

Cognitive symptoms in patients with temporal lobe epilepsy are treatment refractory. In an experimental model of chronic epilepsy, Kahn et al. use chemogenetics to normalize principal cell hyperactivity in the hippocampal dentate gyrus, re-tuning the circuit to rescue memory performance. This approach could represent a novel therapy for cognitive dysfunction.
Keywords: temporal lobe epilepsy, chemogenetics, dentate gyrus, cognitive deficits, circuit-based therapy
Abstract
Temporal lobe epilepsy is associated with significant structural pathology in the hippocampus. In the dentate gyrus, the summative effect of these pathologies is massive hyperexcitability in the granule cells, generating both increased seizure susceptibility and cognitive deficits. To date, therapeutic approaches have failed to improve the cognitive symptoms in fully developed, chronic epilepsy. As the dentate’s principal signalling population, the granule cells’ aggregate excitability has the potential to provide a mechanistically-independent downstream target. We examined whether normalizing epilepsy-associated granule cell hyperexcitability—without correcting the underlying structural circuit disruptions—would constitute an effective therapeutic approach for cognitive dysfunction. In the systemic pilocarpine mouse model of temporal lobe epilepsy, the epileptic dentate gyrus excessively recruits granule cells in behavioural contexts, not just during seizure events, and these mice fail to perform on a dentate-mediated spatial discrimination task. Acutely reducing dorsal granule cell hyperactivity in chronically epileptic mice via either of two distinct inhibitory chemogenetic receptors rescued behavioural performance such that they responded comparably to wild type mice. Furthermore, recreating granule cell hyperexcitability in control mice via excitatory chemogenetic receptors, without altering normal circuit anatomy, recapitulated spatial memory deficits observed in epileptic mice. However, making the granule cells overly quiescent in both epileptic and control mice again disrupted behavioural performance. These bidirectional manipulations reveal that there is a permissive excitability window for granule cells that is necessary to support successful behavioural performance. Chemogenetic effects were specific to the targeted dorsal hippocampus, as hippocampal-independent and ventral hippocampal-dependent behaviours remained unaffected. Fos expression demonstrated that chemogenetics can modulate granule cell recruitment via behaviourally relevant inputs. Rather than driving cell activity deterministically or spontaneously, chemogenetic intervention merely modulates the behaviourally permissive activity window in which the circuit operates. We conclude that restoring appropriate principal cell tuning via circuit-based therapies, irrespective of the mechanisms generating the disease-related hyperactivity, is a promising translational approach.
Introduction
Temporal lobe epilepsy (TLE), the most common form of epilepsy in adults, can develop following an assault to the CNS. Acquired epilepsies particularly are refractory to medical intervention, and these and other intractable epilepsies have higher rates of co-morbidities (Berg et al., 2012). Although treatment focuses on ameliorating seizure events, patients must cope with a host of complicated cognitive co-morbidities that many report are more detrimental to daily life than the seizures (Holmes, 2013). However, the neural mechanisms mediating TLE cognitive impairments are not well understood.
The dentate gyrus is fundamental for cognitive functions in the hippocampus, a structure TLE seizures highly activate and damage. Ordinarily, dentate gyrus granule cells (DGCs) express remarkably sparse firing patterns (Jung and McNaughton, 1993; Chawla et al., 2005). This sparsity enables pattern separation, the ability to recognize and amplify small differences in inputs to avoid confounding overlaps. Extensive work in rodents has shown that compromising the dentate gyrus disrupts spatial discrimination performance (Gilbert et al., 2001; Lee et al., 2005; Clelland et al., 2009; Nakashiba et al., 2012; Kheirbek et al., 2013). Furthermore, with its early location in the trisynaptic circuit, the dentate gyrus can affect firing patterns in downstream hippocampal structures, such as causing the CA3 to express activity consistent with pattern separation instead of its usual pattern completion (Leutgeb et al., 2007; Neunuebel and Knierim, 2012).
Sparse DGC activity has a secondary effect: it limits cortical input to the hippocampus and acts like a ‘gate’, the failure of which may contribute to the excessive cortical-hippocampal activity underlying TLE seizures (Heinemann et al., 1992; Lothman and Stringer, 1992). The dentate gyrus experiences a dramatic increase in excitability in epileptogenesis and chronic epilepsy (Dengler et al., 2017), a phenomenon emerging from many different circuit disruptions that develop in the disease, including interneuron death (de Lanerolle et al., 1989; Obenaus et al., 1993; Kobayashi and Buckmaster, 2003), GABA receptor alterations (Brooks-Kayal et al., 1998; Coulter and Carlson, 2007; Zhang et al., 2007), ectopic adult-born DGCs (Parent et al., 1997), and mossy fibre sprouting (Tauck and Nadler, 1985; Zhang et al., 2012; Supplementary Fig. 1). Furthermore, optogenetically driving or silencing activity in DGCs can incite or truncate seizure events, respectively (Krook-Magnuson et al., 2015).
