

Abstract
Sodium channel β1 subunits modulate α subunit gating and cell surface expression and participate in cell adhesive interactions in vitro. β1(-/-) mice appear ataxic and display spontaneous generalized seizures. In the optic nerve, the fastest components of the compound action potential are slowed and the number of mature nodes of Ranvier is reduced, but Nav1.6, contactin, caspr 1, and Kv1 channels are all localized normally at nodes. At the ultrastructural level, the paranodal septate-like junctions immediately adjacent to the node are missing in a subset of axons, suggesting that β1 may participate in axo-glial communication at the periphery of the nodal gap. Sodium currents in dissociated hippocampal neurons are normal, but Nav1.1 expression is reduced and Nav1.3 expression is increased in a subset of pyramidal neurons in the CA2/CA3 region, suggesting a basis for the epileptic phenotype. Our results show that β1 subunits play important roles in the regulation of sodium channel density and localization, are involved in axo-glial communication at nodes of Ranvier, and are required for normal action potential conduction and control of excitability in vivo.
Keywords: sodium, channel, epilepsy, node, adhesion, β
Introduction
Voltage-gated sodium channels in the brain are composed of a central, pore-forming α subunit and one or two β subunits (Catterall, 2000; Isom, 2001). β1 subunits affect sodium channel gating and cell surface expression when expressed in heterologous cells in vitro. However, the effects of β1 are highly dependent on the experimental system in which they are studied, and different effects on the kinetics and voltage dependence of gating of brain sodium channels are observed on expression in Xenopus oocytes, Chinese hamster lung and ovary cells, and human embryonic kidney cells (Isom et al., 1992, 1995; Qu et al., 2001; Meadows et al., 2002a). These results suggest that effects of β1 on sodium channel gating are dependent on the genetic background and signal transduction pathways present in the cell type used in heterologous expression and emphasize the importance of analyzing the effects of β1 in vivo.
Sodium channels are unique among voltage- and ligand-gated ion channels in that their auxiliary subunits both modulate channel function and serve as cell adhesion molecules (CAMs) through their extracellular Ig-like domain (Isom and Catterall, 1996; Isom, 2001, 2002). β subunits participate in homophilic cell adhesion (Malhotra et al., 2000) and interact with the CAMs contactin and neurofascin-186 (Kazarinova-Noyes et al., 2001; Ratcliffe et al., 2001; McEwen et al., 2004), the extracellular matrix molecules tenascin-R and tenascin-C (Srinivasan et al., 1998; Xiao et al., 1999), and the receptor tyrosine phosphatase β (Ratcliffe et al., 2000). β1 also recruits ankyrin to the plasma membrane at sites of cell-cell contact in response to homophilic cell adhesion (Malhotra et al., 2000, 2002), and this process is regulated by receptor-activated tyrosine phosphorylation of β1 (Malhotra et al., 2002), similar to members of the L1 family of CAMs (Tuvia et al., 1997). These results suggest that, as channel modulators, β subunits play important roles in control of electrical signaling and, as CAMs, β subunits act as critical communication links between extracellular and intracellular signaling molecules. It is now important to determine the functional roles of the novel CAM domains of β subunits in vivo.
β1 is involved in human disease. A mutation (C121W) in the extracellular Ig domain of β1 causes generalized epilepsy with febrile seizures plus type 1 (Wallace et al., 1998, 2002), an inherited human epileptic syndrome. Compared with wild-type β1, C121Wβ1 increased the fraction of available sodium channels at resting membrane potentials and reduced sodium current rundown during high-frequency channel activity (Meadows et al., 2002b). The C121Wβ1 mutation also disrupted homophilic β1-β1 cell adhesion. These potential effects on channel gating and subcellular distribution in vivo may bias neurons toward hyperexcitability and epileptogenesis.
To investigate the effects of loss of β1 function in vivo, we have used gene-targeting methods to produce β1(-/-) mice, and we have analyzed their neuronal phenotypes. β1(-/-) mice exhibit a dramatic neurological phenotype that includes an ataxic gait, spontaneous seizures, and premature death. We find slowing of action potential conduction, reduction in the number of mature nodes of Ranvier, alterations in nodal architecture, loss of sodium channel-contactin interactions, and abnormalities in expression of Nav1.1 and Nav1.3 that may contribute to these phenotypes. Our results show that β1 is required for normal excitability, cell-cell interactions, and action potential conduction in the brain.
Materials and Methods
Materials. PAT-5 embryonic stem (ES) cells were obtained from the University of Michigan Transgenic Core Laboratory (Domino et al., 2001). The pPNT vector was obtained from Richard Mulligan (Children's Hospital, Boston, MA).
Antibodies. Rabbit polyclonal antibodies to β1 (anti-β1ex), β2 (anti-β2ec), and β4 have been described previously (Malhotra et al., 2000; Chen et al., 2002; Yu et al., 2003). A polyclonal anti-β3 subunit antibody was generated to the peptide GKDFLIYEYRNGHQEVESPFQG by Affinity Bioreagents (Golden, CO) and used as described (Maier et al., 2003). Anti Nav1.1, Nav1.2, Nav1.3, Nav1.6, Kv1.1, and Kv1.2 antibodies were obtained from Chemicon (Temecula, CA). Anti-pan-sodium channel antibody was a gift from Dr. J. Trimmer (University of California, Davis, CA). Anti-Caspr 1 antibody was a gift from Dr. E. Peles (Weizmann Institute of Science, Rehovot, Israel). Anti-contactin antibody was a gift from Dr. J. Salzer (New York University School of Medicine, New York, NY). Anti-neurofascin 155 antibody was a gift from Dr. P. Brophy (Edinburgh University, Edinburgh, UK). Anti-ankyrinG antibody was obtained from Zymed Laboratories (South San Francisco, CA).
Generation of β1 (-/-) mice. SCN1B was cloned from a mouse 129 library (Stratagene, La Jolla, CA) using the full-length rat β1 cDNA as a probe. The gene was subsequently mapped and partially sequenced using β1-specific oligonucleotide primers. Approximately 1 kb of the β1 coding sequence [including exon 3 and the first 73 bases of intron 3 that are required for expression of the alternate splice product, β1A (Kazen-Gillespie et al., 2000)] were replaced with the pGKneo cassette inserted in the reverse orientation to preclude the possibility of read-through and to delete both the β1 and β1A transcripts. We created a dual-selection vector containing pGKneo as well as HSV-tk. The 5′ arm contained 1.2 kb of homologous sequence with 5.1 kb of homologous sequence on the 3′ arm. PAT-5 ES cells prepared from the mouse strain 129X1/SvJ (stock number 000691; Jackson Laboratory, Bar Harbor, ME) (Domino et al., 2001) were electroporated, and 300 clones were screened by Southern blot using external 5′ and 3′ probes. A single clone was identified as correctly targeted, subjected to chromosome analysis, and injected into blastocysts. High-percentage chimeric animals were obtained and bred to C57BL/6 mice to produce heterozygous animals. Genotyping was performed by PCR using tail DNA. ES cell electroporation and colony selection, injection of blastocysts, and generation of chimeric mice were provided as a service of the Transgenic Animal Model Core Laboratory at the University of Michigan. All experiments were performed in accordance with the guidelines for animal care of the University of Michigan.
PCR analysis of mouse tail DNA. DNA was prepared from each mouse tail at postnatal days (P) 10-14 using standard methods (Miller et al., 1988). Two sets of primers were used in genotyping. Primers β1KO5′-TCAGTGAAAGAGTGCTTGCCTGAAAATC and β1E3′-TGCCCCGACTACCGTTCCACACCACTC were used for the amplification of the endogenous β1 gene. These primers amplify an 870 bp product. NR′-GCTACTTCCATTTGTCACGTCCTGCAC and β1Flank3′-AGAGAGAATGGAGAATCAAGCCATAG were used to amplify the targeted gene. These primers amplify a 500 bp PCR product. The two primer sets were mixed and used together in a single PCR performed according to the following protocol: an initial denaturation step at 94°C for 2 min, followed by 30 cycles of denaturation at 94°C for 15 sec, annealing at 68°C for 30 sec, and elongation at 72°C for 2 min. The results were analyzed by agarose gel electrophoresis. β1(+/+) DNA showed only the endogenous β1 PCR product. Heterozygous β1(+/-) DNA showed both the endogenous β1 product and the targeted gene product. DNA isolated from mice homozygous for the β1 null mutation, β1 (-/-), showed only the targeted gene PCR product.
Northern blot analysis. Northern blot analysis of total mouse brain RNA was performed as described previously using a digoxigenin-labeled β1 antisense cRNA probe (Isom et al., 1992; Kazen-Gillespie et al., 2000). Total RNA from β1 (+/+), β1 (+/-), and β1 (-/-) littermate mice was purified using Trizol reagent (Life Technologies, Gaithersburg, MD), following the manufacturer's instructions.
Western blot analysis. β1(+/+) and β1(-/-) littermates were killed, at the ages indicated in the figure legends, and brains were immediately removed and stored in ice-cold homogenization buffer. Membranes were prepared as described previously (Isom et al., 1995). Complete protease inhibitor tablets (Roche Molecular Biochemicals, Indianapolis, IN) were included in all solutions. 3H-saxitoxin (Amersham Biosciences, Piscataway, NJ) binding to determine moles of sodium channel per milligram of protein and Western blot analysis were then performed as described previously (Malhotra et al., 2000) to detect sodium channel α, β2, β3, and β4 subunits, ankyrinG, caspr 1, contactin, or neurofascin 155, as indicated in the figure legends. SDS-PAGE gels contained the following percentages of acrylamide: for β2, β3, and β4: 10%; for pan-α and ankyrinG: 4-15% gradient; for contactin, caspr 1, and neurofascin 155: 7.5%.
Analysis of brain sections by immunocytochemistry. P15-P21 β1(+/+) and (-/-) littermate mice were anesthetized with Nembutal and perfused intracardially with a solution of 4% paraformaldehyde in 0.1 m sodium phosphate, pH 7.4 [phosphate buffer (PB)]. The brains were removed and postfixed for 2 hr. The tissue was then sunk in successive solutions of 10 and 30% (w/v) sucrose in PB at 4°C over 72 hr. Sagittal sections (40 μm) of the brain were cut on a sliding microtome and placed in PB until the tissue was processed for immunocytochemistry. Brain sections from β1 (+/+) and β1 (-/-) mice were processed as free-floating sections and were rinsed in 0.1 m Tris buffer, pH 7.4, for 15 min and in 0.1 m TBS, pH 7.4, for 15 min, blocked using 2% avidin in TBS for 30 min, rinsed in TBS for 30 min, blocked using 2% biotin for 30 min, and finally rinsed in TBS for 30 min. The tissue sections were then incubated in purified anti-β1ex antibodies (diluted 1:50), anti-Nav1.1 antibodies (diluted 1:20), or anti-Nav1.3 antibodies (diluted 1:15) for 36 hr at 4°C. All antibodies were diluted in a solution containing 1% NGS in 0.1 m TBS. The tissue sections were rinsed in TBS for 60 min and incubated in biotinylated goat anti-rabbit IgG (diluted 1:300) for 1 hr at 37°C. The tissue sections were rinsed with TBS for 60 min and incubated in avidin D-fluorescein (diluted 1:300) for 1 hr at 37°C. The sections were rinsed in TBS for 10 min, rinsed in Tris buffer for 20 min, and then mounted on gelatin-coated slides, coverslipped with Vectashield (Vector Laboratories, Burlingame, CA), sealed with nail polish, and viewed with an MRC 600 confocal microscope (Bio-Rad, Hercules, CA) located in the W. M. Keck Imaging Facility at the University of Washington.
