2006a; Sandrim et al., 2006c; Pereira et al., 2007; Conen et al., 2008). These discrepancies may be related to consideration limited to only one polymorphism rather than combinations of polymorphisms.
Indeed, the haplotype analysis approach is strongly indicated for the study of candidate genes possibly involved in the development of hypertension (Yagil and Yagil, 2004). Interestingly, haplotypes including g.-786T>C, 4b/4a VNTR and Glu298Asp NOS3 polymorphisms were shown to affect the susceptibility to hypertension (Sandrim et al., 2006a; Sandrim et al., 2006b; Sandrim et al., 2006c; Vasconcellos et al., 2010). Particularly, the C-4b-Glu haplotype was protective, while the C-4b-Asp haplotype increased the susceptibility to hypertension, both in black and in white subjects (Sandrim et al., 2006a), disregarding major differences in the distribution of NOS3 polymorphisms and haplotypes when these two ethnic groups are compared (Tanus-Santos et al., 2001; Marroni et al., 2005).
7.4.2. Hypertensive disorders of pregnancy
Hypertensive disorders of pregnancy affect 3%−5% of pregnancies and are major causes of maternal and neonatal morbidity and mortality (Sandrim et al., 2008a; Sandrim et al., 2010a). Interestingly, studies have observed that the reduction of NO bioavailability may contribute to the increase in blood pressure in important hypertensive disorders of pregnancy, such as gestational hypertension and preeclampsia (Cockell and Poston, 1997; Savvidou et al., 2003; Sandrim et al., 2008b). This evidence led to investigation of the effects of NOS3 polymorphisms on susceptibility to hypertensive disorders of pregnancy and relevant results has been reported (Table 2).
The variant alleles for the g.-786T>C, 4b/4a VNTR and Glu298Asp polymorphisms were associated with preeclampsia or gestational hypertension in some studies (Tempfer et al., 2001; Serrano et al., 2004; Seremak-Mrozikiewicz et al., 2011), and these effects would be related with a possible imbalance in NO production caused by these alleles (Sandrim et al., 2010b). On the other hand, no association of these polymorphisms with hypertensive disorders of pregnancy was reported in other studies (Landau et al., 2004; Serrano et al., 2004; Sandrim et al., 2010b), suggesting an inconsistency of findings when approaching single NOS3 polymorphisms in these diseases. To clarify this question, a recent meta-analysis evaluated studies involving NOS3 polymorphisms in preeclampsia and found that the ‘C’ allele in the promoter and the ‘4a’ allele in intron 4 increase the risk of developing this hypertensive disorder, whereas the Glu298Asp polymorphism is not associated with the disease (Dai et al., 2013). In addition, a study evaluating NOS3 haplotypes in preeclampsia revealed the association of C-4b-Asp haplotype with this disease (Serrano et al., 2004). Another study, in turn, showed a higher frequency of the C-4b-Glu haplotype in healthy pregnant than in pregnant with preeclampsia (Sandrim et al., 2010b). Interestingly, this haplotype was also associated with increased nitrite levels in healthy pregnant, suggesting that the C-4b-Glu haplotype may protect against preeclampsia by increasing NO formation (Sandrim et al., 2010b). Consistent with these findings, a later study included the NOS3 tagSNPs rs743506 and rs7830 in a haplotype analysis involving also the three functional polymorphisms mentioned above and found that the C-4b-Glu-C-G haplotype (regarding to polymorphisms in the promoter, intron 4, exon 7 and the tagSNPs rs743506 and rs7830, respectively) protects against the development of preeclampsia and gestational hypertension (Muniz et al., 2012).
7.4.3. Metabolic disorders
Metabolic disorders such as obesity and diabetes are very common and affect a growing number of subjects (Golden et al., 2009). Importantly, the prevalence of obesity has increased dramatically in the last decades achieving approximately 37% of adults worldwide (Ng et al., 2014). Because NO plays a critical role in regulating the metabolism and body composition, decreased NO bioavailability has been reported in clinical and experimental obesity (Sansbury and Hill, 2014). Interestingly, carriers of the Asp allele for the Glu298Asp NOS3 polymorphism showed larger mean body mass index, waist circumference, and sum of skinfolds (Podolsky et al., 2007), suggesting that variations in NOS3 gene may contribute to the genetic predisposition to obesity. Given that obesity often starts during childhood, the assessment of the genetic susceptibility to obesity in children and adolescents is imperative. In this regard, the 4a4a genotype for the NOS3 polymorphism in intron 4 and the C-T-G-C haplotype, including the NOS3 tagSNPs rs3918226, rs3918188, rs743506 and rs7830, were associated with obesity in children and adolescents (Souza-Costa et al., 2011; de Miranda et al., 2015).
NOS3 polymorphisms also seem to predispose to disorders associated with obesity, such as metabolic syndrome and hypertension (Souza-Costa et al., 2011; Miranda et al., 2013; de Miranda et al., 2015). In fact, the CC genotype for the g.-786T>C polymorphism was associated with metabolic syndrome in children and adolescents (Miranda et al., 2013). In addition, the C-4b-Glu haplotype (including the g.-786T>C, 4b/4a VNTR and Glu298Asp polymorphisms) was more common in boys with metabolic syndrome than in controls (Miranda et al., 2013). Interestingly, this same haplotype was associated with hypertension in obese children and adolescents (Souza-Costa et al., 2011), and has been associated with lower endogenous NO formation in adults of different ethnic backgrounds (Metzger et al., 2007; Metzger et al., 2011).
Impaired NO production was also observed in diabetes mellitus and its complications (Pieper, 1999), thus supporting the relevance of evaluating the contribution of NOS3 polymorphisms to these conditions. There is now evidence that the 4a allele for the 4b/4a VNTR increases the risk for both type 1 and type 2 diabetes mellitus (Galanakis et al., 2008; Mehrab-Mohseni et al., 2011; Jia et al., 2013). Interestingly, this allele was associated with endothelial dysfunction in diabetic patients (Komatsu et al., 2002), suggesting that the 4a allele impairs NO bioavailability, thereby contributing to diabetes susceptibility. Besides the 4a allele, the Asp allele for the Glu298Asp polymorphism was also associated with predisposition to diabetes (Monti et al., 2003; Jia et al., 2013), and this association seems to be particularly important in obese subjects (Bressler et al., 2013). More interestingly, the C-4b-Glu haplotype was described to be protective against type 2 diabetes mellitus (de Syllos et al., 2006).
Chronic hyperglycemia in diabetic patients promotes microvascular complications such as diabetic nephropathy and retinopathy (Creager et al., 2003), and NOS3 polymorphisms may also affect these complications. Indeed, the variant alleles for the g.-786T>C and Glu298Asp polymorphisms were associated with diabetic nephropathy (Shoukry et al., 2012; Kuricova et al., 2013). In addition, although lack of association between NOS3 genotypes and haplotypes and diabetic retinopathy has been consistently reported in type 2 diabetes mellitus (Awata et al., 2004; de Syllos et al., 2006; Ma et al., 2014), the CC genotype for the g.-786T>C polymorphism was associated with this microvascular complication in type 1 diabetes mellitus (Taverna et al., 2005).
7.4.4. Migraine
Migraine is a common neurovascular disorder predominantly affecting women (Victor et al., 2010). Despite the involvement of complex pathogenetic mechanisms in this disease, there is strong evidence that NO plays a pivotal role in migraine pathophysiology (Olesen, 2010). NO is an important mediator in the control of cerebral blood flow and contributes to the activation of nociceptors in the trigeminovascular system during migraine attack (Olesen, 2010).
Variations in NOS3 gene have been implicated in the genetic susceptibility to migraine. In this regard, the variant genotypes for the g.-786T>C polymorphism were associated with migraine susceptibility (Eroz et al., 2014). The ‘AspAsp’ genotype for the Glu298Asp polymorphism was associated with a 2-fold increase in the risk for migraine when compared to controls and a 3-fold increase in the risk of migraine with aura (transient neurologic symptoms preceding a migraine attack) when compared to migraine without aura patients (Borroni et al., 2006). Additionally, the Glu298Asp polymorphism was described to influence also the intensity of pain and the age at the onset of migraine (Eroz et al., 2014). In contrast with these findings, other studies failed to show an association between single NOS3 polymorphisms and migraine (Toriello et al., 2008; Goncalves et al., 2011). Again, it is possible that haplotype analysis would be more appropriated to define genetic contributions to this disease. In fact, although a study failed to show an association between NOS3 haplotypes and migraine (Toriello et al., 2008), another more comprehensive study including variants for the g.-786T>C, g.-665C>T, 4b/4a VNTR, and Glu298Asp polymorphisms, and for the tagSNP rs743506 showed interesting results (Goncalves et al., 2011). The haplotypes C-C-4a-Glu-G and C-C-4b-Glu-G were more commonly found in women with migraine with aura than in women with migraine without aura (Goncalves et al., 2011). Despite the lack of association between NOS3 haplotypes and migraine, this study suggests that NOS3 haplotypes may influence the susceptibility to aura in patients with migraine. Moreover, other genes encoding other isoforms of NO synthases may be relevant to migraine (de et al., 2012), and there is probably major interactions between NOS3 polymorphisms with other genetic variants to increase the susceptibility to migraine (Goncalves et al., 2012).
7.4.5. Erectile dysfunction
NO is essential for relaxation of smooth muscle cells of the corpus cavernosum (Rajfer et al., 1992), and thus impaired NO production contributes to erectile dysfunction, a very common disorder characterized by inability to acquire and maintain sexual intercourse (Hatzimouratidis et al., 2010). Consistent with the importance of NO in erectile dysfunction, variations in NOS3 gene were shown to influence the susceptibility to this disorder. In this context, although lack of association between 4b/4a VNTR and erectile dysfunction was been reported (Erol et al., 2009; Wang et al., 2010), the CC genotype for the g.-786T>C polymorphism was associated with increased risk for erectile dysfunction (Sinici et al., 2010; Safarinejad et al., 2011). Interestingly, this genotype was also associated with earlier onset of erectile dysfunction, suggesting that this is an independent risk factor for endothelial dysfunction in the absence of other risk factors (Sinici et al., 2010). Interestingly, many studies showed significant association between the Asp allele for Glu298Asp polymorphism and erectile dysfunction (Hermans et al., 2012; Lee et al., 2012) (Rosas-Vargas et al., 2004; Erol et al., 2009; Lee et al., 2009; Wang et al., 2010; Safarinejad et al., 2011). While there are no known studies addressing the possible association between NOS3 haplotypes and erectile dysfunction, a recent study reported NOS3 haplotypes affecting the responses to sildenafil, a drug usually prescribed to treat erectile dysfunction (Muniz et al., 2013a). These recent findings suggest important pharmacogenetics implications of NOS3 polymorphisms, which will be discussed below.
7.5. Pharmacogenetics implications of NOS3 polymorphisms
Lack of response or toxicity to usual doses of drugs are well-known problems and cause significant morbidity, mortality, and health-care costs (Lacchini et al., 2010; Haga and LaPointe, 2013). A significant part of these problems is explained by Pharmacogenetics, an interdisciplinary field involving Pharmacology and Genetics, which focuses on how the genetic variations affect drug responses (Evans and McLeod, 2003). Indeed, genetic variation is probably responsible for 20–95% of the variation in individual responses to drugs (Wang et al., 2011). When dealing with drugs that affect NO signaling, polymorphisms in NOS3 gene are major candidates to impact pharmacotherapy (Eisenhardt et al., 2003; Abe et al., 2005; Nagassaki et al., 2006; Peskircioglu et al., 2007; Mason et al., 2012; Muniz et al., 2013a; Silva et al., 2013), and we will briefly discuss some examples below.
