

Abstract
Chemotherapy-induced peripheral neuropathy (CIPN) is a common and debilitating condition associated with a number of chemotherapeutic agents. Drugs commonly implicated in development of CIPN include platinum agents, taxanes, vinca alkaloids, bortezomib, and thalidomide analogues. As response to the same drug can vary between individuals, it is hypothesized that an individual’s specific genetic variants could impact regulation of genes involved in drug pharmacokinetics, ion channel functioning, neurotoxicity, and DNA repair, which in turn affect CIPN development and severity. Variations of other molecular markers may also affect the incidence and severity of CIPN. Hence, the objective of this review is to summarize the known biological (molecular and genomic) predictors of CIPN and discuss means to facilitate progress in this field.
Keywords: chemotherapy-induced peripheral neuropathy, CIPN, neuropathy
Introduction
Chemotherapy induced peripheral neuropathy (CIPN), a common side effect of anti-neoplastic agents, significantly decreases quality of life (QOL) in patients with cancer. CIPN symptoms include numbness, tingling, and pain especially in the hands and feet. This in turn is associated with inability to complete activities of daily living and falls.1 Development of CIPN may lead to dose modifications, decreased patient adherence, and treatment interruptions or discontinuation, thereby potentially impacting oncologic outcomes negatively. Those with CIPN also report increased unemployment and decreased annual income, further demonstrating the negative impact that CIPN can have on patients.2
A meta-analysis involving over 4000 patients estimated CIPN prevalence to be about 68% by the end of the first month of chemotherapy and 30% at 6 months.3 Drugs commonly implicated in development of CIPN include platinum drugs, taxanes, vinca alkaloids, bortezomib, and thalidomide analogues.
Since response to the same drug varies between individuals, it is hypothesized that a patient’s specific genetic variants could impact regulation of genes involved in drug pharmacokinetics, ion channel functioning, neurotoxicity, and DNA repair, which may in turn affect CIPN development and severity. The goal of this manuscript is to summarize the known biological (molecular and genomic) predictors of CIPN and discuss means to facilitate progress in the understanding and eventual management of CIPN.
Demographic and Clinical Predictors of CIPN
While there are established clinical risk factors for CIPN, none accurately predicts the severity of CIPN that an individual patient will have.4 Cumulative dose is a strong risk factor for the development of CIPN with most neurotoxic anti-neoplastic drugs. Patients of older age may be more at risk for developing neurotoxicity,5–7 however other studies have not found age to be associated with greater CIPN incidence.8,9 These differing results may be due to confounding comorbidities. Obese cancer patients with CIPN experience higher levels of neuropathy burden.10 Diabetic patients report a higher grade of CIPN, particularly with taxanes while patients with autoimmune diseases report less severe CIPN.6 African Americans have a higher incidence of CIPN following taxane treatment in comparison to other racial groups.11
Pathophysiology and Biological Predictors of CIPN Table 1
Table 1:
Reported genetic polymorphisms associated with Chemotherapy-Induced Peripheral Neuropathy.
| Agent | Mechanism | Gene (-SNP) | Functional Pathway | Reference |
|---|---|---|---|---|
| Platinum | Interfere with tumor cell proliferation by forming DNA-platinum adducts that accumulate in the DRG and in peripheral neurons.10,13 |
ERCC1 | DNA repair | 13,27 |
| ERCC 2 | DNA repair | 28–30 | ||
| XRCC1 | Cell cycle progression, RNA transcription, DNA repair, and possibly repair after platinum induced damage to DRG |
31,32 | ||
| CCNH | Oxidative damage protection | 33 | ||
| GPX7 | Transporter enzyme | 33 | ||
| ABCC4 | Glutathione | 33 | ||
| Taxanes Paclitaxel |
Stabilization of microtubules and inhibition of depolymerization, forming abnormal microtubule bundles in the cytoplasm and producing mitotic spindle disruption and apoptosis.34 |
CYP2C8 | Metabolizes paclitaxel | 37–41 |
| ABCB1 | Cellular and systemic drug efflux transporter | 41–44 | ||
| TUBB2A | Molecular target for paclitaxel | 44,45 | ||
| FGD4 | Myelin production | 51 | ||
| EPHA4,5,6 | Nervous system development / repair | 41,45, 53–55 | ||
| ARHGEF10 | Neurodevelopment | 58,59 | ||
| Docetaxel | ABCB1 | Cellular and systemic drug efflux transporter | 65–67 | |
| GSTP1 | Drug conjugation and detoxification | 66,68 69 | ||
| VAC14 | Neurodevelopment | 71–74 | ||
| Vinca Alkaloids | Promote disassembly of microtubules by binding to tubulin during S phase and preventing microtubule polymerization, disrupting mitotic spindle formation in M phase and rendering cell unable to divide.74 |
CEP72 | Microtubule formation | 75–79 |
| CYP3A5 | Vincristine metabolism | 80−89 | ||
| Bortezomib | Indirectly polymerize tubulin, causing microtubule stabilization, axonal transport inhibition and G2-M phase cell cycle arrest.93 |
CTLA4 rs4553808 | Immune function | 94 |
| PSMB1 rs1474642 | Drug binding | 94 | ||
| PKNOX1–21q22.3 | Severe bortezomib-IPN | 96 | ||
| CDH13, DCC, TENM3: 4q34.3-rs6552496, 5q14.1- rs12521798, 16q23.3- rs8060632, 18q21.2- rs17748074 |
Unknown: Intergenic |
95 |
||
| Thalidomide | Inhibit production of IL-6 (growth factor), blocks the cell growth-stimulating CD147/MCT1 protein complex from binding to Cereblon, activate apoptotic pathways through caspase 8-mediated cell death, and activates T cells to produce IL-2 augmenting NK-dependent cytotoxicity.97,101 |
ABCA1 | Neuroinflammation, neurodegeneration | 98,100 |
| ICAM1 | Myelinogenesis, nerve regeneration | 98,100 | ||
| PPARD | Promote mitochondrial biogenesis | 98,100 | ||
| SERPINB2 | Proteostasis, neuro-cytoprotection | 98, 100 | ||
| SLC12A6 | K & Cl cotransporter | 98, 100 | ||
| GSTT1 | Drug conjugation and detoxification | 99 |
summarizes the mechanisms of action of chemotherapeutic agents and reported genetic polymorphisms associated with CIPN. The molecular targets and pathways of CIPN are described in Figure 1.