The ability of the dentate gyrus to modify both its own and downstream hippocampal cognitive processes together with its role in seizure propagation make it a powerful potential target for circuit-based therapeutic intervention. This type of intervention is defined as follows: by activating or inhibiting individual circuits in the nervous systems, circuit-based therapies can change the behaviour, fundamental function, or a specific symptom of a disease. Indeed, previous work shows that modifying the dentate gyrus before or as the disease develops, for example blocking ectopic DGCs from forming, can alter disease severity, reducing seizure frequency and allowing for more typical cognitive capabilities (Cho et al., 2015; Hosford et al., 2016). However, no mechanistically specific interventions have rescued cognitive performance in fully-developed chronic epilepsy.
In this study, we sought to intervene at the circuit level, targeting the dentate gyrus’s aggregate output by manipulating DGC activity with designer receptors exclusively activated by designer drugs (DREADDs). As circuit outputs determine behavioural outcomes, circuit excitability provides a potential downstream, mechanistically-independent therapeutic target. Predictably, while wild type animals with sparse DGC activity performed well on a dentate-mediated spatial discrimination task, the pilocarpine-induced TLE mouse model displayed marked deficits. Remarkably, acutely reducing DGC hyperactivity via inhibitory DREADDs was sufficient to rescue behavioural performance in chronically epileptic mice. However, making the epileptic dentate gyrus overly quiescent via elevated DREADD recruitment compromised behavioural performance, suggesting that the DGCs must be tuned appropriately to support information coding. We explored this hypothesis in control animals further, expressing either excitatory or inhibitory DREADDs in dorsal DGCs to determine the consequences of perturbing a properly tuned circuit. Acutely altering DGC recruitment compromised behavioural performance, demonstrating that manipulating principal cell recruitment is sufficient to dictate behavioural outcomes in both health and disease. DREADD-mediated effects were specific to the dorsal dentate gyrus, as a similar hippocampal-independent task and a ventral dentate-dependent behaviour remained unaffected by DREADD activation. Voltage sensitive dye recordings and fos confirmed that DREADDs manipulated DGC responses to afferent inputs. We conclude that appropriately tuned activity in the dentate gyrus is critical for cognitive performance, and that reducing disease-related hyperactivity via circuit-based therapies is a potential translational approach.
Materials and methods
Animals
Animals were singly housed in a biosafety level 2 facility in individually ventilated cages with nestlets and maintained on a 12-h light/dark cycle with target temperature of 20 ± 2°C and with ad libitum food and water. Wild-type C57BL/6 mice from Charles River Laboratories were used for wild-type and pilocarpine-treated groups. FosTRAP × tdTomato transgenic mice were FosCreER mice [c-Fos Cre ERT2 (B6.129(Cg)–Fostm1.1(cre/ERT2)] (Jax #021882) and Lox-tdTomato [B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)] (Jax #007914). These and POMC-Cre mice [B6.FVB-Tg(Pomc-cre)1Lowl/J; Jax #010714] were obtained from Jackson Laboratory. All groups contained 25–50% female subjects; analysis by sex did not reveal significant differences, thus results were pooled. Mice were assigned randomly to experimental groups after balancing for age, sex, and genotype, and were 3–7 months old when assessed. All procedures were approved by the Institutional Animal Care and Use Committee of the Children’s Hospital of Philadelphia.
Pilocarpine-induced status epilepticus model of epilepsy
Mice, 8–9 weeks old, were injected intraperitoneally with scopolamine (1 mg/kg) and with pilocarpine (315 mg/kg) 30 min later to trigger status. Sixty minutes after status onset, diazepam (5 mg/kg) was administered to quell seizure activity. Mice were at least 7 weeks post-status epilepticus before testing began to ensure they were chronically epileptic (Dengler et al., 2017). All included epileptic animals had at least two confirmed spontaneous seizure events.
Viral induction
Control animals were 7–9 weeks old; pilocarpine-treated animals were 1–2-weeks post-status epilepticus. Animals were anaesthetized with isoflurane and received bilateral 200 nl viral injections to the dorsal dentate gyrus (from Bregma AP: −1.7 mm, ML: ±1.0 mm, from dura DV: −1.7 mm). Approximately 5 × 108 infectious particles were infused into each hippocampus. Experiments began at least 3 weeks post-injection to allow full DREADD expression. Bryan Roth provided DREADD constructs. AAV5-CamKIIα-HA-hM3Dq-IRES-mCitrine, AAV5-CamKIIα-HA-hM4Di-IRES-mCitrine, and AAV5-hsyn-DIO-hM3Dq-mcherry were purchased from the UNC Vector Core. AAV5-CamKIIα-GFP and AAV5-hsyn-FLEX-GFP were purchased from the Penn Vector Core. The plasmid for AAV5-CamKIIα-HA-KORD-IRES-mCitrine was purchased from Addgene (#65418) and packaged into AAV5 by the CHOP Research Vector Core.
