

Abstract
Microtubules originate at the centrosome of the neuron and are then released for transport down the axon, in which they can move both anterogradely and retrogradely during axonal growth. It has been hypothesized that these movements occur by force generation against the actin cytoskeleton. To test this, we analyzed the movement, distribution, and orientation of microtubules in neurons pharmacologically depleted of actin filaments. Actin depletion reduced but did not eliminate the anterograde movements and had no effect on the frequency of retrograde movements. Consistent with the idea that microtubules might also move against neighboring microtubules, actin depletion completely inhibited the outward transport of microtubules under experimental conditions of low microtubule density. Interestingly, visualization of microtubule assembly shows that actin depletion actually enhances the tendency of microtubules to align with one another. Such microtubule-microtubule interactions are sufficient to orient microtubules in their characteristic polarity pattern in axons grown overnight in the absence of actin filaments. In fact, microtubule behaviors were only chaotic after actin depletion in peripheral regions of the neuron in which microtubules are normally sparse and hence lack neighboring microtubules with which they could interact. On the basis of these results, we conclude that microtubules are transported against either actin filaments or neighboring microtubules in the anterograde direction but only against other microtubules in the retrograde direction. Moreover, the transport of microtubules against one another provides a surprisingly effective option for the deployment and orientation of microtubules in the absence of actin filaments.
Keywords: microtubule, actin, neuron, axon, microtubule transport, microtubule assembly
Introduction
Neuronal development is dependent on the transport of microtubules from the cell body into and down the length of the axon during its growth (Baas and Buster, 2004). Microtubules are conveyed exclusively into the axon with their plus ends leading, thus establishing their characteristic pattern of polarity orientation (Baas and Ahmad, 1993). Early models posited that the entire microtubule array marches continuously down the axon at slow rates, but more recent studies using live-cell imaging have shown that microtubule transport is actually intermittent, asynchronous, and infrequent (Wang and Brown, 2002). These studies have also established that microtubules are transported individually as rather short polymers, that their transport occurs at rapid rates consistent with known molecular motor proteins, and that their transport occurs in both directions within growing axons. These and related observations have led to the view that microtubule transport is extremely vibrant and plays major roles in the deployment of microtubules into axonal branches and growth cones (Dent et al., 1999; Gallo and Letourneau, 1999), as well as along the length of the axonal shaft (Baas and Buster, 2004). In addition, notable changes in microtubule movements have been shown to occur when the neuron undergoes alterations in morphology, such as during the formation of new axons (Yu et al., 2001) and the retraction of unneeded axons (He et al., 2002), suggesting that microtubule movements are key to orchestrating many aspects of neuronal form.
For a microtubule to move, there must be a molecular motor protein that generates the necessary force, as well as a substrate against which the force is generated. Cytoplasmic dynein is an attractive motor for transporting microtubules into and anterogradely down the axon because it moves toward the minus end of the microtubule (Vallee et al., 2004). Thus, if the cargo-binding domain interacts with a structure of greater resistance to movement than the microtubule, the microtubule will move with its plus end leading. It has been hypothesized that this substrate is the actin cytomatrix, because cytoplasmic dynein is anterogradely transported at the same rate as actin, suggesting a potential association between actin and the cargo-binding domain of the motor (Dillman et al., 1996). In support of this model, suppression of dynein function in cultured neurons curtails the outward transport of microtubules from the centrosome into the axon (Ahmad et al., 1998). The retrograde movement of microtubules is not readily explained by this model, however, and would presumably require a different motor, a different substrate, or both. Retrograde flow of actin filaments fueled by myosin has been shown to corral and sweep microtubules backward within growth cones (Schaefer et al., 2002), and actomyosin contractility is responsible for the massive retrograde movement of microtubules during axonal retraction (Ahmad et al., 2000). However, there are no studies to date on the potential role of actin filaments as substrates for the ongoing anterograde and retrograde movement of individual microtubules within growing axons. In the present study, we sought to investigate this matter.
Materials and Methods
Cell culture. Rat superior cervical ganglia were dissected from postnatal days 0-2 pups with the use of #5 watchmakers forceps and a Zeiss (Oberkochen, Germany) SV6 stereomicroscope, collected in chilled L-15 medium (Invitrogen, Carlsbad, CA), and then treated for 15 min with 2.5 mg/ml collagenase and 45 min with 2.5 mg/ml trypsin (both in 1× PBS and sterilized using a 0.2 micrometer syringe filter). Collagenase and trypsin enzymatic activity was then blocked using a solution of 10% fetal bovine serum in L-15 medium (Invitrogen) warmed to 37°C. Ganglia were triturated in N2 medium three to five times using a sterile, fire-polished Pasteur pipette to produce a single-cell suspension, and cell density was determined using a hemacytometer. In some cases, cells were then transfected with plasmids for the expression of green fluorescent protein (GFP) constructs (for details, see below). Neurons were plated in N2 medium on glass coverslips mounted in the bottom of drilled 35-mm-diameter plastic Petri dishes (commercially available through Mat-Tek, Ashland, MA). Before plating the cells, coverslips were treated for 3 hr with 0.1 mg/ml poly-d-lysine, rinsed extensively with sterile water, incubated overnight at 37°C in water, and air dried for 30 min. Laminin was typically added at some point thereafter to stimulate axonal outgrowth, depending on the particular experiment, as outlined below. Just before imaging, the medium was replaced with prewarmed L-15 plating medium [which has that advantage of maintaining pH in air (He and Baas, 2003)] containing appropriate concentrations of either latrunculin or vehicle.
Fluorescence cytochemistry. For fluorescence microscopy of fixed cells, cultures were simultaneously fixed and extracted (to remove free tubulin) at room temperature for 15 min in a fixation-microtubule stabilizing solution as described previously (He and Baas, 2003). The cells were rinsed three times for 5 min each with PBS, treated twice for 15 min with 10 mg/ml sodium borohydride in PBS to quench aldehyde autofluorescence, and rinsed again for 5 min three times with PBS. The cultures were then blocked with a solution of 10% normal goat serum and 10 mg/ml bovine serum albumin in PBS for 1 hr at room temperature. The fixed neurons were then incubated for 1 hr at room temperature in “staining solution” for microtubules and actin consisting of 1:100 dilution of Cy3-conjugated mouse monoclonal β-tubulin antibody (Sigma-Aldrich, St. Louis, MO) and 1:40 dilution of Alexa Fluor 488-conjugated phalloidin (Molecular Probes, Eugene, OR) in PBS. Cells were rinsed extensively with PBS and mounted in a glycerol-based anti-fade medium for fluorescence-based imaging.
Expression of GFP constructs. Expression in primary neurons of GFP constructs for α tubulin or end-binding protein 3 (EB3) was accomplished using a modified electroporation device called a Nucleofector (Amaxa Biosystems, Cologne, Germany) that delivers DNA directly into the nucleus. We found this approach effective in transfecting 10-20% of the neurons, with expressed proteins detectable within the first several hours after transfection (Karabay et al., 2004). A single-cell suspension (at least one million cells) of rat sympathetic neurons was pelleted in a tabletop centrifuge at 3000 rpm for 5 min, and the supernatant was removed with a Pasteur pipette. The cells were resuspended by gentle trituration in 100 μl of rat Nucleofector solution (Amaxa Biosystems) and then the total volume was added to 10-20 μg of plasmid DNA (in sterile, nuclease-free water) in a transfection cuvette provided by the manufacturer (Amaxa Biosystems). The GFP-EB3 plasmid was obtained as a gift from Niels Galjart (Erasmus Medical Center, Rotterdam, The Netherlands) and is described in detail by Stepanova et al., 2003. The enhanced GFP (EGFP)-tubulin plasmid was purchased from Clontech (Palo Alto, CA). The DNA-cell suspension was mixed by gently shaking the cuvette, which was then placed in the chamber of the Nucleofection device. Cells were transfected using the recommended settings by the manufacturer for primary rat neurons. Immediately after Nucleofection, the cell suspension was transferred to warm N2 medium, and neurons were plated on poly-d-lysine-coated glass as explained above.
Pharmacologic regimens. For most of our experiments, rat sympathetic neurons were treated with vehicle (DMSO) or 2 μm latrunculin-A (Molecular Probes) in culture medium for at least 30 min before fixation or imaging. In many experiments, two stages of differentiation were examined under control or experimental conditions: cells were either cultured on poly-d-lysine-treated glass to examine broad, flat lamellar regions of neuronal cell bodies, or first plated and grown on poly-d-lysine-treated glass for 18-24 hr and then treated with laminin (1:40 final dilution) to examine growth cones and longer axons. For one experiment involving GFP-EB3, the cell was imaged before adding latrunculin for ∼2 min, and then the appropriate volume of latrunculin (final concentration, 2 μm) was pipetted into the culture dish while continuously imaging the same cell for several more minutes. For experiments with axons grown in the presence of latrunculin, neurons were transfected with GFP-EB3 or EGFP-tubulin and plated as usual. Latrunculin was added at a concentration of 100 nm to the cultures 30 min after plating to allow the cells to adhere to the culture surface before depleting the actin cytoskeleton. These cultures were grown overnight, and, the next morning, laminin was added at a final dilution of 1:40 (final concentration, 0.025 mg/ml) to stimulate robust axon outgrowth. Live-cell imaging was performed on these neurons 1-3 hr after addition of laminin to the cultures, after axons had formed. Quantitative fluorescence analyses (see below) revealed that the shorter exposure to the drug at the higher concentration and the longer exposure to the drug at the lower concentration were equally effective at diminishing filamentous actin to virtually undetectable levels.