Figure 1: Molecular Targets and Pathways of CIPN.
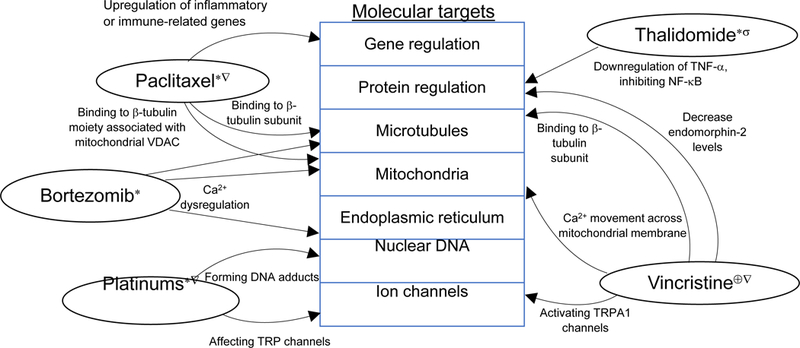
Affected neurons: dorsal root ganglion (∗); sensory neurons (∇); motor neurons (⊕); central projections of primary afferent neurons (σ).
Abbreviations: VDAC – voltage-dependent anion channels; Ca2+ - calcium ions; TRP – transient receptor potential.
Platinum compounds
Platinum compounds interfere with tumor cell proliferation, resulting in damage to non-dividing, post-mitotic peripheral neural tissue. This damage may be associated with sensory neuropathy with anterograde axonal degeneration, first described for cisplatin concentrations in tissue collected posthumously.12,13 Whether this is true for systemic concentrations is less well understood. In addition to chronic neuropathy, acute neuropathy presenting as cold-induced dysesthesia is unique for oxaliplatin and is thought to be related to rapid generation of oxalate metabolites.13
Indirect evidence indicates that neuropathy may be attributable to systemic platinum drug pharmacokinetics (PK). It is suggested that increased fractionation of cisplatin in the bleomycin, etoposide, and cisplatin (BEP) regimen may reduce neurotoxicity with the 3-day regimen producing less acute and late sensory neuropathy than the 5-day with equivalent anti-cancer efficacy.14,15
Preclinical studies on the association between drug transporters and oxaliplatin-induced neuropathy in mice with genetic knockout of the organic cation transporter 2 (OCT2) strongly suggest that platinum accumulation is related to the activity of this transporter,16 suggesting that any indirect association between systemic concentrations and neuropathy would likely have to account for the magnitude of uptake transport into the dorsal root ganglion (DRG). Dasatinib is being investigated for its role in minimizing CIPN as it inhibits the activity of OCT2, which could decrease oxaliplatin uptake in the DRG.17 However, a population-pharmacokinetic model did not detect any association between PK parameters of the parent compound or the free oxaliplatin concentrations and neuropathy incidence;18 other small pilot studies have also not detected a relationship.19,20 Yet, in a randomized trial of reduced glutathione (GSH) co-administration, decreased neuropathy incidence and increased oxaliplatin clearance were reported.21
A number of aberrations identified as potential future therapeutic targets include several single nucleotide polymorphisms (SNPs) such as ERCC1, ERCC2, XRCC1, CCNH, GPX7, and ABCC4, which may play critical roles in neurotoxicity, as described in Table 1 and Figure 1.22–28 One genomically targeted therapeutic intervention focuses on the protein, apurinic/apyrimidinic endonuclease (APE-1). APE-1adaad is critical in the DNA base excision repair pathway and oxidative stress response, thereby mitigating chemotherapy-induced neuronal DNA damage, especially from platinum agents.29 Early phase clinical trials are underway to enhance anti-oxidant effects of APE-1 and inhibit damaging signals such as ERK-1/2.30 Early studies investigating the drug, APX3330, suggest a neuroprotective benefit similar to the genetic APE-1 overexpression. This is achieved without minimizing chemotherapeutic efficacy by reducing redox signaling and improving DNA repair in sensory neurons.29
Taxanes
Taxanes, which are spindle poisons, accumulate in the soma of sensory neurons of dorsal root ganglia. A retrograde “dying back” process is commonly observed, which typically starts at distal nerve endings, and is subsequently associated with Schwann cell, neuronal body, or axonal transport changes. This sequence of alterations is believed to contribute to neurotoxicity. Genomic predictors of paclitaxel and docetaxel associated neuropathy are shown in Table 1.