Drugs
Clozapine N-oxide (CNO; Tocris) was dissolved in 12% dimethyl sulphoxide (DMSO)/saline to create a 10 µM stock, which was diluted in saline to produce appropriate doses (final DMSO <0.5%) (Armbruster et al., 2007; Garner et al., 2012). Salvinorin B (SB; Cayman Chemical) was dissolved in DMSO (25 mg/ml) and diluted to 5 mg/ml in sunflower oil (Sigma); this was diluted further in sunflower oil (Vardy et al., 2015). CNO and SB were administered at 10 ml/kg and 5 ml/kg, respectively. 4-hydroxytamoxifen (4OHT; Sigma) was dissolved in ethanol (20 mg/ml) at 37°C, then diluted in corn oil to 10 mg/ml. The ethanol was evaporated by vacuum under centrifugation to produce 10 mg/ml 4OHT, administered at 5 ml/kg (Guenthner et al., 2013).
Behavioural tests
Before testing, animals were handled for 3–5 min/day for 7 days. Animals were observed for 1 h pre- and post-environmental exposure. Mice displaying prolonged freezing (>10 s) or more pronounced seizure behaviours (Racine stage 2–5) at any point during the observational periods/testing were excluded from analyses to minimize seizure-driven effects on behaviour. Testing occurred in the last 3 h of the light cycle. For DREADD experiments, animals received intraperitoneal injections of saline or CNO or subcutaneous injections of vehicle or SB 1 h prior to all handling sessions and environmental exposures to minimize potential anxiogenic confounds.
Spatial object recognition
This non-aversive dentate-mediated spatial memory test (Kesner et al., 2015) requires mice to learn and remember the positions of three objects in an arena. On training day, animals were exposed to a 50 cm × 30 cm × 30 cm box with orientation markers for 6-min trials. The box was empty for habituation, then identical objects were arranged for three training trials. On test day (24-h later), one object was repositioned (displaced object), changing the spatial configuration. Mice that remember the original configuration spend more time exploring the displaced object compared to the non-displaced objects. Sessions were video recorded and manually scored by a blinded experimenter for object interactions. Object configurations were counterbalanced across subjects. For repeated testing, at least 2 weeks elapsed between tests; new objects, floor textures, and ligand doses were counterbalanced across subjects. Discrimination index calculation:
| (1) |
MATLAB code (Autotyping15.03) provided by Dr David Meaney (University of Pennsylvania Department of Bioengineering) tracked ambulation and generated heat maps of object interaction bouts.
Novel object recognition
This hippocampal-independent task (Oliveira et al., 2010) requires mice to learn and remember object identities. For habituation, animals explored the test arena (45 cm × 28 cm × 30 cm) for 5 min/day for 5 days. On training day, animals explored two identical objects for 15 min. On test day (24 h later), one object was replaced with a novel object. Sessions were video recorded and manually scored by a blinded experimenter for object interactions. Object identities and locations were counterbalanced across subjects. Per cent novel exploration calculation:
| (2) |
Dr Meaney’s MATLAB code tracked ambulation and generated heat maps of object interaction bouts.
Open field
Animals were introduced to the 40 cm × 40 cm × 40 cm open field box for 5 min. Sessions were video recorded and Dr Meaney’s MATLAB code quantified ambulation and thigmotaxis, the measure of the mouse’s tendency to stay close to the walls, indicating anxiety (Simon et al., 1994).
Novel environment exposure
The environment consisted of a clean cage containing a running wheel and hut. One hour into the 2-h exposure, animals received 50 mg/kg 4OHT intraperitoneally. Home cage controls received injections at the same time points.
Voltage sensitive dye slice preparation and recording
Slices were prepared as described in Dengler et al. (2017). Immediately before imaging, slices were stained with 0.0125 mg/ml of di-3-ANEPPDHQ (D36801, Invitrogen) in artificial CSF for 15 min. Slices were imaged in an oxygenated interface chamber (32 ± 1°C) using an 80 × 80 CCD camera recording at a 1-kHz frame rate (NeuroCCD, RedShirt Imaging) and illuminated by a 530 nm Green LED (M530L3-C2, Thorlabs). Trials lasted 1000 ms each, with a 20-s intertrial interval. Amplitude responses were analysed in Igor Pro 6.2 (Wavemetrics, Portland, OR).
Histology
Animals were perfused transcardially with 4% paraformaldehyde/phosphate-buffered saline (PFA/PBS). Brains were submerged in 4% PFA for 12–20 h, then in 30% sucrose/PBS. Thirty micrometre slices from frozen brains were obtained on a cryostat and stored in cryoprotectant. Slices were imaged using an Olympus FV1000 confocal microscope with FluoView 4.1a (Olympus, Center Valley, PA). Viral expression location was visualized by the fluorescent reporter co-expressing with the virus (Supplementary Fig. 2). Only animals that had viral fluorescent expression in the dentate gyrus in both hemispheres were included in analyses. To quantify DGC recruitment, cell counts in each slice were divided by the DGC layer volume/volume expressing fluorescent reporter in that slice (ImageJ, NIH, Bethesda, MD). Slices were averaged within the animal to give an individual’s FosTRAPed (targeted recombinase in active populations) cell density, which was normalized to the control group’s mean.
