

Abstract
The treatment of acute myeloid leukemia (AML) has remained relatively unchanged for the past three to four decades with generally poor outcomes, especially in elderly populations unfit for intensive therapy. Recent advancements, however, have identified several cytogenetic and molecular markers that have not only improved prognostication, but have also led to the development of several new targeted therapies for specific subpopulations. In 2017, the U.S. Food and Drug Administration (FDA) approved four new treatments with indications for FLT3-mutated AML (midostaurin), newly diagnosed or relapsed/refractory CD33+ AML (gemtuzumab ozogamicin), newly diagnosed therapy-related AML or AML with myelodysplasia-related changes (CPX-351), and relapsed/refractory AML with an IDH2 mutation (enasidenib). These newly approved therapies have demonstrated improved response in their target populations in several pivotal clinical trials with some also demonstrating improved overall survival. Additional novel therapies in development for AML include agents that target BCL2, FLT3, IDH1, the ubiquitination pathway, as well as cell therapy using engineered T-cells with chimeric antigen receptors (CAR-T cells). This review provides a summary of the four newly approved therapies for AML, as well as several promising therapies currently in development.
Keywords: Acute Myeloid Leukemia, Molecular, Targeted, Therapy
INTRODUCTION
Acute myeloid leukemia (AML) is a heterogeneous malignant disorder of the myeloid lineage characterized by uncontrolled clonal proliferation of immature cells [1]. Although AML may be found on routine blood cell count testing, definitive diagnosis routinely involves bone marrow biopsy with morphologic, immunohistochemical, flow cytometric, cytogenetic and molecular analyses. Per the 2016 World Health Organization (WHO) update on myeloid disorders, AML is diagnosed by 20% or greater myeloblasts in the peripheral blood or bone marrow [2]. The WHO system classifies AML into several subtypes, including AML with recurrent genetic abnormalities, AML with myelodysplasia-related changes, therapy-related myeloid neoplasms, and AML not otherwise specified. The incidence of AML ranges between 3 to 8 cases per 100,000 adults yearly, with 9 to 17 cases per 100,000 adults aged 65 or greater [3]. Several risk factors for the development of AML have been identified, including antecedent hematologic malignancies, chemotherapy and radiation exposure, and certain congenital disorders [4]. Median age of diagnosis ranges between 67 to 70 years of age. Median overall survival (OS) at 5 years for patients younger than 60 years is approximately 40%, compared to approximately 10% for patients greater than 60 years [1]. In older patients who are unable to undergo leukemia-specific therapy, survival typically ranges from around 4 months with best supportive care to approximately 10 months with lower-intensity therapies such as azacitidine, a hypomethylating agent [5].
Although the molecular heterogeneity of AML has become better appreciated over the past decade, the standard treatment for AML had not changed for the past three to four decades until multiple new drug approvals in 2017. Treatment for AML includes remission induction chemotherapy and post-remission consolidation therapy including chemotherapy and allogeneic hematopoietic stem cell transplantation (allo-HSCT). In eligible adults with newly diagnosed AML, the traditional standard therapy is “7+3” induction chemotherapy with infusional cytarabine for 7 days with 3 days of anthracycline chemotherapy (daunorubicin or idarubicin) [6]. A complete remission (CR) is achieved in 60 to 85% of patients younger than the age of 60, and in 40 to 60% of patients greater than the age of 60 [7]. Patients with a favorable genetic risk profile typically undergo additional consolidation chemotherapy, whereas those with intermediate-risk AML undergo either chemotherapy or allo-HSCT and those with unfavorable-risk AML are recommended for allo-HSCT [8]
Several genetic lesions and morphologic features have been identified as prognostic markers with clinical significance, and importantly, some carry predictive/actionable value (Table 1). During initial diagnostic workup, European LeukemiaNet (ELN) recommends screening for mutations in NPM1, CEBPA, RUNX1, ASXL1 and FLT3 at diagnosis [8]. Mutations in isocitrate dehydrogenase 2 (IDH2) are also often tested for initially, due to their newly actionable status in the relapsed or refractory (R/R) setting. Mutations in NPM1 are found in 25 to 35% of patients with AML, most frequently associated with normal karyotype [1]. Isolated mutations in NPM1 are associated with favorable prognosis [9], and in this population NPM1 has been used as a pilot genetic marker for assessing minimal residual disease after induction [10]. Bi-allelic mutations in CEBPA are associated with a favorable outcome, as well as with familial AML, and are found in 6 to 10% of patients. FLT3-internal tandem duplication (FLT3-ITD) mutations are found in approximately 20% of patients with AML, most often associated with normal cytogenetics, and are associated with an unfavorable outcome in some patients [11]. Patients with a mutation in FLT3-ITD may benefit from allo-HSCT in first CR as well as from tyrosine kinase inhibitors (TKIs) that have either been approved or are in clinical trials. Despite the rapid development of prognostic and predictive molecular characteristics, history and morphologic, immunophenotypic and cytogenetic evaluation at diagnosis remain crucial as there are newly-approved frontline agents shown to have OS benefit with myelodysplasia-related changes or therapy-related AML and AML expressing CD33. This article aims to review the four recently approved AML therapies as well as some in advanced development, as the landscape of AML therapy has changed more in the last year than in many decades prior.