The genetic prediction of paclitaxel-induced peripheral neuropathy has been extensively studied and recently reviewed.31 Though candidate SNPs in cytochrome P450 (CYP) CYP2C8,32 ABCB1,32,33 and TUBB2A33 have been associated with neuropathy, none have been validated for use in clinical care due to inconsistent replication. Strong evidence links paclitaxel PK to the incidence of peripheral neuropathy,34,35 including a randomized clinical trial demonstrating that exposure-guided paclitaxel dosing significantly reduces peripheral neuropathy incidence.36 This pharmacokinetic association likely explains the reported associations for the putatively low-activity CYP2C8*3 variant described above and the increased risk of paclitaxel-induced neuropathy in patients receiving the strong CYP2C8 inhibitor, clopidogrel.37,38 Genome-wide association studies (GWAS) have identified candidates for attempted replication, including SNPs in FGD439 and EPHA5.32 SNPs in several EPHA genes including EPHA4, EPHA5, and EPHA61,5 have been associated with paclitaxel-induced neuropathy, strongly suggesting this gene family is involved in neuropathy predisposition.40 Many SNPs discovered via GWAS have been in genes involved in neurodevelopment, particularly those associated with hereditary neuropathy conditions41 including ARHGEF10.42 Other SNPs reported from GWAS have not been independently replicated.11,43–47 Preclinical studies indicate that nilotinib may reduce paclitaxel-induced peripheral neuropathy through a non-competitive mechanism that allows paclitaxel to effectively attack cancer cells while inhibiting the solute carrier organic anion-transporting polypeptide B2 (OATP1B2). Suppression of OATPIB2 activity was found to minimize peripheral neuropathy.48
Few pharmacogenetic studies have been conducted for docetaxel-induced peripheral neuropathy. Candidate SNPs in ABCB149,50 and GSTP149,51 have been reported, but not yet validated.52 The only completed GWAS reported a variant in VAC14 that increased neuropathy risk and was confirmed to decrease neurite branching in vitro and increase mouse neuropathy sensitivity in VAC14 knockout studies.53,54 There have been no pharmacogenetic studies of neuropathy caused by albumin-bound paclitaxel (nab-paclitaxel) or cabazitaxel.
Vinca alkaloids
Vinca alkaloids, such as vincristine and vinblastine, block microtubule polymerization, consequently disrupting mitotic spindle formation and rendering the cell unable to divide. Mutations in CEP72 and CYP3A5 have been most extensively studied as factors possibly influencing the development of vincristine-induced neuropathy. Studies have demonstrated that SNPs that reduce CEP72 expression may lead to the development of neuropathy in patients on vincristine,55,56 although subsequent studies were unable to consistently reproduce this finding.57,58 Vincristine is primarily metabolized by CYP3A4 and 3A5. Polymorphisms of CYP3A5 are common, with the CYP3A5*1/*1 genotype being associated with expression of CYP3A5 and is more common in African-Americans. The CYP3A5*3/*3 genotype is associated with non-expression of CYP3A5 and is more common in Caucasians.59,60 Early studies suggested a possible association between CYP3A5 genotypes and vincristine-induced peripheral neuropathy.45,61 The CYP3A5*1/*1 genotype is associated with a lower incidence of neuropathy compared to CYP3A5*3/*3 genotype.62 However, others have also reported no effect of CYP3A5 genotype on development of neuropathy.29,63 Early evidence suggests that vincristine PK may be associated with neuropathy27 however, validation has been challenging.59,64
Bortezomib
The proteasome inhibitor, bortezomib, promotes G2-M cell cycle arrest and apoptosis through disruption of the ubiquitin-proteasome pathway which degrades dysfunctional intracellular proteins. Subcutaneous administration of bortezomib has been found to greatly reduce reported bortezomib-induced peripheral neuropathy in comparison to intravenous administration, as well as reduction of other adverse effects.61 Despite the same treatment efficacy, prevalence of peripheral neuropathy in the population treated with subcutaneous injection is 38% in comparison to 53% of those treated with intravenous bortezomib.61 The exact mechanism of bortezomib-induced peripheral neuropathy is unknown. SNPs in CTLA4 rs4553808 and PSMB1 rs1474642 have been reported to be associated with bortezomib-induced neuropathy.65 A GWAS study identified 4 new loci that were associated with bortezomib-induced peripheral neuropathy, found in genes involved in the development and function of the nervous system including CDH13, DCC, and TENM3.63 Another GWAS study identified a gene locus mapping to PKNOX1 and in close proximity to CBS at 21q22.3 that was correlated with the severe bortezomib-induced toxicity.66 However, additional studies to validate these findings are needed prior to establishing these genes for study as therapeutic targets.
Thalidomide
Thalidomide, an immunomodulatory agent, prevents cell proliferation through inhibition of angiogenesis as well as alteration of the immune system through multiple mechanisms including inhibition of interleukin (IL)-6 production, activation of caspase 8–mediated apoptosis, and increased production of IL-2 through T cell activation. Based on a meta-analysis, thalidomide-related peripheral neuropathy has been described in 63.5% of patients.3 Several SNPs have been found to be associated with thalidomide-related peripheral neuropathy: ABCA1, ICAM1, PPARD, SERPINB2, and SLC12A6.67 In addition, a SNP in GSTT1 predicted the frequency of neuropathy.68 Another study was unable to find associations between 300,000 exome SNPs and thalidomide-related peripheral neuropathy.69 Studies of lenalidomide, a sister drug of thalidomide with similar antiangiogenic immunomodulatory mechanisms of action, demonstrate significantly lower incidence of peripheral neuropathy.70 The mechanism is hypothesized to be a pharmacokinetic effect similar to that observed with the platinum compounds; the half maximal inhibitory concentration (IC50) of lenalidomide is almost 500 times less than that of thalidomide (0.4 μmol/L vs. 194 μmol/L, respectively).71 This marked difference reflects the much lower serum concentrations of lenalidomide required for target activity in comparison to thalidomide and highlights the potency of lenalidomide.