Statistical analysis
Statistical analyses were done with MATLAB and Prism 7 (GraphPad). Sample sizes were determined on the basis of calculations performed on empirical data and power analyses. The number of mice used was appropriate to detect behavioural differences with 80% power and α was set at 0.05. Normality was tested with the D’Agostino and Pearson test. Statistical tests were used as indicated in the figure legends.
Data availability
The data supporting this study and the custom-written seizure MATLAB scripts are available from the corresponding author upon reasonable request. The MATLAB behavioural analysis code is available on Dr David Meaney’s website (https://www.seas.upenn.edu/∼molneuro/software.html).
Results
Epileptic animals have hyperactive DGCs
We first examined if pilocarpine mice excessively recruit DGCs in behavioural contexts, not just during seizure events. We used FosTRAP transgenic mice, in which the immediate early gene fos drives expression of CreERT, a tamoxifen-inducible recombinase that recombines a stop cassette upstream of tdTomato (Guenthner et al., 2013). When tamoxifen is present, fos-expressing cells permanently express tdTomato, providing a snapshot view of in vivo activity (Fig. 1A). Mice were exposed to an enriched novel environment (Fig. 1B), which highly activates the dentate gyrus. Halfway through exposure, mice received an injection of 4OHT, tamoxifen’s main metabolite, to constrain the timeline in which cells can be TRAPed (Guenthner et al., 2013). When monitored continually for 2 months with EEG depth electrodes, three pilocarpine mice collectively had 557 seizures; every electrographic seizure was accompanied by a significant behavioural component (Racine scale 3–5; Supplementary Fig. 3). Thus, epileptic FosTRAP animals were video monitored for 12 h before and after testing for any behavioural indication of seizures. Animals exhibiting stereotypical seizure-related behaviours (n = 2) expressed obvious, distinct FosTRAPing compared to epileptic animals that did not exhibit these behaviours (Fig. 1C) and were excluded from statistical analysis. After exploring the novel environment, chronically epileptic animals had significantly more TRAPed DGCs than controls [normalized cell density: control: 1.00 ± 0.21 versus pilocarpine: 3.49 ± 0.04, t(4.284) = 11.53, P = 0.0002, Welch’s t-test; Fig. 1C and D]. Coronal slice recordings examining molecular layer stimulation-evoked responses also revealed increased activity in epileptic tissue compared to controls (Supplementary Fig. 4).
Figure 1.

Epileptic animals have hyperactive DGCs. (A) Schematic of FosTRAP × tdTomato mechanism. Fos drives expression of CreERT so that tdTomato expression only occurs when tamoxifen or its metabolite 4OHT is present to remove the stop codon. (B) Schematic of novel environment protocol. (C) Confocal micrographs of tdTomato expression overlaid the transmitted image to show TRAPed DGCs in control mice (left), epileptic mice (middle), and epileptic mice that displayed seizure-related behaviours while the 4OHT was onboard (right). Scale bar = 318 µm. (D) Cell density analysis per animal normalized to the control group’s mean. Epileptic animals 2–3 months post-status epilepticus recruit significantly more DGCs after exposure to a novel environment [t(4.284) = 11.53, ***P = 0.0002, Welch’s t-test]. Data are represented as mean ± SEM. Each circle represents an animal (number of animals indicated in parentheses).
Reducing DGC hyperactivity in epileptic mice rescues dentate-mediated cognitive performance
To assess how DGC hyperactivity may affect cognition, animals were tested on the non-aversive hippocampal-dependent spatial object recognition (SOR) task (Fig. 2A). Unlike other implementations (Oliveira et al., 2010), three identical objects were used to increase difficulty and to maximize dentate gyrus involvement in the discrimination aspect of the test trial. Wild-type animals successfully discriminated between the displaced object and non-displaced objects (Fig. 2B and C), showing a 38.46 ± 8.1% discrimination index, the difference in time spent exploring the displaced object compared to the non-displaced objects (see ‘Materials and methods’ section for calculation). Pilocarpine animals 2–3 months post-status epilepticus were significantly compromised on this dentate-mediated task [pilocarpine: −9.54 ± 10.9%; F(3,46) = 8.388, P = 0.0001, one-way ANOVA, PWTvPilo = 0.005, Tukey’s multiple comparisons correction; Fig. 2B and C].
Figure 2.