Table 1:
New Targets for Novel AML Therapies
| Target | Frequency in AML | Prognostic Impact | Role in Leukemogenesis |
|---|---|---|---|
| FLT3 (FLT3-ITD/FLT3-TKD)[12] | Mutated in 20-37% (30% ITD and 7% TKD) | Associated with unfavorable outcome in some patients | Leads to production of proteins that circumvent ligandmediated activation, and cause factor-independent growth |
| CD33[13] | Expressed in approximately 85-90% | Outcomes are dependent on coexisting mutations | Not applicable; however, of note, CD33 is highly expressed on most AML blast cells and less on normal hematopoietic cells |
| IDH1, IDH2[12, 14] | IDH1 mutated in 6-16%; IDH2 mutated in 8-19% | Conflicting evidence on prognostic significance | Leads to synthesis of 2-hydroxyglutarate, which results in DNA and histone hypermethylation and ultimately impedes cellular differentiation |
| BCL2[15, 16] | Wide degree of heterogeneity in expression (mean expression of 23% in one study, but as high as 71% in a different study) | Outcomes are dependent on coexisting mutations | Overexpression of gene can lead to impaired cellular apoptosis |
| NEDD8 Activating Enzyme (NAE)[17] | Variable expression in all cell lines including AML stem cells | Unknown impact on outcomes | Leads to protein (including proapoptotic protein) degradation and DNA repair to prevent cellular instability |
| CD123[18, 19] | Expressed in between 44-88% of AML blasts | CD123 expression is associated with high risk disease and disease progression | Overexpression of CD123 may lead to resistance to apoptosis |
Abbreviations: AML, acute myeloid leukemia; FLT3, fms like tyrosine kinase 3; FLT3-ITD, fms like tyrosine kinase 3- internal tandem duplication; FLT3-TKD, fms like tyrosine kinase 3-tyrosine kinase domain; IDH1, isocitrate dehydrogenase 1; IDH2, isocitrate dehydrogenase 2; BCL2, B-cell Lymphoma 2; NEDD8, Neural Precursor Cell Expressed, Developmentally Down-Regulated 8
NEWLY-APPROVED THERAPIES FOR AML
Midostaurin
Midostaurin (Rydapt, Novartis Pharmaceuticals, Inc.) is a multi-targeted kinase inhibitor that was the first of four novel therapies to receive FDA approval for treatment of AML in 2017. As discussed, FLT3 mutations are found in approximately 20-30% of patients with newly diagnosed AML, and are associated with a poor outcome. Most FLT3 mutations are of the internal tandem duplication mutation (ITD) subtype, which is associated with a high relapse rate when there is high mutant to wild-type FLT3 allelic ratio. A smaller subset of FLT3 mutations are of the tyrosine kinase domain (TKD) subtype. Midostaurin was evaluated in a large, international phase III study, the CALGB 10603/RATIFY trial (Table 2). In this randomized, double-blinded, placebo-controlled study, patients younger than age 60 with newly diagnosed FLT3-mutated (both ITD and TKD) AML were randomized to standard 7+3 chemotherapy with either midostaurin or placebo [20]. Patients who achieved CR following induction therapy received four cycles of consolidation therapy with high-dose cytarabine (HiDAC) and midostaurin/placebo maintenance for up to 1 year. Allo-HSCT was performed in 25% of patients who achieved first CR, although this was not mandated by study protocol. The study demonstrated that combination 7+3 chemotherapy and midostaurin led to a significant improvement in overall survival, with a median OS of 74.7 months in the midostaurin group versus 25.6 months (hazard ratio for death 0.78, p=0.009) with placebo. Subgroup analysis between the low allelic ratio ITD, high allelic ratio ITD, and TKD groups did not show any significant difference in OS. The rate of serious adverse effects (AEs) was not significantly different between the two treatment arms, with the exception of a higher rate of grade 3 or greater rash/desquamation and nausea in the midostaurin arm. The most common AEs associated with midostaurin include nausea, vomiting, headache, fever, and febrile neutropenia. As patients in this study were exposed to midostaurin for induction, consolidation and maintenance, the specific benefit and optimal duration of midostaurin maintenance is unclear.
Table 2:
Newly-Approved AML Therapies
| Therapy | Indication | Key Outcomes | Key Adverse Events | Ref# |
|---|---|---|---|---|
| Midostaurin | Newly diagnosed FLT3+ AML | Per the CALGB 10603/RATIFY trial, median OS of 74.7 months in the midostaurin group versus 25.6 months in placebo group | Grade 3 or greater rash/desquamation and nausea | [20] |
| CPX-351 | Newly diagnosed therapy-related AML or AML with myelodysplastic-related changes | Phase III trial comparing CPX-351 with 7+3 chemotherapy showed superior OS (9.56 months versus 5.95 months) in the CPX-351 treatment arm, as well as CR and CRi response rates (47.7% versus 33.3%) | Hemorrhage, neutropenia, hypersensitivity reactions | [30] |
| Gemtuzumab ozogamicin | Newly diagnosed CD33+ AML or R/R CD33+ AML | The 2012 ALFA-0701 study compared low fractionated-dose GO with standard first-line chemotherapy, and found an improvement in median OS in the GO arm (34 months versus 19.2 months, p = 0.046) | Transaminitis, and veno-occlusive disease (rare but severe complication) | [24] |
| Enasidenib | IDH2-mutant R/R AML | CR or CRi in 26.6% of patients, with an additional 12% of patients with partial response for an overall response rate of 38.5%. Median OS was 9.3 months overall, with a median OS of 19.7 months in patients with CR | Hyperleukocytosis, and IDH-inhibitor-associated differentiation syndrome (IDH-DS) | [31] |
Abbreviations: FLT3, fms like tyrosine kinase 3; AML, acute myeloid leukemia; OS, overall survival; CR, complete remission; CRi, complete remission with incomplete hematologic response; R/R, relapsed/refractory; IDH2, isocitrate dehydrogenase 2.
Gemtuzumab ozogamicin (GO)
Gemtuzumab ozogamicin (Mylotarg ; Pfizer, Inc.) is a humanized immunoglobulin antibody directed against CD33 and conjugated with calicheamicin, a cytotoxic antibiotic. CD33 is a transmembrane receptor that is highly expressed on blasts of most patients with AML. GO promotes single- and double-strand breaks in DNA, ultimately leading to cell death. GO initially received accelerated FDA approval for CD33+ AML in 2000 based on CR rates in nonrandomized phase II studies. However, a follow-up phase III study [21] in patients with untreated AML comparing standard 7+3 induction chemotherapy plus GO versus 7+3 alone found a higher mortality rate in the GO arm (5.5% versus 1.4%), without any improvement in CR. This led to the withdrawal of GO from the market in 2010. Subsequent trials, however, showed improved OS rates without increased mortality rates [22, 23]. The phase III randomized ALFA-0701 study investigated low fractionated-dose GO with standard first-line chemotherapy in patients with de novo AML [24], and found an improvement in median OS in the GO arm (34 months versus 19.2 months, p = 0.046). In subgroup analysis, this benefit was limited to patients with favorable-risk and intermediate-risk karyotypes. Subsequent meta-analysis found that among 3,325 patients in five randomized controlled trials, the addition of GO reduced the risk of relapse and improved OS at five years (34.6% versus 30.7%) [25]. Additionally, at six years, survival benefit was significant in patients with favorable-risk (76.3% versus 55.2%) and intermediate-risk (39.4% versus 34.1%) cytogenetics. These studies including the meta-analysis led to reapplication for FDA approval (at the lower, fractionated dosing), which was granted in September 2017 for patients with newly diagnosed CD33+ AML and also for R/R CD33+ AML, based on the MyloFrance-1 study [26] (Table 2). The most common AEs include nausea, vomiting, transaminitis (elevations in AST or ALT), and hemorrhage. Although GO still retains its black box warning for veno-occlusive disease, events using current approved dosing were rare (2%), and can be associated with prior alkylator chemotherapy.