The outward dispersion of microtubules from the cell body was studied using a modification of a pharmacologic regimen that reduces microtubule density and curtails microtubule assembly (Ahmad and Baas, 1995). The day after plating, neurons were treated with laminin for 30-45 min to induce rapid lamellar spreading and neurite outgrowth. The cultures were then treated with nocodozole (10 μg/ml final concentration) for 2 hr to significantly deplete microtubules. One hour into the nocodozole treatment, latrunculin was added to the cultures to deplete actin filaments while keeping the nocodozole concentration constant in the medium. Two latrunculin concentrations, 50 nm and 2 μm, were used to partially or completely deplete the actin filaments, respectively. After 2 hr in nocodozole and 1 hr in latrunculin, the cultures were rinsed two times briefly with prewarmed medium containing vinblastine (50 nm final concentration) and the appropriate latrunculin concentration. After the second rinse, the cultures were incubated with medium containing vinblastine and latrunculin. The neuronal cultures were extracted-fixed at 15 min or 1 hr in vinblastine and fluorescently labeled for microtubules and actin as described above.
Live-cell microtubule transport assay. After transfection with a plasmid for EGFP-tubulin, rat sympathetic neurons were plated as stated previously. The day after plating, cultures were treated with laminin until sufficiently long axons grew (>100 μm in length), at which point they were treated with vehicle (DMSO) or latrunculin as stated previously. Twenty-four to 30 hr after plating, neurons expressing EGFP-tubulin were used for the microtubule transport assay. Individual unbundled axons with clearly identifiable directionality of growth from the cell body were chosen for analysis so that anterograde and retrograde movements could be identified with certainty. The number of photobleached regions per neuron never exceeded two, and only one region within the same axonal branch was bleached. All photobleaching and EGFP-tubulin imaging was performed using an AxioVert 200M inverted microscope (Zeiss) coupled to an Orca II-ER B/W camera (model C4742-98-24ER) equipped with the ER-150 CCD chip (Hamamatsu, Shizouka, Japan). The microscope was mounted on a Micro-g vibration isolation system (Technical Manufacturing Corporation, Peabody, MA) to minimize image distortions caused by sample movement. A 100×, 1.3 numerical aperture Plan Apo oil immersion objective and a GFP filter set (Chroma Technology, Brattleboro, VT) were used for photobleaching and acquiring fluorescent images. The CCD camera and most aspects of the microscope were controlled by Axiovision 4.1 software (Zeiss) running on a Intel Xeon processor-based computer (Fujitsu Siemens, Berlin, Germany) with the Windows XP Professional operating system (Microsoft, Seattle, WA). For photobleaching, a house-modified diaphragm slider was inserted into the light path, which allows for an illumination beam of 20-40 μm width to illuminate the desired axonal region. GFP fluorescence was bleached by continuous, full-strength illumination from a 100 W mercury arc lamp for 60-120 sec in the GFP channel. Time-lapse imaging was initiated at the end of photobleaching, and the diaphragm slider was removed from its compartment during microtubule imaging. The intensity of epifluorescent illumination was attenuated to 20-25% to reduce photobleaching and photodamage during acquisition; 2 × 2 binning was applied to increase signal intensity. Images were acquired at 2 sec intervals, plus a 950 msec exposure time. Total recording time ranged from 9 to 12 min for each axon, which corresponded to 242-301 image cycles. Time-lapse images were saved as “zvi” files and analyzed using Axiovision 4.1 software. Motion analysis was performed only on axons that displayed at least one microtubule that was visible without digital processing. Additional analysis was aided using digital contrast adjustments within the “Image Properties” application of Axiovision 4.1. Pausing was defined as a microtubule that had been moving but stopped translocation (moved <0.137 μm/sec, or 1 pixel/sec at a binning factor of 2) within the photobleached region for at least one cycle of imaging and then proceeded to move once again. Transport velocities and microtubule lengths were calculated using the “measure distance” and “measure length” tools of Axiovision 4.1, respectively. Mean transport velocities were calculated for all observed microtubule transport events (n = 233). Instantaneous velocities were calculated for a randomly chosen subset of the total observed population of microtubules, which represented an equal number of transport events in both control and latrunculin-treated axons (n = 30). Pausing was calculated on the same subset of transport events as was analyzed for instantaneous velocity and is presented as a percentage of the total number of observed instantaneous microtubule movements. Microtubule lengths were calculated using a random subset of the total microtubule population as described for instantaneous velocities (n = 60). Final processing of all images was done using Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA).
Live-cell imaging of GFP-EB3. All GFP-EB3 imaging was performed using identical equipment to that listed previously, except that an Orca-ER Digital CCD Camera (Hamamatsu) and a 100× Plan-Neofluar/1.3 numerical aperture objective (Zeiss) were used to acquire images. Also, the GFP-EB3 imaging system was controlled by Axiovision 3.0 software (Zeiss) running on a Pentium II-based computer (Tekgraf, Alsdorf, Germany) with the Windows 2000 operating system (Microsoft). Excitation of fluorescent molecules and collection of correct emission wavelengths were accomplished with an AttoArc 2 Variable Intensity HBO 100 Arc Lamp Control (Atto Instruments, Rockville, MD), a 100 W mercury vapor lamp, and the appropriate excitation and emission filters and dichroic mirrors (Chroma).
To acquire images for GFP-EB3 time-lapse movies, a neutral density filter (80% reduction in illumination; Chroma) was placed in the illuminating light path to reduce photobleaching and photodamage of the sample. Also, the field diaphragm was adjusted such that only the cell being imaged was illuminated with the excitation light, again reducing photobleaching and photodamage to surrounding cells. Images were acquired at a rate of 1 per second (and one per 4 sec in some cases) for GFP-EB3 time-lapse movies to maximize the amount of information gathered for each microtubule plus-end excursion per set of frames. To maximize the fluorescent signal but minimize exposure times, the CCD camera was set to 2 × 2 binning, allowing for camera integration times of only 150-300 msec/image. Using the Axiovision software, individual frames were assembled into 3-5 min movies (real-time) and then exported as QuickTime movies (Apple Computers, Cupertino, CA) for publication. Frames extracted for still images were exported from Axiovision as “TIFF” image files and processed in Photoshop (Adobe Systems) and/or Canvas (Deneba Systems, Miami, FL). For quantitative analysis, we calculated GFP-EB3 mean excursion velocities and mean excursion durations for both control and actin-depleted neurons. The mean excursion velocity was calculated as the distance moved by each GFP-EB3 comet divided by the time that the comet remained visible. Mean excursion duration was calculated by summing the lifetimes (the time that a GFP-EB3 comet remained visible) of individual comets and dividing this sum by the number of comets observed. All quantification was performed using the Axiovision “measure” module. Raw data were processed, statistical analyses were performed, and graphs and charts were produced using Excel (Microsoft).
Results
A variety of indirect and direct experimental approaches have shown that microtubules are conveyed outward from cell center in many cell types (Ahmad and Baas, 1995; Keating et al., 1997; Abal et al., 2002). In the case of neurons, microtubules are then transported into growing axons (Slaughter et al., 1997), in which they move mainly anterogradely but also retrogradely (Wang and Brown, 2002). To determine the role of actin filaments in these microtubule movements, we examined microtubule distribution, organization, and transport in neurons pharmacologically depleted of filamentous actin with latrunculin. We first sought to determine the effects of latrunculin treatment (2 μm for 30 min) on the actin cytoskeleton of developing rat sympathetic neurons in culture. Figure 1 shows cultured sympathetic neurons, with fluorescently labeled microtubules (Fig. 1B) and actin filaments (Fig. 1A), at an early stage of axonal formation. Bundles of actin filaments are apparent both at the periphery of the lamella and coursing through the lamella (Fig. 1A). Microtubules follow closely along these actin bundles in some instances (Fig. 1A,B, arrows), which is consistent with the idea that microtubules could use the actin bundles as substrates for their transport into nascent axons. Figure 2 shows neurons before the initiation of axonal outgrowth (Fig. 2A-D) and after axonal outgrowth (Fig. 2E-H); examples are shown of control neurons and similar neurons after latrunculin treatment. Particular attention was given to growth cones and the peripheral regions of lamellae, because they normally display the thickest and most prominent bundles of actin filaments.
Figure 1.