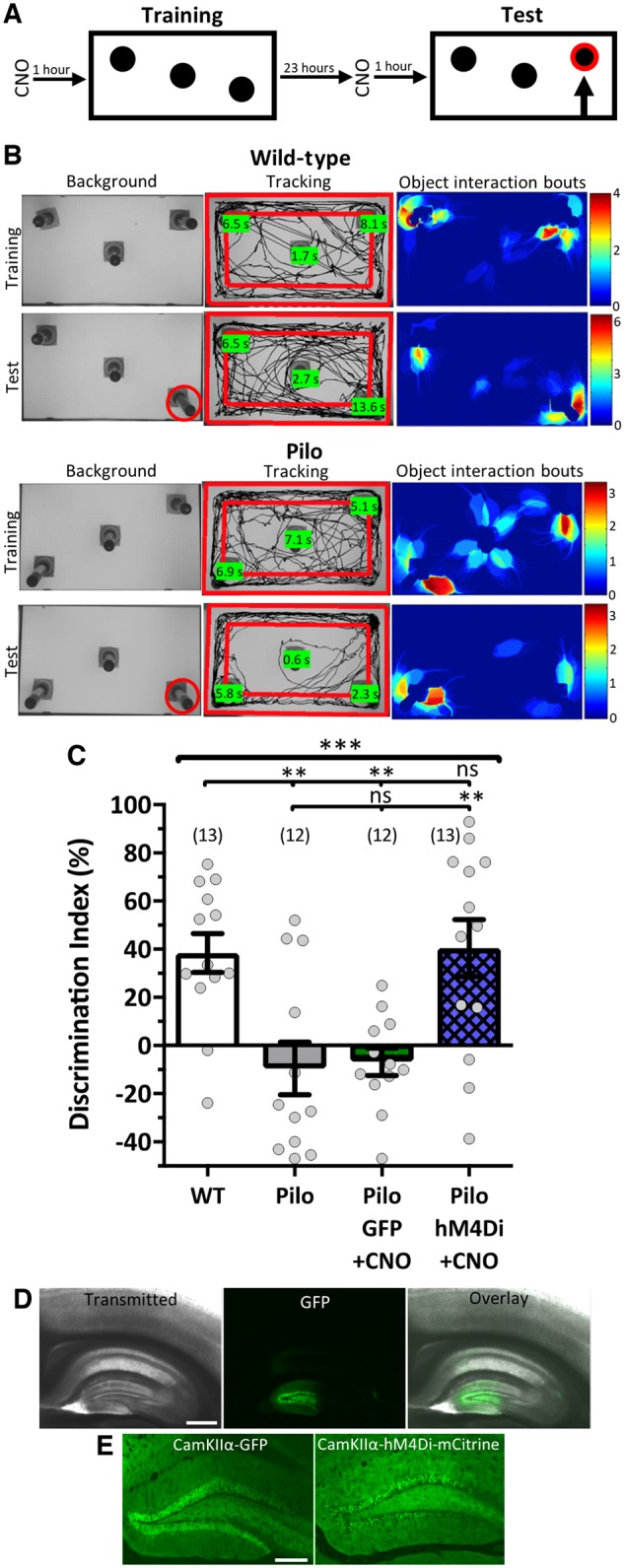
Reducing DGC hyperactivity via inhibitory DREADDs rescues performance. (A) Schematic of SOR task. (B) Representative images of SOR exploration for a wild-type (top) and a pilocarpine (bottom) mouse during their third training trial and their test trial. Background images show the object configuration with the displaced object marked in red for the test trial. Tracking images show the mouse’s total exploration during the trial with the total interaction time with each object quantified in the green boxes. The object interaction bouts images are heat maps of the cumulative time (s) the animal spent in each location when it was interacting with an object. The wild-type mouse shows an increased preference for the displaced object during its test trial while the Pilo mouse does not. (C) Quantified SOR performance showing that hM4Di recruitment rescues discrimination performance in epileptic mice [F(3,46) = 8.388, P = 0.0001, one-way ANOVA with Tukey’s multiple comparisons correction]. CNO = 1.5 mg/kg. (D and E) Representative confocal micrographs of the fluorescent reporter co-expressing with the viral vectors. (D) Micrographs (4×) showing viral expression was localized to the dentate gyrus. Scale bar = 500 µm. (E) Micrographs (10×) of reporter for the CamKIIα-GFP (left) and CamKIIα-hM4Di (right). Scale bar = 200 µm. Data are represented as mean ± SEM. Each circle represents an animal (number of animals indicated in parentheses). nsP > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001.
To investigate if a circuit-based therapy (i.e. reducing DGC hyperactivity) could improve behavioural performance, we bilaterally injected inhibitory DREADDs (CamKIIα-hM4Di) or GFP-expressing vector into the dorsal DGs of epileptic mice (Fig. 2D and E). hM4Di DREADDs are mutated muscarinic acetylcholine receptors that act through G-protein coupling receptor (GPCR) signalling cascades, in this case inhibitory Gi cascades, to affect neuronal activity upon activation by an otherwise inert ligand, traditionally CNO (Armbruster et al., 2007). The CamKIIα promoter restricted DREADD expression to glutamatergic neurons. Activating inhibitory DREADDs with CNO (1.5 mg/kg) significantly improved performance in epileptic animals (Pilo+hM4Di: 40.52 ± 11.8%) such that they discriminated comparably to wild-type mice (PWTvPilo+hM4Di = 0.999, Tukey’s correction; Fig. 2C). In contrast, GFP-expressing epileptic animals treated with CNO (1.5 mg/kg) performed equivalently to the initial epileptic group (Pilo + GFP: −6.73 ± 5.7%, PPilov Pilo+GFP = 0.997, Tukey’s correction; Fig. 2C); therefore, potential off-target ligand effects did not affect performance. We concluded that reducing dentate gyrus hyperactivity in epileptic mice rescued the animals’ ability to distinguish the displaced object.