CPX-351 (Liposomal daunorubicin and cytarabine)
CPX-351 (Vyxeos ; Jazz Pharmaceuticals) is a liposomal formulation of cytarabine and daunorubicin (the standard agents in 7+3 induction chemotherapy) in a fixed, synergistic 5:1 molar ratio. Prior pre-clinical studies had demonstrated that molar ratios of cytarabine and daunorubicin between 1:1 and 10:1 were synergistic, whereas lower ratios were antagonistic [27]. Although CPX-351 is not a targeted therapy, the fixed 5:1 molar ratio and liposomal packaging demonstrated more effective delivery and cytotoxicity to AML blasts. A multicenter, randomized phase II trial compared CPX-351 with investigator’s choice of chemotherapy for patients with AML in first relapse [28]. The study demonstrated an improvement in the rate of CR or CR with incomplete hematologic recovery (CRi) in the CPX-351 arm (49.4% versus 40.9%) as well as improvement in median OS in the CPX-351 arm (8.5 months versus 6.3 months). A randomized phase II trial in 2014 comparing CPX-351 to 7+3 chemotherapy in first-line treatment of AML in older patients did not demonstrate a statistically significant difference in OS between the study’s two arms; however, response rate was improved (57.6% versus 31.6%, p = 0.06) in patients with secondary AML [29]. This latter phase II trial led to a phase III trial comparing CPX-351 versus 7+3 chemotherapy in patients 60-75 years of age with high-risk secondary AML (untreated therapy-related AML, antecedent myelodysplastic syndrome or CMML, or AML with myelodysplasia-related changes), and confirmed superior OS (9.56 months versus 5.95 months, p = 0.005) in the CPX-351 arm, as well as CR/CRi rates (47.7% versus 33.3%, p = 0.016) [30]. Severe AEs were similar in both treatment arms. CPX-351 subsequently received FDA approval through Priority Review and Breakthrough Therapy designations for newly diagnosed therapy-related AML or AML with myelodysplasia-related changes in August 2017 (Table 2).
Enasidenib
Enasidenib (Idhifa, Agios Pharmaceuticals, Inc. and Celgene Corporation) is a selective inhibitor of mutated IDH2. Mutations in IDH2 occur in approximately 10% of patients with AML [4], and lead to the synthesis of 2-hydroxyglutarate, which results in DNA and histone hypermethylation and impaired cellular differentiation. Enasidenib received FDA approval in August 2017 for IDH2-mutant R/R AML at a dosing of 100 mg orally once per day (Table 2). In the first-in-human phase I/II study published in 2017, enasidenib at the approved dose produced CR/(CRi) in 26.6% of patients with R/R AML [31]. An additional 12% of patients experienced either a partial response (PR) or achieved a morphologic leukemia-free state (where blasts are not increased but no recovery of any cell lineage has occurred), corresponding to an overall response rate (ORR) of 38.5%. Median time to best response was 3.7 months. Median OS was 9.3 months (95% CI 8.2 to 10.9 months) overall, with a median OS of 19.7 months in patients who achieved CR. Of note, individuals with clinical responses did not have a reduction in IDH2 mutant allele burden, reflecting a shift from undifferentiated to differentiated clonal hematopoiesis. The most common AEs from enasidenib included nausea and indirect hyperbilirubinemia. Hyperleukocytosis secondary to en masse differentiation was also seen infrequently (<4%), and was managed with hydroxyurea. The most distinct AE was IDH-inhibitor-associated differentiation syndrome (IDH-DS), which occurred in approximately 10% of patients in this study. IDH-DS typically occurred within 3-4 months of treatment initiation, and did not have a pathognomonic sign or symptom. IDH-DS typically presented with new or worsening dyspnea or hypoxemia, new or worsening pulmonary infiltrates on radiographs, new or worsening peripheral edema, and worsening renal function [32]. As there is no available confirmatory laboratory testing for IDH-DS, careful clinical evaluation for suspected IDH-HS is warranted. Based off the study authors’ experience, recommended treatment for IDH-DS is corticosteroids (e.g. dexamethasone 10 mg every 12 hours) started immediately, and to briefly hold enasidenib until there is improvement in symptoms.
THERAPEUTICS IN DEVELOPMENT FOR AML
Several novel anti-AML agents are in various stages of development. Examples are highlighted in Table 3 and described in the following sections.
Table 3:
Select Novel AML Therapies in Clinical Trials
| Therapy | Type | Target | Development Status |
Potential Patient Population |
|---|---|---|---|---|
| Venetoclax | Small molecule inhibitor | BCL2 | Randomized phase III | Newly diagnosed ineligible for standard induction |
| Sorafenib | Small molecule kinase inhibitor | Multikinase including FLT3 | Randomized phase II | FLT3-mutated newly diagnosed, R/R, and possible maintenance |
| Gilteritinib | Small molecule kinase inhibitor | FLT3-ITD/FLT3-TKD/AXL | Randomized phase III | FLT3-mutated, both newly diagnosed and R/R |
| Crenolanib | Small molecule kinase inhibitor | FLT3-ITD/FLT3-TKD | Randomized phase III | FLT3-mutated, both newly diagnosed and R/R |
| Quizartinib | Small molecule kinase inhibitor | FLT3-ITD | Randomized phase III | FLT3-mutated, both newly diagnosed and R/R |
| Ivosidenib | Small molecule | IDH1 | Randomized Phase III | Newly diagnosed IDH1-mutated ineligible for intensive therapy |
| Pevonedistat | Small molecule inhibitor | NAE | Randomized phase III | Newly diagnosed low blast count(20-30%) |
| CAR-T cells | Adoptive cell therapy | CD33/CD123/others | Phase I | R/R |
| Flotetuzumab | DART | CD123 | Phase I | R/R |
Abbreviations: BCL2, B-cell Lymphoma 2; FLT3-ITD, fms like tyrosine kinase 3- internal tandem duplication; FLT3-TKD, fms like tyrosine kinase 3- tyrosine kinase domain; IDH1, isocitrate dehydrogenase 1; NAE, Nedd8-activating enzyme; CAR-T, chimeric antigen receptor T-cells; DART, dual-affinity re-targeting antibody.