Microtubules align with actin filament bundles during early axogenesis. Shown are images of neurons fixed and stained for fluorescence visualization of actin filaments (A) and microtubules (B) during early axonal outgrowth. Large lamellae developed in which microtubules and actin bundles formed along the axes of presumed future axons. Microtubules and actin bundles were seen to colocalize in some cases (see arrows in A and B). Scale bar, 10 μm.
Figure 2.
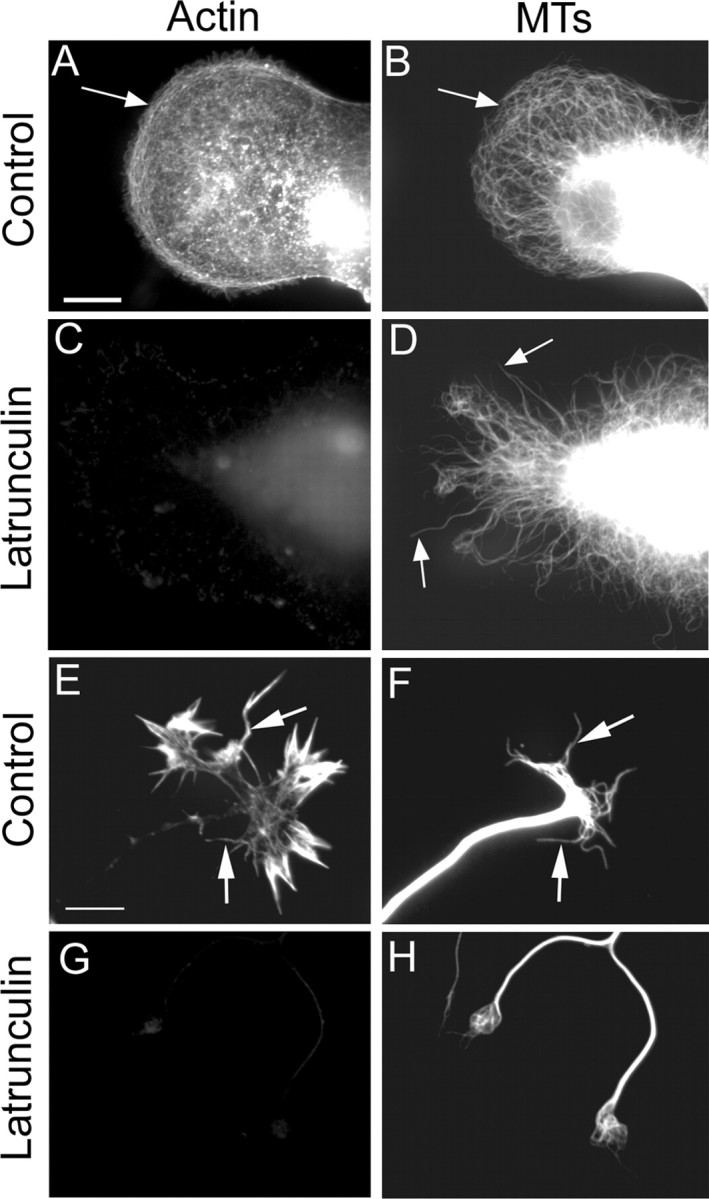
Actin depletion alters peripheral microtubule organization. Shown are images of neurons fixed and stained for fluorescence visualization of actin filaments (left column) and microtubules (right column) before axonal outgrowth (A-D) and after significant axonal outgrowth had occurred (E-H). Before axogenesis, neurons developed broad lamellae in which microtubules extended to the periphery (B) but did not invade the actin-rich cortical region (A). Treatment for 30 min with latrunculin depleted virtually all of the filamentous actin (C), which resulted in the extension of microtubules to the edge of the lamella (D). Note that microtubules also fail to curve back on themselves after actin depletion. Control neurons showed dense bundles of actin filaments occupying the peripheral regions of growth cones and filopodia (E). Individual microtubules (F) were seen to align with some of these actin bundles (arrows in E). After actin depletion (G, H), the growth cones typically collapsed into rounded bulbs, with microtubules swirling in a disorganized manner within the bulb (H). Arrows point to actin bundles in A and E and to microtubules (MTs) in B, D, and F. Scale bar, 10 μm.
Before axonal formation, actin filaments are present throughout the cell body and lamella, and there is a particularly dense array of aligned cortical actin filaments at the periphery of the lamella (Fig. 2A) (Yu et al., 2001). These actin filaments tend to run parallel to the edge of the lamella. Microtubules splay into the lamella from deeper within the cell body but curve back on themselves as they encounter this cortical layer of actin (Fig. 2B). After actin depletion by latrunculin (Fig. 2C), the microtubules straighten somewhat and extend to the edge of the lamella, no longer impeded by the cortical actin (Fig. 2D). As reported previously (Tang and Goldberg, 2000), microtubules within the growth cones of control neurons are tightly bundled as they enter the central region of the cone from the axonal shaft but splay apart in the peripheral region, in which they can be seen to align with actin bundles as they invade filopodia (Fig. 2E,F). After actin depletion with latrunculin, the microtubules within the growth cone collapsed into a disorganized knot-like mass (Fig. 2G,H). Microtubule bundles within the axonal shaft were not detectably altered by the latrunculin treatment. Axonal length was unaffected, except for the collapse of the growth cone. The latrunculin treatment resulted in the complete loss of any detectable actin filaments, reducing phalloidin staining by >99% compared with control neurons (Fig. 2C,G; quantification not shown). Low levels of fluorescence observed after latrunculin treatment did not appear filamentous and hence was presumably background.
Effects of actin depletion on microtubule transport in the axon
To directly test the role of the actin cytoskeleton in transporting microtubules within the axon, we used the photobleach method of Wang and Brown (2002), except that we expressed EGFP-tubulin rather than injecting rhodamine-tubulin. An example of a moving microtubule is shown in Figure 3A. Latrunculin was added to cultures 7-10 hr after initiating axonal outgrowth with laminin. Incubations in latrunculin were from 30-90 min, with no difference in the results observed at longer exposure times verses shorter exposure times. In controls (0.1% DMSO vehicle), the majority of microtubule transport was observed in the anterograde direction, with an anterograde to retrograde frequency ratio of ∼2:1 (anterograde, 65.2%; retrograde, 34.8%; n = 138). As a separate control, a population of transfected but untreated neurons were also observed and showed no difference from treatment with vehicle (data not shown). After latrunculin treatment, the total number of anterograde microtubule transport events was significantly reduced in actin-depleted axons compared with control axons from 0.885 to 0.508 transport events/min, respectively (p < 0.001; χ2 test). The reduced frequency of anterograde microtubule movements resulted in a marked change in the ratio of anterograde to retrograde movements, which dropped to ∼1:1 in actin-depleted axons (anterograde, 51.6%; retrograde, 48.4%; n = 95). Interestingly, retrograde transport frequency remained unchanged after actin depletion and displayed frequencies of 0.476 and 0.478 events/min in control and latrunculin-treated neurons, respectively (Fig. 3B).
Figure 3.

Effects of actin depletion on microtubule transport in the axon. A, Time-lapse images reveal a microtubule moving in the anterograde direction through the photobleached region. Brightness and contrast were adjusted to best reveal the moving microtubule. Red arrows mark the leading and trailing ends of the microtubule. B, Analysis of microtubule transport events demonstrated that the frequency (events per minute) of anterograde transport was significantly reduced in actin-depleted axons (*p < 0.001;χ2 test), whereas the frequency of retrograde transport was not significantly affected. C, Histogram of microtubule lengths displays no significant difference between control and latrunculin-treated neurons. D, Histogram depicting the mean velocity distributions of both anterograde and retrograde microtubule transport in control and latrunculin-treated neurons. A significant increase in mean velocity was detected in both anterograde and retrograde microtubule movements (p < 0.005 and p < 0.001, respectively; two-tailed t test). E, Histogram showing the distributions of the instantaneous velocities of microtubules chosen randomly from the total observed microtubule population shown in D. No significant difference was detected between the instantaneous velocities of moving microtubules between control and actin-depleted axons. F, The combined data from D and E depicting microtubule velocity change as a function of transport directionality shows a significant increase in the mean microtubule transport velocities that is bidirectional (*p < 0.01; two-tailed t test). A small but insignificant increase is seen in comparing control and latrunculin-treated instantaneous velocities. Scale bar, 5 μm.