As previously discussed in Section 6.2, statins increase NOS3 expression and NO production (Laufs and Liao, 1998; Liao and Laufs, 2005). Curiously, NOS3 polymorphisms modulate the upregulation of NOS3 and NO formation by statins. For example, statins increased NOS3 mRNA levels to higher concentrations in cultured endothelial cells with CC genotype for the g.-786T>C polymorphism than endothelial cells with the TT genotype (Abe et al., 2005). This effect was associated with increased transcriptional activity, higher mRNA stability, and reduced expression of repressor protein RPA-1 (Abe et al., 2005). Consistent with these cell experiments, a clinical study showed similar effects in healthy subjects (Nagassaki et al., 2006). Treatment with a low dose of atorvastatin significantly increased NO bioavailability in subjects carrying the CC genotype, but not in subjects carrying the TT genotype (Nagassaki et al., 2006). Another study showed that simvastatin treatment increased nitrite levels (a maker of endogenous NO formation) to a higher level in obese women with the CC genotype for the g.-786T>C polymorphism than in those with the TT genotype (Andrade et al., 2013).
NOS3 polymorphisms also modify the effects of some antihypertensive drugs (Mason et al., 2012; Silva et al., 2013). One of the most prescribed antihypertensive drugs are the angiotensin-converting enzyme (ACE) inhibitors (von Lueder and Krum, 2013). Their antihypertensive effects involve vasodilation and improved endothelial function resulting from ACE inhibition, which reduces angiotensin II formation and enhances bradykinin levels (Mentz et al., 2013). Bradykinin, in turn, stimulates receptors on endothelial cells causing NOS3 activation, NO release and vasodilation (Linz et al., 1999). This is important because a recent pharmacogenetic study in hypertensive patients treated with the ACE inhibitor enalapril (Silva et al., 2013) showed that the “TC” or “CC” genotypes and the “C” allele for the g.-786T>C polymorphism were more commonly found in patients with good responses to this drug than in those patients classified as poor responders (Silva et al., 2013). Another example indicating similar effects is a study showing that the angiotensin II receptor blocker (ARB) olmesartan enhanced NO release from endothelial cells homozygous for C allele of this polymorphism to a higher extent than that found in heterozygous cells (Mason et al., 2012).
The pharmacogenetics of erectile dysfunction (Lacchini and Tanus-Santos, 2014) is also influenced by NOS3 polymorphisms (Muniz et al., 2013a). The phosphodiesterase type 5 (PDE-5) inhibitors such as sildenafil are widely used to treat erectile dysfunction because the inhibition of this enzyme increases tissue cGMP concentrations, especially when NO signaling is impaired (Lacchini and Tanus-Santos, 2014). While subjects homozygous for the “Asp” allele of the Glu298Asp polymorphism were reported as less responsive to sildenafil than ancestral allele carriers (Eisenhardt et al., 2003), there is some controversy with respect to this issue (Peskircioglu et al., 2007; Muniz et al., 2013a). However, improved responses to sildenafil were observed in patients with erectile dysfunction carrying the “C” and the “4a” alleles compared with patients carrying the “T” and “4b” alleles for the g.-786T>C and 4b/4a VNTR polymorphisms, respectively (Peskircioglu et al., 2007; Muniz et al., 2013a). Interestingly, these findings are consistent with another study showing that reduced plasma nitrite concentrations predict better responses to sildenafil (Muniz et al., 2013b). In addition, the haplotype analysis showed that NOS3 haplotypes including the g.-786T>C, 4b/4a VNTR and Glu298Asp polymorphisms affect the responses to sildenafil (Muniz et al., 2013a).
8. Concluding remarks and future perspectives
The endothelium-derived relaxing factor was identified more than three decades ago as NO, and thereafter many physiological and biochemical investigations have proved the crucial importance of this molecule for the cardiovascular homeostasis. In the vasculature, the dominant NO synthase is NOS3, which produces NO to promote vasodilation and to control blood pressure. For this reason, reduced NOS3 expression and activity result in impaired NO production leading to a variety of disease conditions. This seems to be the case of functional NOS3 polymorphisms, which affect NOS3 expression or activity, and have been associated with many diseases, as briefly discussed in the present article. Genotypes or haplotypes for NOS3 polymorphisms are now recognized as genetic markers of increased risk for developing cardiovascular diseases, which may also affect the response to drugs and other procedures used to treat cardiovascular diseases.
While a number of studies support an important contribution of NOS3 polymorphisms particularly to cardiovascular phenotypes, some studies failed to confirm significant clinical and pharmacogenetic implications. These inconsistences may be related to interethnic differences in the distributions of NOS3 polymorphisms tested, or to the analysis of single genetic polymorphisms, without taking into consideration the interactions between different makers within NOS3. Moreover, the inconsistences may be attributed to heterogeneous phenotypes. In this case, variability in the etiology and mechanisms involved may explain the differences in observed phenotype, thereby decreasing the possibility of successfully detecting associations between NOS3 polymorphisms with diseases or drug response. Therefore, it is crucial that subjects are carefully phenotyped and maybe surrogate markers of disease should also be evaluated.
Given the inconsistent data, further studies are necessary to clarify the functional and clinical implications of NOS3 haplotypes. The development of biomarkers that accurately predict a given phenotype is a challenging possibility and will probably require large and comprehensive studies, with replication of findings in different populations. Effort should be made to assess the effects of NOS3 haplotypes or combinations of genetic variants on biochemical parameters that reflect NOS3 gene expression and enzyme activity more precisely, either under different disease conditions or after drug treatment. This approach may be helpful in improving our understanding how genetic contribution involving NOS3 may be relevant to cardiovascular diseases and to optimize cardiovascular drug therapy.
Highlights.
Nitric oxide (NO) is very important in physiology and pathophysiology.
In the cardiovascular system, most NO is synthesized by endothelial NO synthase (NOS3).
The NOS3 gene exhibits a number of polymorphic sites.
Some NOS3 polymorphisms have functional and clinical implications.
NOS3 polymorphisms affect disease susceptibility and drug responses.
Acknowledgments
This review and the corresponding Gene Wiki article are written as part of the Cardiac Gene Wiki Review series--a series resulting from a collaboration between the journal GENE, the Gene Wiki Initiative, and the BD2K initiative. The Cardiac Gene Wiki Initiative is supported by National Institutes of Health (GM089820 and GM114833). Additional support for Gene Wiki Reviews is provided by Elsevier, the publisher of GENE. The authors would like to thank Fundaçao de Amparo a Pesquisa do Estado de Sao Paulo (FAPESP) and Conselho Nacional de Desenvolvimento Cientifico e Tecnologico (CNPq) for financial support. The corresponding Gene Wiki entry for this review can be found here: https://en.wikipedia.org/wiki/Endothelial_NOS
List of abbreviations:
- 3’-UTR
3’-mRNA untranslated region
- 5’-UTR
5’-mRNA untranslated region
- ACE
angiotensin-converting enzyme
- ARB
angiotensin II receptor blocker
- BH4
tetrahydrobiopterin
- cGMP
cyclic guanosine monophosphate
- NOS3
endotelial nitric oxide synthase
- FAD
flavin adenine dinucleotide
- FMN
flavin mononucleotide
- NOS2
inducible nitric oxide synthase
- IRAG
phosphorylates 1,4,5 inositoltriphosphate receptor-associated cGMP kinase substrate
- MAZ
MYC-associated zinc finger protein
- NADPH
nicotinamide adenine dinucleotide phosphate
- NOS1
neuronal nitric oxide synthase
- NO
nitric oxide
- PDE-5
phosphodiesterase type 5
- PRD
positive regulatory domain
- PKG-1
protein kinase G-1
- RPA1
replication protein A1
- SERCA
sarcoplasmic reticulum ATPase
- sirRNA
small interference RNA
- SNP
single nucleotide polymorphism
- sGC
soluble guanylate cyclase
- TGF-β1
transforming growth factor β1
- VNTR
variable number of tandem repeats
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare no conflict of interest.