Venetoclax
Venetoclax is an oral targeted small molecule that inhibits BCL2, an important anti-apoptotic protein expressed in AML [33]. Venetoclax was approved in April 2016 for use in chronic lymphocytic lymphoma (CLL) with a 17p deletion after one prior therapy, and is undergoing clinical trial evaluation in many other hematologic neoplasms, including AML. Initial activity was modest (ORR 19%) as monotherapy in a nonrandomized phase II trial in R/R AML [34], but a subsequent nonrandomized phase II study combining venetoclax with hypomethylating agents azacitidine or decitabine in the upfront elderly population unfit for intensive chemotherapy showed an ORR of 67%. Primary AEs were cytopenias, as is seen in CLL. Only 5 of 145 patients died within 1 month of starting drug [35]. Based on these results and other phase II trials [36, 37], two phase III randomized clinical trials are under way in adults with AML ineligible for standard induction chemotherapy, one () evaluating venetoclax with azacitidine versus azacitidine alone, the other () studying venetoclax with low-dose cytarabine (LDAC) vs LDAC alone. These combinations may offer this population much more efficacious options, considering traditionally poor overall outcomes.
FLT3 Inhibitors
In addition to midostaurin, other FLT3 inhibitors are undergoing evaluation for use in AML. Sorafenib is a multi-kinase inhibitor currently approved for use in advanced hepatocellular carcinoma, renal cell carcinoma, and differentiated thyroid cancer. Preclinical data and earlyphase clinical trials suggested some activity in AML, which led to the German SORAML randomized phase II trial [38]. In 267 patients with previously untreated AML, the study compared the addition of sorafenib versus placebo to standard induction and consolidation chemotherapy and then sorafenib or placebo to 12 months of maintenance. Patients were not tested or selected for FLT3 mutations as part of the study protocol. Median event-free survival (EFS) was prolonged with sorafenib compared to placebo (21 months vs 9 months), with a significantly longer 3-year EFS (40% vs 22%, HR 0.64, p = 0.013), although adverse events of fever, diarrhea, bleeding, cardiac events, and hand-foot-skin reactions were increased. Nonrandomized data also suggests improved outcomes compared to historical controls with sorafenib added to induction and consolidation chemotherapy in those with FLT3-ITD and FLT3-TKD mutations [39] and in the relapsed setting after allo-HSCT [40].
Quizartinib is a more selective and potent TKI that inhibits FLT3-wild type and FLT3-ITD activity, but not FLT3-TKD [41]. An open-label phase II trial evaluated quizartinib monotherapy in two cohorts of patients with FTL3-ITD positive or negative R/R AML after second-line chemotherapy or allo-HSCT [42, 43]. The majority of the patients in both cohorts were FLT3-ITD positive. The two cohorts demonstrated promising efficacy of single-agent quizartinib with a composite CR rate (CRc, defined as the combination of CR, CR with incomplete platelet recovery and CRi) of 44-50% in R/R FLT3-ITD positive AML. Common AEs of quizartinib include myelosupression, QT prolongation, nausea, and diarrhea. Two phase III trials are currently investigating quizartinib, QuANTUM-First () which compares standard induction chemotherapy in addition to quizartinib or placebo in newly-diagnosed FTL3-ITD AML patients, and QuANTUM-R (), studying single-agent quizartinib versus salvage chemotherapy in R/R FTL3-ITD positive AML.
Gilteritinib, an inhibitor of both FLT3 and related kinase AXL, has shown promising response data in early phase trials and is now being studied in four phase III trials. In the first-in-human phase I/II study, 252 patients with R/R AML were enrolled, 25% had received prior TKI and 63% had FLT3 mutations. Diarrhea (16%) and fatigue (15%) were the most common treatment-related AEs. ORR was 12% in FLT3-wild-type patients, but was 52% in FLT3-mutated patients receiving ≥80 mg. This enriched group had a median OS of 31 weeks and median duration of response of 20 weeks [44]. Current phase III trials include the ADMIRAL study () in FLT3-mutated R/R AML compared to salvage chemotherapy. Gilteritinib is also being evaluated in the maintenance setting for FLT3-ITD-mutated patients after allo-HSCT compared to placebo (), and also in those patients not proceeding with transplant (). In addition, it is being studied in newly diagnosed patients with a FLT3 mutation not eligible for intensive induction chemotherapy in a three-arm study, comparing azacitidine alone, gilteritinib alone, and combination azacitidine and gilteritinib ().
Crenolanib is another second-generation FLT3 inhibitor which has activity in preclinical studies including against resistance mutations that develop with other FLT3 inhibitor therapy [45]. In a single-center open-label phase II study of R/R AML patients with either FLT3-ITD or FLT3-TKD mutations, crenolanib was studied as a single agent. Out of 34 evaluable patients, 13 were FLT3 TKI-naïve, and 21 had progressed on prior FLT3-TKI, and 10 had progressed after Allo-SCT. ORR was 47%, with 23% of the TKI-naïve cohort achieving CRi (5% in the prior TKI cohort). The primary toxicities were abdominal pain and nausea, with no deaths attributed to therapy [46]. Preliminary data from a phase II trial () evaluating crenolanib with 7+3 induction and consolidation in newly-diagnosed FLT3-mutated patients, similar to midostaurin’s approved indication, was presented at the ASH 2017 annual meeting. Out of 29 patients, 21 (72%) achieved CR after one induction cycle, three more achieved CR with second induction, high-dose cytarabine, and allo-HSCT. At a median followup of 14 months, one patient out of the 24 achieving CR died of post-transplant complications, and only two relapsed [47]. Crenolanib is now being studied in two randomized phase III studies, one comparing directly to midostaurin in the newly diagnosed setting with induction chemotherapy (), and the other in the R/R setting combined with salvage chemotherapy ().
