
Fig. 1.
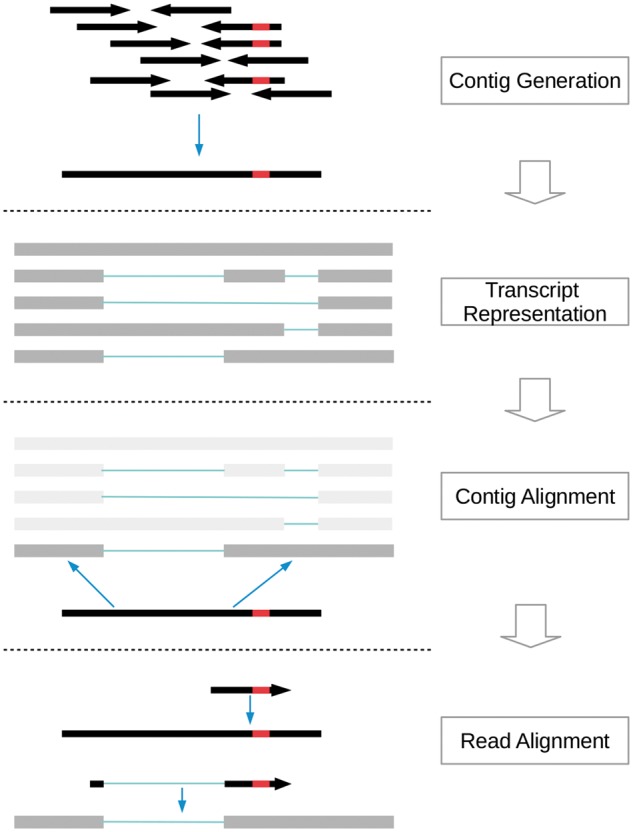
Overview of ABRA2 workflow. Reads overlapping a genomic window of 400 bp are extracted. Contigs are generated using a variety of mechanisms including localized assembly, identification of substantially soft clipped reads, placement of observed indels in localized reference representations and input known indels. Reference representations of putative transcripts are generated using annotated splice junctions as well as unannotated splice junctions observed in the original read alignments. Contigs are exhaustively mapped to each transcript/reference representation using semi-global alignment and the single best alignment is identified. Reads are then aligned back to the contigs using a simple seed and extend approach. If a read unambiguously aligns more closely to a contig than the reference, then the read alignment is updated based upon the contig’s alignment to the reference