References
- Abe K, Nakayama M, Yoshimura M, Nakamura S, Ito T, Yamamuro M, Sakamoto T, Miyamoto Y, Yoshimasa Y, Saito Y, Nakao K, Yasue H and Ogawa H, 2005. Increase in the transcriptional activity of the endothelial nitric oxide synthase gene with fluvastatin: a relation with the −786T>C polymorphism. Pharmacogenet Genomics 15, 329–336. [DOI] [PubMed] [Google Scholar]
- Abu-Soud HM, Feldman PL, Clark P and Stuehr DJ, 1994. Electron transfer in the nitric-oxide synthases. Characterization of L-arginine analogs that block heme iron reduction. J Biol Chem 269, 32318–32326. [PubMed] [Google Scholar]
- Abu-Soud HM, Ichimori K, Presta A and Stuehr DJ, 2000. Electron transfer, oxygen binding, and nitric oxide feedback inhibition in endothelial nitric-oxide synthase. J Biol Chem 275, 17349–17357. [DOI] [PubMed] [Google Scholar]
- Albina JE, Abate JA and Henry WL Jr., 1991. Nitric oxide production is required for murine resident peritoneal macrophages to suppress mitogen-stimulated T cell proliferation. Role of IFN-gamma in the induction of the nitric oxide-synthesizing pathway. J Immunol 147, 144–148. [PubMed] [Google Scholar]
- Albrecht EW, Stegeman CA, Heeringa P, Henning RH and van Goor H, 2003. Protective role of endothelial nitric oxide synthase. J Pathol 199, 8–17. [DOI] [PubMed] [Google Scholar]
- Alderton WK, Cooper CE and Knowles RG, 2001. Nitric oxide synthases: structure, function and inhibition. Biochem J 357, 593–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alioua A, Tanaka Y, Wallner M, Hofmann F, Ruth P, Meera P and Toro L, 1998. The large conductance, voltage-dependent, and calcium-sensitive K+ channel, Hslo, is a target of cGMP-dependent protein kinase phosphorylation in vivo. J Biol Chem 273, 32950–32956. [DOI] [PubMed] [Google Scholar]
- Amaral JH, Montenegro MF, Pinheiro LC, Ferreira GC, Barroso RP, Costa-Filho AJ and Tanus-Santos JE, 2013. TEMPOL enhances the antihypertensive effects of sodium nitrite by mechanisms facilitating nitrite-derived gastric nitric oxide formation. Free Radic Biol Med 65, 446–455. [DOI] [PubMed] [Google Scholar]
- Andrade VL, Sertorio JT, Eleuterio NM, Tanus-Santos JE, Fernandes KS and Sandrim VC, 2013. Simvastatin treatment increases nitrite levels in obese women: modulation by T(−786)C polymorphism of eNOS. Nitric Oxide 33, 83–87. [DOI] [PubMed] [Google Scholar]
- Awata T, Neda T, Iizuka H, Kurihara S, Ohkubo T, Takata N, Osaki M, Watanabe M, Nakashima Y, Sawa T, Inukai K, Inoue I, Shibuya M, Mori K, Yoneya S and Katayama S, 2004. Endothelial nitric oxide synthase gene is associated with diabetic macular edema in type 2 diabetes. Diabetes Care 27, 2184–2190. [DOI] [PubMed] [Google Scholar]
- Balla G, Jacob HS, Balla J, Rosenberg M, Nath K, Apple F, Eaton JW and Vercellotti GM, 1992. Ferritin: a cytoprotective antioxidant strategem of endothelium. J Biol Chem 267, 18148–18153. [PubMed] [Google Scholar]
- Balligand JL, Kelly RA, Marsden PA, Smith TW and Michel T, 1993. Control of cardiac muscle cell function by an endogenous nitric oxide signaling system. Proc Natl Acad Sci U S A 90, 347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baran I, 1996. Calcium and cell cycle progression: possible effects of external perturbations on cell proliferation. Biophys J 70, 1198–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP, 2009. MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjafield AV and Morris BJ, 2000. Association analyses of endothelial nitric oxide synthase gene polymorphisms in essential hypertension. Am J Hypertens 13, 994–998. [DOI] [PubMed] [Google Scholar]
- Benjamin N, Dutton JA and Ritter JM, 1991. Human vascular smooth muscle cells inhibit platelet aggregation when incubated with glyceryl trinitrate: evidence for generation of nitric oxide. Br J Pharmacol 102, 847–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian K, Doursout MF and Murad F, 2008. Vascular system: role of nitric oxide in cardiovascular diseases. J Clin Hypertens (Greenwich) 10, 304–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borroni B, Rao R, Liberini P, Venturelli E, Cossandi M, Archetti S, Caimi L and Padovani A, 2006. Endothelial nitric oxide synthase (Glu298Asp) polymorphism is an independent risk factor for migraine with aura. Headache 46, 1575–1579. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Hwang PM, Glatt CE, Lowenstein C, Reed RR and Snyder SH, 1991. Cloned and expressed nitric oxide synthase structurally resembles cytochrome P-450 reductase. Nature 351, 714–718. [DOI] [PubMed] [Google Scholar]
- Bressler J, Pankow JS, Coresh J and Boerwinkle E, 2013. Interaction between the NOS3 gene and obesity as a determinant of risk of type 2 diabetes: the Atherosclerosis Risk in Communities study. PLoS One 8, e79466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan AJ, 1999. Hsp90’s secrets unfold: new insights from structural and functional studies. Trends Cell Biol 9, 262–268. [DOI] [PubMed] [Google Scholar]
- Cardillo C, Kilcoyne CM, Cannon RO 3rd and Panza JA, 1998. Racial differences in nitric oxide-mediated vasodilator response to mental stress in the forearm circulation. Hypertension 31, 1235–1239. [DOI] [PubMed] [Google Scholar]
- Cardon LR and Bell JI, 2001. Association study designs for complex diseases. Nat Rev Genet 2, 91–99. [DOI] [PubMed] [Google Scholar]
- Chan Y, Fish JE, D’Abreo C, Lin S, Robb GB, Teichert AM, Karantzoulis-Fegaras F, Keightley A, Steer BM and Marsden PA, 2004. The cell-specific expression of endothelial nitric-oxide synthase: a role for DNA methylation. J Biol Chem 279, 35087–35100. [DOI] [PubMed] [Google Scholar]
- Chaudhury A, Chander P and Howe PH, 2010. Heterogeneous nuclear ribonucleoproteins (hnRNPs) in cellular processes: Focus on hnRNP E1’s multifunctional regulatory roles. Rna 16, 1449–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Castranova V, Shi X and Demers LM, 1999a. New insights into the role of nuclear factor-kappaB, a ubiquitous transcription factor in the initiation of diseases. Clin Chem 45, 7–17. [PubMed] [Google Scholar]
- Chen Z, Peng IC, Sun W, Su MI, Hsu PH, Fu Y, Zhu Y, DeFea K, Pan S, Tsai MD and Shyy JY, 2009. AMP-activated protein kinase functionally phosphorylates endothelial nitric oxide synthase Ser633. Circ Res 104, 496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, Power DA, Ortiz de Montellano PR and Kemp BE, 1999b. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett 443, 285–289. [DOI] [PubMed] [Google Scholar]
- Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL Jr., Jones DW, Materson BJ, Oparil S, Wright JT Jr. and Roccella EJ, 2003. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. Jama 289, 2560–2572. [DOI] [PubMed] [Google Scholar]
- Cockcroft JR, 2005. Exploring vascular benefits of endothelium-derived nitric oxide. Am J Hypertens 18, 177S–183S. [DOI] [PubMed] [Google Scholar]
- Cockell AP and Poston L, 1997. Flow-mediated vasodilatation is enhanced in normal pregnancy but reduced in preeclampsia. Hypertension 30, 247–251. [DOI] [PubMed] [Google Scholar]
- Conen D, Glynn RJ, Buring JE, Ridker PM and Zee RY, 2008. Association of renin-angiotensin and endothelial nitric oxide synthase gene polymorphisms with blood pressure progression and incident hypertension: prospective cohort study. J Hypertens 26, 1780–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke GE, Doshi A and Binkley PF, 2007. Endothelial nitric oxide synthase gene: prospects for treatment of heart disease. Pharmacogenomics 8, 1723–1734. [DOI] [PubMed] [Google Scholar]
- Cornwell TL, Arnold E, Boerth NJ and Lincoln TM, 1994. Inhibition of smooth muscle cell growth by nitric oxide and activation of cAMP-dependent protein kinase by cGMP. Am J Physiol 267, C1405–1413. [DOI] [PubMed] [Google Scholar]
- Corson MA, James NL, Latta SE, Nerem RM, Berk BC and Harrison DG, 1996. Phosphorylation of endothelial nitric oxide synthase in response to fluid shear stress. Circ Res 79, 984–991. [DOI] [PubMed] [Google Scholar]
- Cortese-Krott MM and Kelm M, 2014. Endothelial nitric oxide synthase in red blood cells: key to a new erythrocrine function? Redox Biol 2, 251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox A, Camp NJ, Nicklin MJ, di Giovine FS and Duff GW, 1998. An analysis of linkage disequilibrium in the interleukin-1 gene cluster, using a novel grouping method for multiallelic markers. Am J Hum Genet 62, 1180–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven PA and DeRubertis FR, 1978. Restoration of the responsiveness of purified guanylate cyclase to nitrosoguanidine, nitric oxide, and related activators by heme and hemeproteins. Evidence for involvement of the paramagnetic nitrosyl-heme complex in enzyme activation. J Biol Chem 253, 8433–8443. [PubMed] [Google Scholar]
- Crawford DC and Nickerson DA, 2005. Definition and clinical importance of haplotypes. Annu Rev Med 56, 303–320. [DOI] [PubMed] [Google Scholar]
- Creager MA, Luscher TF, Cosentino F and Beckman JA, 2003. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: Part I. Circulation 108, 1527–1532. [DOI] [PubMed] [Google Scholar]
- Dai B, Liu T, Zhang B, Zhang X and Wang Z, 2013. The polymorphism for endothelial nitric oxide synthase gene, the level of nitric oxide and the risk for pre-eclampsia: a meta-analysis. Gene 519, 187–193. [DOI] [PubMed] [Google Scholar]
- Davis ME, Grumbach IM, Fukai T, Cutchins A and Harrison DG, 2004. Shear stress regulates endothelial nitric-oxide synthase promoter activity through nuclear factor kappaB binding. J Biol Chem 279, 163–168. [DOI] [PubMed] [Google Scholar]
- de Miranda JA, Lacchini R, Belo VA, Lanna CM, Sertorio JT, Luizon MR and Tanus-Santos JE, 2015. The effects of endothelial nitric oxide synthase tagSNPs on nitrite levels and risk of hypertension and obesity in children and adolescents. J Hum Hypertens 29, 109–114. [DOI] [PubMed] [Google Scholar]
- de OSMT, Goncalves FM, Martins-Oliveira A, Speciali JG, Dach F, Lacchini R and Tanus-Santos JE, 2012. Inducible nitric oxide synthase haplotype associated with migraine and aura. Mol Cell Biochem 364, 303–308. [DOI] [PubMed] [Google Scholar]
- de Syllos RW, Sandrim VC, Lisboa HR, Tres GS and Tanus-Santos JE, 2006. Endothelial nitric oxide synthase genotype and haplotype are not associated with diabetic retinopathy in diabetes type 2 patients. Nitric Oxide 15, 417–422. [DOI] [PubMed] [Google Scholar]