Ivosidenib (AG120)
Isocitrate dehydrogenase 1 (IDH1) is mutated in approximately 9-16% of AML patients who are most often cytogenetically normal. IDH1 mutations have thus far found to be mutually exclusive with IDH2 mutations [48, 49]. Ivosidenib (AG120, Agios Pharmaceuticals) is a novel IDH1 inhibitor in development, and has been studied in a phase I trial presented at the 2017 ASH annual meeting [50]. In this first-in-human safety study in mutant IDH1 advanced hematologic malignancies including R/R AML, 258 patients received ivosidenib including dose escalation (78 patients) and dose expansion (180 patients) phases. The highest frequency AEs were diarrhea (33%), leukocytosis (30%), nausea (30%), fatigue (29%), febrile neutropenia (25%), dyspnea (24%), anemia (23%), and QT prolongation (23%), most of these being low grade and unrelated to therapy. Differentiation syndrome (DS), as seen with IDH2 inhibitor enasidenib, was seen in 29 patients (11.2%), but no deaths occurred due to DS. Regarding efficacy, ORR was 41.6%, including 21.6% achieving CR and 8.8% with CR with partial hematologic recovery (CRh). Median duration of CR+CRh was 8.2 months. Ivosidenib is currently being studied in a global randomized phase III trial () in untreated IDH1-mutated AML combined with azacitidine or placebo.
Pevonedistat (MLN4924)
Further understanding of AML biology has revealed defects in the ubiquitination/protein degradation system including a process called neddylation, essential for some ubiquitin ligases and subsequent degradation of their substrates [51, 52]. Pevonidostat is a first-in-class inhibitor of the NEDD8-activating enzyme that works by causing accumulation of otherwise-degraded proteins with antiproliferative effects [17]. After preclinical experiments demonstrated increased reactive oxygen species and apoptosis in AML cell lines and animal models when combined with azacitidine [53, 54], pevonidostat was studied in phase I clinical trials, first in a monotherapy safety study which observed a few CR and PR in a heavily pretreated population [55], and then in a phase Ib study in combination with azacitidine in older AML patients who had not received cytotoxic chemotherapy [56]. Here, dose-limiting toxicities were elevated bilirubin and transaminases, but were reversible without clinical sequelae. The ORR in combination with azacitidine was 60% with a CR/CRi rate of 47% that was not influenced by disease status (de novo versus secondary), blast count or cytogenetic risk. After a median follow-up of 16.4 months, median OS was seven months and 12-month survival was 45%. Based on these results, a randomized phase III study of pevonedistat and azacitidine versus azacitidine for patients with treatment naïve higher risk MDS, chronic myelomonocytic leukemia and low blast count AML (up to 30% blasts) is ongoing ().
Adoptive Cell Therapy
Novel cell therapies using engineered T-cells with chimeric antigen receptors (CAR-T cells) are now approved for B-cell acute lymphoblastic lymphoma and R/R large B-cell lymphoma. In AML, preclinical studies have used targets such as CD33 and CD123 with efficacy in vitro [57, 58], but the heterogeneous nature of AML makes specificity and on-target, off-tumor effects an issue in vivo. Wang, et al. reported treatment of a single patient with refractory AML with CD33-directed CAR-T cell therapy, without any conditioning chemotherapy [59]. Within two weeks, the patient’s bone marrow blast percentage decreased from >50% before infusion to <6%. AEs included cytokine-release syndrome (CRS) and worsened cytopenias. Etanercept, an anti-TNF-alpha agent, was administered for anti-inflammatory effects. Unfortunately, despite initial clinical response, blast percentage rose to 70% at 9 weeks with retention of CD33 expression, suggesting an escape mechanism other than CAR target loss. Two phase I studies in China in R/R AML are currently ongoing, with the first studying autologous CD33-directed CAR-Ts (), and the second study using allogeneic CAR-T therapy ().CD123 is another promising target in AML, as it is expressed on AML blasts and leukemic stem cells, but minimally on hematopoietic stem cells [18]. Several other phase I CAR-T trials are currently accruing patients using CD123, CD123/CD28 or CD123/CD33 combinations, among other targets. Overall, major challenges remain regarding elucidation of the best targets or combinations of, CRS, and co-stimulatory molecules that improve efficacy while balancing toxic off-tumor effects. The ideal preparative regimens are also yet to be defined.
Flotetuzumab
Flotetuzumab is a dual-affinity re-targeting antibody (DART), which engages both CD123-expressing AML cells and T-cells via CD3. The dual-targeting antibody-based approach is similar in principle to bispecific T-cell engagers such as blinatumomab, which is currently approved for B-ALL. Preliminary data from a phase I study using flotetuzumab in R/R AML and high-risk MDS was reported at ASH 2017 [60]. Forty-five patients were enrolled, of which 89% had AML. A lower lead-in dose of flotetuzumab was given for the first week to help mitigate CRS. Drug-related grade 3 or worse AEs were experienced in 44% of patients, the most common toxicity being CRS/infusion-related reaction in 76% of all patients (any grade). Fourteen patients were treated with the maximum tolerated dose and schedule or greater and completed at least one cycle. Six of these (43%) had an objective response (3 CR, 1 CRi, 1 MLFS, 1 PR), two more had stable disease, and correlative studies showed an increase in T-cell activating cytokine levels and CD8-positive T-cells in the bone marrow.