Limitations of Current Studies
Biomarker discovery studies of CIPN have several limitations, the primary being a lack of an objectively assessable, universally accepted CIPN phenotype.72 Varying methods to define the phenotypes, such as clinician-assessed NCI CTCAE and grading classifications have complicated the comparison of multiple study findings with one another. In addition, discordance across GWAS studies is a significant limitation.72 Collapsing different phenotypes into a single definition may limit the ability to identify genetic predictor of any single phenotype. For instance, it is likely that a genetic predictor of neuropathic pain is distinct from a predictor of sensory versus motor neuropathy.73 Failing to adjust for clinical (particularly cumulative dose received) or environmental differences may have impeded the precision of reported findings.
Data from patients treated with combination therapies rather than single agent may have contributed to the inconsistent results across studies. While some SNPs may predispose patients to CIPN regardless of the neurotoxic agent, each class of agents, and perhaps each agent within that class is likely to have independent risk factors. As one illustrative example, analyses have combined patients taking paclitaxel or docetaxel and included SNPs in CYP2C8, which is only involved in paclitaxel metabolism but not docetaxel.74 Labeling how all taxanes are involved with CYP2C8 would not be appropriate.
A well-powered sample size is the cornerstone to the generation of valuable genetic association studies, granting the study enough power to confirm the correlations. Currently, most studies are retrospective in nature and are limited to the fixed sample size of the prospective study from which they were conducted, leading to underpowered analyses. Further, because a smaller number of toxicities occur in studies with smaller sample sizes, the ability to produce extensive data on toxicity for further analysis is restricted.72 An additional factor to inspect is the genetic ancestry of the study population, reflected by distinct allele frequencies in the variations being investigated. Since these frequencies are due to the ancestral history of the populations, they must be accounted for in the association analyses. Otherwise, observed differences between those experiencing neuropathy and those who are not may simply be due to ancestral composition of the compared groups rather than differences in chronic toxicities.
The reporting of genetic association studies should be as transparent as possible. The STrengthening and the REporting of Genetic Association Studies (STREGA) recommendations promote the transparency, excellence, and thoroughness of genetic association study reporting.75 Studies should also provide essential information such as quality error and call rates as they may have a significant effect on the ability to detect linkage or association.
Novel analytical approaches
Besides traditional approaches, analyses of gene expression (i.e. transcriptomics) or differential gene expression, protein expression (i.e. proteomics), or biochemical metabolite concentrations (i.e. metabolomics) may be useful for predicting future neuropathy occurrence, but data on applying these techniques to measure toxicity from cancer treatments is scarce.76 Discovery-phase candidates from proteomic and metabolomic analyses require independent validation prior to translation into clinical practice.
Recent advances in human stem cell technology have allowed for increasingly detailed in vitro studies of CIPN. Adult human somatic cells (often skin fibroblasts or lymphocytes) can be reprogrammed into induced pluripotent stem cells (iPSCs),77 which then may differentiate into specific cell types of interest. Neurons produced from these stem cell differentiation protocols allow researchers to harness human biology and genetics, including within specific patient populations such as patients with CIPN.
Future Directions
To mitigate CIPN’s detrimental impact on patient quality of life, a clear understanding of the molecular pathways underlying its development and natural history is necessary. This understanding will support the development of targeted clinical strategies.78 Building on the foundational work done to date, the growth of novel research platforms will allow for new ways of interrogating these pathways. Table 2 provides a list of recommendations for future genetic studies of CIPN, which aim to ensure the translation of research findings into clinical decision-making. However, none of these biomarkers are currently ready to be translated into routine practice. To continue to make progress on understanding predictors of CIPN and predictors of response to various therapeutic strategies, the following strategies will be helpful: new methodological approaches to study genomics and molecular pathways, data sharing and real world data, as well as multidisciplinary funding and collaboration.
Table 2.
Methodological Recommendations for Future Research Studies on Biological Predictors of CIPN
| ✓Distinguishing the various phenotypes of CIPN (motor vs. sensory vs. neuropathic pain), in order to support consistency in the classification of “cases” across studies; studies may also want to target high-risk patients (e.g. patients who develop peripheral severe neuropathy after first few doses of treatment). ✓Longitudinal assessment of CIPN, including pre-treatment assessment. ✓Prospective research design is ideal, to ensure the collection of known relevant clinical factors including cumulative dose, drug exposure, diabetes, race. ✓Studies should clearly report the justification of genomic predictors interrogated. ✓Consistent reporting of the process used for genotyping. ✓Collaboration between study centers to increase sample size and confirm the generalizability of findings. ✓Collaboration between patients, clinicians and translational researchers will support the application of innovative methods to address clinically meaningful outcomes. ✓Development and testing of interventions targeted to implicated pathways. |
New methodological approaches from complex data analysis techniques can be applied to the intersecting effects of symptom biology. For instance, cluster analyses may include impact of symptoms on the condition, manifestation of toxicities, and interaction of toxicities on the overall condition. Dissection of symptoms to underlying biology using genome-wide approaches can yield information on polymorphisms that may affect innate risk and response, as well as adaptive variability in gene expression resulting in an individually dynamic pathobiology. Although genomic (genotyping, gene expression, and epigenetic) approaches are useful for dissecting effects and interactions of the tumor, treatment, and host susceptibility factors, these studies must involve a large number of subjects. As methodology improves power with smaller sample sizes, these studies may be linked with those conducted to examine primary mechanisms and toxicities.79
To additionally make progress in CIPN research, clinical research networks can be developed into data repositories where variable interactions such as pharmacogenomics, pharmacoproteomics, gene expression/proteomic changes in human specimens, and patient-reported outcomes can be linked to clinical phenotypes. This will ultimately move the field towards population-based rather than clinic-specific research, while encouraging standardization of data measures. Such networks are exemplified by NIH initiatives using public-private partnering mechanisms to provide publically available resources such as PhenX (the Phenotyping and Exposures project) and PROMIS (Patient Reported Outcomes Measurement Information System).