- Dekker RJ, van Soest S, Fontijn RD, Salamanca S, de Groot PG, VanBavel E, Pannekoek H and Horrevoets AJ, 2002. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Kruppel-like factor (KLF2). Blood 100, 1689–1698. [DOI] [PubMed] [Google Scholar]
- Denninger JW and Marletta MA, 1999. Guanylate cyclase and the .NO/cGMP signaling pathway. Biochim Biophys Acta 1411, 334–350. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R and Zeiher AM, 1999. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399, 601–605. [DOI] [PubMed] [Google Scholar]
- Dinerman JL, Dawson TM, Schell MJ, Snowman A and Snyder SH, 1994. Endothelial nitric oxide synthase localized to hippocampal pyramidal cells: implications for synaptic plasticity. Proc Natl Acad Sci U S A 91, 4214–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudzinski DM, Igarashi J, Greif D and Michel T, 2006. The regulation and pharmacology of endothelial nitric oxide synthase. Annu Rev Pharmacol Toxicol 46, 235–276. [DOI] [PubMed] [Google Scholar]
- Durante W, Kroll MH, Christodoulides N, Peyton KJ and Schafer AI, 1997. Nitric oxide induces heme oxygenase-1 gene expression and carbon monoxide production in vascular smooth muscle cells. Circ Res 80, 557–564. [DOI] [PubMed] [Google Scholar]
- Eisenhardt A, Sperling H, Hauck E, Porst H, Stief C, Rubben H, Muller N and Siffert W, 2003. ACE gene I/D and NOS3 G894T polymorphisms and response to sildenafil in men with erectile dysfunction. Urology 62, 152–157. [DOI] [PubMed] [Google Scholar]
- Ellis G, Adatia I, Yazdanpanah M and Makela SK, 1998. Nitrite and nitrate analyses: a clinical biochemistry perspective. Clin Biochem 31, 195–220. [DOI] [PubMed] [Google Scholar]
- Erol B, Bozdogan G, Akduman B, Dursun A, Bozdogan S, Onem K and Mungan A, 2009. eNOS gene intron 4 VNTR and exon 7-G894T polymorphisms in Turkish men with erectile dysfunction: a case control study. J Sex Med 6, 1423–1429. [DOI] [PubMed] [Google Scholar]
- Eroz R, Bahadir A, Dikici S and Tasdemir S, 2014. Association of endothelial nitric oxide synthase gene polymorphisms (894G/T, −786T/C, G10T) and clinical findings in patients with migraine. Neuromolecular Med 16, 587–593. [DOI] [PubMed] [Google Scholar]
- Evans WE and McLeod HL, 2003. Pharmacogenomics--drug disposition, drug targets, and side effects. N Engl J Med 348, 538–549. [DOI] [PubMed] [Google Scholar]
- Ferreira LB, Mendes-Junior CT, Wiezel CE, Luizon MR and Simoes AL, 2006. Genomic ancestry of a sample population from the state of Sao Paulo, Brazil. Am J Hum Biol 18, 702–705. [DOI] [PubMed] [Google Scholar]
- Fish JE and Marsden PA, 2006. Endothelial nitric oxide synthase: insight into cell-specific gene regulation in the vascular endothelium. Cell Mol Life Sci 63, 144–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish JE, Matouk CC, Yeboah E, Bevan SC, Khan M, Patil K, Ohh M and Marsden PA, 2007. Hypoxia-inducible expression of a natural cis-antisense transcript inhibits endothelial nitric-oxide synthase. J Biol Chem 282, 15652–15666. [DOI] [PubMed] [Google Scholar]
- Fleming I, 2010. Molecular mechanisms underlying the activation of eNOS. Pflugers Arch 459, 793–806. [DOI] [PubMed] [Google Scholar]
- Fleming I and Busse R, 1999. Signal transduction of eNOS activation. Cardiovasc Res 43, 532–541. [DOI] [PubMed] [Google Scholar]
- Fleming I and Busse R, 2003. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am J Physiol Regul Integr Comp Physiol 284, R1–12. [DOI] [PubMed] [Google Scholar]
- Fleming I, Fisslthaler B, Dimmeler S, Kemp BE and Busse R, 2001. Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin-dependent endothelial nitric oxide synthase activity. Circ Res 88, E68–75. [DOI] [PubMed] [Google Scholar]
- Forstermann U and Munzel T, 2006. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation 113, 1708–1714. [DOI] [PubMed] [Google Scholar]
- Forstermann U, Pollock JS, Schmidt HH, Heller M and Murad F, 1991. Calmodulin-dependent endothelium-derived relaxing factor/nitric oxide synthase activity is present in the particulate and cytosolic fractions of bovine aortic endothelial cells. Proc Natl Acad Sci U S A 88, 1788–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis SH, Busch JL, Corbin JD and Sibley D, 2010. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev 62, 525–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukai T, Siegfried MR, Ushio-Fukai M, Cheng Y, Kojda G and Harrison DG, 2000. Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. J Clin Invest 105, 1631–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton D, Gratton JP and Sessa WC, 2001. Post-translational control of endothelial nitric oxide synthase: why isn’t calcium/calmodulin enough? J Pharmacol Exp Ther 299, 818–824. [PubMed] [Google Scholar]
- Galanakis E, Kofteridis D, Stratigi K, Petraki E, Vazgiourakis V, Fragouli E, Mamoulakis D, Boumpas DT and Goulielmos GN, 2008. Intron 4 a/b polymorphism of the endothelial nitric oxide synthase gene is associated with both type 1 and type 2 diabetes in a genetically homogeneous population. Hum Immunol 69, 279–283. [DOI] [PubMed] [Google Scholar]
- Garcia-Cardena G, Fan R, Shah V, Sorrentino R, Cirino G, Papapetropoulos A and Sessa WC, 1998. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature 392, 821–824. [DOI] [PubMed] [Google Scholar]
- Garcia-Cardena G, Fan R, Stern DF, Liu J and Sessa WC, 1996. Endothelial nitric oxide synthase is regulated by tyrosine phosphorylation and interacts with caveolin-1. J Biol Chem 271, 27237–27240. [DOI] [PubMed] [Google Scholar]
- Garcin ED, Bruns CM, Lloyd SJ, Hosfield DJ, Tiso M, Gachhui R, Stuehr DJ, Tainer JA and Getzoff ED, 2004. Structural basis for isozyme-specific regulation of electron transfer in nitric-oxide synthase. J Biol Chem 279, 37918–37927. [DOI] [PubMed] [Google Scholar]
- Gladwin MT, Schechter AN, Kim-Shapiro DB, Patel RP, Hogg N, Shiva S, Cannon RO, Kelm M, Wink D.a., Espey MG, Oldfield EH, Pluta RM, Freeman B.a., Lancaster JR, Feelisch M and Lundberg JO, 2005. The emerging biology of the nitrite anion. Nature chemical biology 1, 308–314. [DOI] [PubMed] [Google Scholar]
- Godfrey V, Chan SL, Cassidy A, Butler R, Choy A, Fardon T, Struthers A and Lang C, 2007. The functional consequence of the Glu298Asp polymorphism of the endothelial nitric oxide synthase gene in young healthy volunteers. Cardiovasc Drug Rev 25, 280–288. [DOI] [PubMed] [Google Scholar]
- Golden SH, Robinson KA, Saldanha I, Anton B and Ladenson PW, 2009. Clinical review: Prevalence and incidence of endocrine and metabolic disorders in the United States: a comprehensive review. J Clin Endocrinol Metab 94, 1853–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves FM, Luizon MR, Speciali JG, Martins-Oliveira A, Dach F and Tanus-Santos JE, 2012. Interaction among nitric oxide (NO)-related genes in migraine susceptibility. Mol Cell Biochem 370, 183–189. [DOI] [PubMed] [Google Scholar]
- Goncalves FM, Martins-Oliveira A, Speciali JG, Luizon MR, Izidoro-Toledo TC, Silva PS, Dach F and Tanus-Santos JE, 2011. Endothelial nitric oxide synthase haplotypes associated with aura in patients with migraine. DNA Cell Biol 30, 363–369. [DOI] [PubMed] [Google Scholar]
- Haga SB and LaPointe NM, 2013. The potential impact of pharmacogenetic testing on medication adherence. Pharmacogenomics J 13, 481–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzimouratidis K, Amar E, Eardley I, Giuliano F, Hatzichristou D, Montorsi F, Vardi Y and Wespes E, 2010. Guidelines on male sexual dysfunction: erectile dysfunction and premature ejaculation. Eur Urol 57, 804–814. [DOI] [PubMed] [Google Scholar]
- Herce-Pagliai C, Kotecha S and Shuker DE, 1998. Analytical methods for 3-nitrotyrosine as a marker of exposure to reactive nitrogen species: a review. Nitric Oxide 2, 324–336. [DOI] [PubMed] [Google Scholar]
- Hermans MP, Ahn SA and Rousseau MF, 2012. eNOS [Glu298Asp] polymorphism, erectile function and ocular pressure in type 2 diabetes. Eur J Clin Invest 42, 729–737. [DOI] [PubMed] [Google Scholar]
- Horowitz A, Menice CB, Laporte R and Morgan KG, 1996. Mechanisms of smooth muscle contraction. Physiol Rev 76, 967–1003. [DOI] [PubMed] [Google Scholar]
- Hyndman ME, Parsons HG, Verma S, Bridge PJ, Edworthy S, Jones C, Lonn E, Charbonneau F and Anderson TJ, 2002. The T-786-->C mutation in endothelial nitric oxide synthase is associated with hypertension. Hypertension 39, 919–922. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ, Buga GM, Wei LH, Bauer PM, Wu G and del Soldato P, 2001. Role of the arginine-nitric oxide pathway in the regulation of vascular smooth muscle cell proliferation. Proc Natl Acad Sci U S A 98, 4202–4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignarro LJ, Buga GM, Wood KS, Byrns RE and Chaudhuri G, 1987. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A 84, 9265–9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD and Roman RJ, 1992. Nitric oxide modulates vascular tone in preglomerular arterioles. Hypertension 19, 770–774. [DOI] [PubMed] [Google Scholar]
- Inoue N, Venema RC, Sayegh HS, Ohara Y, Murphy TJ and Harrison DG, 1995. Molecular regulation of the bovine endothelial cell nitric oxide synthase by transforming growth factor-beta 1. Arterioscler Thromb Vasc Biol 15, 1255–1261. [DOI] [PubMed] [Google Scholar]
- Jachymova M, Horky K, Bultas J, Kozich V, Jindra A, Peleska J and Martasek P, 2001. Association of the Glu298Asp polymorphism in the endothelial nitric oxide synthase gene with essential hypertension resistant to conventional therapy. Biochem Biophys Res Commun 284, 426–430. [DOI] [PubMed] [Google Scholar]
- Jaenisch R and Bird A, 2003. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33 Suppl, 245–254. [DOI] [PubMed] [Google Scholar]
- Jia Z, Zhang X, Kang S and Wu Y, 2013. Association of endothelial nitric oxide synthase gene polymorphisms with type 2 diabetes mellitus: a meta-analysis. Endocr J 60, 893–901. [DOI] [PubMed] [Google Scholar]
- Jones DS, Andrawis NS and Abernethy DR, 1999. Impaired endothelial-dependent forearm vascular relaxation in black Americans. Clin Pharmacol Ther 65, 408–412. [DOI] [PubMed] [Google Scholar]
- Joshi MS, Mineo C, Shaul PW and Bauer JA, 2007. Biochemical consequences of the NOS3 Glu298Asp variation in human endothelium: altered caveolar localization and impaired response to shear. Faseb J 21, 2655–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju H, Zou R, Venema VJ and Venema RC, 1997. Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. J Biol Chem 272, 18522–18525. [DOI] [PubMed] [Google Scholar]