Analysis of sciatic and optic nerves by immunocytochemistry. P17-P19 β1(+/+) and β1(-/-) littermate mice were anesthetized and perfused intracardially as described previously (Chen et al., 2002). Sciatic nerves were dissected, desheathed, and teased into single fibers with collagenase/dispase (3.5 mg/ml; Sigma, St. Louis, MO). In most cases, axons were teased on 2% gelatin-coated slices (Superfrost Plus; Fisher Scientific, Houston, TX) and postfixed in 4% paraformaldehyde in PB for 10 min. In some preparations, nerves were fixed before teasing; results were identical in both procedures. Optic nerves were dissected and postfixed for 20 min. Nerves were then rinsed with PB, cryoprotected with 10% sucrose overnight, frozen in optimal cutting temperature compound on dry ice, and then stored at -80°C. The tissues were cryosectioned into 5 μm slices, allowed to air dry, and stored at -20°C. Both sciatic and optic nerves were washed with PB three times and then blocked in PB containing 0.3% Triton X-100 and 10% goat serum for 1 hr at room temperature. The tissues were typically first incubated overnight with primary antibody, followed by a secondary goat anti-rabbit or goat anti-mouse antibody coupled with Alexa 488 (green) or Alexa 594 (red) at a 1:500 dilution (Molecular Probes, Eugene, OR). Between each antibody step, the tissues were washed three times with PB for 10 min. The tissues were then allowed to air dry and were mounted on slides using an anti-fade mounting medium (Gel/Mount; Fisher Scientific). Images were collected using an LSM 510 confocal microscope (Zeiss, Oberkochen, Germany) mounted on an Axiovert 100M inverted microscope (Zeiss) in the Microscopy and Image Analysis Laboratory at the University of Michigan.
Analysis of compound action potentials. Optic and sciatic nerves were dissected and placed in Locke's solution consisting of (in mm) 154 NaCl, 5.6 KCl, 2 CaCl2, 5 d-glucose, and 10 HEPES, pH 7.4. Nerves were then transferred to a recording chamber that was continuously perfused, oxygenated, and temperature regulated. For stimulation and recording of action potentials, each end of the nerve was drawn into a suction electrode (Stys et al., 1991). The stimulus was adjusted to ∼10% above the level required for a maximum response. Compound action potentials (CAPs) were amplified, digitized, recorded, and analyzed on a laboratory computer. For conduction velocity measurements, the time-to-peak amplitude of the CAP was measured from the onset of the nerve stimulus. The conduction velocity was calculated as the length of the nerve divided by the time-to-peak amplitude. Optic nerve CAPs were analyzed by fitting each sweep manually to the sum of three Gaussian curves. Starting values for amplitude, width, and time-to-peak of each Gaussian were made by fitting each component of the CAP to a third-order polynomial. The fit was then refined on the screen by altering one parameter at a time. The resulting time-to-peak amplitudes did not differ significantly from those estimated directly from the original records. Student's t tests were applied to the data.
Voltage-clamp analysis of hippocampal neurons. Hippocampal neurons from young littermate mice (16-18 d postnatal) were acutely isolated using standard procedures (Kay and Wong, 1987; Surmeier et al., 1991; Cantrell et al., 1996). In brief, mice were decapitated under halothane anesthesia. Brains were removed quickly, iced, and blocked before slicing. Slices (400-500 μm) were cut and transferred to a low-calcium, HEPES-buffered saline solution containing (in mm) 140 Na isethionate, 2 KCl, 4 MgCl2, 0.1 CaCl2, 23 glucose, and 15 HEPES, pH 7.4, at 300-305 mOsm/l. Slices were incubated for 1-6 hr in NaHCO3-buffered Earle's balanced salt solution (Sigma) bubbled with 95% O2 and 5% CO2, pH 7.4, at 300-305 mOsm/l. Single slices were then moved into the low-calcium buffer, and with the aid of a dissecting microscope, regions of hippocampus were removed and placed in a treatment chamber containing protease type XIV (1.5 mg/ml; Sigma) in HEPES-buffered HBSS (Sigma) at 35°C, pH 7.4, at 300-305 mOsm/l. After 20-30 min of enzyme treatment, the tissue was rinsed several times in the low-calcium buffer and dissociated mechanically, using a series of fire-polished Pasteur pipettes. The cell suspension was plated into a 35 mm tissue culture dish (Corning, Corning, NY) containing 1 ml of HEPES-buffered phosphate-free HBSS saline and mounted on the stage of an inverted microscope. After allowing the cells to settle for ∼5 min, the solution bathing the cells was changed to external recording solution (Cantrell et al., 1996). Sodium currents in pyramidally shaped hippocampal neurons were recorded in the whole-cell mode. The external recording solution consisted of (in mm) 20 NaCl, 10 HEPES, 1 MgCl2, 1 CdCl2, 60 CsCl, and 150 glucose (pH 7.3 with CsOH; 300-305 mOsm/l). The internal solution consisted of (in mm) 179 N-methyl d-glucamine, 40 HEPES, 4 MgCl2, 10 EGTA, 40 NaCl, 25 phosphocreatine, 2 ATP, 0.2 GTP, and 0.1 leupeptin (pH 7.2 with H2S04; 270-275mOsm/l). Electrode resistances were typically 3-5 MΩ in the bath. Recordings were obtained using an Axopatch 1C patch clamp (Axon Instruments, Foster City, CA). Series resistance compensation (70-80%) was used routinely. Data were filtered at 10 kHz.
Transmission electron microscopy. P19 β1 (+/+) and β1 (-/-) littermates (nine sciatic nerve samples, seven optic nerve samples, and three spinal cord samples from each genotype) were anesthetized and perfused with 0.1 m cacodylate buffer, pH 7.4, followed by cardiac perfusion containing 2.5% glutaraldehyde/2.0% paraformaldehyde in the same buffer. After perfusion, optic nerves, sciatic nerves, and spinal cords were dissected carefully and placed in fresh fixative for 2 hr, rinsed, and then postfixed in 1% osmium tetroxide containing 0.75% potassium ferrocyanide with gentle agitation for 2 hr at room temperature. The samples were then washed well in several changes of buffer, dehydrated in ascending strengths of ethanol, and embedded in Spurr's (Spurr, 1969) epoxy resin or Embed 812 epoxy resin. Semithin (0.5 μm) sections were stained with 1% toluidine blue for morphological examination and orientation. Ultrathin sections (70 nm) were stained in uranyl acetate/lead-citrate and examined using a Philips CM-100 TEM in the Microscopy and Image Analysis Laboratory at the University of Michigan. Images were recorded digitally using a 1.6 Megaplus camera system (Eastman Kodak, Rochester, NY) operated using AMT software (Advanced Microscopy Techniques, Danvers, MA). The resulting images were imported into Adobe Photoshop and digitally made into plates.
Numbers of myelinated axons in optic nerves were determined in one ultrathin cross-section from each of three β1(+/+) and β1(-/-) mice. Fifty evenly distributed, non-overlapping fields from each optic nerve section were photographed at 7900×. Each field represented 108 μm2 of endoneurial area. Myelinated axons were defined as axons wrapped by two or more layers of myelin. The total numbers of myelinated axons per field were counted and reported as the mean ± SEM. The incidence of degenerating axons was determined in the same ultrathin sections. Degenerating axons were defined as collapsed or delaminated myelinated axons containing membranous debris. Entire cross-sectional areas were examined at 3400×. Degenerated axons were counted and reported as mean ± SEM.
Electrocorticographic recordings. Silver wire electrodes (0.005 inch diameter) soldered to a microminiature connector were implanted bilaterally into the subdural space over the frontal and parietal cortex of mice under Avertin anesthesia several days before recording. Simultaneous cortical activity and behavioral video/electrographic monitoring was performed using a digital electroencephalograph (Stellate Systems, Montreal, Canada) from five β1(-/-) and five β1(+/+) mice moving freely in the test cage for prolonged periods, including sleep (Noebels, 1984). Seizure behavior was observed directly and annotated on all recordings.
Results
Gene-targeting of SCN1B
SCN1B was cloned from a mouse 129 library (Stratagene) using the full-length rat β1 cDNA as a probe (Isom et al., 1992). The gene was subsequently mapped and partially sequenced using β1-specific oligonucleotide primers. Approximately 1 kb of the β1 coding sequence [including exon 3 and the first 73 bases of intron 3 that are required for expression of the alternate splice product, β1A (Kazen-Gillespie et al., 2000)] were replaced with the pGKneo cassette inserted in the reverse orientation to preclude the possibility of read-through and to delete both the β1 and β1A transcripts (Fig. 1A). The amino acid sequence of β1 is shown, including exon 3 (gray), the transmembrane domain (underlined), and the cysteine residues that define the Ig fold domain (asterisks) (Fig. 1B). We created a dual-selection vector containing pGKneo as well as HSV-tk (Fig. 1A). The 5′ arm contained 1.2 kb of homologous sequence with 5.1 kb of homologous sequence on the 3′ arm. PAT-5 ES cells prepared from the mouse strain 129X1/SvJ (stock number 000691; Jackson Laboratory) (Domino et al., 2001) were electroporated, and 300 clones were screened by Southern blot using external 5′ and 3′ probes (Fig. 1C). Digestion with KpnI, followed by Southern blotting with the 5′ probe, yielded a 4.8 kb endogenous band and a 3.8 kb targeted band. A similar experiment using the 3′ probe yielded a 5.9 kb endogenous band and a 7.7 kb targeted band. A single clone was identified as correctly targeted (clone 79) and was injected into blastocysts. High-percentage chimeric animals were obtained and bred to produce heterozygous animals. Tail DNA samples were analyzed using a PCR strategy (Fig. 1D). Northern blot analysis of the brains of β1 (+/+), β1 (+/-), and β1 (-/-) mice showed that β1 mRNA is significantly reduced in the heterozygotes and completely absent in the homozygotes (Fig. 1E). β1(-/-) mice were born in an approximate Mendelian ratio of β1 (+/+) 366/β1 (+/-) 717/β1(-/-) 258, demonstrating nearly normal survival during fetal development. The present study was performed on F2 hybrid mice obtained from breeding 129 germ line chimeras to the inbred strain C57BL/6 as described (Silva et al., 1997).
Figure 1.
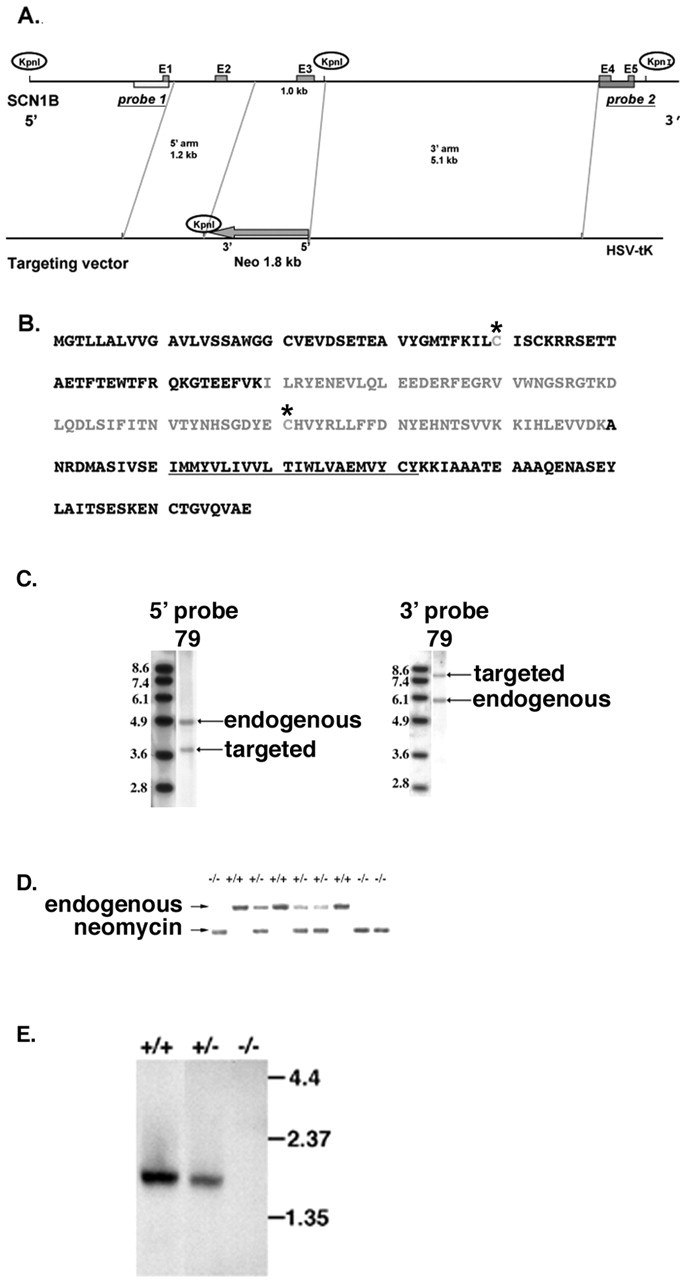
Generation of β1 (-/-) mice by homologous recombination. A, SCN1B targeting vector. B, Amino acid sequence of β1. Gray, Exon 3; underline, transmembrane domain; asterisks, cysteine residues that define the Ig fold domain. C, Southern blot analysis of ES cell clones. Digestion with KpnI, followed by Southern blotting with the 5′ probe, yielded a 4.8 kb endogenous band and a 3.8 kb targeted band (left). A similar experiment using the 3′ probe yielded a 5.9 kb endogenous band and a 7.7 kb targeted band (right). D, PCR analysis of tail DNA isolated from β1 (+/+), (+/-), and (-/-) mice, as indicated. The arrows indicate positions of endogenous and targeted (neomycin) PCR products. E, Northern blot analysis of total brain RNA isolated from β1 (+/+), (+/-), and (-/-) mice, as indicated. Molecular weight markers are shown in kilobases.