As discussed previously, many different circuit properties permanently change in the dentate gyrus over the course of epilepsy development and into chronic epilepsy. In short, the chronically epileptic dentate gyrus is a different, structurally damaged circuit from the one found in controls, making this rescue surprising in its effectiveness. However, these results suggest that the disrupted epileptic dentate gyrus retains its underlying ability to code information. We hypothesized that there is bell curve relationship between DGC recruitment (the x-axis) and behavioural performance (the y-axis; Supplementary Fig. 5).
Epileptic animals fail to discriminate when the dentate gyrus is overly quiescent
If this hypothesis is correct, reducing DGC hyperactivity to a point would be beneficial, but too much reduction would again prohibit behavioural performance. However, to achieve this effect, we would require a high CNO dose. Recent work invalidates this approach as Gomez et al. (2017) showed that back-metabolized clozapine, produced in response to higher CNO concentrations, likely would have off-target effects on endogenous receptors. To avoid this complication, we used the kappa-opioid receptor inhibitory DREADD (KORD), whose ligand salvinorin B (SB) has not demonstrated off-target effects (Vardy et al., 2015).
Epileptic animals injected bilaterally with the CamKIIα-KORD were tested on SOR (Fig. 3A) in response to three treatments: vehicle, 1.5 mg/kg SB, and 3 mg/kg SB (Fig. 3B). The mice failed to discriminate when treated with vehicle (−4.891 ± 9.1%), reflecting the expected disease-related behavioural deficit. When treated with the ‘moderate’ SB dose (1.5 mg/kg), KORD mice recapitulated the SOR performance rescue observed in the hM4Di cohort (Pilo+KORD+SB 1.5: 28.92 ± 8.8%). However, with ‘high’ SB (3 mg/kg), the mice failed to discriminate (Pilo+KORD+SB 3: −9.745 ± 13.0%). Within this one cohort, we recreated the hypothesized bell curve [F(1.29,12.91) = 4.759, P = 0.041, repeated measures one-way ANOVA with Geisser-Greenhouse’s correction; Fig. 3B, Supplementary Fig. 5B].
Figure 3.

DGC tuning dictates SOR performance. (A) Schematic of SOR task with drug treatment timeline. (B and C) SOR performance. (B) Pilo+KORD mice were tested on SOR under multiple treatment conditions. SB at 1.5 mg/kg rescued performance while 3 mg/kg SB compromised performance [F(1.29,12.91) = 4.759, P = 0.041, repeated measures one-way ANOVA with Geisser-Greenhouse’s correction and Tukey’s multiple comparisons correction]. (C) Decreasing [left, CNO = 1.5 mg/kg, t(22) = 2.991, P = 0.007, Student’s t-test] or increasing [right, CNO = 0.3 mg/kg, t(22) = 2.893, P = 0.008, Student’s t-test] DGC activity in control animals disrupted discrimination. (D) Representative confocal micrographs (10 x) of fluorescent reporter co-expressing with the CamKIIα-GFP (left), CamKIIα-KORD (middle left), CamKIIα-hM4Di (middle right), and CamKIIα-hM3Dq (right) viral vectors. Scale bar = 200 µm. Data are represented as mean ± SEM. Each circle represents an animal (number of animals indicated in parentheses). nsP > 0.05, *P < 0.05, **P < 0.01.
A potential confound in these findings is that DREADDs could be blocking pathological, seizure activity. However, if behavioural recovery was a function of seizure suppression, we would expect a more positive association between behavioural performance and dentate gyrus quiescence: the quieter the dentate gyrus, the better it can prevent seizure-related activity. Instead, we found further reducing DGC activity compromised performance, suggesting possible seizure suppression was not the critical factor. Moreover, several mice had seizures after inhibitory DREADDs were activated (Pilo+hM4Di+CNO mice n = 3, Pilo+KORD+SB n = 1), suggesting that acute recruitment of these focally expressed receptors was insufficient to suppress the multifocal seizure events that the systemic pilocarpine epilepsy model exhibits. Animals experiencing seizures during testing had identifiable behavioural metrics and were excluded from further analyses (Supplementary Fig. 6). Finally, we implanted CamKIIα-KORD-expressing mice and recorded them for 2 months to establish a baseline and to examine if seizures persisted during KORD recruitment; mice continued to have seizure clusters while receiving daily 3 mg/kg SB injections (Supplementary Fig. 7).
These data demonstrate that DREADD effects are dose-dependent and reversible. Furthermore, these results support our hypothesis that there is a permissive window of excitability in which the dentate gyrus can code properly. If an appropriate number of DGCs are recruited, behavioural outcomes are successful, even in the face of significant local circuit structural changes (Supplementary Fig. 5).