With regard to the velocities of the moving microtubules, we measured mean velocities for all observed transport events (n = 233) for the entire duration of the movement and instantaneous velocities every 2.95 sec for a randomly chosen subset (n = 60) of the total observed transport events (see Materials and Methods). The same subset of moving microtubules evaluated for instantaneous velocities was compared for microtubule length variations (n = 60) among control and latrunculin-treated neurons, but no significant difference was observed (Fig. 3C). Comparing the mean transport velocities of control and actin-depleted axons, there was a small but statistically significant increase in the average velocities of both anterograde (p < 0.005; two-tailed t test) and retrograde movements (p < 0.001; two-tailed t test) (Fig. 3D). When comparing the instantaneous velocities of a subset of the microtubule population, no significant difference was observed between control and latrunculin-treated neurons (Fig. 3E). Additional analysis of instantaneous transport velocities revealed that microtubules showed a slight decrease in the percentage of time spent pausing in latrunculin-treated cells (10.7%) compared with vehicle-treated controls (11.1%). This minor variation seems unlikely to account for the significantly augmented mean velocities observed in actin-depleted axons. Microtubule pausing was defined as occurring when a moving microtubule stopped for at least one cycle of imaging (2.95 sec) and then proceeded to move again. This criterion allowed for the identification of intermittent pausing during observed bouts of movement but is likely an underestimation of the total percentage of microtubule pausing. The inability to observe very short bouts of pausing as well as sustained pausing throughout the observation period would have remained undetectable under this experimental regimen. Nevertheless, the data lead to the conclusion that small instantaneous velocity increases in actin-depleted neurons become significant increases in velocity when measured over the total distance of microtubule translocation. Figure 3F displays the mean change in the microtubule transport velocities depicted in Figure 3, D and E. Although transport velocities can be seen to vary considerably, on average, there is an inverse correlation between actin depletion and transport velocity that is statistically significant regardless of transport directionality (p < 0.01; two-tailed t test).
Microtubules fail to move outward when actin filaments are depleted and microtubule density is low
The fact that actin depletion partially but not totally inhibited the transport of microtubules within the axon indicates that, although actin is one substrate for microtubule movements, it is not the only substrate. The most reasonable possibility for an alternative substrate would be other microtubules, given that microtubules can move against one another during mitosis (Baas, 1999) and have been shown to track along one another in other cell types (Krylyshkina et al., 2003). The ideal test of this hypothesis would be to deplete microtubule levels within the axon to see whether their transport is compromised when they no longer have nearby neighbors against which to move. However, even prolonged treatment with anti-microtubule drugs does not deplete microtubules within the axonal shaft sufficiently to achieve this goal (Baas and Black, 1990). Therefore, we instead used a modification of a pharmacologic regimen in which a small population of microtubules has been shown to relocate from cell center to cell periphery within 15-60 min (Ahmad and Baas, 1995; Ahmad et al., 1998) (see Materials and Methods). With this regimen, we can reduce the density of microtubules to a point at which their capacity to encounter one another is severely limited. Unlike our original studies using this regimen, here we permitted neurons to grow elaborate lamellae (that began to pinch off into nascent axons) before drug treatment. Microtubules were only partially depolymerized within the central region of the cell body during nocodozole treatment but were completely depolymerized in more peripheral regions, after which vinblastine was added to curtail further assembly (shown schematically in Fig. 4A). Without latrunculin, the microtubules rapidly scattered to the peripheral regions in which they invaded filopodial extensions of the lamellae and nascent axons (Fig. 4B). A low dose of latrunculin (50 nm) partially depleted actin filaments, but microtubules were still able to move distally (Fig. 4D), where they colocalized with the remaining actin filaments (Fig. 4C). However, with the higher dose of latrunculin (2 μm), actin filaments were completely absent (Fig. 4E), and microtubules remained close to cell center, never appearing within the peripheral regions of the neuron (Fig. 4F). Interestingly, the microtubules also appeared twisted and contorted under these conditions. Under these culture conditions, we commonly observed growth cone collapse but only minor alterations in the expanse of broad lamella and no change in axonal length. These results, which were also obtained when actin filaments were disrupted with Cytochalasin B (data not shown), demonstrate that, when microtubules do not have abundant neighboring microtubules, they can use actin filaments as effective substrates for transport. However, when actin is depleted while microtubule density is low, microtubules are no longer able to move outward, consistent with the hypothesis that microtubules use either actin filaments or other microtubules as their transport substrate.
Figure 4.
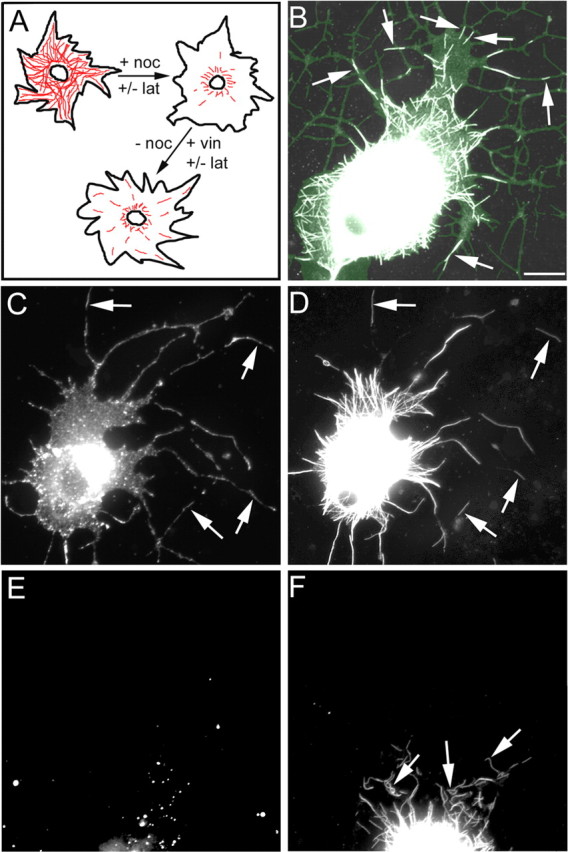
Outward transport of microtubules requires actin filaments when microtubule density is low. A is a schematic of the drug treatment regimen used in this experiment. Microtubules are drawn in red. lat, Latrunculin; noc, nocodozole; vin, vinblastine. B shows a control neuron (actin filaments present) after a 15 min exposure to vinblastine (after treatment with and removal of nocodozole) in which short microtubules (white) were seen to invade distal regions in which actin filaments (green) were also localized. Arrows in B denote several examples of microtubules that have moved outward to the cell periphery and into processes. C and D show fluorescently labeled actin filaments and microtubules, respectively, in a neuron that has been partially depleted of actin filaments using a lower drug concentration. Microtubules invade peripheral regions of the neuron in which actin filaments remain. Arrows in C and D denote regions in which microtubules and actin filaments colocalized. E and F show phalloidin staining and microtubules, respectively, in a neuron that has been completely depleted of actin filaments. Short microtubules were seen in the cytoplasm (F), but they were not transported to the cell periphery and many appeared curved and not outwardly oriented as in control cells (see B). Arrows in F denote examples of mal-oriented and/or curved microtubules. Scale bar, 10 μm.
Observations on microtubule behaviors in the presence and absence of actin filaments using a GFP construct for EB3, a microtubule plus-end tracking protein.
Microtubules undergo a variety of behaviors other than transport. In fact, it appears that the vast majority of microtubules do not undergo concerted transport, given that transport appears to be limited to only the very short microtubules. One potential function for the longer non-moving microtubules is to act as a substrate for the movement of the short microtubules. To better appreciate the totality of microtubule behaviors in control and actin-depleted neurons, we observed bouts of microtubule assembly in neurons induced to express GFP-EB3. EB3 is a member of a newly described family of microtubule-related proteins called +TIPS (plus-end tracking proteins), which associate with the plus end of the microtubule during bouts of assembly (Nakagawa et al., 2000; Carvalho et al., 2003). Because the associated +TIPS rapidly lose association with the microtubule as new assembly occurs at the plus end, the GFP fluorescence typically appears in the shape of a comet, with the trailing end toward the minus end of the microtubule. Stepanova et al. (2003) recently used GFP-EB3 to observe bouts of microtubule assembly throughout various compartments of cultured neurons. Figure 5 (and movie 1, available at www.jneurosci.org as supplemental material) shows a live-cell experiment on a GFP-EB3-expressing neuron with a broad lamella (before axonal outgrowth). In this experiment, we were able to view the same neuron before and after the addition of latrunculin. In all other experiments involving GFP-EB3, image acquisition began after the addition of latrunculin. Before the addition of the drug, microtubules were mostly constrained to the central region of the cell body (Fig. 5A). When latrunculin was added to the culture, the GFP-EB3 protein appeared to dissociate from the plus ends of the microtubules for a few seconds (Fig. 5B), but the association was rapidly restored (Fig. 5C). Shortly after adding the drug, the edges of the lamella retracted slightly, and the plus ends of microtubules were seen to invade more peripheral regions of the lamella (Fig. 5C,D). In this and other control cell bodies, we observed an outward streaming of a high proportion of microtubules from a site near cell center, presumably the centrosome. Before drug treatment, 58% of the GFP-EB3 excursions occurred from the cell center outward to the cell periphery, whereas 40% moved across the cell and 2% traveled inward from the cell edge to the center (Fig. 5E,I). After drug treatment, there was an increase in these outward excursions to 65%, as well as a small increase in inward excursions to 4% and a corresponding diminution in excursions going across the cell to 31% (Fig. 5F,I). Figure 5, E (before actin depletion) and F (after actin depletion), are semi-schematics of the same cell shown in Figure 5A-D, in which the arrowheads denote both the direction and location of GFP-EB3-labeled microtubule plus ends. There was a small but statistically significant increase in the mean velocity of the GFP-EB3 excursions from 0.18 ± 0.01 μm/sec before actin depletion to 0.21 ± 0.01 μm/sec after actin depletion (p < 0.05; two-tailed t test; n > 50 excursions before and after treatment) and also a significant and more dramatic increase in the mean duration of the excursions from 9.6 ± 0.59 to 16.3 ± 2.45 sec (p < 0.005; two-tailed t test) before and after actin depletion, respectively (Fig. 5G,H). Collectively, these observations indicate that microtubule behaviors in these regions of the cell body and lamella show increases in speed, duration, and outward directionality of the EB3 excursions after actin depletion.