β1 (-/-) mice exhibit defects in growth, control of movement, and life span
β1(-/-) mice have an ataxic gait, exhibit frequent spontaneous generalized seizures and growth retardation (Fig. 2A), and die at approximately P19.9 (mean) (n = 177; SEM, 0.16; SD, 2.1). The earliest day of death recorded for the β1 (-/-) mice (according to genotyping performed between P10 and P14) was P13, and the latest day of death was P26.
Figure 2.
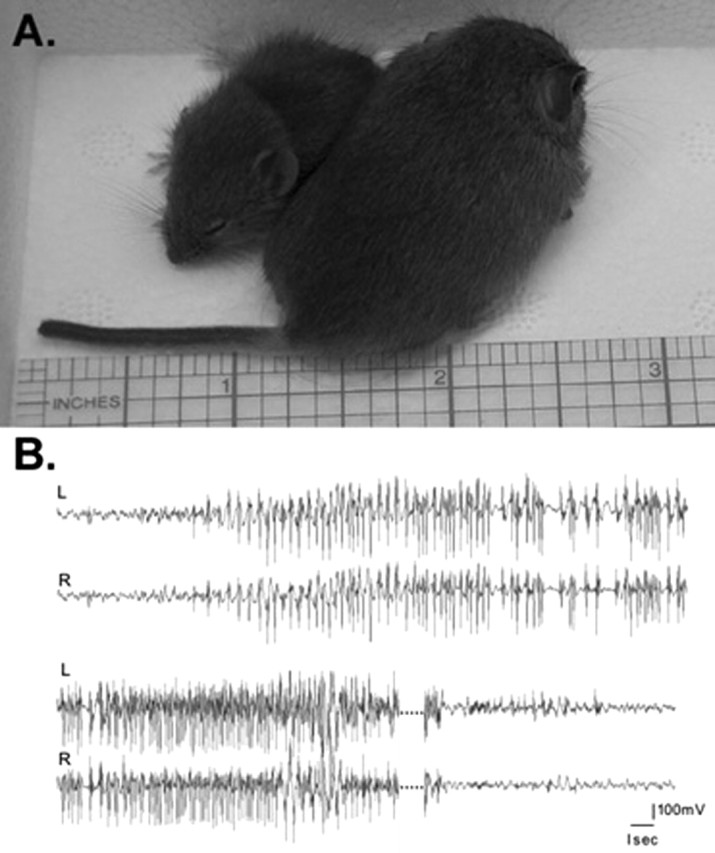
β1(-/-) mice show growth retardation and spontaneous seizures. A, Growth retardation in β1 (-/-) mice.β1 (-/-) (left) and β1 (+/+) littermates at P19. The scale is shown in inches. B, Generalized myoclonic seizure in a β1 (-/-) mouse. Electrocorticographic monitoring of P19 β1 (-/-) mice reveals abnormal interictal discharges and seizures during waking behavior. A spontaneous seizure episode of 55 sec duration displayed here shows abrupt bilateral onset and steady acceleration in the frequency of cortical spike discharges, with a sudden return to normal cortical activity at the termination of the seizure. The dotted lines represent 16 sec of continuous seizure activity.
Beginning at approximately P10, β1 (-/-) mice exhibit frequent, severe seizure episodes lasting from seconds to minutes at random intervals during both day and night. Seizure episodes are then followed by refractory periods during which the mice are immobile. The electroclinical correlation of these episodes was further explored with chronic video/electrographic recordings of freely moving mice. The baseline cortical activity of the mice showed periods of normal, low-amplitude desynchronized EEG with the occasional appearance of interictal discharges. A spontaneous bilateral myoclonic seizure lasting ∼55 sec was captured, revealing the abrupt initiation of a high-voltage, rhythmic polyspike discharge, terminating abruptly without post-ictal depression (Fig. 2B). During this electrographic seizure, the mouse was tossed to its side and moved chaotically while showing continuous, clonic, jerking of all four limbs, followed by a return to normal behavior on termination of the seizure. β1 (-/-) mice are frequently separated from the rest of the litter and are rarely fed by the mother. Postmortem examinations of the β1 (-/-) mice showed impacted esophagi, possibly attributable to enteric nervous system impairments. Thus, the mice receive very little nutrition, and this is likely to contribute to their premature deaths. Here, we have analyzed the cellular and molecular changes caused by deletion of the β1 subunit gene that may underlie the ataxia and seizures of β1 (-/-) mice.
Western blot analysis showed that neither α (as measured with a pan-sodium channel antibody), β2, β3, nor β4 proteins were upregulated in whole-brain membrane preparations as a result of the β1 null mutation (Fig. 3A). No consistent changes in the expression levels of contactin, caspr 1, neurofascin 155, or ankyrinG were observed in brain homogenates of β1(-/-) mice compared with wild type (Fig. 3B). The lower immunoreactive band visible in the neurofascin 155 Western blot is thought to be the 140 kDa isoform of neurofascin, an alternate splice product (Davis et al., 1993). This protein is also unchanged in the β1 (-/-) mice. These results suggest that no major compensatory changes in the levels of proteins that interact with β1 subunits or proteins that are critical to the axo-glial apparatus have occurred as a result of the β1 null mutation.
Figure 3.
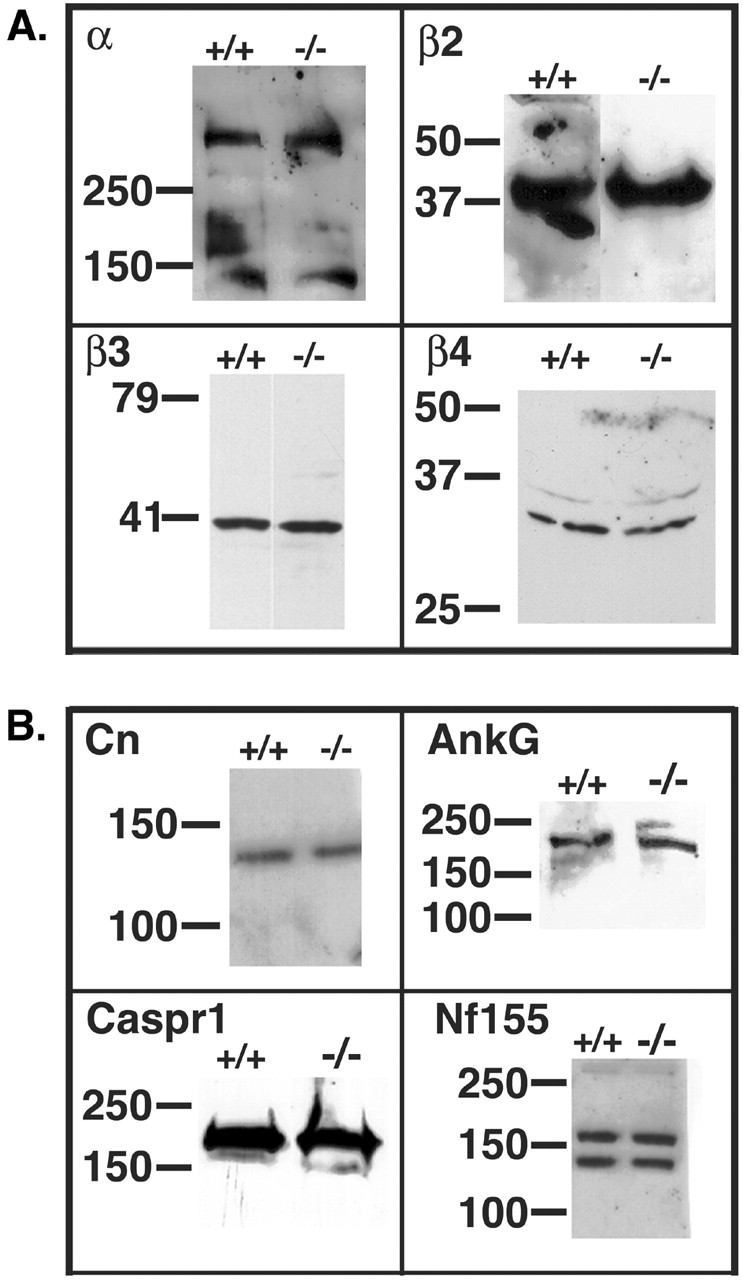
Western blot analysis of brain membranes from β1 (-/-) mice. A, Sodium channel subunits are not upregulated in β1 (-/-) brain membranes. Equal aliquots of β1 (-/-) and β1 (+/+) mouse brain membranes were analyzed by Western blot using anti-pan-α, anti-β2, anti-β3, or anti-β4 antibodies (as indicated), followed by HRP-conjugated goat anti-rabbit IgG. Molecular weight markers are shown in kilodaltons. B, Western blot analysis of proteins involved in the axo-glial apparatus. Thirty micrograms of β1 (-/-) or β1 (+/+) mouse brain membranes were analyzed by Western blot using primary antibodies, as indicated, followed by HRP-conjugated goat anti-rabbit IgG. Dilutions of primary antibodies were: contactin (Cn), 1:2000; Caspr 1,1:500; neurofascin 155 (Nf155), 1:2500; ankyrinG (AnkG), 1:250. Molecular weight markers are shown in kilodaltons.
Molecular structure of central and peripheral nodes of Ranvier in β1 (-/-) mice
The apparent ataxic gait and seizures in the β1 (-/-) mice suggested that control of action potential generation in cell bodies and/or action potential propagation in axons may be impaired. To test this hypothesis, we investigated the structure and function of myelinated nerves and the excitability and distribution of sodium channels in neuronal cell bodies of β1 (-/-) mice. To investigate whether the lack of β1 subunits affected the molecular structures of central and peripheral nerve nodes of Ranvier, we performed fluorescent immunocytochemical analyses of optic and sciatic nerves isolated from β1 (+/+) and β1 (-/-) littermates. Figure 4 shows that sodium channels detected with a broad-specificity antibody (Pan), caspr 1, contactin, and Kv1.2 are localized normally in β1 (-/-) sciatic nerves compared with β1 (+/+). β1 subunits are concentrated with sodium channel α subunits in the nodal gap in β1 (+/+) mice (Fig. 4A). β1 staining is absent in sciatic nerves isolated from β1 (-/-) mice (Fig. 4B), but sodium channel α subunits are still present (Fig. 4D). Caspr 1 and contactin (Fig. 4C-F) are colocalized in the paranode, and Kv1.2 channels are localized to the juxtaparanodal region in both genotypes (Fig. 4G,H).
Figure 4.