- Jung SB, Kim CS, Naqvi A, Yamamori T, Mattagajasingh I, Hoffman TA, Cole MP, Kumar A, Dericco JS, Jeon BH and Irani K, 2010. Histone deacetylase 3 antagonizes aspirin-stimulated endothelial nitric oxide production by reversing aspirin-induced lysine acetylation of endothelial nitric oxide synthase. Circ Res 107, 877–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karantzoulis-Fegaras F, Antoniou H, Lai SL, Kulkarni G, D’Abreo C, Wong GK, Miller TL, Chan Y, Atkins J, Wang Y and Marsden PA, 1999. Characterization of the human endothelial nitric-oxide synthase promoter. J Biol Chem 274, 3076–3093. [DOI] [PubMed] [Google Scholar]
- Kato N, Sugiyama T, Morita H, Nabika T, Kurihara H, Yamori Y and Yazaki Y, 1999. Lack of evidence for association between the endothelial nitric oxide synthase gene and hypertension. Hypertension 33, 933–936. [DOI] [PubMed] [Google Scholar]
- Kleinbongard P, Dejam A, Lauer T, Jax T, Kerber S, Gharini P, Balzer J, Zotz RB, Scharf RE, Willers R, Schechter AN, Feelisch M and Kelm M, 2006. Plasma nitrite concentrations reflect the degree of endothelial dysfunction in humans. Free Radic Biol Med 40, 295–302. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Kawagishi T, Emoto M, Shoji T, Yamada A, Sato K, Hosoi M and Nishizawa Y, 2002. ecNOS gene polymorphism is associated with endothelium-dependent vasodilation in Type 2 diabetes. Am J Physiol Heart Circ Physiol 283, H557–561. [DOI] [PubMed] [Google Scholar]
- Koppenol WH, 1998. The basic chemistry of nitrogen monoxide and peroxynitrite. Free Radic Biol Med 25, 385–391. [DOI] [PubMed] [Google Scholar]
- Kubes P, Kanwar S, Niu XF and Gaboury JP, 1993. Nitric oxide synthesis inhibition induces leukocyte adhesion via superoxide and mast cells. Faseb J 7, 1293–1299. [DOI] [PubMed] [Google Scholar]
- Kubes P, Suzuki M and Granger DN, 1991. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci U S A 88, 4651–4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuricova K, Tanhauserova V, Pacal L, Bartakova V, Brozova L, Jarkovsky J and Kankova K, 2013. NOS3 894G>T polymorphism is associated with progression of kidney disease and cardiovascular morbidity in type 2 diabetic patients: NOS3 as a modifier gene for diabetic nephropathy? Kidney Blood Press Res 38, 92–98. [DOI] [PubMed] [Google Scholar]
- Kuzkaya N, Weissmann N, Harrison DG and Dikalov S, 2003. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric-oxide synthase. J Biol Chem 278, 22546–22554. [DOI] [PubMed] [Google Scholar]
- Lacchini R, Silva PS and Tanus-Santos JE, 2010. A pharmacogenetics-based approach to reduce cardiovascular mortality with the prophylactic use of statins. Basic Clin Pharmacol Toxicol 106, 357–361. [DOI] [PubMed] [Google Scholar]
- Lacchini R and Tanus-Santos JE, 2014. Pharmacogenetics of erectile dysfunction: navigating into uncharted waters. Pharmacogenomics 15, 1519–1538. [DOI] [PubMed] [Google Scholar]
- Lai PF, Mohamed F, Monge JC and Stewart DJ, 2003. Downregulation of eNOS mRNA expression by TNFalpha: identification and functional characterization of RNA-protein interactions in the 3’UTR. Cardiovasc Res 59, 160–168. [DOI] [PubMed] [Google Scholar]
- Lamas S, Marsden PA, Li GK, Tempst P and Michel T, 1992. Endothelial nitric oxide synthase: molecular cloning and characterization of a distinct constitutive enzyme isoform. Proc Natl Acad Sci U S A 89, 6348–6352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau R, Xie HG, Dishy V, Wood AJ, Stein CM and Smiley RM, 2004. No association of the Asp298 variant of the endothelial nitric oxide synthase gene with preeclampsia. Am J Hypertens 17, 391–394. [DOI] [PubMed] [Google Scholar]
- Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE and Harrison DG, 2003. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 111, 1201–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laufs U and Liao JK, 1998. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem 273, 24266–24271. [DOI] [PubMed] [Google Scholar]
- Laumonnier Y, Nadaud S, Agrapart M and Soubrier F, 2000. Characterization of an upstream enhancer region in the promoter of the human endothelial nitric-oxide synthase gene. J Biol Chem 275, 40732–40741. [DOI] [PubMed] [Google Scholar]
- Lee YC, Huang SP, Liu CC, Yang YH, Yeh HC, Li WM, Wu WJ, Wang CJ, Juan YS, Huang CN, Hour TC, Chang CF and Huang CH, 2012. The association of eNOS G894T polymorphism with metabolic syndrome and erectile dysfunction. J Sex Med 9, 837–843. [DOI] [PubMed] [Google Scholar]
- Lee YC, Wu WJ, Liu CC, Wang CJ, Li WM, Huang CH, Yeh HC, Ke HL and Huang SP, 2009. The associations among eNOS G894T gene polymorphism, erectile dysfunction, and benign prostate hyperplasia-related lower urinary tract symptoms. J Sex Med 6, 3158–3165. [DOI] [PubMed] [Google Scholar]
- Lefer AM, Scalia R and Lefer DJ, 2001. Vascular effects of HMG CoA-reductase inhibitors (statins) unrelated to cholesterol lowering: new concepts for cardiovascular disease. Cardiovasc Res 49, 281–287. [DOI] [PubMed] [Google Scholar]
- Levinsson A, Olin AC, Bjorck L, Rosengren A and Nyberg F, 2014. Nitric oxide synthase (NOS) single nucleotide polymorphisms are associated with coronary heart disease and hypertension in the INTERGENE study. Nitric Oxide 39, 1–7. [DOI] [PubMed] [Google Scholar]
- Li H, Horke S and Forstermann U, 2014. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 237, 208–219. [DOI] [PubMed] [Google Scholar]
- Li H, Wallerath T and Forstermann U, 2002. Physiological mechanisms regulating the expression of endothelial-type NO synthase. Nitric Oxide 7, 132–147. [DOI] [PubMed] [Google Scholar]
- Liao JK and Laufs U, 2005. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol 45, 89–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebhaber SA, 1997. mRNA stability and the control of gene expression. Nucleic Acids Symp Ser, 29–32. [PubMed]
- Lin MI, Fulton D, Babbitt R, Fleming I, Busse R, Pritchard KA Jr. and Sessa WC, 2003. Phosphorylation of threonine 497 in endothelial nitric-oxide synthase coordinates the coupling of L-arginine metabolism to efficient nitric oxide production. J Biol Chem 278, 44719–44726. [DOI] [PubMed] [Google Scholar]
- Linz W, Wohlfart P, Scholkens BA, Malinski T and Wiemer G, 1999. Interactions among ACE, kinins and NO. Cardiovasc Res 43, 549–561. [DOI] [PubMed] [Google Scholar]
- Lisanti MP, Scherer PE, Tang Z and Sargiacomo M, 1994. Caveolae, caveolin and caveolin-rich membrane domains: a signalling hypothesis. Trends Cell Biol 4, 231–235. [DOI] [PubMed] [Google Scholar]
- List BM, Klosch B, Volker C, Gorren AC, Sessa WC, Werner ER, Kukovetz WR, Schmidt K and Mayer B, 1997. Characterization of bovine endothelial nitric oxide synthase as a homodimer with down-regulated uncoupled NADPH oxidase activity: tetrahydrobiopterin binding kinetics and role of haem in dimerization. Biochem J 323 ( Pt 1), 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Garcia-Cardena G and Sessa WC, 1995. Biosynthesis and palmitoylation of endothelial nitric oxide synthase: mutagenesis of palmitoylation sites, cysteines-15 and/or −26, argues against depalmitoylation-induced translocation of the enzyme. Biochemistry 34, 12333–12340. [DOI] [PubMed] [Google Scholar]
- Liu J and Sessa WC, 1994. Identification of covalently bound amino-terminal myristic acid in endothelial nitric oxide synthase. J Biol Chem 269, 11691–11694. [PubMed] [Google Scholar]
- Liu J, Wang L, Liu Y, Wang Z, Li M, Zhang B, Wang H, Liu K and Wen S, 2015. The association between endothelial nitric oxide synthase gene G894T polymorphism and hypertension in Han Chinese: a case-control study and an updated meta-analysis. Ann Hum Biol 42, 184–194. [DOI] [PubMed] [Google Scholar]
- Lopez-Jaramillo P, Gonzalez MC, Palmer RM and Moncada S, 1990. The crucial role of physiological Ca2+ concentrations in the production of endothelial nitric oxide and the control of vascular tone. Br J Pharmacol 101, 489–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luizon MR, Izidoro-Toledo TC, Simoes AL and Tanus-Santos JE, 2009. Endothelial nitric oxide synthase polymorphisms and haplotypes in Amerindians. DNA Cell Biol 28, 329–334. [DOI] [PubMed] [Google Scholar]
- Luizon MR, Metzger IF, Lacchini R and Tanus-Santos JE, 2012. Endothelial nitric oxide synthase polymorphism rs3918226 associated with hypertension does not affect plasma nitrite levels in healthy subjects. Hypertension 59, e52; author reply e53. [DOI] [PubMed] [Google Scholar]
- Luo S, Lei H, Qin H and Xia Y, 2014. Molecular mechanisms of endothelial NO synthase uncoupling. Curr Pharm Des 20, 3548–3553. [DOI] [PubMed] [Google Scholar]
- Ma ZJ, Chen R, Ren HZ, Guo X, Guo J and Chen LM, 2014. Association between eNOS 4b/a polymorphism and the risk of diabetic retinopathy in type 2 diabetes mellitus: a meta-analysis. J Diabetes Res 2014, 549747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maines MD, 1997. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol 37, 517–554. [DOI] [PubMed] [Google Scholar]
- Maron BA and Michel T, 2012. Subcellular localization of oxidants and redox modulation of endothelial nitric oxide synthase. Circ J 76, 2497–2512. [DOI] [PubMed] [Google Scholar]
- Marroni AS, Metzger IF, Souza-Costa DC, Nagassaki S, Sandrim VC, Correa RX, Rios-Santos F and Tanus-Santos JE, 2005. Consistent interethnic differences in the distribution of clinically relevant endothelial nitric oxide synthase genetic polymorphisms. Nitric Oxide 12, 177–182. [DOI] [PubMed] [Google Scholar]
- Marsden PA, Heng HH, Scherer SW, Stewart RJ, Hall AV, Shi XM, Tsui LC and Schappert KT, 1993. Structure and chromosomal localization of the human constitutive endothelial nitric oxide synthase gene. J Biol Chem 268, 17478–17488. [PubMed] [Google Scholar]
- Mason RP and Cockcroft JR, 2006. Targeting nitric oxide with drug therapy. J Clin Hypertens (Greenwich) 8, 40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason RP, Jacob RF, Kubant R, Jacoby A, Louka F, Corbalan JJ and Malinski T, 2012. Effects of angiotensin receptor blockers on endothelial nitric oxide release: the role of eNOS variants. Br J Clin Pharmacol 74, 141–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrab-Mohseni M, Tabatabaei-Malazy O, Hasani-Ranjbar S, Amiri P, Kouroshnia A, Bazzaz JT, Farahani-Shrhabi M, Larijani B and Amoli MM, 2011. Endothelial nitric oxide synthase VNTR (intron 4 a/b) polymorphism association with type 2 diabetes and its chronic complications. Diabetes Res Clin Pract 91, 348–352. [DOI] [PubMed] [Google Scholar]