Figure 5.

Live-cell visualization with GFP-EB3 of microtubule behaviors in neurons depleted of actin filaments before axon formation. A-D are still images extracted from a time-lapse movie of a single GFP-EB3-expressing neuronal cell body with a large lamellar region. See movie 1 (available at www.jneurosci.org as supplemental material). Arrows in A, C, and D denote GFP-EB3 comets. Before depletion of filamentous actin, microtubules were mostly constrained to the central region of the cell body, presumably attributable to the actin-rich lamellae found in the periphery (A). At the time of drug addition to the culture, the GFP-EB3 appeared to temporarily dissociate from the plus ends of the microtubules (B). After actin filaments were depleted, plus ends of microtubules were seen to invade more peripheral regions of the neuron (C, D). Before drug treatment, ∼60% of the excursions occurred from the cell center outward to the cell periphery, whereas most of the others occurred across the cell (E, I). After drug treatment, there was an increase in these outward excursions as well as a small increase in inward excursions and a corresponding diminution in excursions going across the cell (F, I). Arrowheads in E and F indicate the direction of GFP-EB3 excursions, and red indicates inward directionality. There was also a small increase in the velocity and a greater increase in the duration of GFP-EB3 excursions after actin depletion (G, H). Blue bars indicate control data, and red bars indicate experimental (actin-depleted) data in G-I. Scale bar, 5 μm.
We next examined the axonal shaft and growth cones. We found that the mean velocity of GFP-EB3 excursions was slightly decreased from 0.20 ± 0.02 μm/sec in control axons to 0.17 ± 0.01 μm/sec in axons depleted of actin filaments (p < 0.01; two-tailed t test) (Fig. 6C). Also, as was the case for cell bodies, we found a significant increase in the mean GFP-EB3 excursion duration from 5.6 ± 0.77 sec in control axons to 11.4 ± 1.5 sec within axons after actin depletion (p < 0.005; two-tailed t test) (Fig. 6D; also see images A, B). The polarity orientation of microtubules (as assessed by the directionality of the comet movements) was essentially unchanged within axons depleted of actin filaments when compared with control axons, in which, in both cases, >95% of the GFP-EB3 excursions were forward (plus end distal) within the axon (Fig. 6E). With regard to growth cones, the GFP-EB3 imaging allowed us to visualize plus ends of microtubules splaying out from the axonal shaft region into the central region of the growth cone and often invading filopodia (Fig. 6F-H). However, when actin filaments were depleted, the microtubule plus ends tumbled chaotically within the collapsed growth cones (Fig. 6K), unlike the outward microtubule growth seen in controls. The mean velocity of GFP-EB3 excursions was significantly increased from 0.21 ± 0.04 μm/sec in control growth cones to 0.43 ± 0.01 μm/sec in growth cones depleted of actin filaments (p < 0.001; two-tailed t test; n > 5 growth cones) (Fig. 6I). Also, the mean excursion duration of GFP-EB3 comets was significantly decreased from 16.3 ± 1.9 sec in control growth cones to 4.1 ± 0.55 sec in actin-depleted growth cones (p < 0.001; two-tailed t test) (Fig. 6J). These results indicate that actin filaments are required for the proper configuration and behavior of microtubules within growth cones but show no evidence for such a dependency within the axonal shaft.
Figure 6.
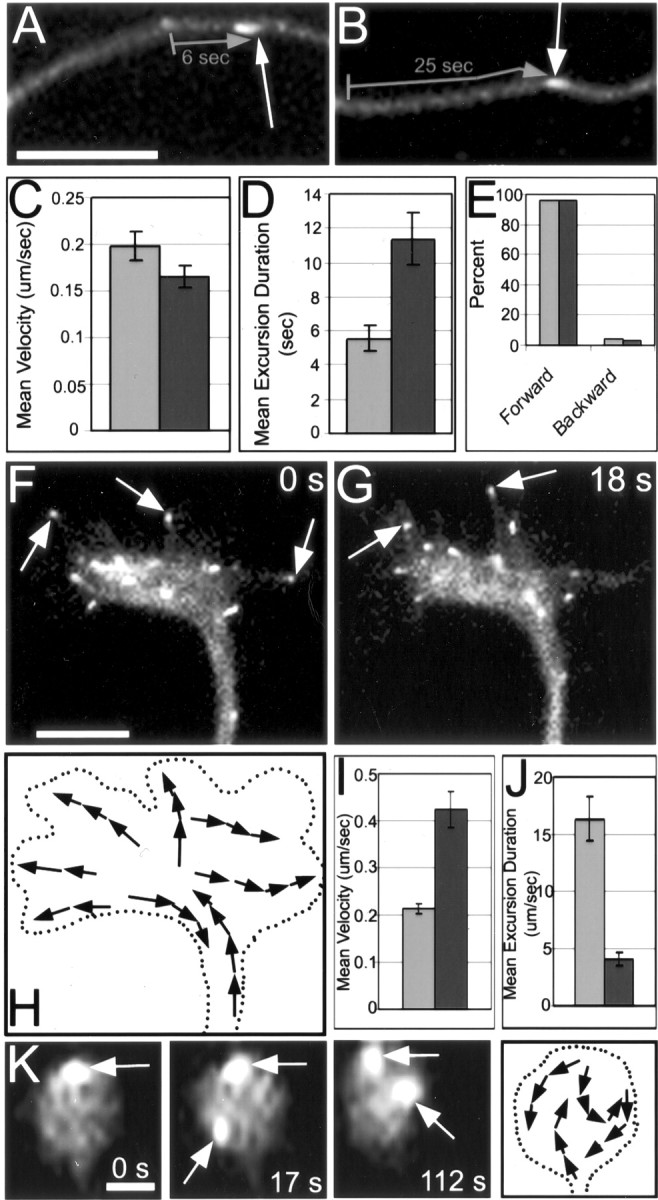
Live-cell visualization with GFP-EB3 of microtubule behaviors in axons and growth cones depleted of actin filaments. Neurons that had been transfected to express GFP-EB3 were permitted to grow axons for several hours before treatment with latrunculin. A and B show still images extracted from time-lapse movies of control and actin-depleted axons, respectively. White arrows in A and B point out GFP-EB3 comets, and gray arrows denote the length and duration of examples of GFP-EB3 excursions. The mean velocity of GFP-EB3 excursions in actin-depleted axons was slightly decreased (C, darker bar; lighter bars are controls). The mean duration of the excursions was dramatically increased (D, darker bar). Directionality of the excursions was unchanged after depletion of actin filaments, with >95% of microtubules showing forward excursions (E; lighter bars are controls, and darker bars are actin-depleted data). F and G are images of control growth cones extracted from a time-lapse movie in which GFP-EB3 comets (see arrows) were seen to move outward and invade filopodia. The first three panels of K shows till images extracted from a time-lapse movie of a growth cone after actin depletion showing chaotic movements of GFP-EB3 comets, which indicate highly disorganized microtubules (arrows point out several comets). Also, GFP-EB3 excursion velocity was significantly increased and excursion duration was notably decreased in growth cones depleted of actin filaments (I, J, darker bars). The schematics in H and the last panel of K show the paths and directions of representative individual plus-end excursions in control and drug-treated growth cones, respectively. Scale bars: A, B, F, G, 5 μm; K, 1 μm.
Microtubules track along other microtubules in the absence of actin filaments
In our studies on neurons before and during early phases of axogenesis, it was our general impression that, after actin depletion, microtubule behaviors became disorganized as they approached the leading edge of the lamella and that the microtubules farther from the leading edge tended to follow the paths of preexisting microtubule bundles that appeared as weakly fluorescent rib-like structures in our movies. To examine both of these issues in more detail, we analyzed actin-depleted neurons in which we could visualize both the GFP-EB3 comets as well as the lengths of the microtubules with particular clarity in both the leading edge and regions contiguous to it. One of these cells is shown in Figure 7 (and movie 2, available at www.jneurosci.org as supplemental material). Panels in the left column of Figure 7 are the conventional fluorescence images, and panels in the right column are inverse images that show the lengths of the microtubules (with weak fluorescence) better than the conventional images. At the cell edge, microtubule trajectories were clearly disorganized and appeared to tumble in similar manner to those within growth cones depleted of actin filaments (Fig. 7, arrowheads in top two panels). In the adjacent region farther from the leading edge (closer to the nucleus), microtubules were seen to follow identical trajectories as other microtubules, as shown by frames from the time-lapse movie that captured multiple GFP-EB3 comets colinear with one another (sets of arrows in each panel). The inverse images demonstrated more clearly that the common trajectories were attributable to underlying preexisting microtubules that act as guides. Preexisting microtubules were identified by their faint fluorescence, which was dissimilar to neurofilament distribution, or any internal membrane distribution observed by electron microscopy (Yu et al., 2001). Similar colinear excursions were commonly observed in neurons depleted of actin filaments but were uncommon (although not absent) in control neurons. These observations demonstrate that microtubules undergoing bouts of assembly can and do track along other preexisting microtubules and that this phenomenon is accentuated when actin filaments are depleted.