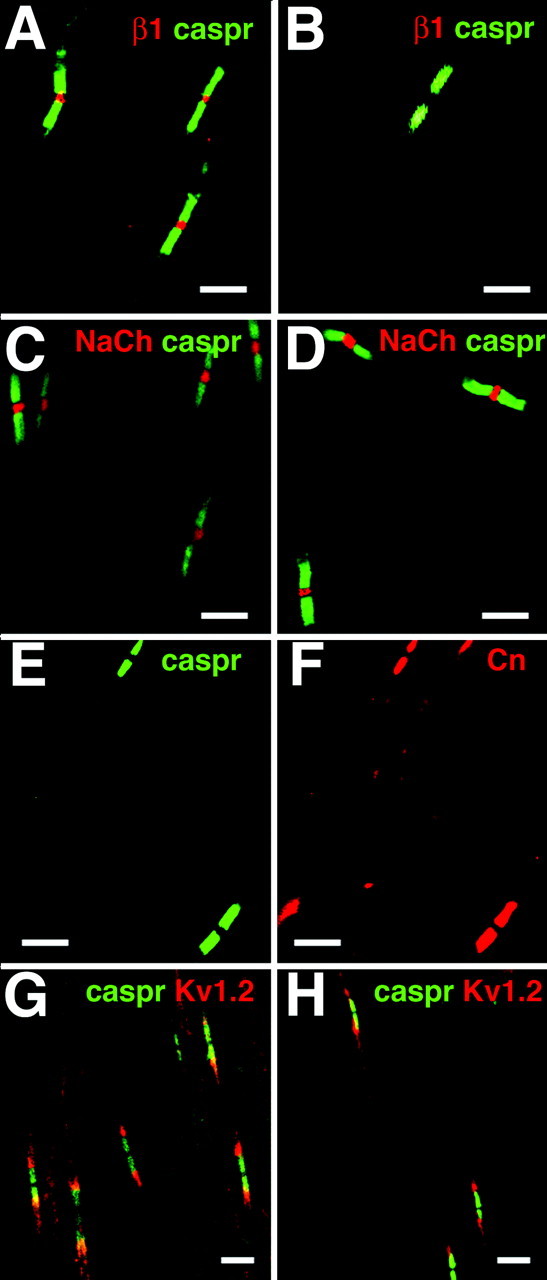
Sciatic nerve nodes of Ranvier in β1 (-/-) mice. Sciatic nerves were isolated from β1 (+/+) and β1 (-/-) P17-P19 mice and prepared for immunocytochemistry, as described in Materials and Methods. Immunofluorescent staining obtained from antibodies directed against pan-sodium channels, caspr 1, contactin, and Kv1.2 appeared to be unchanged in the mutant mice compared with control. A, β1 (+/+) mice: β1 subunits (red), caspr 1 (green). B, β1 (-/-) mice: β1 subunits (red), caspr 1 (green). There is no staining for β1 subunits. C,β1(+/+) mice: pan-sodium channels (red), caspr 1 (green). D,β1(-/-) mice: pan-sodium channels (red), caspr 1 (green). E,β1(-/-) mice: caspr 1 (green). F,β1(-/-) mice (identical section as in E): contactin (red). G, β1 (+/+) mice: caspr 1 (green) and Kv1.2 (red). H, β1 (-/-) mice: caspr 1 (green) and Kv1.2 (red). Scale bars, 10 μm.
A similar result was obtained for nodes of Ranvier in optic nerves (Fig. 5). We found no detectable changes in localization of sodium channels (Fig. 5A), caspr 1 (Fig. 5A-C), Kv1.1 channels (Fig. 5B), or contactin (Fig. 5E,F) in the β1(-/-) animals compared with β1 (+/+) littermate controls (data not shown). In addition, the predominant sodium channel subtype at central nodes (Fig. 5C) and peripheral nodes (data not shown) at P17-P19 is Nav1.6 in both genotypes, as expected from previous work (Boiko et al., 2001). Thus, at the level of resolution obtained using fluorescent confocal microscopy, there are no detectable aberrations in the molecular structure of the node of Ranvier or the axo-glial complex as a result of deletion of the β1 gene. In all experiments, however, we observed significantly fewer nodes of Ranvier in the β1 (-/-) optic nerve compared with β1(+/+). This issue is addressed in detail below.
Figure 5.

Optic nerve nodes of Ranvier in β1 (-/-) mice. Optic nerves were isolated from β1 (+/+) and β1 (-/-) P17-P19 mice and prepared for immunocytochemistry, as described in Materials and Methods. Immunofluorescent staining obtained from antibodies directed against pan-sodium channels, caspr 1, contactin, and Kv1.1 appeared to be unchanged in the mutant mice compared with control. A, β1(-/-) mice: pan-sodium channels (green), caspr 1 (red). B, β1(-/-) mice: caspr 1 (green), Kv1.1 (red). C, β1(-/-) mice: Nav1.6 (red), caspr 1 (green). D, β1(-/-) mice: pan-sodium channels (green). E, β1 (-/-) mice: contactin (red) (same slide as in D). F, β1(-/-) mice: merged images of D and E. Scale bar: A-F, 5 μm. G, Sodium channels and contactin do not associate in the absence of β1 subunits. Whole-brain homogenates from β1 (+/+) or β1 (-/-) mice, as indicated, were immunoprecipitated with anti-contactin antibody or nonimmune IgG [lane IgG, containing β1 (+/+) homogenate], and immunoblotted with anti pan-sodium channel antibody, as described in Materials and Methods. Molecular weight markers are shown in kilodaltons. RB, Nonimmunoprecipitated aliquot of rat brain homogenate containing 500 fmol of sodium channel included as a positive Western blot control.
We showed previously that contactin and sodium channels colocalize at optic nerve nodes of Ranvier and that, in a heterologous expression system, this interaction is dependent on the presence of β1 subunits (Kazarinova-Noyes et al., 2001; McEwen et al., 2004). Contactin and sodium channel α subunits associate in brain homogenates of β1 (+/+) mice (Fig. 5G), as expected, and these two molecules remain colocalized at nodes of Ranvier in the β1 null mutant (Fig. 5D-F). Despite their colocalization, however, contactin and sodium channels do not associate in β1 (-/-) mice (Fig. 5G). Immunoprecipitation of β1 (+/+) brain homogenates with anti-contactin antibody revealed the presence of a sodium channel immunoreactive band at ∼260 kDa on Western blot analysis and, as shown previously (Kazarinova-Noyes et al., 2001), this represents a subpopulation of the total sodium channel pool (Fig. 5G). Identical treatment of brain homogenates prepared from β1 (-/-) littermates showed that contactin and sodium channels do not associate, confirming our previous observation and demonstrating that this interaction occurs in vivo.
CAPs in β1 (-/-) mice
CAPs were recorded from optic and sciatic nerves at P15-P16 using suction electrodes for stimulation and recording. Conduction velocities were measured for the various components of the CAP at temperatures from 25 to 37°C. In the sciatic nerve (data not shown), conduction velocity in the fastest myelinated axons was not significantly different in β1(-/-) mice [5.2 ± 0.6 m/sec (25°C) and 8.7 ± 1.1 m/sec (37°C)] compared with β1 (+/+) littermates [6.7 ± 0.7 m/sec (25°C) and 10.6 ± 1.2 m/sec (37°C)]. A component attributable to C-fibers was readily seen and measured in the sciatic nerve CAP when the stimulus strength was raised sufficiently. C-fiber velocities also did not differ significantly in β1(-/-) animals [0.26 ± 0.02 (25°C) and 0.50 ± 0.03 m/sec (37°C)] versus β1 (+/+) [0.27 ± 0.01 m/sec (25°C) and 0.49 ± 0.01 m/sec (37°C)]. Thus, axonal conduction was normal in the PNS of β1 (-/-) mice. In contrast, clear functional distinctions were seen in the CNS. The CAP from optic nerves (Fig. 6) consists typically of three components, thought to represent three classes of retinal ganglion cells. Retinal ganglion cells are classified in numerous ways in each species. In one scheme, there are W, X, and Y, with Y cells the largest and fastest and W cells the smallest and slowest (Fukuda, 1977). In the mouse, physiologically classified X and Y are alternatively known morphologically as II and I, respectively (Doi et al., 1995). Peak 1 in our experiments was difficult to resolve because the length of the optic nerve was short at the ages available. The CAPs were fitted to the sum of three Gaussian curves for analysis, as shown in Figure 6A-D, and conduction velocities were calculated (Fig. 6E). The two fastest components (peaks 1 and 2) were significantly slower in the β1 (-/-) nerves at both 25 and 37°C, whereas for peak 3 the difference in conduction velocity was minimal (p = 0.04 at 25°C). Thus, there is a substantial functional deficit in rapidly conducting fibers in the optic nerve, and similar defects in other nerve tracts may contribute to the ataxia of these animals and their failure to survive past approximately P20.
Figure 6.
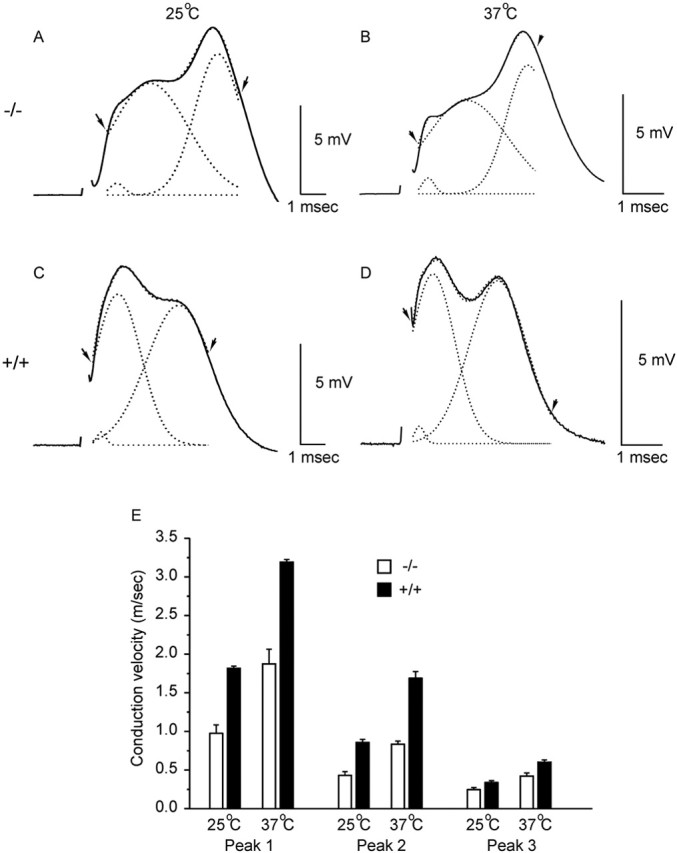
Conduction velocities in the optic nerve. A-D show CAPs at P15-P17 from β1 (-/-) mice (A, B) and β1 (+/+) littermates (C, D). Records were taken at room temperature (A,C; 25 ±1 °C) and at 37 °C (B, D). The solid lines illustrate the original sweeps. CAPs were fitted to the sum of three Gaussian curves between the arrows, and the fit is superimposed (dotted line just discernable). Also shown are the three individual curves (dotted lines) from which times to peak were measured. E, Bar graph showing conduction velocities for each component (mean ± SEM). Differences between β1 (-/-) (open bars) and β1 (+/+) (filled bars) were significant for peaks 1 and 2 (p ≤ 0.005 at both temperatures) and borderline for peak 3 (p = 0.04 at 25°C).
β1 (-/-) mice have fewer nodes of Ranvier in optic nerves
We also investigated the number of mature nodes of Ranvier in optic nerve using immunofluorescence localization of sodium channels and caspr 1 to mark the nodal and paranodal regions, respectively (Fig. 7A). Nodes of Ranvier are defined by a region of high-sodium channel density in the nodal gap, bordered by caspr 1-positive paranodes. As shown in Figure 7A, individual nodes had a normal appearance in both β1 (-/-) and β1 (+/+) littermates, but there were fewer such sites in the null mutant. For sites showing both caspr 1 and sodium channels, we found 20.9 ± 0.9 sites per field of view (FOV) for β1 (+/+) optic nerves versus 14.9 ± 1.1 sites per FOV for β1 (-/-) optic nerves (Fig. 7B) [mean ± SEM; p = 0.0001, Student's t test; total FOV: β1 (+/+), 33; β1 (-/-), 31; total sites counted: β1 (+/+), 689; β1, (-/-) 463]. Ultrastructural cross-sectional analysis of optic nerves isolated from β1 (+/+) and β1 (-/-) mice showed no significant differences in optic nerve myelinated axonal number per square micrometer between the two genotypes (Table 1; Fig. 7C), however, β1 (-/-) mice displayed a greater number of degenerating axons per optic nerve cross-section (p = 0.067, Student's t test) (Table 2; Fig. 7C), indicating that some axonal loss may be taking place by P19. An example of a degenerating axon in the β1(-/-) optic nerve is shown in Figure 8H. Taken together, these data are consistent with the conduction data presented in Figure 6, suggesting that certain classes of retinal ganglion cells are adversely affected by the β1 null mutation and that node formation and action potential conduction in the axons projecting from these cells is compromised.