CONCLUSIONS
In summary, 2017 has been a pivotal year for AML clinicians, researchers, and patients. As understanding of AML biology improves, particularly its molecular underpinnings, novel prognostic and targetable subpopulations are being defined (Figure 1). The four newly-approved therapies have changed the standard of care for their respective populations. For those newly diagnosed, fit for intensive therapy and with favorable and intermediate-risk cytogenetic and molecular features, the addition of gemtuzumab ozogomicin improves overall survival when added to 7+3 chemotherapy. For fit patients with a FLT3 mutation, midostaurin significantly improves median OS when added to induction chemotherapy. In newly diagnosed, fit older patients with secondary AML or therapy-related AML, liposomal daunorubicin and cytarabine improves OS and response rate compared to 7+3 with similar rates of AEs. For younger/fit patients without the above features, traditional 7+3 remains the standard upfront therapy. For newly diagnosed unfit patients not eligible for intensive chemotherapy, hypomethylating agents remain the standard of care, however several emerging therapeutics are currently in clinical trial testing utilizing novel targets. In the R/R setting, enasidenib now offers a noncytotoxic option for patients with IDH2 mutations via its differentiating effect. Venetoclax targets BCL2 and is in late phase combination trials for patients not eligible for induction. Multi-kinase and specific, potent inhibitors of FLT3, including sorafenib, crenolanib, gilteritinib, and quizartinib, are also in late phase trials. Some may be best suited for adjunct upfront usage with chemotherapy like midostaurin, while others may have single agent activity in the R/R setting. Adoptive cell therapy and antibody-based immunotherapy are novel approaches earlier in development that aim to improve anti-leukemia T-cell function. Certainly, the AML landscape is now already dramatically changed, and much promise awaits in the years to come.
Figure 1.
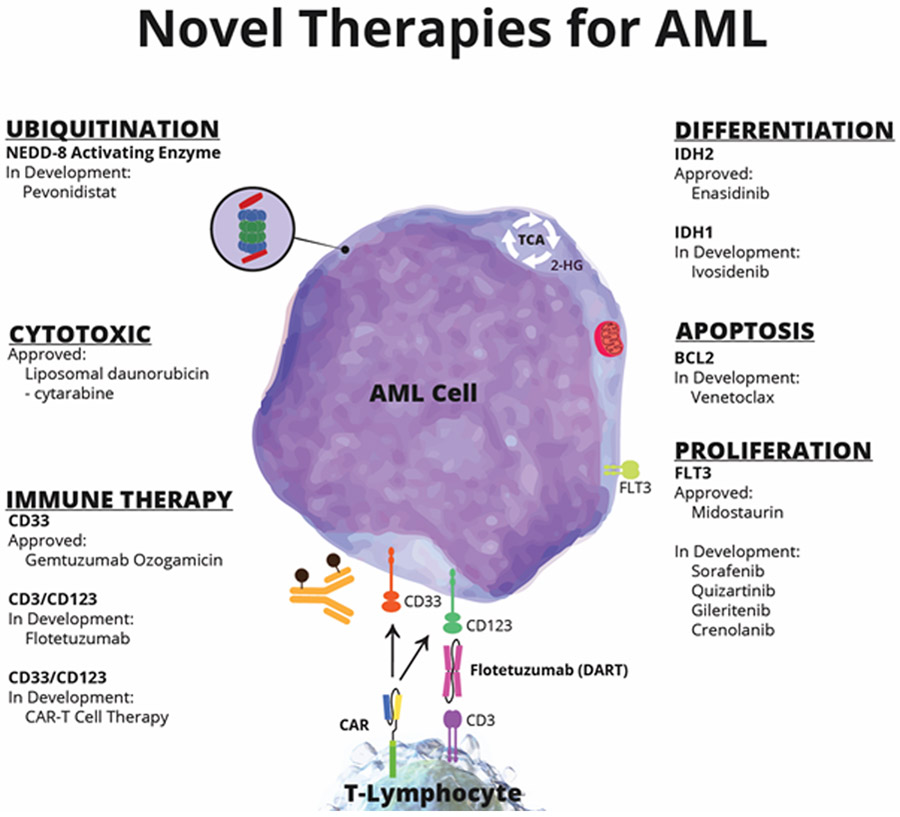
Novel AML Targets and Therapies
Abbreviations: NEDD-8, neural precursor cell expressed, developmentally down-regulated 8-activating enzyme; IDH, isocitrate dehydrogenase; BCL2, B-cell Lymphoma 2; FLT3, fms like tyrosine kinase 3; CAR, chimeric antigen receptor; DART, dual-affinity re-targeting antibody.
Acknowledgements
Dr. Jonas received support by the National Institutes of Health National Cancer Institute (K12 CA138464-04).
Footnotes
Conflicts of Interest
Davis – None.
Benjamin – None.
Jonas – consultant for Rigel, AbbVie, Amgen; advisory board member for Celgene; independent response review committee for Tolero; research funding to his institution from AbbVie, Daiichi Sankyo, Pharmacyclics, Genentech/Roche, Glycomimetics, Esanex, Kalobios, Incyte.