Lastly, joint funding of proposals with a variety of funding agencies such as the National Cancer Institute, National Institute of Neurological Disorders and Stroke, and other research funders with overlapping missions has been considered an approach to leverage funds. Other examples of recent facilitation of larger scientific endeavors include: a growing investment in clinical research networks such as Patient Based Research Networks; the linking of Clinical and Translational Study Award-supported academic sites; and complementary electronic resources, such as Clinical Research Networks. These encourage the expansion of current clinical research networks by conducting studies across multiple research sites. Collaboration across sites through these mechanisms makes it feasible to increase sample size, increase generalizability, facilitate standardized data collection methods, and promote scientific exchange across programs and study sites.
Conclusions
A number of clinical and genetic predictors have been identified, yet we are still unable to adequately prevent or treat CIPN. Future work is needed to develop a CIPN risk model where drug, genomic, and clinical data are incorporated to better understand the risk of various neurotoxic therapies. In summary, advances in research methodologies, new technologies, and creative partnering relationships enhance the feasibility of these proposed strategies through efficiency in conduct as well as economy of funding.
Acknowledgments
Acknowledgments of people, grants, funds, etc. should be placed in a separate section on the title page. The names of funding organizations should be written in full.
National Institutes of Health/National Cancer Institute (R01CA211887)
National Institutes of Health/National Cancer Institute (R01CACA189947)
References
- 1.Gewandter JS, Fan L, Magnuson A, et al. Falls and functional impairments in cancer survivors with chemotherapy-induced peripheral neuropathy (CIPN): a University of Rochester CCOP study. Support Care Cancer. 2013;21(7):2059–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miaskowski C, Mastick J, Paul SM, et al. Impact of chemotherapy-induced neurotoxicities on adult cancer survivors’ symptom burden and quality of life. J Cancer Surviv. 2018;12(2):234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seretny M, Currie GL, Sena ES, et al. Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: A systematic review and meta-analysis. Pain. 2014;155(12):2461–2470. [DOI] [PubMed] [Google Scholar]
- 4.Molassiotis A, Cheng HL, Leung KT, et al. Risk factors for chemotherapy-induced peripheral neuropathy in patients receiving taxane- and platinum-based chemotherapy. Brain Behav. 2019:e01312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bulls HW, Hoogland AI, Kennedy B, et al. A longitudinal examination of associations between age and chemotherapy-induced peripheral neuropathy in patients with gynecologic cancer. Gynecol Oncol. 2019;152(2):310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hershman DL, Till C, Wright JD, et al. Comorbidities and Risk of Chemotherapy-Induced Peripheral Neuropathy Among Participants 65 Years or Older in Southwest Oncology Group Clinical Trials. J Clin Oncol. 2016;34(25):3014–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raphael MJ, Fischer HD, Fung K, et al. Neurotoxicity Outcomes in a Population-based Cohort of Elderly Patients Treated With Adjuvant Oxaliplatin for Colorectal Cancer. Clin Colorectal Cancer. 2017;16(4):397–404 e391. [DOI] [PubMed] [Google Scholar]
- 8.Argyriou AA, Polychronopoulos P, Koutras A, et al. Is advanced age associated with increased incidence and severity of chemotherapy-induced peripheral neuropathy? Support Care Cancer. 2006;14(3):223–229. [DOI] [PubMed] [Google Scholar]
- 9.Nurgalieva Z, Xia R, Liu CC, Burau K, Hardy D, Du XL. Risk of chemotherapy-induced peripheral neuropathy in large population-based cohorts of elderly patients with breast, ovarian, and lung cancer. Am J Ther. 2010;17(2):148–158. [DOI] [PubMed] [Google Scholar]
- 10.Cox-Martin E, Trahan LH, Cox MG, Dougherty PM, Lai EA, Novy DM. Disease burden and pain in obese cancer patients with chemotherapy-induced peripheral neuropathy. Supportive Care in Cancer. 2017;25(6):1873–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schneider BP, Li L, Radovich M, et al. Genome-Wide Association Studies for Taxane-Induced Peripheral Neuropathy in ECOG-5103 and ECOG-1199. Clin Cancer Res. 2015;21(22):5082–5091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gregg RW, Molepo JM, Monpetit VJ, et al. Cisplatin neurotoxicity: the relationship between dosage, time, and platinum concentration in neurologic tissues, and morphologic evidence of toxicity. J Clin Oncol. 1992;10(5):795–803. [DOI] [PubMed] [Google Scholar]
- 13.Starobova H, Vetter I. Pathophysiology of Chemotherapy-Induced Peripheral Neuropathy. Front Mol Neurosci. 2017;10:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Wit R, Roberts JT, Wilkinson PM, et al. Equivalence of three or four cycles of bleomycin, etoposide, and cisplatin chemotherapy and of a 3- or 5-day schedule in good-prognosis germ cell cancer: a randomized study of the European Organization for Research and Treatment of Cancer Genitourinary Tract Cancer Cooperative Group and the Medical Research Council. J Clin Oncol. 2001;19(6):1629–1640. [DOI] [PubMed] [Google Scholar]