Figure 7.
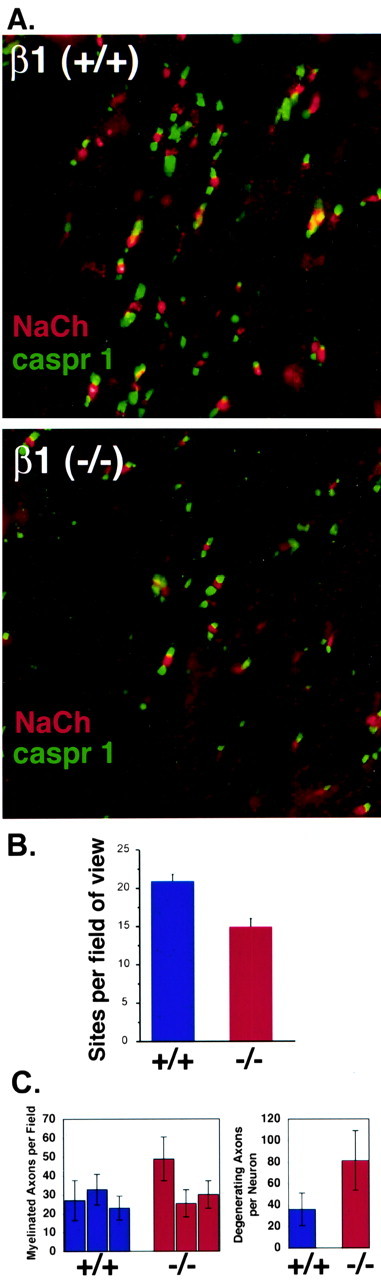
β1 (-/-) mice have fewer optic nerve nodes of Ranvier. A, Optic nerves of P17-P19 β1 (-/-) and β1 (+/+) littermates were cryosectioned and stained for pan-sodium channels (red) and caspr 1 (green). The panels in A show typical FOV. Nodes of Ranvier were defined by a region of high-sodium channel density in the nodal gap, bordered by caspr 1-positive paranodes. Individual nodes had a normal appearance in both β1 (-/-) and β1 (+/+) animals, but there were fewer such sites in the null mutant. B, Over 30 FOV were analyzed for both β1 (-/-) (red bar) and β1 (+/+) (blue bar) mice, and the results are summarized in the bar graph. β1 (-/-) optic nerves had significantly fewer nodes than β1 (+/+) (p < 0.001). C, Left, Ultra structural analysis of optic nerve cross-sections shows similar numbers of myelinated axons per unit area for β1 (+/+) (blue bars) and β1 (-/-) (red bars). However, a greater percentage of myelinated axons per nerve cross-section in β1 (-/-) optic nerves (right, red bar) appear to be degenerating (p = 0.067, Student's t test). Error bars in both panels represent SDs.
Table 1.
Myelinated axons per 108 μm2 in the optic nerve
|
Animal |
Myelinated axons per 108 μm2 in optic nerve |
|---|---|
| β1 (+/+) 1102-1 | 27 ± 1.5 |
| β1 (+/+) 3122-1 | 32 ± 1.2 |
| β1 (+/+) 3196 | 23 ± 0.9 |
| β1 (−/−) 1102-1 | 49 ± 1.7 |
| β1 (−/−) 3121-1 | 25 ± 1.0 |
| β1 (−/−) 3148 |
30 ± 1.0 |
Ultrathin optic nerve cross-sections were prepared from β1 (+/+) and β1 (−/−) mice and analyzed as described in Materials and Methods. Fifty evenly distributed, nonoverlapping fields, each representing 108 μm2 of endoneurial area, were photographed at 7900X. The total numbers of myelinated axons per field were counted and reported as mean ± SEM. The average numbers of axons in each genotype were not significantly different as assessed by Student's t test.
Table 2.
Axonal degeneration in β1 (−/−) optic nerves
|
Animal |
Number of degenerating axons per cross-section of optic nerve |
|---|---|
| β1 (+/+) 1102-1 | 52 |
| β1 (+/+) 3122-1 | 34 |
| β1 (+/+) 3196 | 22 |
| Mean ± SEM | 36 ± 8.7 |
| β1 (−/−) 1102-1 | 85 |
| β1 (−/−) 3121-1 | 107 |
| β1 (−/−) 3148 | 52 |
| Mean ± SEM |
81 ± 16; p = 0.067 |
Ultrathin optic nerve cross-sections were prepared from β1 (+/+) and β1 (−/−) mice and analyzed as described in Materials and Methods. Degenerating axons were defined as collapsed or delaminated myelinated axons containing membranous debris. Entire cross-sectional areas were examined at 3400X. Degenerated axons were counted and reported as mean ± SEM. p value was determined using Student's t test (SigmaPlot).
Figure 8.
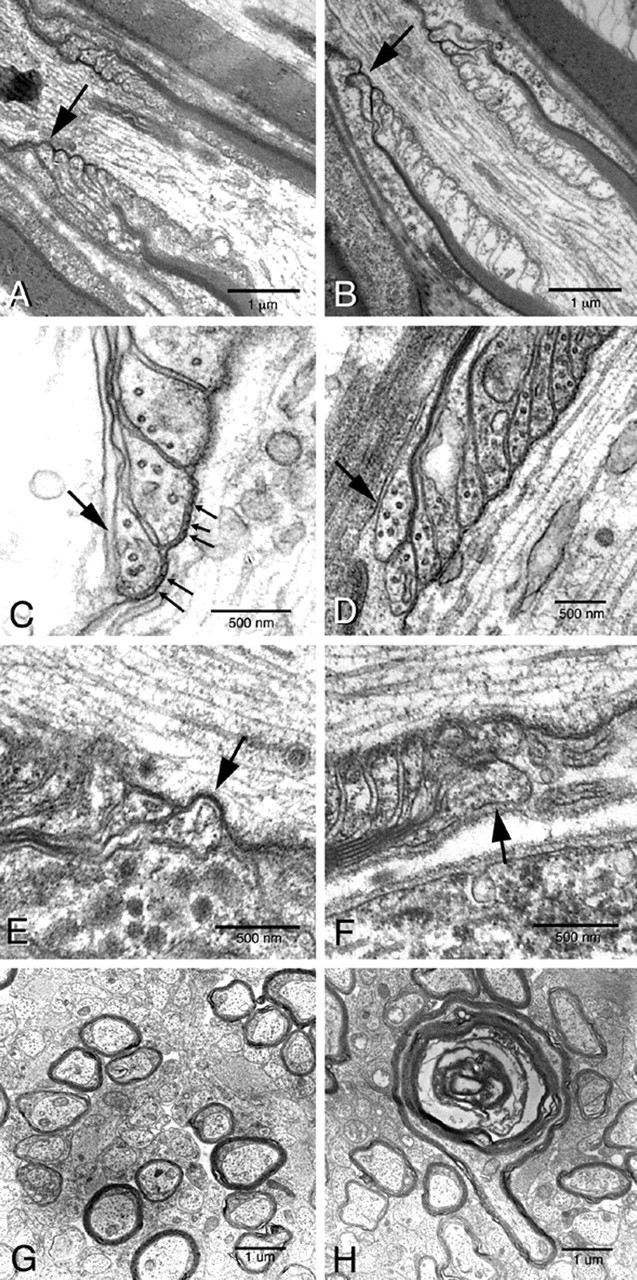
β1 (-/-) mice show disruptions in nodal architecture in the CNS and PNS. Sciatic nerves, optic nerves, and spinal cords were isolated from P19 β1 (-/-) and β1 (+/+) littermates and prepared for transmission electron microscopy as described in Materials and Methods. A, E, β1 (+/+) sciatic nerve. B, F, β1 (-/-) sciatic nerve. C, β1 (+/+) spinal cord. D,β1 (-/-) spinal cord. G, Optic nerve cross-section fromβ1 (+/+) mouse. H, Optic nerve cross-section from β1(-/-) mouse showing an example of a degenerating axon. Large arrows indicate normal paranodal myelin loops [for β1 (+/+)] or paranodal loops that are everted or pulled away [for β1 (-/-)]. Small arrows indicate electron dense transverse bands. Scale bars are as indicated for each image.
Disruptions of nodal architecture
To investigate the effect of the β1 null mutation on nodal and paranodal architecture, we performed ultrastructural analyses of longitudinal sections of β1 (+/+) and β1 (-/-) myelinated spinal cord and sciatic nerves at P19 (Fig. 8). In both β1 (+/+) and β1 (-/-) mice, the nodal and paranodal regions were clearly discernable, although we consistently observed fewer nodes in the β1(-/-) mice compared with β1(+/+), in agreement with data presented above. In both genotypes, the paranodal myelin loops and electron dense transverse bands, thought to represent sites of direct contact between the axolemma and the terminal myelin loops, were present. However, in ∼20-30% of nodes of β1 (-/-) mice, the paranodal myelin loop located adjacent to the nodal gap in β1 (-/-) mice lacked transverse bands, and the loop itself appeared to be everted away from the axon (Fig. 8B,D,F, arrows). In contrast, these same loops were normal in every node examined in the β1 (+/+) mice (Fig. 8A,C,E, arrows). These results suggest that sodium channel β1 subunits are involved in the formation or maintenance of the axo-glial junctions at the periphery of the nodal gap in axons in both the CNS and PNS. Because the paranodal loops other than the loop nearest the node appear to attach to the axolemma with normal, electron-dense transverse bands, we propose that β1 subunits may play a role in the septate-like junctional cell adhesive complex closest to the nodal gap but not in adjacent complexes.
To investigate the fine structure of the myelin sheath, we performed ultrastructural analyses of cross-sections of spinal cord, sciatic nerves, and optic nerves from β1 (+/+) and β1 (-/-) mice at P19. Multiple myelin lamellae encircling individual axons were observed in transverse sections of both genotypes (data not shown). Morphometric analysis showed that the g value (the ratio of axon diameter to fiber diameter) was within the normal range for mouse myelinated axons (0.64-0.73) (Little and Heath, 1994) in all cases except for β1(-/-) spinal cord, which was 0.76 (Table 3). g values for optic nerve were not significantly different for β1 (-/-) compared with β1 (+/+). However, g values for sciatic nerve and spinal cord were significantly higher in the β1 (-/-) mice (0.67 vs 0.72 in sciatic nerve, p = 0.006; 0.69 vs 0.76 in spinal cord, p = 0.003), suggesting that some demyelination or dysmyelination may have occurred. More extensive analysis using mice with defined genetic background will be required to determine the physiological importance of these small but reproducible changes in myelination.
Table 3.
Morphometric analysis of β1 (−/−) and β1 (+/+) myelinated nerves
|
Parameter |
+/+ |
−/− |
p |
|---|---|---|---|
| Sciatic nerve | |||
| Axon diameter (μm) | 1.93 (0.08) | 2.00 (0.12) | |
| Fiber diameter (μm) | 2.88 (0.13) | 2.77 (0.15) | |
| Ratio (g value) | 0.67 (0.12) | 0.72 (0.01) | 0.006 |
| Number | 42 | 43 | |
| Spinal cord | |||
| Axon diameter (μm) | 1.06 (0.06) | 1.06 (0.09) | |
| Fiber diameter (μm) | 1.54 (0.08) | 1.37 (0.10) | |
| Ratio (g value) | 0.69 (0.02) | 0.76 (0.01) | 0.003 |
| Number | 67 | 66 | |
| Optic nerve | |||
| Axon diameter (μm) | 0.84 (0.04) | 0.70 (0.03) | |
| Fiber diameter (μm) | 1.13 (0.04) | 0.98 (0.03) | |
| Ratio (g value) | 0.73 (0.01) | 0.70 (0.01) | 0.013 |
| Number |
68 |
87 |
|
Data are presented as mean (SE). p values were determined using Student's t test (SigmaPlot).