Manipulating DGC activity compromises SOR performance in control animals
To explore further if circuit tuning determines behavioural performance, we sought to manipulate DGC activity in control animals. Presumably, a normal dentate gyrus is already tuned appropriately. Thus, we expected to degrade behavioural performance by modulating the circuit to be hypo- or hyperactive (Supplementary Fig. 5C). We expressed either inhibitory DREADD CamKIIα-hM4Di, excitatory DREADD CamKIIα-hM3Dq, or CamKIIα-GFP bilaterally in the dorsal dentate gyri of control animals (Fig. 3D) and tested them on the SOR task. One procedural difference was we only administered CNO before the test trail (Fig. 3A). Previous work demonstrated the DGCs’ importance in acquiring new spatial environments (Kheirbek et al., 2013), so we focused on how DGC tuning may affect the SOR’s discrimination component.
The modulation that restored performance in a hyperactive epileptic dentate gyrus should compromise performance in an optimally tuned, normal dentate gyrus. Therefore, we gave control CamKIIα-hM4Di-expressing mice the same CNO dose that Pilo+hM4Di animals received (1.5 mg/kg). While GFP-expressing mice preferred the displaced object (35.84 ± 6.5%), CamKIIα-hM4Di mice failed to distinguish [3.61 ± 8.6%; GFP + 1.5 mg/kg versus hM4Di+1.5 mg/kg: t(22) = 2.991, P = 0.007, Student’s t-test; Fig. 3C].
We also modulated the control dentate gyrus using excitatory DREADDs. While GFP-expressing mice treated with CNO (0.3 mg/kg) preferred the displaced object (31.32 ± 8.8%), CamKIIα-hM3Dq activation disrupted discrimination, recreating the behavioural deficit observed in the epileptic population [hM3Dq+CNO: −14.55 ± 13.2%, GFP+0.3 mg/kg versus hM3Dq+0.3 mg/kg: t(22) = 2.893, P = 0.008, Student’s t-test; Fig. 3C]. Neither the GFP+0.3 mg/kg CNO group nor the GFP+1.5 mg/kg CNO group were significantly different from the original wild-type cohort [F(2,34) = 0.2111, P = 0.811, one-way ANOVA], so changes in SOR performance were not due to off-target CNO effects. Thus, acutely manipulating DGC excitability—without inducing other structural changes to the circuit—is sufficient to compromise cognitive performance (Supplementary Fig. 5C).
DREADD effects are specific to the dorsal dentate gyrus
To examine if DREADD effects were circuit-specific, we tested the same cohorts on the novel object recognition task, which is similar in nature to SOR, but is hippocampal-independent (Oliveira et al., 2010; Fig. 4A and B). While wild-type animals preferentially investigated the novel object (67.39 ± 3.1%), epileptic animals failed to distinguish, spending equal time exploring both objects [Pilo: 48.24 ± 5.2%; F(4,56) = 4.234, P = 0.005, one-way ANOVA; Fig. 4B and C]. These results are not surprising as the systemic pilocarpine epilepsy model also disrupts cortical structures. Similarly, both GFP- and hM4Di-expressing epileptic animals failed to distinguish when treated with 1.5 mg/kg CNO (Pilo+GFP: 49.03 ± 3.7%, Pilo+hM4Di: 44.81 ± 4.1%), nor did the KORD-expressing epileptic mice treated with 3 mg/kg SB (47.63 ± 6.4%). Furthermore, neither increasing nor decreasing DGC activity during the test trial in control mice affected their preference for the novel object [GFP+0.3 mg/kg versus hM3Dq+0.3 mg/kg: t(22) = 0.1066, P = 0.916, Student’s t-test; GFP+1.5 mg/kg versus hM4Di+1.5 mg/kg: t(22) = 0.7774, P = 0.445, Student’s t-test; Fig. 4D]. These data demonstrate that DREADD-mediated effects were hippocampal-specific.
Figure 4.

DREADD effects are specific to the hippocampus. DREADD manipulations of the dentate gyrus in epileptic and control animals do not affect novel object recognition performance, showing DREADD effects are structurally specific. (A) Schematic of task. (B) Representative images of exploration for a wild-type (top) and a pilocarpine (bottom) mouse during their test trial. Background images show the object configuration with the novel object marked in red. Tracking images show the mouse’s total exploration during the trial with the total interaction time with each object quantified in the green boxes. The object interaction bouts images are heat maps of the cumulative time (s) the animal spent in each location when it was interacting with an object. The wild-type mouse shows an increased preference for the novel object while the Pilo mouse does not differentiate. (C) While Pilo mice display a deficit on the task compared to wild-type (WT) mice, DREADDs do not affect task performance [F(4,56) = 4.234, P = 0.005, one-way ANOVA with Tukey’s multiple comparisons correction]. CNO (1.5 mg/kg) or SB (3 mg/kg) were administered before the training and test trials. (D) DREADDs do not affect task performance in control mice [left, t(22) = 0.1066, P = 0.916, Student’s t-test; right, t(22) = 0.7774, P = 0.445, Student’s t-test]. Control mice received saline before the training trial and CNO (left, 0.3 mg/kg; right, 1.5 mg/kg) before the test trial. Data are represented as mean ± SEM. Each circle represents an animal (number of animals indicated in parentheses). nsP > 0.05, *P < 0.05, **P < 0.01.