Figure 7.
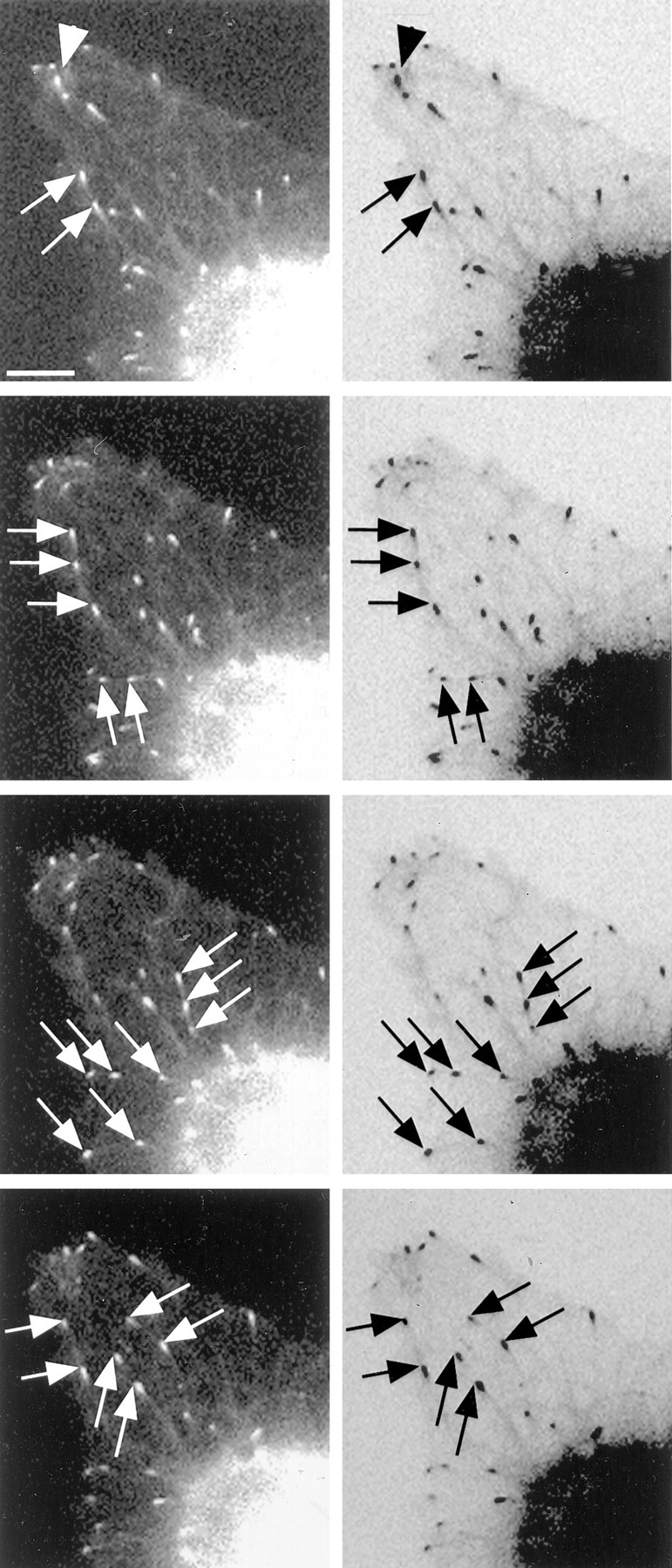
Microtubules track along other microtubules after actin depletion. Shown in this figure are still images from a time-lapse movie of a region of a lamella of a neuron depleted of actin after being transfected to express GFP-EB3 as in Figure 5. Panels on the right are inverse images that show the lengths of the microtubules (with weak fluorescence) better than the conventional images. At the cell edge, microtubule trajectories were clearly disorganized and gave the impression of tumbling chaotically (region of arrowheads in top 2 panels). See movie 2 (available at www.jneurosci.org as supplemental material). In the central region, microtubules were seen to follow identical trajectories as other microtubules, as shown by individual frames that capture multiple GFP-EB3 comets colinear with one another (sets of arrows). The inverse images demonstrated that the common trajectories follow along underlying preexisting microtubules that acted as tracks. Similar colinear excursions were not as commonly observed in control neurons. Scale bar, 5 μm.
Polarity orientation of microtubules in axons grown in the absence of actin filaments
The establishment of correct microtubule polarity orientation in axons is thought to result from the transport of microtubules by cytoplasmic dynein against the actin cytoskeleton (Dillman et al., 1996; Pfister, 1999). If microtubules absolutely require actin tracks for their orderly transport, we would expect that microtubules would be unable to achieve their uniformly plus end distal polarity pattern in axons that had developed after the depletion of actin filaments. To test this, neurons that had been induced to express GFP-EB3 were cultured overnight in the presence of latrunculin to inhibit the formation of a filamentous actin network and then induced to grow axons the following day by the addition of laminin in the presence of the drug (for schematic of treatment regimen, see Fig. 8A). Under these conditions, virtually all of the neurons extended axons, but the axons were notably different from control axons with regard to their length and morphology. The experimental axons were approximately half as long as control axons, were at least twice as broad, and lacked discernable growth cones at their tips. Significantly more GFP-EB3 “comets” could be seen across the axonal width (Fig. 8B-D) (and movie 3, available at www.jneurosci.org as supplemental material). The tips of the axons appeared as club-like regions, dense in microtubule plus ends (Fig. 8B, arrowhead). The mean velocity of GFP-EB3 excursions in the experimental axons was indistinguishable from controls at 0.18 ± 0.01 μm/sec (p > 0.05; two-tailed t test; n > 5) (Fig. 8F). The mean duration of GFP-EB3 excursions between control and actin-depleted axons increased from 9.6 ± 1.6 to 12.7 ± 1.7 sec (Fig. 8G). The directionality of the GFP-EB3 excursions is an indicator of the polarity orientation of the microtubule, so we calculated the percentage of forward and backward GFP-EB3 excursions in both control axons and axons extended in the absence of filamentous actin. In seven control axons, the proportion of forward excursions was 96.3 ± 3%, suggesting that such backward movements are rare in sympathetic neurons grown in this manner. The percentage of forward excursions was 85.3 ± 3% in 10 experimental axons, indicating that microtubule polarity orientation is slightly flawed in axons depleted of actin. Thus, microtubules are able to achieve the correct or nearly correct pattern of polarity orientation as control axons in the absence of actin filaments (Fig. 8E,H). These results demonstrate that, even in the absence of actin filaments, the transport of microtubules against one another is sufficient for them to achieve their correct (or nearly correct) pattern of polarity orientation.
Figure 8.
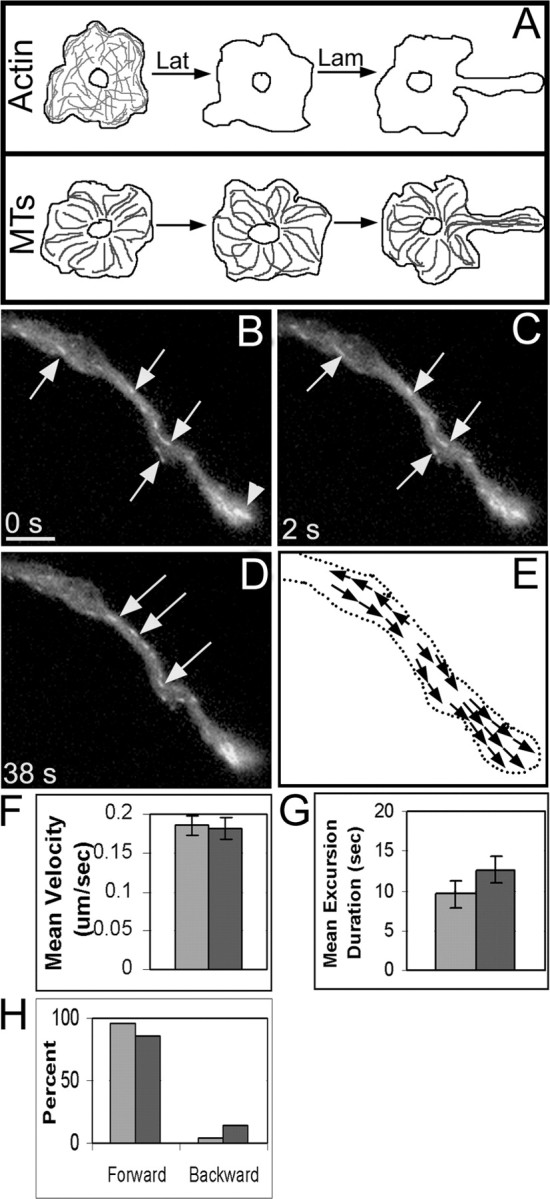
Directionality of GFP-EB3 excursions in axons grown in the absence of actin filaments indicates uniform or nearly uniform microtubule polarity orientation. Neurons were cultured overnight in the presence of latrunculin to inhibit the formation of a filamentous actin network and then induced to grow axons the following day in the presence of the drug (for treatment regimen, see A). Axons extended in the absence of actin filaments were approximately half as long as control axons and were thicker in diameter. More GFP-EB3 “comets” were seen across the axon width (arrows in B-D). See movie 3 (available at www.jneurosci.org as supplemental material). The growth cone was essentially absent in the drug-treated axon, and instead appeared as a club-like region, dense in microtubule ends, at the distal end of the growing axon (arrowhead region denoted in B). The mean velocity was unchanged compared with control axons, whereas the duration of GFP-EB3 excursions increased (F, G; darker bars are actin-depleted data, and lighter bars are control data). The percentage of forward excursions was only slightly lower than in control axons, indicating that microtubules in the axon achieved uniform (or nearly uniform) plus-end distal polarity orientation in the absence of actin filaments (H). Scale bar, 5 μm. Lam, Laminin; Lat, latrunculin; MT, microtubule.