- Mentz RJ, Bakris GL, Waeber B, McMurray JJ, Gheorghiade M, Ruilope LM, Maggioni AP, Swedberg K, Pina IL, Fiuzat M, O’Connor CM, Zannad F and Pitt B, 2013. The past, present and future of renin-angiotensin aldosterone system inhibition. Int J Cardiol 167, 1677–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger IF, Ishizawa MH, Rios-Santos F, Carvalho WA and Tanus-Santos JE, 2011. Endothelial nitric oxide synthase gene haplotypes affect nitrite levels in black subjects. Pharmacogenomics J 11, 393–399. [DOI] [PubMed] [Google Scholar]
- Metzger IF, Sertorio JT and Tanus-Santos JE, 2007. Modulation of nitric oxide formation by endothelial nitric oxide synthase gene haplotypes. Free Radic Biol Med 43, 987–992. [DOI] [PubMed] [Google Scholar]
- Metzger IF, Sertorio JTC and Tanus-Santos JE, 2006. Relationship between systemic nitric oxide metabolites and cyclic GMP in healthy male volunteers. Acta Physiol, in Press. [DOI] [PubMed]
- Metzger IF, Souza-Costa DC, Marroni AS, Nagassaki S, Desta Z, Flockhart DA and Tanus-Santos JE, 2005. Endothelial nitric oxide synthase gene haplotypes associated with circulating concentrations of nitric oxide products in healthy men. Pharmacogenet Genomics 15, 565–570. [DOI] [PubMed] [Google Scholar]
- Michel T, Li GK and Busconi L, 1993. Phosphorylation and subcellular translocation of endothelial nitric oxide synthase. Proc Natl Acad Sci U S A 90, 6252–6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda JA, Belo VA, Souza-Costa DC, Lanna CM and Tanus-Santos JE, 2013. eNOS polymorphism associated with metabolic syndrome in children and adolescents. Mol Cell Biochem 372, 155–160. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, Saito Y, Kajiyama N, Yoshimura M, Shimasaki Y, Nakayama M, Kamitani S, Harada M, Ishikawa M, Kuwahara K, Ogawa E, Hamanaka I, Takahashi N, Kaneshige T, Teraoka H, Akamizu T, Azuma N, Yoshimasa Y, Yoshimasa T, Itoh H, Masuda I, Yasue H and Nakao K, 1998. Endothelial nitric oxide synthase gene is positively associated with essential hypertension. Hypertension 32, 3–8. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, Saito Y, Nakayama M, Shimasaki Y, Yoshimura T, Yoshimura M, Harada M, Kajiyama N, Kishimoto I, Kuwahara K, Hino J, Ogawa E, Hamanaka I, Kamitani S, Takahashi N, Kawakami R, Kangawa K, Yasue H and Nakao K, 2000. Replication protein A1 reduces transcription of the endothelial nitric oxide synthase gene containing a −786T-->C mutation associated with coronary spastic angina. Hum Mol Genet 9, 2629–2637. [DOI] [PubMed] [Google Scholar]
- Moncada S and Higgs A, 1993. The L-arginine-nitric oxide pathway. N Engl J Med 329, 2002–2012. [DOI] [PubMed] [Google Scholar]
- Montenegro MF, Amaral JH, Pinheiro LC, Sakamoto EK, Ferreira GC, Reis RI, Marçal DMO, Pereira RP and Tanus-Santos JE, 2011. Sodium nitrite downregulates vascular NADPH oxidase and exerts antihypertensive effects in hypertension. Free radical biology & medicine [DOI] [PubMed]
- Monti LD, Barlassina C, Citterio L, Galluccio E, Berzuini C, Setola E, Valsecchi G, Lucotti P, Pozza G, Bernardinelli L, Casari G and Piatti P, 2003. Endothelial nitric oxide synthase polymorphisms are associated with type 2 diabetes and the insulin resistance syndrome. Diabetes 52, 1270–1275. [DOI] [PubMed] [Google Scholar]
- Mun GI and Boo YC, 2012. A regulatory role of Kruppel-like factor 4 in endothelial argininosuccinate synthetase 1 expression in response to laminar shear stress. Biochem Biophys Res Commun 420, 450–455. [DOI] [PubMed] [Google Scholar]
- Muniz JJ, Lacchini R, Rinaldi TO, Nobre YT, Cologna AJ, Martins AC and Tanus-Santos JE, 2013a. Endothelial nitric oxide synthase genotypes and haplotypes modify the responses to sildenafil in patients with erectile dysfunction. Pharmacogenomics J 13, 189–196. [DOI] [PubMed] [Google Scholar]
- Muniz JJ, Lacchini R, Sertorio JT, Jordao AA Jr., Nobre YT, Tucci S Jr., Martins AC and Tanus-Santos JE, 2013b. Low nitric oxide bioavailability is associated with better responses to sildenafil in patients with erectile dysfunction. Naunyn Schmiedebergs Arch Pharmacol 386, 805–811. [DOI] [PubMed] [Google Scholar]
- Muniz L, Luizon MR, Palei AC, Lacchini R, Duarte G, Cavalli RC, Tanus-Santos JE and Sandrim VC, 2012. eNOS tag SNP haplotypes in hypertensive disorders of pregnancy. DNA Cell Biol 31, 1665–1670. [DOI] [PubMed] [Google Scholar]
- Murad F, Mittal CK, Arnold WP, Katsuki S and Kimura H, 1978. Guanylate cyclase: activation by azide, nitro compounds, nitric oxide, and hydroxyl radical and inhibition by hemoglobin and myoglobin. Adv Cyclic Nucleotide Res 9, 145–158. [PubMed] [Google Scholar]
- Nagassaki S, Sertorio JT, Metzger IF, Bem AF, Rocha JB and Tanus-Santos JE, 2006. eNOS gene T-786C polymorphism modulates atorvastatin-induced increase in blood nitrite. Free Radic Biol Med 41, 1044–1049. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Yasue H, Yoshimura M, Shimasaki Y, Kugiyama K, Ogawa H, Motoyama T, Saito Y, Ogawa Y, Miyamoto Y and Nakao K, 1999. T-786-->C mutation in the 5’-flanking region of the endothelial nitric oxide synthase gene is associated with coronary spasm. Circulation 99, 2864–2870. [DOI] [PubMed] [Google Scholar]
- Nejatizadeh A, Kumar R, Stobdan T, Goyal AK, Sikdar S, Gupta M, Javed S and Pasha MA, 2008. Endothelial nitric oxide synthase gene haplotypes and circulating nitric oxide levels significantly associate with risk of essential hypertension. Free Radic Biol Med 44, 1912–1918. [DOI] [PubMed] [Google Scholar]
- Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, Mullany EC, Biryukov S, Abbafati C, Abera SF, Abraham JP, Abu-Rmeileh NM, Achoki T, AlBuhairan FS, Alemu ZA, Alfonso R, Ali MK, Ali R, Guzman NA, Ammar W, Anwari P, Banerjee A, Barquera S, Basu S, Bennett DA, Bhutta Z, Blore J, Cabral N, Nonato IC, Chang JC, Chowdhury R, Courville KJ, Criqui MH, Cundiff DK, Dabhadkar KC, Dandona L, Davis A, Dayama A, Dharmaratne SD, Ding EL, Durrani AM, Esteghamati A, Farzadfar F, Fay DF, Feigin VL, Flaxman A, Forouzanfar MH, Goto A, Green MA, Gupta R, Hafezi-Nejad N, Hankey GJ, Harewood HC, Havmoeller R, Hay S, Hernandez L, Husseini A, Idrisov BT, Ikeda N, Islami F, Jahangir E, Jassal SK, Jee SH, Jeffreys M, Jonas JB, Kabagambe EK, Khalifa SE, Kengne AP, Khader YS, Khang YH, Kim D, Kimokoti RW, Kinge JM, Kokubo Y, Kosen S, Kwan G, Lai T, Leinsalu M, Li Y, Liang X, Liu S, Logroscino G, Lotufo PA, Lu Y, Ma J, Mainoo NK, Mensah GA, Merriman TR, Mokdad AH, Moschandreas J, Naghavi M, Naheed A, Nand D, Narayan KM, Nelson EL, Neuhouser ML, Nisar MI, Ohkubo T, Oti SO, Pedroza A, et al. , 2014. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 384, 766–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu W and Qi Y, 2011. An updated meta-analysis of endothelial nitric oxide synthase gene: three well-characterized polymorphisms with hypertension. PLoS One 6, e24266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen J, 2010. Nitric oxide-related drug targets in headache. Neurotherapeutics 7, 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira-Paula GH, Lacchini R and Tanus-Santos JE, 2014. Inducible nitric oxide synthase as a possible target in hypertension. Curr Drug Targets 15, 164–174. [DOI] [PubMed] [Google Scholar]
- Pagidipati NJ and Gaziano TA, 2013. Estimating deaths from cardiovascular disease: a review of global methodologies of mortality measurement. Circulation 127, 749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda K, Rosenfeld RJ, Ghosh S, Meade AL, Getzoff ED and Stuehr DJ, 2002. Distinct dimer interaction and regulation in nitric-oxide synthase types I, II, and III. J Biol Chem 277, 31020–31030. [DOI] [PubMed] [Google Scholar]
- Parra FC, Amado RC, Lambertucci JR, Rocha J, Antunes CM and Pena SD, 2003. Color and genomic ancestry in Brazilians. Proc Natl Acad Sci U S A 100, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira TV, Rudnicki M, Cheung BM, Baum L, Yamada Y, Oliveira PS, Pereira AC and Krieger JE, 2007. Three endothelial nitric oxide (NOS3) gene polymorphisms in hypertensive and normotensive individuals: meta-analysis of 53 studies reveals evidence of publication bias. J Hypertens 25, 1763–1774. [DOI] [PubMed] [Google Scholar]
- Peskircioglu L, Atac FB, Erdem SR, Deveci S, Verdi H and Ozkardes H, 2007. The association between intron 4 VNTR, E298A and IVF 23+10 G/T polymorphisms of ecNOS gene and sildenafil responsiveness in patients with erectile dysfunction. Int J Impot Res 19, 149–153. [DOI] [PubMed] [Google Scholar]
- Pesole G, Mignone F, Gissi C, Grillo G, Licciulli F and Liuni S, 2001. Structural and functional features of eukaryotic mRNA untranslated regions. Gene 276, 73–81. [DOI] [PubMed] [Google Scholar]
- Piazza M, Futrega K, Spratt DE, Dieckmann T and Guillemette JG, 2012. Structure and dynamics of calmodulin (CaM) bound to nitric oxide synthase peptides: effects of a phosphomimetic CaM mutation. Biochemistry 51, 3651–3661. [DOI] [PubMed] [Google Scholar]
- Pieper GM, 1999. Enhanced, unaltered and impaired nitric oxide-mediated endothelium-dependent relaxation in experimental diabetes mellitus: importance of disease duration. Diabetologia 42, 204–213. [DOI] [PubMed] [Google Scholar]
- Pinheiro LC, Montenegro MF, Amaral JH, Ferreira GC, Oliveira AM and Tanus-Santos JE, 2012. Increase in gastric pH reduces hypotensive effect of oral sodium nitrite in rats. Free Radic Biol Med 53, 701–709. [DOI] [PubMed] [Google Scholar]
- Podolsky RH, Barbeau P, Kang HS, Zhu H, Treiber FA and Snieder H, 2007. Candidate genes and growth curves for adiposity in African- and European-American youth. Int J Obes (Lond) 31, 1491–1499. [DOI] [PubMed] [Google Scholar]