- 15.Nichols CR, Williams SD, Loehrer PJ, et al. Randomized study of cisplatin dose intensity in poor-risk germ cell tumors: a Southeastern Cancer Study Group and Southwest Oncology Group protocol. J Clin Oncol. 1991;9(7):1163–1172. [DOI] [PubMed] [Google Scholar]
- 16.Sprowl JA, Ciarimboli G, Lancaster CS, et al. Oxaliplatin-induced neurotoxicity is dependent on the organic cation transporter OCT2. Proc Natl Acad Sci U S A. 2013;110(27):11199–11204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hucke A, Ciarimboli G. The Role of Transporters in the Toxicity of Chemotherapeutic Drugs: Focus on Transporters for Organic Cations. J Clin Pharmacol. 2016;56 Suppl 7:S157–172. [DOI] [PubMed] [Google Scholar]
- 18.Chalret du Rieu Q, White-Koning M, Picaud L, et al. Population pharmacokinetics of peritoneal, plasma ultrafiltrated and protein-bound oxaliplatin concentrations in patients with disseminated peritoneal cancer after intraperitoneal hyperthermic chemoperfusion of oxaliplatin following cytoreductive surgery: correlation between oxaliplatin exposure and thrombocytopenia. Cancer Chemother Pharmacol. 2014;74(3):571–582. [DOI] [PubMed] [Google Scholar]
- 19.Shord SS, Bernard SA, Lindley C, et al. Oxaliplatin biotransformation and pharmacokinetics: a pilot study to determine the possible relationship to neurotoxicity. Anticancer Res. 2002;22(4):2301–2309. [PubMed] [Google Scholar]
- 20.Ishibashi K, Okada N, Miyazaki T, Sano M, Ishida H. Effect of calcium and magnesium on neurotoxicity and blood platinum concentrations in patients receiving mFOLFOX6 therapy: a prospective randomized study. Int J Clin Oncol. 2010;15(1):82–87. [DOI] [PubMed] [Google Scholar]
- 21.Albers JW, Chaudhry V, Cavaletti G, Donehower RC. Interventions for preventing neuropathy caused by cisplatin and related compounds. Cochrane Database Syst Rev. 2014(3):CD005228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao Y, Zhang G, Wang P, et al. Clinical significance of UGT1A1 polymorphism and expression of ERCC1, BRCA1, TYMS, RRM1, TUBB3, STMN1 and TOP2A in gastric cancer. BMC Gastroenterol. 2017;17(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang X, Jiang LP, Yin Y, Wang YD. XRCC1 and XPD genetic polymorphisms and clinical outcomes of gastric cancer patients treated with oxaliplatin-based chemotherapy: a meta-analysis. Tumour Biol. 2014;35(6):5637–5645. [DOI] [PubMed] [Google Scholar]
- 24.Whitehouse CJ, Taylor RM, Thistlethwaite A, et al. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell. 2001;104(1):107–117. [DOI] [PubMed] [Google Scholar]
- 25.Savas S, Kim DY, Ahmad MF, Shariff M, Ozcelik H. Identifying functional genetic variants in DNA repair pathway using protein conservation analysis. Cancer Epidemiol Biomarkers Prev. 2004;13(5):801–807. [PubMed] [Google Scholar]
- 26.Song X, Wang S, Hong X, et al. Single nucleotide polymorphisms of nucleotide excision repair pathway are significantly associated with outcomes of platinum-based chemotherapy in lung cancer. Sci Rep. 2017;7(1):11785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Custodio A, Moreno-Rubio J, Aparicio J, et al. Pharmacogenetic predictors of severe peripheral neuropathy in colon cancer patients treated with oxaliplatin-based adjuvant chemotherapy: a GEMCAD group study. Ann Oncol. 2014;25(2):398–403. [DOI] [PubMed] [Google Scholar]
- 28.Johnson C, Pankratz VS, Velazquez AI, et al. Candidate pathway-based genetic association study of platinum and platinum-taxane related toxicity in a cohort of primary lung cancer patients. J Neurol Sci. 2015;349(1–2):124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelley MR, Wikel JH, Guo C, et al. Identification and Characterization of New Chemical Entities Targeting Apurinic/Apyrimidinic Endonuclease 1 for the Prevention of Chemotherapy-Induced Peripheral Neuropathy. J Pharmacol Exp Ther. 2016;359(2):300–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schloss J, Colosimo M, Vitetta L. New Insights into Potential Prevention and Management Options for Chemotherapy-Induced Peripheral Neuropathy. Asia Pac J Oncol Nurs. 2016;3(1):73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frederiks CN, Lam SW, Guchelaar HJ, Boven E. Genetic polymorphisms and paclitaxel- or docetaxel-induced toxicities: A systematic review. Cancer Treat Rev. 2015;41(10):935–950. [DOI] [PubMed] [Google Scholar]
- 32.Boora GK, Kanwar R, Kulkarni AA, et al. Testing of candidate single nucleotide variants associated with paclitaxel neuropathy in the trial NCCTG N08C1 (Alliance). Cancer Med. 2016;5(4):631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abraham JE, Guo Q, Dorling L, et al. Replication of genetic polymorphisms reported to be associated with taxane-related sensory neuropathy in patients with early breast cancer treated with Paclitaxel. Clin Cancer Res. 2014;20(9):2466–2475. [DOI] [PubMed] [Google Scholar]
- 34.Hertz D, Kidwell K, K. V, D. S, NL. H. Association of systemic paclitaxel concentrations with severity and progression of paclitaxel-induced peripheral neuropathy. San Antonio Breast Cancer Symposium 2017 2017. [Google Scholar]
- 35.Mielke S, Sparreboom A, Steinberg SM, et al. Association of Paclitaxel pharmacokinetics with the development of peripheral neuropathy in patients with advanced cancer. Clin Cancer Res. 2005;11(13):4843–4850. [DOI] [PubMed] [Google Scholar]