Discussion
Significant progress has been made in recent years on the mechanisms of microtubule transport within neurons. The original view, articulated two decades ago, held that microtubules are transported continuously and coherently down the axon in the anterograde direction at rates much slower than would be consistent with known motor proteins (Lasek, 1986). However, studies involving small photobleached regions of the microtubule array failed to detect any such slow coherent movement of the bleached zone (Lim et al., 1989). When a longer zone was bleached and images were acquired more frequently, it became apparent that short microtubules move through the zone intermittently and asynchronously at the rapid rates of known motor proteins (Wang and Brown, 2002). In addition, at least in growing axons, the movements were observed to occur retrogradely as well as anterogradely. Based on these observations, a new model has emerged in which the vast majority of the microtubules in the axon are motionless at any given moment, whereas a small number of microtubules move very rapidly. The average rate of microtubule transport is slow because it includes non-moving as well as moving microtubules.
A potential role for actin filaments in the transport of microtubules was first suggested on the basis of the fact that most of the cytoplasmic dynein that moves anterogradely down the axon does so at the same average rate that actin filaments are transported (Dillman et al., 1996). On this basis, it was proposed that the cargo-binding domain of cytoplasmic dynein interfaces with the actin cytomatrix, via dynactin, thus leaving the motor domain available for microtubules to slide along. According to this model, because the actin cytomatrix has a greater resistance to backward movement than the microtubule has to forward movement, the microtubule moves forward. This concept is attractive because the directionality of the dynein motor can explain why microtubules move into the axon with plus ends leading (Vallee and Bloom, 1991; Ahmad et al., 1998) and because dynein-driven force interactions between microtubules and actin filaments are known to occur in other cellular events, such as mitosis and migration (Baas, 1999). In addition, there are numerous proteins that have been reported to promote the alignment and interaction of microtubules and actin filaments (Bearer and Reese, 1999; Ozer and Halpain, 2000; Zmuda and Rivas, 2000; Lee and Kolodziej, 2002; Lee and Van Vactor, 2003), which could assist in properly configuring the filaments for them to polymerize along one another (Rochlin et al., 1999; Dent and Kalil, 2001) or slide relative to one another. Finally, we have shown that inhibition of dynein-driven forces can enhance the contractility of the actomyosin system within the axon, which is consistent with removing the drag normally imposed by the motor attempting to move the microtubules (Ahmad et al., 2000).
To directly test whether actin filaments are the substrate against which microtubules move, we used the approach of Wang and Brown (2002) to view microtubule movements in the axon after pharmacologic depletion of actin filaments. The results show a sizeable but not complete diminution in anterograde microtubule movements but no diminution whatsoever in the retrograde movements. Although we cannot dismiss the possibility that a small number of actin filaments might have resisted depolymerization, the marked difference in response between anterograde and retrograde movements suggests that the former is at least partially dependent on actin as a substrate, whereas the latter is not. The model described above for cytoplasmic dynein could not account for the backward movement of microtubules in the axon, and hence it makes sense that the retrograde movements would not be impeded by the depletion of actin filaments.
In a recent study also using the photobleach approach, the movement of tubulin-containing structures along longer microtubules was directly observed, and the movement was documented to occur in both directions (Ma et al., 2004). However, the authors argued that the moving structures in their study were actually not microtubules at all but rather some non-microtubule organelle that contains tubulin. Their argument was based on their observations that (1) movement occurred along microtubules rather than actin filaments, (2) the moving structures sometimes bended, and (3) the moving structures were somewhat dimmer in their fluorescence intensity than the long microtubules. We believe the first two arguments are invalid because microtubule movement against other microtubules has certainly been documented in other cellular events, such as mitosis and migration (see above), and because there are numerous documented cases of short microtubules bending (Yu and Baas, 1994; Gallo and Letourneau, 1999) (Fig. 4F). We are uncertain why these authors found there to be a discrepancy in fluorescence intensity between their moving structures and the longer microtubules, but it is noteworthy that, after almost two decades of live-cell imaging of microtubules in other cell types (Sammak and Borisy, 1988), no such moving tubulin-containing, non-microtubule organelles have been observed. In additional support of this argument, we recently obtained data showing that anterograde, but not retrograde, transport of these moving structures is compromised in dynein-depleted neurons, which is consistent with the moving structures being microtubules rather than non-microtubule organelles (Y. He, K. A. Myers, and P. W. Baas, unpublished observations).
To investigate further the possibility of microtubules being transported against either actin filaments or neighboring microtubules, we used a modification of a pharmacologic assay developed previously by our laboratory for studying the outward movement of microtubules from the cell center. In this assay, microtubule assembly is suppressed and the total levels of microtubules are severely reduced, thus limiting the availability of neighboring microtubules that might serve as transport substrates. Indeed, we found that microtubules failed to move outward under these conditions when actin filaments were depleted. Interestingly, in cases in which actin filaments were only partially depleted, there was a near perfect alignment of any microtubule that appeared distal to cell center with the remaining actin filaments.
Additional insights were provided by observations on microtubule assembly behaviors in neurons expressing GFP-EB3, which is a neuron-specific member of a family of proteins that track with the plus end of the microtubule during bouts of polymerization (Stepanova et al., 2003). Based on numerous studies showing a dependency of microtubules on actin for their organization in the growth cone (Schaefer et al., 2002; Zhou et al., 2002; Gordon-Weeks, 2004), we anticipated the possibility that depleting actin would result in chaotic microtubule behaviors and disorganization of microtubules throughout all regions of the neuron. Interestingly, however, we observed microtubule behaviors and organization that were quite orderly within the cell body and axonal shaft after actin depletion. In fact, the percentage of outwardly oriented microtubules in the cell body actually increased after actin depletion, as did the rate and duration of the assembly excursions. Only in the peripheral regions of the lamellae and growth cone did microtubule behaviors become notably chaotic after actin depletion. We suspect that this is because these regions do not contain preexisting microtubule scaffolding against which invading microtubules could align in the absence of actin filaments. In particularly flat regions of lamellae, microtubules could be seen to align with one another after actin depletion, with multiple microtubules showing identical trajectories, a phenomenon that was rarely observed in control neurons. Most notably, axons grown out after the depletion of actin filaments contained microtubules of nearly uniform polarity orientation. This suggests that the motor proteins that orient microtubules in the axon are able to do so without pushing against actin filaments. Thus, not only do microtubules have an option of moving against other microtubules, but this option is enhanced when actin is deficient and is sufficient for generating orderly behavior and orientation of microtubules in all but the most peripheral regions of the developing neuron.
On the basis of the results presented here, we conclude that retrograde microtubule transport occurs independently of actin filaments, with microtubules presumably moving along other microtubules. Anterograde microtubule transport occurs against either actin filaments or neighboring microtubules, depending on the availability of each of these two potential substrates. Given that cytoplasmic dynein can generate forces between neighboring microtubules as well as between microtubules and actin filaments (Vallee et al., 2004), cytoplasmic dynein could certainly fuel both types of anterograde transport. Nevertheless, it is apparent that other motors, presumably kinesins, are also involved. This would be essential, in particular, for the retrograde movements, which do not appear to be dynein dependent. The kinesin-related protein termed Eg5 may be a good candidate for the retrograde motor, given that inhibition of Eg5 causes a transient increase in the rate of axonal growth that would be consistent with an increase in the ratio of anterograde to retrograde microtubule movements (Haque et al., 2004). Certain kinesins might also contribute to the anterograde transport, because we have shown that the kinesin-related protein termed CHO2 can cause microtubules to move anterogradely into axon-like processes (Sharp et al., 1997). We look forward to future studies of the particular motor proteins involved in transporting axonal microtubules and of the functional roles played by the particular movements that are indeed actin dependent.