- Pollock JS, Forstermann U, Mitchell JA, Warner TD, Schmidt HH, Nakane M and Murad F, 1991. Purification and characterization of particulate endothelium-derived relaxing factor synthase from cultured and native bovine aortic endothelial cells. Proc Natl Acad Sci U S A 88, 10480–10484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postberg J, Kanders M, Forcob S, Willems R, Orth V, Hensel KO, Weil PP, Wirth S and Jenke AC, 2015. CpG signalling, H2A.Z/H3 acetylation and microRNA-mediated deferred self-attenuation orchestrate foetal NOS3 expression. Clin Epigenetics 7, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J and Fulton D, 2013. Post-translational regulation of endothelial nitric oxide synthase in vascular endothelium. Front Physiol 4, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radomski MW, Palmer RM and Moncada S, 1987. Comparative pharmacology of endothelium-derived relaxing factor, nitric oxide and prostacyclin in platelets. Br J Pharmacol 92, 181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafikov R, Fonseca FV, Kumar S, Pardo D, Darragh C, Elms S, Fulton D and Black SM, 2011. eNOS activation and NO function: structural motifs responsible for the posttranslational control of endothelial nitric oxide synthase activity. J Endocrinol 210, 271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajfer J, Aronson WJ, Bush PA, Dorey FJ and Ignarro LJ, 1992. Nitric oxide as a mediator of relaxation of the corpus cavernosum in response to nonadrenergic, noncholinergic neurotransmission. N Engl J Med 326, 90–94. [DOI] [PubMed] [Google Scholar]
- Raman CS, Li H, Martasek P, Kral V, Masters BS and Poulos TL, 1998. Crystal structure of constitutive endothelial nitric oxide synthase: a paradigm for pterin function involving a novel metal center. Cell 95, 939–950. [DOI] [PubMed] [Google Scholar]
- Recalcati S, Taramelli D, Conte D and Cairo G, 1998. Nitric oxide-mediated induction of ferritin synthesis in J774 macrophages by inflammatory cytokines: role of selective iron regulatory protein-2 downregulation. Blood 91, 1059–1066. [PubMed] [Google Scholar]
- Robb GB, Carson AR, Tai SC, Fish JE, Singh S, Yamada T, Scherer SW, Nakabayashi K and Marsden PA, 2004. Post-transcriptional regulation of endothelial nitric-oxide synthase by an overlapping antisense mRNA transcript. J Biol Chem 279, 37982–37996. [DOI] [PubMed] [Google Scholar]
- Robinson LJ and Michel T, 1995. Mutagenesis of palmitoylation sites in endothelial nitric oxide synthase identifies a novel motif for dual acylation and subcellular targeting. Proc Natl Acad Sci U S A 92, 11776–11780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Vargas H, Coral-Vazquez RM, Tapia R, Borja JL, Salas RA and Salamanca F, 2004. Glu298Asp endothelial nitric oxide synthase polymorphism is a risk factor for erectile dysfunction in the Mexican Mestizo population. J Androl 25, 728–732. [DOI] [PubMed] [Google Scholar]
- Safarinejad MR, Khoshdel A, Shekarchi B, Taghva A and Safarinejad S, 2011. Association of the T-786C, G894T and 4a/4b polymorphisms of the endothelial nitric oxide synthase gene with vasculogenic erectile dysfunction in Iranian subjects. BJU Int 107, 1994–2001. [DOI] [PubMed] [Google Scholar]
- Sakoda T, Hirata K, Kuroda R, Miki N, Suematsu M, Kawashima S and Yokoyama M, 1995. Myristoylation of endothelial cell nitric oxide synthase is important for extracellular release of nitric oxide. Mol Cell Biochem 152, 143–148. [DOI] [PubMed] [Google Scholar]
- Salvemini D and Cuzzocrea S, 2002. Superoxide, superoxide dismutase and ischemic injury. Curr Opin Investig Drugs 3, 886–895. [PubMed] [Google Scholar]
- Salvi E, Kutalik Z, Glorioso N, Benaglio P, Frau F, Kuznetsova T, Arima H, Hoggart C, Tichet J, Nikitin YP, Conti C, Seidlerova J, Tikhonoff V, Stolarz-Skrzypek K, Johnson T, Devos N, Zagato L, Guarrera S, Zaninello R, Calabria A, Stancanelli B, Troffa C, Thijs L, Rizzi F, Simonova G, Lupoli S, Argiolas G, Braga D, D’Alessio MC, Ortu MF, Ricceri F, Mercurio M, Descombes P, Marconi M, Chalmers J, Harrap S, Filipovsky J, Bochud M, Iacoviello L, Ellis J, Stanton AV, Laan M, Padmanabhan S, Dominiczak AF, Samani NJ, Melander O, Jeunemaitre X, Manunta P, Shabo A, Vineis P, Cappuccio FP, Caulfield MJ, Matullo G, Rivolta C, Munroe PB, Barlassina C, Staessen JA, Beckmann JS and Cusi D, 2012. Genomewide association study using a high-density single nucleotide polymorphism array and case-control design identifies a novel essential hypertension susceptibility locus in the promoter region of endothelial NO synthase. Hypertension 59, 248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvi E, Kuznetsova T, Thijs L, Lupoli S, Stolarz-Skrzypek K, D’Avila F, Tikhonoff V, De Astis S, Barcella M, Seidlerova J, Benaglio P, Malyutina S, Frau F, Velayutham D, Benfante R, Zagato L, Title A, Braga D, Marek D, Kawecka-Jaszcz K, Casiglia E, Filipovsky J, Nikitin Y, Rivolta C, Manunta P, Beckmann JS, Barlassina C, Cusi D and Staessen JA, 2013. Target sequencing, cell experiments, and a population study establish endothelial nitric oxide synthase (eNOS) gene as hypertension susceptibility gene. Hypertension 62, 844–852. [DOI] [PubMed] [Google Scholar]
- Sanchez de Miguel L, Alonso J, Gonzalez-Fernandez F, de la Osada J, Monton M, Rodriguez-Feo JA, Guerra JI, Arriero MM, Rico L, Casado S and Lopez-Farre A, 1999. Evidence that an endothelial cytosolic protein binds to the 3’-untranslated region of endothelial nitric oxide synthase mRNA. J Vasc Res 36, 201–208. [DOI] [PubMed] [Google Scholar]
- Sandrim VC, Coelho EB, Nobre F, Arado GM, Lanchote VL and Tanus-Santos JE, 2006a. Susceptible and protective eNOS haplotypes in hypertensive black and white subjects. Atherosclerosis 186, 428–432. [DOI] [PubMed] [Google Scholar]
- Sandrim VC, de Syllos RW, Lisboa HR, Tres GS and Tanus-Santos JE, 2006b. Endothelial nitric oxide synthase haplotypes affect the susceptibility to hypertension in patients with type 2 diabetes mellitus. Atherosclerosis 189, 241–246. [DOI] [PubMed] [Google Scholar]
- Sandrim VC, de Syllos RW, Lisboa HR, Tres GS and Tanus-Santos JE, 2007. Influence of eNOS haplotypes on the plasma nitric oxide products concentrations in hypertensive and type 2 diabetes mellitus patients. Nitric Oxide 16, 348–355. [DOI] [PubMed] [Google Scholar]
- Sandrim VC, Palei AC, Cavalli RC, Araujo FM, Ramos ES, Duarte G and Tanus-Santos JE, 2008a. eNOS haplotypes associated with gestational hypertension or preeclampsia. Pharmacogenomics 9, 1467–1473. [DOI] [PubMed] [Google Scholar]
- Sandrim VC, Palei AC, Luizon MR, Izidoro-Toledo TC, Cavalli RC and Tanus-Santos JE, 2010a. eNOS haplotypes affect the responsiveness to antihypertensive therapy in preeclampsia but not in gestational hypertension. Pharmacogenomics J 10, 40–45. [DOI] [PubMed] [Google Scholar]
- Sandrim VC, Palei AC, Metzger IF, Gomes VA, Cavalli RC and Tanus-Santos JE, 2008b. Nitric oxide formation is inversely related to serum levels of antiangiogenic factors soluble fms-like tyrosine kinase-1 and soluble endogline in preeclampsia. Hypertension 52, 402–407. [DOI] [PubMed] [Google Scholar]
- Sandrim VC, Palei AC, Sertorio JT, Cavalli RC, Duarte G and Tanus-Santos JE, 2010b. Effects of eNOS polymorphisms on nitric oxide formation in healthy pregnancy and in pre-eclampsia. Mol Hum Reprod 16, 506–510. [DOI] [PubMed] [Google Scholar]
- Sandrim VC, Yugar-Toledo JC, Desta Z, Flockhart DA, Moreno H Jr. and Tanus-Santos JE, 2006c. Endothelial nitric oxide synthase haplotypes are related to blood pressure elevation, but not to resistance to antihypertensive drug therapy. J Hypertens 24, 2393–2397. [DOI] [PubMed] [Google Scholar]
- Sansbury BE and Hill BG, 2014. Regulation of obesity and insulin resistance by nitric oxide. Free Radic Biol Med 73, 383–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhanam L, Lim HK, Miriel V, Brown T, Patel M, Balanson S, Ryoo S, Anderson M, Irani K, Khanday F, Di Costanzo L, Nyhan D, Hare JM, Christianson DW, Rivers R, Shoukas A and Berkowitz DE, 2007. Inducible NO synthase dependent S-nitrosylation and activation of arginase1 contribute to age-related endothelial dysfunction. Circ Res 101, 692–702. [DOI] [PubMed] [Google Scholar]
- Sase K and Michel T, 1995. Expression of constitutive endothelial nitric oxide synthase in human blood platelets. Life Sci 57, 2049–2055. [DOI] [PubMed] [Google Scholar]
- Savvidou MD, Hingorani AD, Tsikas D, Frolich JC, Vallance P and Nicolaides KH, 2003. Endothelial dysfunction and raised plasma concentrations of asymmetric dimethylarginine in pregnant women who subsequently develop pre-eclampsia. Lancet 361, 1511–1517. [DOI] [PubMed] [Google Scholar]
- Schlossmann J, Ammendola A, Ashman K, Zong X, Huber A, Neubauer G, Wang GX, Allescher HD, Korth M, Wilm M, Hofmann F and Ruth P, 2000. Regulation of intracellular calcium by a signalling complex of IRAG, IP3 receptor and cGMP kinase Ibeta. Nature 404, 197–201. [DOI] [PubMed] [Google Scholar]
- Schneider MP, Erdmann J, Delles C, Fleck E, Regitz-Zagrosek V and Schmieder RE, 2000. Functional gene testing of the Glu298Asp polymorphism of the endothelial NO synthase. J Hypertens 18, 1767–1773. [DOI] [PubMed] [Google Scholar]
- Schubeler D, Lorincz MC, Cimbora DM, Telling A, Feng YQ, Bouhassira EE and Groudine M, 2000. Genomic targeting of methylated DNA: influence of methylation on transcription, replication, chromatin structure, and histone acetylation. Mol Cell Biol 20, 9103–9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searles CD, 2006. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression. Am J Physiol Cell Physiol 291, C803–816. [DOI] [PubMed] [Google Scholar]
- Searles CD, Miwa Y, Harrison DG and Ramasamy S, 1999. Posttranscriptional regulation of endothelial nitric oxide synthase during cell growth. Circ Res 85, 588–595. [DOI] [PubMed] [Google Scholar]
- Seremak-Mrozikiewicz A, Drews K, Barlik M, Sieroszewski P, Grzeskowiak E and Mrozikiewicz P, 2011. The significance of −786T > C polymorphism of endothelial NO synthase (eNOS) gene in severe preeclampsia. J Matern Fetal Neona 24, 432–436. [DOI] [PubMed] [Google Scholar]
- Serrano NC, Casas JP, Diaz LA, Paez C, Mesa CM, Cifuentes R, Monterrosa A, Bautista A, Hawe E, Hingorani AD, Vallance P and Lopez-Jaramillo P, 2004. Endothelial NO synthase genotype and risk of preeclampsia: a multicenter case-control study. Hypertension 44, 702–707. [DOI] [PubMed] [Google Scholar]
- Shaul PW, Smart EJ, Robinson LJ, German Z, Yuhanna IS, Ying Y, Anderson RG and Michel T, 1996. Acylation targets emdothelial nitric-oxide synthase to plasmalemmal caveolae. J Biol Chem 271, 6518–6522. [DOI] [PubMed] [Google Scholar]
- Shinozaki K, Nishio Y, Okamura T, Yoshida Y, Maegawa H, Kojima H, Masada M, Toda N, Kikkawa R and Kashiwagi A, 2000. Oral administration of tetrahydrobiopterin prevents endothelial dysfunction and vascular oxidative stress in the aortas of insulin-resistant rats. Circ Res 87, 566–573. [DOI] [PubMed] [Google Scholar]