- 36.Joerger M, von Pawel J, Kraff S, et al. Open-label, randomized study of individualized, pharmacokinetically (PK)-guided dosing of paclitaxel combined with carboplatin or cisplatin in patients with advanced non-small-cell lung cancer (NSCLC). Ann Oncol. 2016;27(10):1895–1902. [DOI] [PubMed] [Google Scholar]
- 37.Agergaard K, Mau-Sorensen M, Stage TB, et al. Clopidogrel-Paclitaxel Drug-Drug Interaction: A Pharmacoepidemiologic Study. Clin Pharmacol Ther. 2017;102(3):547–553. [DOI] [PubMed] [Google Scholar]
- 38.Matsuo M, Ito H, Takemura Y, et al. Increased risk of paclitaxel-induced peripheral neuropathy in patients using clopidogrel: a retrospective pilot study. J Anesth. 2017;31(4):631–635. [DOI] [PubMed] [Google Scholar]
- 39.Lam SW, Frederiks CN, van der Straaten T, Honkoop AH, Guchelaar HJ, Boven E. Genotypes of CYP2C8 and FGD4 and their association with peripheral neuropathy or early dose reduction in paclitaxel-treated breast cancer patients. Br J Cancer. 2016;115(11):1335–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilkinson DG. Multiple roles of EPH receptors and ephrins in neural development. Nature reviews Neuroscience. 2001;2(3):155–164. [DOI] [PubMed] [Google Scholar]
- 41.Chhibber A, Mefford J, Stahl EA, et al. Polygenic inheritance of paclitaxel-induced sensory peripheral neuropathy driven by axon outgrowth gene sets in CALGB 40101 (Alliance). Pharmacogenomics J. 2014;14(4):336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boora GK, Kulkarni AA, Kanwar R, et al. Association of the Charcot-Marie-Tooth disease gene ARHGEF10 with paclitaxel induced peripheral neuropathy in NCCTG N08CA (Alliance). J Neurol Sci. 2015;357(1–2):35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schneider BP, Li L, Miller K, et al. Genetic associations with taxane-induced neuropathy by a genome-wide association study (GWAS) in E5103. ASCO Meeting Abstracts. 2011;29(15_suppl):1000. [Google Scholar]
- 44.Bergmann TK, Vach W, Feddersen S, et al. GWAS-based association between RWDD3 and TECTA variants and paclitaxel induced neuropathy could not be confirmed in Scandinavian ovarian cancer patients. Acta Oncol. 2012:1–3. [DOI] [PubMed] [Google Scholar]
- 45.Kulkarni AA, Boora G, Kanwar R, et al. RWDD3 and TECTA variants not linked to paclitaxel induced peripheral neuropathy in North American trial Alliance N08C1. Acta Oncol. 2015;54(8):1227–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schneider BP, Lai D, Shen F, et al. Charcot-Marie-Tooth gene, SBF2, associated with taxane-induced peripheral neuropathy in African Americans. Oncotarget. 2016;7(50):82244–82253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sucheston-Campbell LE, Clay-Gilmour AI, Barlow WE, et al. Genome-wide meta-analyses identifies novel taxane-induced peripheral neuropathy-associated loci. Pharmacogenetics and genomics. 2018;28(2):49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leblanc AF, Sprowl JA, Alberti P, et al. OATP1B2 deficiency protects against paclitaxel-induced neurotoxicity. J Clin Invest. 2018;128(2):816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eckhoff L, Feddersen S, Knoop AS, Ewertz M, Bergmann TK. Docetaxel-induced neuropathy: a pharmacogenetic case-control study of 150 women with early-stage breast cancer. Acta Oncol. 2015;54(4):530–537. [DOI] [PubMed] [Google Scholar]
- 50.Kus T, Aktas G, Kalender ME, et al. Polymorphism of CYP3A4 and ABCB1 genes increase the risk of neuropathy in breast cancer patients treated with paclitaxel and docetaxel. OncoTargets and therapy. 2016;9:5073–5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Rossum AGJ, Kok M, McCool D, et al. Independent replication of polymorphisms predicting toxicity in breast cancer patients randomized between dose-dense and docetaxel-containing adjuvant chemotherapy. Oncotarget. 2017;8(69):113531–113542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marsh S, Paul J, King CR, Gifford G, McLeod HL, Brown R. Pharmacogenetic assessment of toxicity and outcome after platinum plus taxane chemotherapy in ovarian cancer: the Scottish Randomised Trial in Ovarian Cancer. J Clin Oncol. 2007;25(29):4528–4535. [DOI] [PubMed] [Google Scholar]
- 53.Hertz DL, Owzar K, Lessans S, et al. Pharmacogenetic Discovery in CALGB (Alliance) 90401 and Mechanistic Validation of a VAC14 Polymorphism that Increases Risk of Docetaxel-Induced Neuropathy. Clin Cancer Res. 2016;22(19):4890–4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lenk GM, Szymanska K, Debska-Vielhaber G, et al. Biallelic Mutations of VAC14 in Pediatric-Onset Neurological Disease. Am J Hum Genet. 2016;99(1):188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stock W, Diouf B, Crews KR, et al. An Inherited Genetic Variant in CEP72 Promoter Predisposes to Vincristine-Induced Peripheral Neuropathy in Adults With Acute Lymphoblastic Leukemia. Clin Pharmacol Ther. 2017;101(3):391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wright GEB, Amstutz U, Drogemoller BI, et al. Pharmacogenomics of Vincristine-Induced Peripheral Neuropathy Implicates Pharmacokinetic and Inherited Neuropathy Genes. Clin Pharmacol Ther. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gutierrez-Camino A, Martin-Guerrero I, Lopez-Lopez E, et al. Lack of association of the CEP72 rs924607 TT genotype with vincristine-related peripheral neuropathy during the early phase of pediatric acute lymphoblastic leukemia treatment in a Spanish population. Pharmacogenet Genomics. 2016;26(2):100–102. [DOI] [PubMed] [Google Scholar]