References
- 1.Dohner H, Weisdorf DJ and Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med. 2015; 373:1136–1152 doi: 10.1056/NEJMra1406184 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016; 127:2391–2405 doi: 10.1182/blood-2016-03-643544 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Kayser S and Levis MJ. Advances in targeted therapy for acute myeloid leukaemia. Br J Haematol. 2018; 180:484–500 doi: 10.1111/bjh.15032 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med. 2016; 374:2209–2221 doi: 10.1056/NEJMoa1516192 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009; 10:223–232 doi: 10.1016/S1470-2045(09)70003-8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roboz GJ. Current treatment of acute myeloid leukemia. Curr Opin Oncol. 2012; 24:711–719 doi: 10.1097/CCO.0b013e328358f62d (2012). [DOI] [PubMed] [Google Scholar]
- 7.Sekeres MA. Treatment of older adults with acute myeloid leukemia: state of the art and current perspectives. Haematologica. 2008; 93:1769–1772 doi: 10.3324/haematol.2008.000497 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017; 129:424–447 doi: 10.1182/blood-2016-08-733196 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y, He P, Liu F, et al. Prognostic significance of NPM1 mutations in acute myeloid leukemia: A meta-analysis. Mol Clin Oncol. 2014; 2:275–281 doi: 10.3892/mco.2013.222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ivey A, Hills RK, Simpson MA, et al. Assessment of Minimal Residual Disease in Standard-Risk AML. N Engl J Med. 2016; 374:422–433 doi: 10.1056/NEJMoa1507471 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Kayser S and Levis MJ. FLT3 tyrosine kinase inhibitors in acute myeloid leukemia: clinical implications and limitations. Leuk Lymphoma. 2014; 55:243–255 doi: 10.3109/10428194.2013.800198 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012; 366:1079–1089 doi: 10.1056/NEJMoa1112304 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Propris MS, Raponi S, Diverio D, et al. High CD33 expression levels in acute myeloid leukemia cells carrying the nucleophosmin (NPM1) mutation. Haematologica. 2011; 96:1548–1551 doi: 10.3324/haematol.2011.043786 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DiNardo CD, Ravandi F, Agresta S, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol. 2015; 90:732–736 doi: 10.1002/ajh.24072 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kornblau SM, Thall PF, Estrov Z, et al. The prognostic impact of BCL2 protein expression in acute myelogenous leukemia varies with cytogenetics. Clin Cancer Res. 1999; 5:1758–17661999). [PubMed] [Google Scholar]
- 16.Campos L, Rouault JP, Sabido O, et al. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood. 1993; 81:3091–30961993). [PubMed] [Google Scholar]
- 17.Swords RT, Kelly KR, Smith PG, et al. Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood. 2010; 115:3796–3800 doi: 10.1182/blood-2009-11-254862 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Testa U, Pelosi E and Frankel A. CD 123 is a membrane biomarker and a therapeutic target in hematologic malignancies. Biomark Res. 2014; 2:4 doi: 10.1186/2050-7771-2-4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graf M, Hecht K, Reif S, et al. Expression and prognostic value of hemopoietic cytokine receptors in acute myeloid leukemia (AML): implications for future therapeutical strategies. Eur J Haematol. 2004; 72:89–106 doi: 10.1046/j.0902-4441.2003.00184.x (2004). [DOI] [PubMed] [Google Scholar]
- 20.Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med. 2017; 377:454–464 doi: 10.1056/NEJMoa1614359 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petersdorf SH, Kopecky KJ, Slovak M, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood. 2013; 121:4854–4860 doi: 10.1182/blood-2013-01-466706 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burnett AK, Russell NH, Hills RK, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy improves survival in older patients with acute myeloid leukemia. J Clin Oncol. 2012; 30:3924–3931 doi: 10.1200/JCO.2012.42.2964 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Burnett AK, Hills RK, Milligan D, et al. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: results of the MRC AML15 trial. J Clin Oncol. 2011; 29:369–377 doi: 10.1200/JCO.2010.31.4310 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Castaigne S, Pautas C, Terre C, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet. 2012; 379:1508–1516 doi: 10.1016/S0140-6736(12)60485-1 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014; 15:986–996 doi: 10.1016/S1470-2045(14)70281-5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taksin AL, Legrand O, Raffoux E, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia. 2007; 21:66–71 doi: 10.1038/sj.leu.2404434 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Mayer LD, Harasym TO, Tardi PG, et al. Ratiometric dosing of anticancer drug combinations: controlling drug ratios after systemic administration regulates therapeutic activity in tumor-bearing mice. Mol Cancer Ther. 2006; 5:1854–1863 doi: 10.1158/1535-7163.MCT-06-0118 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Cortes JE, Goldberg SL, Feldman EJ, et al. Phase II, multicenter, randomized trial of CPX-351 (cytarabine:daunorubicin) liposome injection versus intensive salvage therapy in adults with first relapse AML. Cancer. 2015; 121:234–242 doi: 10.1002/cncr.28974 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lancet JE, Cortes JE, Hogge DE, et al. Phase 2 trial of CPX-351, a fixed 5:1 molar ratio of cytarabine/daunorubicin, vs cytarabine/daunorubicin in older adults with untreated AML. Blood. 2014; 123:3239–3246 doi: 10.1182/blood-2013-12-540971 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lancet JE, Uy GL, Cortes JE, et al. Final results of a phase III randomized trial of CPX-351 versus 7+3 in older patients with newly diagnosed high risk (secondary) AML. Journal of Clinical Oncology. 2016; 34:7000–7000 doi: 10.1200/JCO.2016.34.15_suppl.7000 (2016). [DOI] [Google Scholar]
- 31.Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017; 130:722–731 doi: 10.1182/blood-2017-04-779405 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fathi AT, DiNardo CD, Kline I, et al. Differentiation Syndrome Associated With Enasidenib, a Selective Inhibitor of Mutant Isocitrate Dehydrogenase 2: Analysis of a Phase 1/2 Study. JAMA Oncol. 2018; doi: 10.1001/jamaoncol.2017.4695 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Konopleva M, Zhao S, Hu W, et al. The anti-apoptotic genes Bcl-X(L) and Bcl-2 are over-expressed and contribute to chemoresistance of non-proliferating leukaemic CD34+ cells. Br J Haematol. 2002; 118:521–5342002). [DOI] [PubMed] [Google Scholar]