Footnotes
This work was supported by two grants (P.W.B.) and a postdoctoral training grant (T.P.H.) from National Institutes of Health. We thank Niels Galjart for providing the GFP-EB3 construct and for ongoing advice and assistance.
Correspondence should be addressed to Peter W. Baas, Department of Neurobiology and Anatomy, Drexel University College of Medicine, 2900 Queen Lane, Philadelphia, PA 19129. E-mail: peter.w.baas@drexel.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2411291-11$15.00/0
T.P.H. and K.A.M. contributed equally to this work.
References
- Abal M, Piel M, Bouckson-Castaing V, Mogensen M, Sibarita J, Bornens M (2002) Microtubule release from the centrosome in migrating cells. J Cell Biol 159: 731-737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad FJ, Baas PW (1995) Microtubules released from the neuronal centrosome are transported into the axon. J Cell Sci 108: 2761-2769. [DOI] [PubMed] [Google Scholar]
- Ahmad FJ, Echeverri CJ, Vallee RB, Baas PW (1998) Cytoplasmic dynein and dynactin are required for the transport of microtubules into the axon. J Cell Biol 140: 391-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad FJ, Hughey J, Wittmann T, Hyman A, Greaser M, Baas PW (2000) Motor proteins regulate force interactions between microtubules and microfilaments in the axon. Nat Cell Biol 2: 276-280. [DOI] [PubMed] [Google Scholar]
- Baas PW (1999) Microtubules and neuronal polarity: lessons from mitosis. Neuron 22: 23-31. [DOI] [PubMed] [Google Scholar]
- Baas PW, Ahmad FJ (1993) The transport properties of axonal microtubules establish their polarity orientation. J Cell Biol 120: 1427-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas PW, Black MM (1990) Individual microtubules in the axon consist of domains that differ in both composition and stability. J Cell Biol 111: 495-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas PW, Buster DW (2004) Slow axonal transport and the genesis of neuronal morphology. J Neurobiol 58: 3-17. [DOI] [PubMed] [Google Scholar]
- Bearer EL, Reese TS (1999) Association of actin filaments with axonal microtubule tracts. J Neurocytol 28: 85-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho P, Tirnauer JS, Pellman D (2003) Surfing on microtubule ends. Trends Cell Biol 13: 229-237. [DOI] [PubMed] [Google Scholar]
- Dent EW, Kalil K (2001) Axon branching requires interactions between dynamic microtubules and actin filaments. J Neurosci 21: 9757-9769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent EW, Callaway JL, Szebenyi G, Baas PW, Kalil K (1999) Reorganization and movement of microtubules in axonal growth cones and developing interstitial branches. J Neurosci 19: 8894-8908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillman III JF, Dabney LP, Karki S, Paschal BM, Holzbaur EL, Pfister KK (1996) Functional analysis of dynactin and cytoplasmic dynein in slow axonal transport. J Neurosci 16: 6742-6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo G, Letourneau PC (1999) Different contributions of microtubule dynamics and transport to the growth of axons and collateral sprouts. J Neurosci 19: 3860-3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon-Weeks PR (2004) Microtubules and growth cone function. J Neurobiol 58: 70-83. [DOI] [PubMed] [Google Scholar]
- Haque SA, Hasaka TP, Brooks AD, Lobanov PV, Baas PW (2004) Monastrol, a prototype anti-cancer drug that inhibits a mitotic kinesin, induces rapid bursts of axonal outgrowth from cultured postmitotic neurons. Cell Motil Cytoskeleton 58: 10-16. [DOI] [PubMed] [Google Scholar]
- He Y, Baas PW (2003) Growing and working with peripheral neurons. Methods Cell Biol 71: 17-35. [DOI] [PubMed] [Google Scholar]
- He Y, Yu W, Baas PW (2002) Microtubule reconfiguration during axonal retraction induced by nitric oxide. J Neurosci 22: 5982-5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karabay A, Yu W, Solowska JM, Baird DH, Baas PW (2004) Axonal growth is sensitive to the levels of Katanin, a protein that severs microtubules. J Neurosci 24: 5778-5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating TJ, Peloquin JG, Rodionov VI, Momcilovic D, Borisy GG (1997) Microtubule release from the centrosome. Proc Natl Acad Sci USA 94: 5078-5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krylyshkina O, Anderson KI, Kaverina I, Upmann I, Manstein DJ, Small JV, Toomre DK (2003) Nanometer targeting of microtubules to focal adhesions. J Cell Biol 161: 853-859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasek RJ (1986) Polymer sliding in axons. J Cell Sci Suppl 5: 161-179. [DOI] [PubMed] [Google Scholar]
- Lee H, Van Vactor D (2003) Neurons take shape. Curr Biol 13: R152-R161. [DOI] [PubMed] [Google Scholar]
- Lee S, Kolodziej PA (2002) Short Stop provides an essential link between f-actin and microtubules during axon extension. Development 129: 1195-1204. [DOI] [PubMed] [Google Scholar]
- Lim SS, Sammak PJ, Borisy GG (1989) Progressive and spatially differentiated stability of microtubules in developing neuronal cells. J Cell Biol 109: 253-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Shakiryanova D, Vardya I, Popov SV (2004) Quantitative analysis of microtubule transport in growing nerve processes. Curr Biol 14: 725-730. [DOI] [PubMed] [Google Scholar]
- Nakagawa H, Koyama K, Murata Y, Morito M, Akiyama T, Nakamura Y (2000) EB3, a novel member of the EB1 family preferentially expressed in the central nervous system, binds to a CNS-specific APC homologue. Oncogene 19: 210-221. [DOI] [PubMed] [Google Scholar]
- Ozer RS, Halpain S (2000) Phosphorylation-dependent localization of microtubule-associated protein MAP2c to the actin cytoskeleton. Mol Biol Cell 11: 3573-3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister KK (1999) Cytoplasmic dynein and microtubule transport in the axon: the actin connection. Mol Neurobiol 20: 81-91. [DOI] [PubMed] [Google Scholar]
- Rochlin MW, Dailey ME, Bridgman PC (1999) Polymerizing microtubules activate site-directed F-actin assembly in nerve growth cones. Mol Biol Cell 10: 2309-2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sammak PJ, Borisy GG (1988) Direct observation of microtubule dynamics in living cells. Nature 332: 724-726. [DOI] [PubMed] [Google Scholar]
- Schaefer AW, Kabir N, Forscher P (2002) Filopodia and actin arcs guide the assembly and transport of two populations of microtubules with unique dynamic parameters in neuronal growth cones. J Cell Biol 158: 139-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp DJ, Kuriyama R, Essner R, Baas PW (1997) Expression of a minus-end-directed motor protein induces Sf9 cells to form axon-like processes with uniform microtubule polarity orientation. J Cell Sci 110: 2373-2380. [DOI] [PubMed] [Google Scholar]
- Slaughter T, Wang J, Black MM (1997) Microtubule transport from the cell body into the axons of growing neurons. J Neurosci 17: 5807-5819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanova T, Slemmer J, Hoogenraad CC, Lansbergen G, Dortland B, De Zeeuw CI, Grosveld F, van Cappellen G, Akhmanova A, Galjart N (2003) Visualization of microtubule growth in cultured neurons via the use of EB3-GFP (End-Binding Protein 3-Green Fluorescent Protein). J Neurosci 23: 2655-2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Goldberg DJ (2000) Bundling of microtubules in the growth cone induced by laminin. Mol Cell Neurosci 15: 303-313. [DOI] [PubMed] [Google Scholar]
- Vallee RB, Bloom GS (1991) Mechanisms of fast and slow axonal transport. Annu Rev Neurosci 14: 59-92. [DOI] [PubMed] [Google Scholar]
- Vallee RB, Williams JC, Varma D, Barnhart LE (2004) Dynein: an ancient motor protein involved in multiple modes of transport. J Neurobiol 58: 189-200. [DOI] [PubMed] [Google Scholar]
- Wang L, Brown A (2002) Rapid movement of microtubules in axons. Curr Biol 12: 1496-1501. [DOI] [PubMed] [Google Scholar]
- Yu W, Baas PW (1994) Changes in microtubule number and length during axon differentiation. J Neurosci 14: 2818-2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Ling C, Baas PW (2001) Microtubule reconfiguration during axogenesis. J Neurocytol 30: 861-875. [DOI] [PubMed] [Google Scholar]
- Zhou F, Waterman-Storer CM, Cohan CS (2002) Focal loss of actin bundles causes microtubule redistribution and growth cone turning. J Cell Biol 157: 839-849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmuda JF, Rivas RJ (2000) Actin disruption alters the localization of tau in the growth cones of cerebellar granule neurons. J Cell Sci 113: 2797-2809. [DOI] [PubMed] [Google Scholar]