- Shoukry A, Shalaby SM, Abdelazim S, Abdelazim M, Ramadan A, Ismail MI and Fouad M, 2012. Endothelial nitric oxide synthase gene polymorphisms and the risk of diabetic nephropathy in type 2 diabetes mellitus. Genet Test Mol Biomarkers 16, 574–579. [DOI] [PubMed] [Google Scholar]
- Silacci P, Formentin K, Bouzourene K, Daniel F, Brunner HR and Hayoz D, 2000. Unidirectional and oscillatory shear stress differentially modulate NOS III gene expression. Nitric Oxide 4, 47–56. [DOI] [PubMed] [Google Scholar]
- Silva PS, Fontana V, Luizon MR, Lacchini R, Silva WA Jr., Biagi C and Tanus-Santos JE, 2013. eNOS and BDKRB2 genotypes affect the antihypertensive responses to enalapril. Eur J Clin Pharmacol 69, 167–177. [DOI] [PubMed] [Google Scholar]
- Sinici I, Guven EO, Serefoglu E and Hayran M, 2010. T-786C polymorphism in promoter of eNOS gene as genetic risk factor in patients with erectile dysfunction in Turkish population. Urology 75, 955–960. [DOI] [PubMed] [Google Scholar]
- Souza-Costa DC, Belo VA, Silva PS, Sertorio JT, Metzger IF, Lanna CM, Machado MA and Tanus-Santos JE, 2011. eNOS haplotype associated with hypertension in obese children and adolescents. Int J Obes (Lond) 35, 387–392. [DOI] [PubMed] [Google Scholar]
- Stephens M, Smith NJ and Donnelly P, 2001. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68, 978–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez Y, Fernandez-Hernando C, Pober JS and Sessa WC, 2007. Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circ Res 100, 1164–1173. [DOI] [PubMed] [Google Scholar]
- Sun HX, Zeng DY, Li RT, Pang RP, Yang H, Hu YL, Zhang Q, Jiang Y, Huang LY, Tang YB, Yan GJ and Zhou JG, 2012. Essential role of microRNA-155 in regulating endothelium-dependent vasorelaxation by targeting endothelial nitric oxide synthase. Hypertension 60, 1407–1414. [DOI] [PubMed] [Google Scholar]
- Sun K and Lai EC, 2013. Adult-specific functions of animal microRNAs. Nat Rev Genet 14, 535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surks HK, Mochizuki N, Kasai Y, Georgescu SP, Tang KM, Ito M, Lincoln TM and Mendelsohn ME, 1999. Regulation of myosin phosphatase by a specific interaction with cGMP- dependent protein kinase Ialpha. Science 286, 1583–1587. [DOI] [PubMed] [Google Scholar]
- Tanus-Santos JE and Casella-Filho A, 2007. Endothelial nitric oxide synthase polymorphisms and susceptibility to hypertension: genotype versus haplotype analysis. Hypertension 49, E1; author reply E2. [DOI] [PubMed] [Google Scholar]
- Tanus-Santos JE, Desai M, Deak LR, Pezzullo JC, Abernethy DR, Flockhart DA and Freedman JE, 2002. Effects of endothelial nitric oxide synthase gene polymorphisms on platelet function, nitric oxide release, and interactions with estradiol. Pharmacogenetics 12, 407–413. [DOI] [PubMed] [Google Scholar]
- Tanus-Santos JE, Desai M and Flockhart DA, 2001. Effects of ethnicity on the distribution of clinically relevant endothelial nitric oxide variants. Pharmacogenetics 11, 719–725. [DOI] [PubMed] [Google Scholar]
- Taverna MJ, Elgrably F, Selmi H, Selam JL and Slama G, 2005. The T-786C and C774T endothelial nitric oxide synthase gene polymorphisms independently affect the onset pattern of severe diabetic retinopathy. Nitric Oxide 13, 88–92. [DOI] [PubMed] [Google Scholar]
- Tempfer CB, Dorman K, Deter RL, O’Brien WE and Gregg AR, 2001. An endothelial nitric oxide synthase gene polymorphism is associated with preeclampsia. Hypertens Pregnancy 20, 107–118. [DOI] [PubMed] [Google Scholar]
- Thomas BN, Thakur TJ, Yi L, Guindo A, Diallo DA and Ott J, 2013. Extensive ethnogenomic diversity of endothelial nitric oxide synthase (eNOS) polymorphisms. Gene Regul Syst Bio 7, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GD, Zhang W and Victor RG, 2001. Nitric oxide deficiency as a cause of clinical hypertension: promising new drug targets for refractory hypertension. Jama 285, 2055–2057. [DOI] [PubMed] [Google Scholar]
- Thomas SR, Chen K and Keaney JF Jr., 2002. Hydrogen peroxide activates endothelial nitric-oxide synthase through coordinated phosphorylation and dephosphorylation via a phosphoinositide 3-kinase-dependent signaling pathway. J Biol Chem 277, 6017–6024. [DOI] [PubMed] [Google Scholar]
- Toriello M, Oterino A, Pascual J, Castillo J, Colas R, Alonso-Arranz A, Ruiz-Alegria C, Quintela E, Monton F and Ruiz-Lavilla N, 2008. Lack of association of endothelial nitric oxide synthase polymorphisms and migraine. Headache 48, 1115–1119. [DOI] [PubMed] [Google Scholar]
- Tsujita Y, Baba S, Yamauchi R, Mannami T, Kinoshita M, Yamamoto R, Katsuya T, Higaki J, Ogihara T, Ogata J and Iwai N, 2001. Association analyses between genetic polymorphisms of endothelial nitric oxide synthase gene and hypertension in Japanese: The Suita Study. J Hypertens 19, 1941–1948. [DOI] [PubMed] [Google Scholar]
- Ulker S, McKeown PP and Bayraktutan U, 2003. Vitamins reverse endothelial dysfunction through regulation of eNOS and NAD(P)H oxidase activities. Hypertension 41, 534–539. [DOI] [PubMed] [Google Scholar]
- Uwabo J, Soma M, Nakayama T and Kanmatsuse K, 1998. Association of a variable number of tandem repeats in the endothelial constitutive nitric oxide synthase gene with essential hypertension in Japanese. Am J Hypertens 11, 125–128. [DOI] [PubMed] [Google Scholar]
- Vallance P, Collier J and Moncada S, 1989. Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet 2, 997–1000. [DOI] [PubMed] [Google Scholar]
- Vasconcellos V, Lacchini R, Jacob-Ferreira AL, Sales ML, Ferreira-Sae MC, Schreiber R, Nadruz W and Tanus-Santos JE, 2010. Endothelial nitric oxide synthase haplotypes associated with hypertension do not predispose to cardiac hypertrophy. DNA Cell Biol 29, 171–176. [DOI] [PubMed] [Google Scholar]
- Verhaar MC, Westerweel PE, van Zonneveld AJ and Rabelink TJ, 2004. Free radical production by dysfunctional eNOS. Heart 90, 494–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victor TW, Hu X, Campbell JC, Buse DC and Lipton RB, 2010. Migraine prevalence by age and sex in the United States: a life-span study. Cephalalgia 30, 1065–1072. [DOI] [PubMed] [Google Scholar]
- Villanueva C and Giulivi C, 2010. Subcellular and cellular locations of nitric oxide synthase isoforms as determinants of health and disease. Free Radic Biol Med 49, 307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarreal G Jr., Zhang Y, Larman HB, Gracia-Sancho J, Koo A and Garcia-Cardena G, 2010. Defining the regulation of KLF4 expression and its downstream transcriptional targets in vascular endothelial cells. Biochem Biophys Res Commun 391, 984–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virag L, Szabo E, Gergely P and Szabo C, 2003. Peroxynitrite-induced cytotoxicity: mechanism and opportunities for intervention. Toxicol Lett 140–141, 113–124. [DOI] [PubMed] [Google Scholar]
- von Lueder TG and Krum H, 2013. RAAS inhibitors and cardiovascular protection in large scale trials. Cardiovasc Drugs Ther 27, 171–179. [DOI] [PubMed] [Google Scholar]
- Walford G and Loscalzo J, 2003. Nitric oxide in vascular biology. J Thromb Haemost 1, 2112–2118. [DOI] [PubMed] [Google Scholar]
- Wang J, Dudley D and Wang XL, 2002. Haplotype-specific effects on endothelial NO synthase promoter efficiency: modifiable by cigarette smoking. Arterioscler Thromb Vasc Biol 22, e1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JL, Wang HG, Gao HQ, Zhai GX, Chang P and Chen YG, 2010. Endothelial nitric oxide synthase polymorphisms and erectile dysfunction: a meta-analysis. J Sex Med 7, 3889–3898. [DOI] [PubMed] [Google Scholar]
- Wang L, McLeod HL and Weinshilboum RM, 2011. Genomics and drug response. N Engl J Med 364, 1144–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattanapitayakul SK, Mihm MJ, Young AP and Bauer JA, 2001. Therapeutic implications of human endothelial nitric oxide synthase gene polymorphism. Trends Pharmacol Sci 22, 361–368. [DOI] [PubMed] [Google Scholar]
- White RE, Lee AB, Shcherbatko AD, Lincoln TM, Schonbrunn A and Armstrong DL, 1993. Potassium channel stimulation by natriuretic peptides through cGMP-dependent dephosphorylation. Nature 361, 263–266. [DOI] [PubMed] [Google Scholar]
- Yagil Y and Yagil C, 2004. Candidate genes, association studies and haplotype analysis in the search for the genetic basis of hypertension. J Hypertens 22, 1255–1258. [DOI] [PubMed] [Google Scholar]
- Yan L, Kang M, Qin Z, Zhang W, Li Y and Ou H, 2013. An intronic miRNA regulates expression of the human endothelial nitric oxide synthase gene and proliferation of endothelial cells by a mechanism related to the transcription factor SP-1. PLoS One 8, e70658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Xu JR, Liu XM, Yang Y, Na XF, Li M and Wang YJ, 2013. Polymorphisms of rs1799983 (G>T) and rs1800780 (A>G) of the eNOS gene associated with susceptibility to essential hypertension in the Chinese Hui ethnic population. Genet Mol Res 12, 3821–3829. [DOI] [PubMed] [Google Scholar]
- Yang L, Liu G, Zakharov SI, Bellinger AM, Mongillo M and Marx SO, 2007. Protein kinase G phosphorylates Cav1.2 alpha1c and beta2 subunits. Circ Res 101, 465–474. [DOI] [PubMed] [Google Scholar]
- Yang W, Ando J, Korenaga R, Toyo-oka T and Kamiya A, 1994. Exogenous nitric oxide inhibits proliferation of cultured vascular endothelial cells. Biochem Biophys Res Commun 203, 1160–1167. [DOI] [PubMed] [Google Scholar]
- Zhang MX, Zhang C, Shen YH, Wang J, Li XN, Chen L, Zhang Y, Coselli JS and Wang XL, 2008a. Effect of 27nt small RNA on endothelial nitric-oxide synthase expression. Mol Biol Cell 19, 3997–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MX, Zhang C, Shen YH, Wang J, Li XN, Zhang Y, Coselli J and Wang XL, 2008b. Biogenesis of short intronic repeat 27-nucleotide small RNA from endothelial nitric-oxide synthase gene. J Biol Chem 283, 14685–14693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Min W and Sessa WC, 1995. Functional analysis of the human endothelial nitric oxide synthase promoter. Sp1 and GATA factors are necessary for basal transcription in endothelial cells. J Biol Chem 270, 15320–15326. [DOI] [PubMed] [Google Scholar]
- Zhou XB, Ruth P, Schlossmann J, Hofmann F and Korth M, 1996. Protein phosphatase 2A is essential for the activation of Ca2+-activated K+ currents by cGMP-dependent protein kinase in tracheal smooth muscle and Chinese hamster ovary cells. J Biol Chem 271, 19760–19767. [DOI] [PubMed] [Google Scholar]