- 34.Konopleva M, Pollyea DA, Potluri J, et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016; 6:1106–1117 doi: 10.1158/2159-8290.CD-16-0313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DiNardo CD, Pratz KW, Letai A, et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol. 2018; 19:216–228 doi: 10.1016/S1470-2045(18)30010-X (2018). [DOI] [PubMed] [Google Scholar]
- 36.Pratz K, Pollyea D, Jonas B, et al. SAFETY AND EFFICACY OF VENETOCLAX (VEN) IN COMBINATION WITH DECITABINE OR AZACITIDINE IN TREATMENT-NAIVE, ELDERLY PATIENTS (>= 65 YEARS) WITH ACUTE MYELOID LEUKEMIA (AML). Haematologica. FERRATA STORTI FOUNDATION VIA GIUSEPPE BELLI 4, 27100; PAVIA, ITALY; 2017:175–176. [Google Scholar]
- 37.Wei A, Strickland S, Roboz G, et al. UPDATED SAFETY AND EFFICACY RESULTS OF PHASE 1/2 STUDY OF VENETOCLAX PLUS LOW-DOSE CYTARABINE IN TREATMENT-NAIVE ACUTE MYELOID LEUKEMIA PATIENTS AGED>= 65 YEARS AND UNFIT FOR STANDARD INDUCTION THERAPY. Haematologica. FERRATA STORTI FOUNDATION VIA GIUSEPPE BELLI 4, 27100; PAVIA, ITALY; 2017:176–176. [Google Scholar]
- 38.Rollig C, Serve H, Huttmann A, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015; 16:1691–1699 doi: 10.1016/S1470-2045(15)00362-9 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Uy GL, Mandrekar SJ, Laumann K, et al. A phase 2 study incorporating sorafenib into the chemotherapy for older adults with FLT3-mutated acute myeloid leukemia: CALGB 11001. Blood Adv. 2017; 1:331–340 doi: 10.1182/bloodadvances.2016003053 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rautenberg C, Nachtkamp K, Dienst A, et al. Sorafenib and azacitidine as salvage therapy for relapse of FLT3-ITD mutated AML after allo-SCT. Eur J Haematol. 2017; 98:348–354 doi: 10.1111/ejh.12832 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Levis M Quizartinib in acute myeloid leukemia. Clin Adv Hematol Oncol. 2013; 11:586–5882013). [PMC free article] [PubMed] [Google Scholar]
- 42.Cortes JE, Perl AE, Dombret H, et al. Final results of a phase 2 open-label, monotherapy efficacy and safety study of quizartinib (AC220) in patients≥ 60 years of age with FLT3 ITD positive or negative relapsed/refractory acute myeloid leukemia. Am Soc Hematology; 2012. [Google Scholar]
- 43.Levis MJ, Perl AE, Dombret H, et al. Final results of a phase 2 open-label, monotherapy efficacy and safety study of quizartinib (AC220) in patients with FLT3-ITD positive or negative relapsed/refractory acute myeloid leukemia after second-line chemotherapy or hematopoietic stem cell transplantation. Am Soc Hematology; 2012. [Google Scholar]
- 44.Perl AE, Altman JK, Cortes JE, et al. Final results of the Chrysalis trial: a first-in-human phase 1/2 dose-escalation, dose-expansion study of gilteritinib (ASP2215) in patients with relapsed/refractory acute myeloid leukemia (R/R AML). Am Soc Hematology; 2016. [Google Scholar]
- 45.Galanis A, Ma H, Rajkhowa T, et al. Crenolanib is a potent inhibitor of FLT3 with activity against resistance-conferring point mutants. Blood. 2014; 123:94–100 doi: 10.1182/blood-2013-10-529313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Randhawa JK, Kantaijian HM, Borthakur G, et al. Results of a phase II study of crenolanib in relapsed/refractory acute myeloid leukemia patients (Pts) with activating FLT3 mutations. Am Soc Hematology; 2014. [Google Scholar]
- 47.Wang ES, Tallman MS, Stone RM, et al. Low Relapse Rate in Younger Patients≤ 60 Years Old with Newly Diagnosed FLT3-Mutated Acute Myeloid Leukemia (AML) Treated with Crenolanib and Cytarabine/Anthracycline Chemotherapy. Am Soc Hematology; 2017. [Google Scholar]
- 48.Marcucci G, Maharry K, Wu YZ, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2010; 28:2348–2355 doi: 10.1200/JCO.2009.27.3730 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patel KP, Ravandi F, Ma D, et al. Acute myeloid leukemia with IDH1 or IDH2 mutation: frequency and clinicopathologic features. Am J Clin Pathol. 2011; 135:35–45 doi: 10.1309/AJCPD7NR2RMNQDVF (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DiNardo CD, De Botton S, Stein EM, et al. Ivosidenib (AG-120) in Mutant IDH1 AML and Advanced Hematologic Malignancies: Results of a Phase 1 Dose Escalation and Expansion Study. Am Soc Hematology; 2017. [Google Scholar]
- 51.Chiba T and Tanaka K. Cullin-based ubiquitin ligase and its control by NEDD8-conjugating system. Curr Protein Pept Sci. 2004; 5:177–1842004). [DOI] [PubMed] [Google Scholar]
- 52.Hershko A The ubiquitin system for protein degradation and some of its roles in the control of the cell division cycle. Cell Death Differ. 2005; 12:1191–1197 doi: 10.1038/sj.cdd.4401702 (2005). [DOI] [PubMed] [Google Scholar]
- 53.Smith PG, Traore T, Grossman S, et al. Azacitidine/decitabine synergism with the NEDD8-activating enzyme inhibitor MLN4924 in pre-clinical AML models. Am Soc Hematology; 2011. [Google Scholar]
- 54.Traore T, Milhollen M and Grossman S. Synergistic combination of MLN4924, an investigational small molecule inhibitor of NEDD8-activating enzyme (NAE), with azacitidine, a hypomethylating agent, in pre-clinical AML cancer models. Hematologica. 2012; 97:4352012). [Google Scholar]
- 55.Swords RT, Watts J, Erba HP, et al. Expanded safety analysis of pevonedistat, a first-in-class NEDD8-activating enzyme inhibitor, in patients with acute myeloid leukemia and myelodysplastic syndromes. Blood Cancer J. 2017; 7:e520 doi: 10.1038/bcj.2017.1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Swords RT, Coutre S, Maris MB, et al. Results of a clinical study of pevonedistat (pev), a first-in-class NEDD8-activating enzyme (NAE) inhibitor, combined with azacitidine (aza) in older patients (pts) with acute myeloid leukemia (AML). Am Soc Hematology; 2016. [Google Scholar]
- 57.Gill S, Tasian SK, Ruella M, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014; 123:2343–2354 doi: 10.1182/blood-2013-09-529537 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kenderian SS, Ruella M, Shestova O, et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. 2015; 29:1637–1647 doi: 10.1038/leu.2015.52 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang QS, Wang Y, Lv HY, et al. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. 2015; 23:184–191 doi: 10.1038/mt.2014.164 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Uy GL, Godwin J, Rettig MP, et al. Preliminary Results of a Phase 1 Study of Flotetuzumab, a CD123 x CD3 Bispecific Dart® Protein, in Patients with Relapsed/Refractory Acute Myeloid Leukemia and Myelodysplastic Syndrome. Am Soc Hematology; 2017. [Google Scholar]