Sodium currents in acutely dissociated hippocampal neurons
β1 subunits exert subtle effects on sodium channel gating that differ with the heterologous expression system in which they are studied (see above). To assess the effects of the absence of β1 subunits in vivo, we compared sodium currents recorded in acutely dissociated hippocampal neurons from β1(+/+) and β1 (-/-) mice using low-sodium extracellular solution to ensure precise voltage-clamp control (Fig. 9). The magnitude of peak sodium current recorded at -10 mV was unaffected in the β1 (-/-) mice [β1(+/+): 1.52 ± 0.20 nA, n = 8; β1(-/-): 1.36 ± 0.11 nA, n = 11]. Rates of activation and inactivation also were unaffected by the absence of the β1 subunit. The time course of inward currents at -30 mV or outward currents at +20 mV were similar in the two mouse strains. Finally, the voltage dependence of channel activation and steady-state inactivation were not significantly different in dissociated neurons from β1(+/+) and β1 (-/-) mice. These results indicate either that β1 subunits have little effect on the functional properties of sodium channels in hippocampal pyramidal neurons or that the other β subunits can substitute for them in this brain region.
Figure 9.
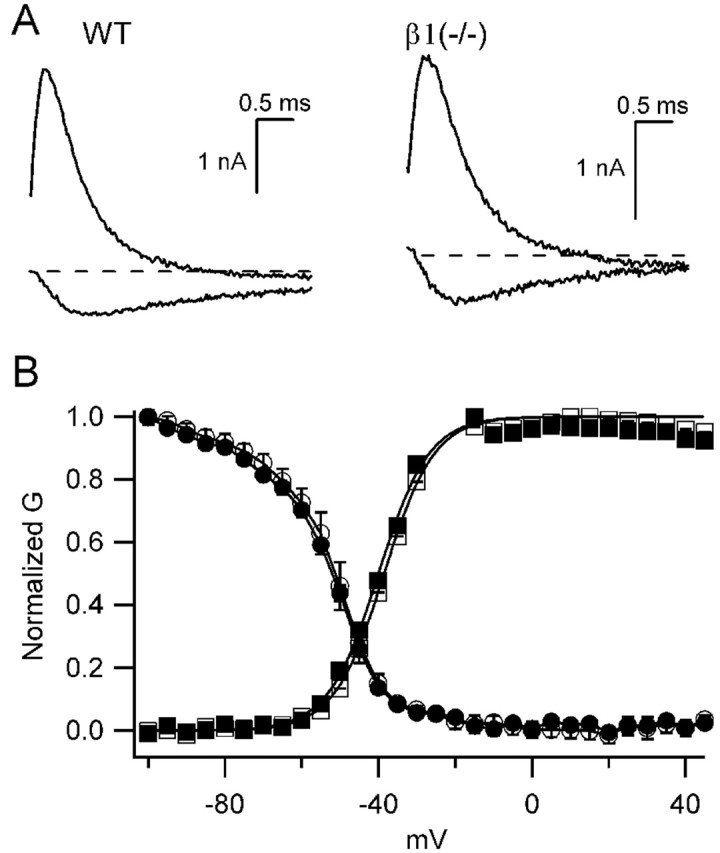
Voltage-dependent properties of β1 (+/+) and β1 (-/-) mice. A, Sodium current traces in response to depolarizations to -30 mV (inward current) or +20 mV (outward current) from a holding potential of -80 mV from β1 (+/+) (left) or β1 (-/-) (right) mice. B, Mean normalized conductance-voltage curves (squares) and inactivation curves (circles) for wild-type (WT; open symbols) and β1 (-/-) (filled symbols) mice. To generate conductance-voltage relationships, 20 msec depolarizations from a holding potential of -80 mV to the indicated potentials were applied. Peak current versus voltage curves were measured and fit with[(V - Vrev) × Gmax]/{1 + exp[(V - V.5)/k]}, where V was the test pulse voltage, Vrev, the reversal potential, V.5 the half activation voltage, and k a slope factor. Normalized conductance-voltage curves for each experiment were determined as 1/{1 + exp[(V - V.5)/k]}, and the means ± SEM of these curves are plotted. Inactivation curves were measured using a 20 msec long prepulse to the indicated potentials, followed by a test depolarization to +10 mV. Mean normalized peak test pulse current is plotted versus prepulse potential.
Sodium channel expression patterns are changed in β1 (-/-) mice
Despite our electrophysiological results showing no changes in the properties of sodium currents in dissociated pyramidal neurons from the whole hippocampus, the possibility remained that subsets of neurons in the hippocampus or elsewhere might have altered sodium channel expression or localization. To test this possibility, we investigated the expression patterns of sodium channels in the CNSs of β1 (-/-) and β1 (+/+) mice using anti-peptide antibodies raised against Nav1.1, Nav1.2, Nav1.3, or Nav1.6 α subunits (Fig. 10). As expected, staining for the β1 subunit of sodium channels was absent throughout the brains of β1 (-/-) mice compared with β1 (+/+) (Fig. 10A,B). Immunolocalization of β1 subunits in the CA1, CA3, and dentate gyrus regions of the hippocampus in wild-type mice was in agreement with previous studies in the rat (Gorter et al., 2002). We found no detectable changes in the expression patterns of Nav1.2 or Nav1.6 in the β1 (-/-) mice compared with β1 (+/+) animals (data not shown). In contrast, there were detectable changes in the expression patterns of Nav1.1 and Nav1.3 in β1(-/-) mice compared with β1(+/+) in a subset of neurons in the hippocampus. Nav1.1 and Nav1.3 expression in other brain areas was not detectably different from wild type. Nav1.1 expression in β1 (+/+) animals was present uniformly in the cell bodies of pyramidal neurons of CA1-CA3, in interneurons of the hippocampus, and in the granule cells of the dentate gyrus (Fig. 10C). In contrast, the expression of Nav1.1 was absent in a subset of CA3/CA2 pyramidal neurons and in granule cells located in the outer leaflet of the dentate gyrus of β1 (-/-) mice (Fig. 10D). In addition, fewer interneurons were immunostained for Nav1.1 in β1(-/-) mice. Expression of Nav1.3 is normally absent from neurons located in the hippocampus of adult wild-type mice (Fig. 10E). In contrast, in β1(-/-) mice there was significantly increased expression of Nav1.3 in scattered interneurons located in the CA1-CA3 regions (Fig. 10F) and in the dentate gyrus (Fig. 10G). Sections incubated in the absence of primary antibody were devoid of signal (Fig. 10H), further confirming the specificity of the immunostaining. These results show striking and unexpected changes of sodium channel expression in the hippocampus and dentate gyrus, two brain regions that are critically involved in generation of epileptic seizures.
Figure 10.
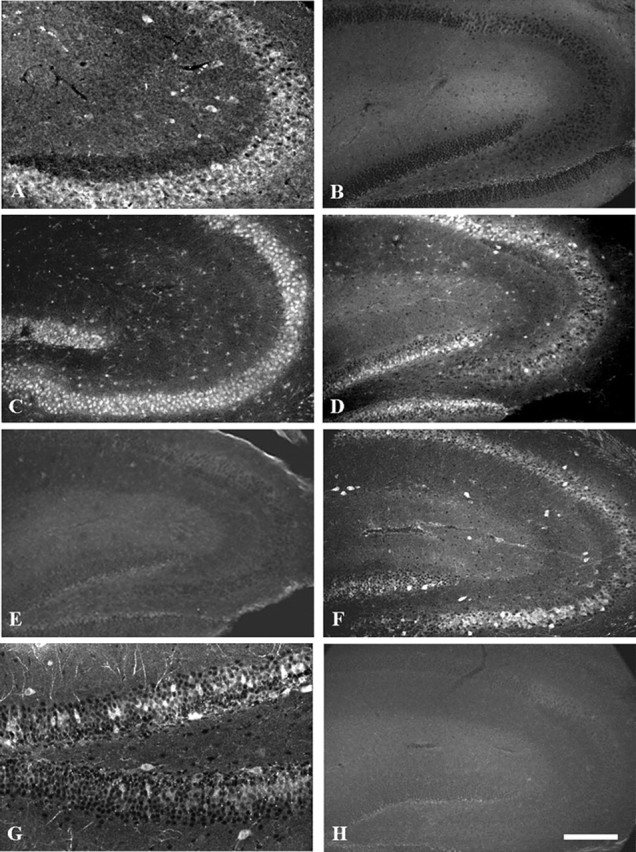
β1 (-/-) mice show abnormal expression of Nav 1.1 and Nav 1.3 in the hippocampus. A, β1 (+/+) mice stained with anti-β1ex. B, β1 (-/-) mice illustrating the lack of anti-β1ex staining and the specificity of the null mutation. C, β1 (+/+) mice stained with anti-Nav1.1. D,β1(-/-) mice stained with anti-Nav1.1 illustrating lack of staining of many neurons in the CA2/CA3 region and in the outer leaflet of the dentate gyrus compared with controls (C). E, β1 (+/+) mice stained with anti-Nav1.3. F, β1 (-/-) mice stained with anti-Nav1.3 illustrating increased staining in interneurons of the hippocampus. G, Higher magnification of the dentate gyrus labeled with anti-Nav 1.3 antibodies illustrating increased staining in neurons located in this region of β1 (-/-) mice. H, Tissue section incubated with no primary antibodies to illustrate the specificity of antibody staining. Scale bars, 200 μm.
Discussion
Sodium channel β1 subunits are multifunctional molecules that participate in ion channel modulation and cell adhesion in vitro (Isom, 2001). In the present study, we showed that the absence of β1 subunits in vivo resulted in a dramatic phenotype that includes an ataxic gait and spontaneous seizures beginning at approximately P10-P12. β1 (-/-) mice are born in approximate Mendelian ratios and develop normally until P10-P12, suggesting that this CNS phenotype is likely attributable to the absence of β1 itself and not the embryonic splice variant β1A in the brain (Kazen-Gillespie et al., 2000). Our results provide insights into the cellular and molecular changes caused by deletion of the β1 subunits, which may contribute to the ataxia and seizures observed in these animals.
Alterations in the structure and function of myelinated nerves
Sodium channels and contactin colocalize at CNS nodes of Ranvier in vivo, and contactin enhances sodium channel cell surface expression through its association with β1 subunits in vitro (Kazarinova-Noyes et al., 2001). Formation of the sodium channel-contactin complex is dependent on the presence of β1 when measured in transfected cells (Kazarinova-Noyes et al., 2001; McEwen et al., 2004). Contactin and sodium channels colocalize but do not associate in the CNS of β1(-/-) mice, demonstrating that β1 is critical for formation of this complex in vivo. We propose that the observed reduction in mature nodes of Ranvier and action potential conduction velocity in β1 (-/-) mice is attributable to the loss of interaction of contactin with the sodium channel complex resulting in reductions in sodium channel cell surface expression in the axonal membrane. Comparison of the β1(-/-) CNS phenotype with that observed in contactin (-/-) mice (Kazarinova-Noyes and Shrager, 2002) revealed some interesting similarities that suggest a functional interaction between these two molecules in vivo. Similar to β1(-/-) mice, contactin (-/-) mice are ataxic and die by P18 (Berglund et al., 1999). Contactin (-/-) mice exhibited a reduction in the number of nodes of Ranvier in the optic nerve, slowed action potential conduction because of loss of the fast component of the CAP, distorted morphology of nodal sodium channel clusters, and the absence of caspr 1 clusters or transverse bands. It was proposed that loss of β1-contactin interactions in contactin (-/-) mice could result in decreased levels of nodal sodium channels, fewer mature nodes, and slowed action potential conduction (Kazarinova-Noyes and Shrager, 2002). Disruption of ion channel clustering in contactin (-/-) mice is likely attributable to the disruption of paranodal myelin, particularly the transverse bands, which are essentially normal in β1(-/-) mice.