- 58.Zgheib NK, Ghanem KM, Tamim H, et al. Genetic polymorphisms in candidate genes are not associated with increased vincristine-related peripheral neuropathy in Arab children treated for acute childhood leukemia: a single institution study. Pharmacogenetics and genomics. 2018;28(8):189–195. [DOI] [PubMed] [Google Scholar]
- 59.Moore AS, Norris R, Price G, et al. Vincristine pharmacodynamics and pharmacogenetics in children with cancer: a limited-sampling, population modelling approach. Journal of paediatrics and child health. 2011;47(12):875–882. [DOI] [PubMed] [Google Scholar]
- 60.Skiles JL, Chiang C, Li CH, et al. CYP3A5 genotype and its impact on vincristine pharmacokinetics and development of neuropathy in Kenyan children with cancer. Pediatr Blood Cancer. 2018;65(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moreau P, Pylypenko H, Grosicki S, et al. Subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma: a randomised, phase 3, non-inferiority study. Lancet Oncol. 2011;12(5):431–440. [DOI] [PubMed] [Google Scholar]
- 62.Egbelakin A, Ferguson MJ, MacGill EA, et al. Increased risk of vincristine neurotoxicity associated with low CYP3A5 expression genotype in children with acute lymphoblastic leukemia. Pediatric blood & cancer. 2011;56(3):361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Campo C, da Silva Filho MI, Weinhold N, et al. Bortezomib-induced peripheral neuropathy: A genome-wide association study on multiple myeloma patients. Hematol Oncol. 2018;36(1):232–237. [DOI] [PubMed] [Google Scholar]
- 64.Guilhaumou R, Solas C, Bourgarel-Rey V, et al. Impact of plasma and intracellular exposure and CYP3A4, CYP3A5, and ABCB1 genetic polymorphisms on vincristine-induced neurotoxicity. Cancer Chemother Pharmacol. 2011;68(6):1633–1638. [DOI] [PubMed] [Google Scholar]
- 65.Favis R, Sun Y, van de Velde H, et al. Genetic variation associated with bortezomib-induced peripheral neuropathy. Pharmacogenetics and genomics. 2011;21(3):121–129. [DOI] [PubMed] [Google Scholar]
- 66.Magrangeas F, Kuiper R, Avet-Loiseau H, et al. A Genome-Wide Association Study Identifies a Novel Locus for Bortezomib-Induced Peripheral Neuropathy in European Patients with Multiple Myeloma. Clin Cancer Res. 2016;22(17):4350–4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johnson DC, Corthals SL, Walker BA, et al. Genetic factors underlying the risk of thalidomide-related neuropathy in patients with multiple myeloma. J Clin Oncol. 2011;29(7):797–804. [DOI] [PubMed] [Google Scholar]
- 68.Cibeira MT, de Larrea CF, Navarro A, et al. Impact on response and survival of DNA repair single nucleotide polymorphisms in relapsed or refractory multiple myeloma patients treated with thalidomide. Leuk Res. 2011;35(9):1178–1183. [DOI] [PubMed] [Google Scholar]
- 69.Garcia-Sanz R, Corchete LA, Alcoceba M, et al. Prediction of peripheral neuropathy in multiple myeloma patients receiving bortezomib and thalidomide: a genetic study based on a single nucleotide polymorphism array. Hematol Oncol. 2017;35(4):746–751. [DOI] [PubMed] [Google Scholar]
- 70.Richardson PG, Schlossman RL, Weller E, et al. Immunomodulatory drug CC-5013 overcomes drug resistance and is well tolerated in patients with relapsed multiple myeloma. Blood. 2002;100(9):3063–3067. [DOI] [PubMed] [Google Scholar]
- 71.Anderson KC. Lenalidomide and thalidomide: mechanisms of action--similarities and differences. Semin Hematol. 2005;42(4 Suppl 4):S3–8. [DOI] [PubMed] [Google Scholar]
- 72.Ng T, Chan M, Khor CC, Ho HK, Chan A. The genetic variants underlying breast cancer treatment-induced chronic and late toxicities: a systematic review. Cancer Treat Rev. 2014;40(10):1199–1214. [DOI] [PubMed] [Google Scholar]
- 73.Themistocleous AC, Crombez G, Baskozos G, Bennett DL. Using stratified medicine to understand, diagnose, and treat neuropathic pain. Pain. 2018;159 Suppl 1:S31–S42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hertz DL, Roy S, Jack J, et al. Genetic heterogeneity beyond CYP2C8*3 does not explain differential sensitivity to paclitaxel-induced neuropathy. Breast Cancer Res Treat. 2014;145(1):245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Little J, Higgins JP, Ioannidis JP, et al. STrengthening the REporting of Genetic Association Studies (STREGA): an extension of the STROBE statement. PLoS Med. 2009;6(2):e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Backshall A, Sharma R, Clarke SJ, Keun HC. Pharmacometabonomic profiling as a predictor of toxicity in patients with inoperable colorectal cancer treated with capecitabine. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17(9):3019–3028. [DOI] [PubMed] [Google Scholar]
- 77.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. [DOI] [PubMed] [Google Scholar]
- 78.Hu S, Huang KM, Adams EJ, Loprinzi CL, Lustberg MB. Recent developments of novel pharmacologic therapeutics for prevention of chemotherapy-induced peripheral neuropathy. Clin Cancer Res. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.deAndres-Galiana EJ, Fernandez-Martinez JL, Sonis ST. Sensitivity analysis of gene ranking methods in phenotype prediction. J Biomed Inform. 2016;64:255–264. [DOI] [PubMed] [Google Scholar]