Tenascin-R and sodium channel β1 subunits are present at CNS nodes of Ranvier, and both molecules interact with contactin (Pesheva et al., 1993; Xiao et al., 1996). Transfected fibroblasts expressing β1 subunits are repelled from a tenascin-R substrate, and a recombinant amino terminal epidermal growth factor-like domain of tenascin-R increases peak sodium current expressed in Xenopus oocytes with no change in gating kinetics (Xiao et al., 1999). Like β1 (-/-) and contactin (-/-) animals, tenascin-R (-/-) mice exhibit slowed optic nerve conduction velocities (Weber et al., 1999). Thus, all three molecules may form a functional complex that modulates sodium channel function in the axonal membrane at CNS nodes of Ranvier.
Saltatory conduction in myelinated axons is dependent on the precise localization of voltage-gated sodium and potassium channels in the nodes of Ranvier and juxtaparanodal regions, respectively (Vabnick and Shrager, 1998; Girault and Peles, 2002). Paranodal septate-like junctions between the axon and the myelin sheath (visible as transverse bands by electron microscopy) composed of the CAMs contactin, caspr 1, and possibly neurofascin 155 act as diffusion barriers to ion channel movement. Although these septate-like junctions are not obligatory for sodium channel clustering, their disruption using gene-targeting experiments in mice results in the displacement of voltage-gated potassium channels from the juxtaparanode into the paranodal region, a slight broadening of the nodal gap containing voltage-gated sodium channels, and action potential conduction abnormalities (Dupree et al., 1998, 1999; Bhat et al., 2001; Boyle et al., 2001; Poliak et al., 2001; Rosenbluth et al., 2003). The molecular composition and arrangement of the axo-glial apparatus appears to be normal in the absence of β1 subunits at the level of confocal microscopy. Sodium channel targeting to nodes of Ranvier also appears to be unaffected by the loss of β1 subunits, because Nav1.6 is the predominant sodium channel isoform detected in CNS and PNS nodes at P17-P19 in both wild-type and mutant animals. Ultrastructural analysis of central and peripheral nerve nodes of Ranvier in β1 (-/-) mice revealed impairments in the axo-glial junctions adjacent to the nodal gap in a subset of axons. Transverse bands were present between the axon and the myelin loops throughout the paranode, except for the region immediately adjacent to the node of Ranvier where the myelin loops were everted or pulled away from the axolemma in a subset of affected axons. We propose that normally in the CNS, β1-contactin complexes located on the border of the nodal gap may interact in trans with CAMs (e.g., neurofascin 155) present on the myelin membrane to secure the perimeter of the node, ensuring the formation of a high-resistance barrier to the flow of ionic current. In the PNS, β1 subunits may interact with axonal CAMs other than contactin, such as neurofascin-186 or sodium channel β2 subunits, to participate in septate-like junction formation with the myelin membrane. Alternatively, β1 subunits may function as CAMs on oligodendrocyte or Schwann cell membranes and thus contribute to the septate-like junctions at the nodal border via homophilic interactions with axonal β1 subunits. In support of this hypothesis, we identified β1 subunit protein expression in oligodendrocytes by both immunocytochemical and Western blot analyses (M. J. Detrow and L. L. Isom, unpublished observations) (Kaplan et al., 2001). Overall, these alterations in structure and function of the myelinated nerves are sufficient to contribute to the ataxia observed in the β1(-/-) mice.
Alterations in sodium channel expression
Sodium channel electrophysiological properties appear to be normal in dissociated hippocampal pyramidal neurons isolated from β1 (-/-) mice, and the majority of β1 (-/-) hippocampal neurons have a normal sodium channel distribution as assessed by immunocytochemistry. Although we observed no compensatory change in the level of β2, β3, or β4 subunits in the brain, it is possible that these subunits were able to substitute for β1 subunits in modulating sodium channel function in most hippocampal neurons, resulting in no change in the average properties of dissociated neurons from this brain region. In contrast to these electrophysiological results with dissociated hippocampal neurons, we found that sodium channel expression is significantly altered in a subset of cell bodies of CA2/CA3 pyramidal neurons and interneurons in the hippocampus of β1 (-/-) mice as assessed by immunocytochemistry. Nav1.1 expression is primarily downregulated and replaced by expression of Nav1.3, a channel that is not normally present in this region in the adult brain. Pyramidal neurons in the CA3 region of the hippocampus have a propensity for generation of abnormal electrical activity and bursts of action potentials leading to epileptiform activity (McCormick and Contreras, 2001), and neurons in the CA2-CA3 boundary are most vulnerable to neurodegeneration in experimental models of epilepsy (Friedman et al., 1994). Thus, alterations in sodium channel expression or localization in this area may be amplified into large changes in overall excitability. For example, transgenic expression of a gain-of-function Nav1.2 channel mutant throughout the brain resulted in focal seizure activity in the hippocampus and extensive cell loss within the hippocampal areas CA1, CA2, and CA3 (Kearney et al., 2001). Ectopic expression of Nav1.3 in this region would be predicted to result in hyperexcitability as well. It has been suggested, based on heterologous expression systems, that neurons expressing Nav1.3 may have reduced thresholds and/or relatively high frequencies of firing attributable to the rapid recovery from inactivation and the slow closed-state inactivation kinetics observed for this channel (Cummins et al., 2001). A significant upregulation of Nav1.3 mRNA has been observed in the human epileptic hippocampus (Whitaker et al., 2001), although it is unclear whether this change plays a causal role or is secondary to the seizures.
It is clear that the level of Nav1.1 expression in the brain is critical and even partial reductions in expression levels result in dramatic changes in excitability. De novo truncation mutations resulting in haplo-insufficiency of Nav1.1 channels have been shown to cause familial severe myoclonic epilepsy of infancy (Ohmori et al., 2002; Sugawara et al., 2002; Claes et al., 2003; Gennaro et al., 2003). Nav1.1 (-/-) mice exhibit a severely epileptic phenotype and death by P16, and Nav1.1 (+/-) littermates also have seizures and a shortened life span (F. Yu, M. Mantegazza, R. E. Westenbroek, T. Scheuer, and W. A. Catterall, unpublished observations). In contrast, Nav1.2a (+/-) mice (Planells-Cases et al., 2000) and Nav1.6 (+/-) mice (Burgess et al., 1995) display a normal behavioral phenotype. Thus, neuronal excitability appears to be exquisitely sensitive to Nav1.1 expression. We propose that selective decreases in Nav1.1 expression and increases in Nav1.3 expression in a subset of hippocampal neurons in response to the loss of β1 subunits may contribute to the generation of epileptiform activity and underlie, at least in part, the observed seizure phenotype in β1 (-/-) mice.
Footnotes
This work was supported by Research Grant RG-2882 from the National Multiple Sclerosis Society (L.L.I.); National Science Foundation Research Grant IBN-9734462 (L.L.I.); Grant 001654 from the Michigan Life Sciences Corridor (L.L.I.); National Institutes of Health Research Grants NS25704 (W.A.C.), NS17965 (P. S.), NS043067 (D.P.M.), and NS29709 (J.L.N.); and University of Michigan Grants 5P30CA46592, 5P60DK20572-22S2, 5P30DK34933, 5T32HD07505-03, GM07767-26, and 5T32HD07505-03. We thank Richard Mulligan for the pPNT plasmid. We thank Matthew Koopmann, Christy Avery, and Audrey Speelman for expert technical assistance and Dr. Miriam Meisler for critical reading of this manuscript.
Correspondence should be addressed to Dr. Lori L. Isom, Department of Pharmacology, The University of Michigan, 1301 Medical Science Research Building III, 1150 West Medical Center Drive, Ann Arbor, MI48109-0632. E-mail: lisom@umich.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/244030-13$15.00/0
References
- Berglund EO, Murai KK, Fredette B, Sekerkova G, Marturano B, Weber L, Mugnaini E, Ranscht B (1999) Ataxia and abnormal cerebellar microorganization in mice with ablated contactin gene expression. Neuron 24: 739-750. [DOI] [PubMed] [Google Scholar]
- Bhat MA, Rios JC, Lu Y, Garcia-Fresco GP, Ching W, St Martin M, Li J, Einheber S, Chesler M, Rosenbluth J, Salzer JL, Bellen HJ (2001) Axonglia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron 30: 369-383. [DOI] [PubMed] [Google Scholar]
- Boiko T, Rasband M, Levinson S, Caldwell J, Mandel G, Trimmer J, Matthews G (2001) Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron 30: 91-104. [DOI] [PubMed] [Google Scholar]
- Boyle MET, Berglund EO, Murai KK, Weber L, Peles E, Ranscht B (2001) Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron 30: 385-397. [DOI] [PubMed] [Google Scholar]
- Burgess DL, Kohrman DC, Galt J, Plummer NW, Jones JM, Spear B, Meisler MH (1995) Mutation of a new sodium channel gene, Scn8a, in the mouse mutant “motor endplate disease.” Nat Genet 10: 461-465. [DOI] [PubMed] [Google Scholar]
- Cantrell AR, Ma JY, Scheuer T, Catterall WA (1996) Muscarinic modulation of sodium current by activation of protein kinase C in rat hippocampal neurons. Neuron 16: 1019-1026. [DOI] [PubMed] [Google Scholar]
- Catterall WA (2000) From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26: 13-25. [DOI] [PubMed] [Google Scholar]
- Chen C, Bharucha B, Chen Y, Westenbroek RE, Brown A, Malhotra JD, Jones D, Avery C, Gillespie III PJ, Kazen-Gillespie KA, Saunders TL, Macdonald RL, Ransom B, Scheuer T, Catterall WA, Isom LL (2002) Reduced sodium channel density, altered voltage dependence of inactivation, and increased susceptibility to seizures in mice lacking sodium channel β2 subunits. Proc Natl Acad Sci USA 99: 17072-17077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes L, Ceulemans B, Audenaert D, Smets K, Lofgren A, Del-Favero J, Ala-Mello S, Basel-Vanagaite L, Plecko B, Raskin S, Thiry P, Wolf NI, Van Broeckhoven C, De Jonghe P (2003) De novo SCN1A mutations are a major cause of severe myoclonic epilepsy of infancy. Hum Mutat 21: 615-621. [DOI] [PubMed] [Google Scholar]
- Cummins TR, Aglieco F, Renganathan M, Herzog RI, Dib-Hajj SD, Waxman SG (2001) Nav1.3 sodium channels: rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J Neurosci 21: 5952-5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JQ, McLaughlin T, Bennett V (1993) Ankyrin-binding proteins related to nervous system cell adhesion molecules: candidates to provide transmembrane and intercellular connections in adult brain. J Cell Biol 121: 121-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi M, Uji Y, Yamamura H (1995) Morphological classification of retinal ganglion cells in mice. J Comp Neurol 356: 368-386. [DOI] [PubMed] [Google Scholar]
- Domino SE, Zhang L, Gillespie PJ, Saunders TL, Lowe JB (2001) Deficiency of reproductive tract alpha(1,2)fucosylated glycans and normal fertility in mice with targeted deletions of the FUT1 or FUT2 alpha(1,2)fucosyltransferase locus. Mol Cell Biol 21: 8336-8345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupree JL, Coeztee T, Blight A, Suzuki K, Popko B (1998) Myelin galactolipids are essential for proper node of Ranvier formation in the CNS. J Neurosci 18: 1642-1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupree JL, Girault JA, Popko B (1999) Axo-glial interactions regulate the localization of axonal paranodal proteins. J Cell Biol 147: 1145-1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman LK, Pellegrini-Giampietro DE, Sperber EF, Bennett MV, Moshe SL, Zukin RS (1994) Kainate-induced status epilepticus alters glutamate and GABAA receptor gene expression in adult rat hippocampus: an in situ hybridization study. J Neurosci 14: 2697-2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda Y (1977) A three-group classification of rat retinal ganglion cells: histological and physiological studies. Brain Res 119: 327-334. [DOI] [PubMed] [Google Scholar]
- Gennaro E, Veggiotti P, Malacarne M, Madia F, Cecconi M, Cardinali S, Cassetti A, Cecconi I, Bertini E, Bianchi A, Gobbi G, Zara F (2003) Familial severe myoclonic epilepsy of infancy: truncation of Nav1.1 and genetic heterogeneity. Epileptic Disord 5: 21-25. [PubMed] [Google Scholar]
- Girault JA, Peles E (2002) Development of nodes of Ranvier. Curr Opin Neurobiol 12: 476-485. [DOI] [PubMed] [Google Scholar]
- Gorter JA, van Vliet EA, Lopes da Silva FH, Isom LL, Aronica E (2002) Sodium channel beta1-subunit expression is increased in reactive astrocytes in a rat model for mesial temporal lobe epilepsy. Eur J Neurosci 16: 360-364. [DOI] [PubMed] [Google Scholar]
- Isom LL (2001) Sodium channel β subunits: anything but auxiliary. The Neuroscientist 7: 42-54. [DOI] [PubMed] [Google Scholar]
- Isom LL (2002) The role of sodium channels in cell adhesion. Front Biosci 7: 12-23. [DOI] [PubMed] [Google Scholar]
- Isom LL, Catterall WA (1996) Na+ channel subunits and Ig domains. Nature 383: 307-308. [DOI] [PubMed] [Google Scholar]
- Isom LL, De Jongh KS, Patton DE, Reber BFX, Offord J, Charbonneau H, Walsh K, Goldin AL, Catterall WA (1992) Primary structure and functional expression of the β1 subunit of the rat brain sodium channel. Science 256: 839-842. [DOI] [PubMed] [Google Scholar]
- Isom LL, Scheuer T, Brownstein AB, Ragsdale DS, Murphy BJ, Catterall WA (1995) Functional co-expression of the beta 1 and type IIA alpha subunits of sodium channels in a mammalian cell line. J Biol Chem 270: 3306-3312. [DOI] [PubMed] [Google Scholar]
- Kaplan MR, Cho M-H, Ullian EM, Isom LL, Levinson SR, Barres BA (2001) Differential control of clustering of the sodium channels Nav1.2 and Nav1.6 at developing CNS nodes of Ranvier. Neuron 30: 105-119. [DOI] [PubMed] [Google Scholar]
- Kay AR, Wong RKS (1987) Calcium current activation kinetics in isolated pyramidal neurones of the CA1 region of the mature guinea-pig hippocampus. J Physiol (Lond) 392: 603-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazarinova-Noyes K, Shrager P (2002) Molecular constituents of the node of Ranvier. Mol Neurobiol 26: 167-182. [DOI] [PubMed] [Google Scholar]
- Kazarinova-Noyes K, Malhotra JD, McEwen DP, Mattei LN, Berglund EO, Ranscht B, Levinson SR, Schachner M, Shrager P, Isom LL, Xiao Z-C (2001) Contactin associates with Na+ channels and increases their functional expression. J Neurosci 21: 7517-7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazen-Gillespie KA, Ragsdale DS, D'Andrea MR, Mattei LN, Rogers KE, Isom LL (2000) Cloning, localization, and functional expression of sodium channel β1A subunits. J Biol Chem 275: 1079-1088. [DOI] [PubMed] [Google Scholar]
- Kearney JA, Plummer NW, Smith MR, Kapur J, Cummins TR, Waxman SG, Goldin AL, Meisler MH (2001) A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Neuroscience 102: 307-317. [DOI] [PubMed] [Google Scholar]
- Little GJ, Heath JW (1994) Morphometric analysis of axons myelinated during adult life in the mouse superior cervical ganglion. J Anat 184: 387-398. [PMC free article] [PubMed] [Google Scholar]
- Maier SK, Westenbroek RE, Yamanushi TT, Dobrzynski H, Boyett MR, Catterall WA, Scheuer T (2003) An unexpected requirement for brain-type sodium channels for control of heart rate in the mouse sinoatrial node. Proc Natl Acad Sci USA 100: 3507-3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra JD, Kazen-Gillespie K, Hortsch M, Isom LL (2000) Sodium channel β subunits mediate homophilic cell adhesion and recruit ankyrin to points of cell-cell contact. J Biol Chem 275: 11383-11388. [DOI] [PubMed] [Google Scholar]
- Malhotra JD, Koopmann MC, Kazen-Gillespie KA, Fettman N, Hortsch M, Isom LL (2002) Structural requirements for interaction of sodium channel β1 subunits with ankyrin. J Biol Chem 277: 26681-26688. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Contreras D (2001) On the cellular and network bases of epileptic seizures. Annu Rev Physiol 63: 815-846. [DOI] [PubMed] [Google Scholar]
- McEwen DP, Meadows LS, Chen C, Thyagarajan V, Isom LL (2004) Sodium channel β1 subunit-mediated modulation of Nav1.2 currents and cell surface density is dependent on interactions with contactin and ankyrin. J Biol Chem, in press. [DOI] [PubMed]
- Meadows LS, Chen YH, Powell AJ, Clare JJ, Ragsdale DS (2002a) Functional modulation of human brain Nav1.3 sodium channels, expressed in mammalian cells, by auxiliary beta 1, beta 2 and beta 3 subunits. Neuroscience 114: 745-753. [DOI] [PubMed] [Google Scholar]
- Meadows LS, Malhotra J, Loukas A, Thyagarajan V, Kazen-Gillespie KA, Koopman MC, S Kriegler, Isom LL, Ragsdale DS (2002b) Functional and biochemical analysis of a sodium channel β1 subunit mutation responsible for generalized epilepsy with febrile seizures plus type 1. J Neurosci 22: 10699-10709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16: 1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noebels JL (1984) A single gene error of noradrenergic axon growth synchronizes central neurones. Nature 310: 409-411. [DOI] [PubMed] [Google Scholar]
- Ohmori I, Ouchida M, Ohtsuka Y, Oka E, Shimizu K (2002) Significant correlation of the SCN1A mutations and severe myoclonic epilepsy in infancy. Biochem Biophys Res Commun 295: 17-23. [DOI] [PubMed] [Google Scholar]
- Pesheva P, Gennarini G, Gordis C, Schachner M (1993) The F3/F11 cell adhesion molecule mediates the repulsion of neurons by the extracellular matrix glycoprotein J1-160/180. Neuron 10: 69-72. [DOI] [PubMed] [Google Scholar]
- Planells-Cases R, Caprini M, Zhang J, Rockenstein EM, Rivera RR, Murre C, Masliah E, Montal M (2000) Neuronal death and perinatal lethality in voltage-gated sodium channel αII-deficient mice. Biophys J 78: 2878-2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak S, Gollan L, Salomon D, Berglund EO, Ohara R, Ranscht B, Peles E (2001) Localization of Caspr2 in myelinated nerves depends on axon-glia interactions and the generation of barriers along the axon. J Neurosci 21: 7568-7575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Curtis R, Lawson D, Gilbride K, Ge P, DiStefano PS, Silos-Santiago I, Catterall WA, Scheuer T (2001) Differential modulation of sodium channel gating and persistent sodium currents by the beta1, beta2, and beta3 subunits. Mol Cell Neurosci 18: 570-580. [DOI] [PubMed] [Google Scholar]
- Ratcliffe CF, Qu Y, McCormick KA, Tibbs VC, Dixon JE, Scheuer T, Catterall WA (2000) A sodium channel signaling complex: modulation by associated receptor protein tyrosine phosphatase β. Nat Neurosci 3: 437-444. [DOI] [PubMed] [Google Scholar]
- Ratcliffe CF, Westenbroek RE, Curtis R, Catterall WA (2001) Sodium channel beta1 and beta3 subunits associate with neurofascin through their extracellular immunoglobulin-like domain. J Cell Biol 154: 427-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluth J, Dupree JL, Popko B (2003) Nodal sodium channel domain integrity depends on the conformation of the paranodal junction, not on the presence of transverse bands. Glia 41: 318-325. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Simpson EM, Takahashi JS, Lipp H-P, Nakanishi S, Wehner JM, Giese KP, Tully T, Abel T, Chapman PF, Fox K, Grant S, Itohara S, Lathe R, Mayford M, McNamara JO, Morris RJ, Picciotto M, Roder J, Shin H-S, Slesinger PA, Storm DR, Stryker MP, Tonegawa S, Wang Y, Wolfer DP (1997) Mutant mice and neuroscience: recommendations concerning genetic background. Banbury Conference on genetic background in mice. Neuron 19: 755-759. [DOI] [PubMed] [Google Scholar]
- Spurr AR (1969) A low-viscosity epoxy resin embedding medium for electron microscopy. J Ultrastruct Res 26: 31-43. [DOI] [PubMed] [Google Scholar]
- Srinivasan J, Schachner M, Catterall WA (1998) Interaction of voltage-gated sodium channels with the extracellular matrix molecules tenascin-C and tenascin-R. Proc Natl Acad Sci USA 95: 15753-15757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys P, Ransom BR, Waxman SG (1991) Compound action potential of nerve recorded by suction electrode: theoretical and experimental analysis. Brain Res 546: 18-32. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Mazaki-Miyazaki E, Fukushima K, Shimomura J, Fujiwara T, Hamano S, Inoue Y, Yamakawa K (2002) Frequent mutations of SCN1A in severe myoclonic epilepsy in infancy. Neurology 58: 1122-1124. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Stefani A, Foehring RC, Kitai ST (1991) Developmental regulation of a slowly-inactivating potassium conductance in rat neostriatal neurons. Neurosci Lett 122: 41-46. [DOI] [PubMed] [Google Scholar]
- Tuvia S, Garver TD, Bennett V (1997) The phosphorylation state of the FIGQY tyrosine of neurofascin determines ankyrin-binding activity and patterns of cell segregation. Proc Natl Acad Sci USA 94: 12957-12962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabnick I, Shrager P (1998) Ion channel redistribution and function during development of the myelinated axon. J Neurobiol 37: 80-96. [PubMed] [Google Scholar]
- Wallace RH, Wang DW, Singh R, Scheffer IE, George Jr AL, Phillips HA, Saar K, Reis A, Johnson EW, Sutherland GR, Berkovic SF, Mulley JC (1998) Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat Genet 19: 366-370. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Scheffer IE, Parasivam G, Barnett S, Wallace GB, Sutherland GR, Berkovic SF, Mulley JC (2002) Generalized epilepsy with febrile seizures plus: mutation of the sodium channel subunit SCN1B. Neurology 58: 1426-1429. [DOI] [PubMed] [Google Scholar]
- Weber P, Bartsch U, Rasband MN, Czaniera R, Lang Y, Bluethmann H, Margolis RU, Levinson SR, Shrager P, Montag D, Schachner M (1999) Mice deficient for tenascin-R display alterations of the extracellular matrix and decreased axonal conduction velocities in the CNS. J Neurosci 19: 4245-4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker WR, Faull RL, Dragunow M, Mee EW, Emson PC, Clare JJ (2001) Changes in the mRNAs encoding voltage-gated sodium channel types II and III in human epileptic hippocampus. Neuroscience 106: 275-285. [DOI] [PubMed] [Google Scholar]
- Xiao ZC, Taylor J, Montag D, Rougon G, Schachner M (1996) Distinct effects of recombinant tenascin-R domains in neuronal cell functions and identification of the domain interacting with the neuronal recognition molecule F3/11. Eur J Neurosci 8: 766-782. [DOI] [PubMed] [Google Scholar]
- Xiao Z-C, Ragsdale DS, Malhorta JD, Mattei LN, Braun PE, Schachner M, Isom LL (1999) Tenascin-R is a functional modulator of sodium channel β subunits. J Biol Chem 274: 26511-26517. [DOI] [PubMed] [Google Scholar]
- Yu FH, Westenbroek RE, Silos-Santiago I, McCormick KA, Lawson D, Ge P, Ferriera H, Lilly J, DiStefano PS, Catterall WA, Scheuer T, Curtis R (2003) Sodium channel beta4, a new disulfide-linked auxiliary subunit with similarity to beta2. J Neurosci 23: 7577-7585. [DOI] [PMC free article] [PubMed] [Google Scholar]