In addition to its role in cognition, the dentate gyrus critically contributes to anxiety regulation, specifically, the ventral dentate (Kheirbek et al., 2013). To examine if DREADD effects acted across the septotemporal axis, we analysed the SOR habituation trial as an open field test to measure anxiety. Epileptic mice spent significantly more time in the chamber’s outer edges compared to wild-type mice [F(3,41) = 7.011, P = 0.0006, one-way ANOVA with Tukey’s correction], suggesting these animals were more anxious (Fig. 5A and B) and corroborating previous reports of anxiety in TLE models (Gröticke et al., 2007). Notably, the epileptic groups were consistently anxious across treatments (Fig. 5B and C). As control cohorts received saline before their SOR habituation/training (Fig. 3A), we tested them on a novel open field to examine DREADD specificity (Fig. 5D). There was no significant change in thigmotaxis across groups [F(4,56) = 1.769, P = 0.148, one-way ANOVA; Fig. 5D and E], suggesting no effect on anxiety. Therefore, the dorsally expressed DREADDs specifically modulated dorsal hippocampal functions without affecting the ventral hippocampus.
Figure 5.

DREADD effects are regionally specific within the hippocampus. DREADDs do not affect behavioural measures of anxiety, showing dorsal hippocampal specificity. (A) Representative images of open field analysis performed on the SOR habituation trial. Left images show ambulation tracking with boundary for anxiety analysis. Right images show heat maps of ambulation tracking depicting the cumulative time (s) the animal spent in each location. (B and C) Thigmotaxis analysis. While epileptic animals display anxiogenic behaviour, DREADDs do not affect performance [B: F(3,41) = 7.011, P = 0.0006; C: F(1.30, 12.96) = 1.502, P = 0.251]. (D) Representative images of open field analysis for control animals in a novel open field arena with their respective CNO doses. (E) Thigmotaxis analysis [F(4,56) = 1.769, P = 0.148]. Statistical comparisons were done with either an ordinary one-way ANOVA (B and E) or a repeated measures one-way ANOVA with Geisser-Greenhouse correction (C), all with Tukey’s multiple comparisons correction. Data are represented as mean ± SEM. Each circle represents an animal (number of animals indicated in parentheses). nsP > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001. WT = wild-type.
Additionally, we analysed total ambulation during the SOR habituation trial to examine if there were disease-, DREADD-, or ligand-related effects on locomotion. All epileptic groups were significantly more active than wild-type mice, but were not significantly different from each other no matter their treatment (Supplementary Fig. 8A and B). Importantly, this disease-related hyperactivity did not affect how much time epileptic groups spent interacting with SOR objects compared to wild-type mice (Supplementary Fig. 9A), so it did not compromise SOR performance.
Confirming DREADD modulation of dentate gyrus activity
To assess how DREADDs affected dentate gyrus activity, we wanted to assess the whole circuit’s function, not just the activity of individual neurons. We used voltage sensitive dye imaging with a CCD camera, which has high spatial and temporal resolution, allowing us to visualize the location and magnitude of synaptic membrane voltage changes in an acute slice in real time. It is particularly well suited to our goals as we can image the whole dentate gyrus at once for its response to afferent inputs, and it does not involve dialysing cell contents during recordings (as evident during patch clamp measurements), avoiding the potential disruption in GPCR-dependent DREADD effects. Coronal slices from Pilo+hM4Di animals were recorded in artificial CSF and then in 10 µM CNO (Armbruster et al., 2007; Krashes et al., 2011; Marchant et al., 2016) to provide within subject controls, accounting for differences in individual disease severity and viral expression levels. KORD-expressing slices were recorded in artificial CSF and 200 nM SB (Vardy et al., 2015; Marchant et al., 2016). Molecular layer stimulation-evoked responses in the DGC layer were significantly reduced following bath application of ligand in both the hM4Di-expressing slices [t(4) = 4.768, P = 0.009, paired t-test; Fig. 6A–E], as well as the KORD-expressing slices [t(4) = 6.186, P = 0.004, paired t-test; Fig. 6F–J]. CamKIIα-hM3Dq activation in control slices via 10 µM CNO significantly increased the stimulation response in the DGC and molecular layer [t(2) = 7.652, P = 0.017, paired t-test; Fig. 6K–O], confirming that DREADDs were affecting the dentate gyrus as expected.
Figure 6.

Voltage sensitive dye imaging confirms DREADD-mediated changes in activity. (A) Representative micrograph from the CCD camera of the recording setup and analysis region of interest in blue. Scale bar = 425 µm. (B and C) Per cent ΔF/F0 maximal response of the hM4Di-expressing epileptic slice in artificial CSF (aCSF) (B) and in 10 µM CNO (C). (D) Representative traces of ΔF/F0 (%) in artificial CSF and CNO. (E) Quantified ΔF/F0 (%) in aCSF and in CNO [t(4) = 4.768, P = 0.009]. F–J are comparable to A–E, examining KORD effects in epileptic tissue. (J) Quantified ΔF/F0 (%) in artificial CSF and in 200 nM SB [t(4) = 6.186, P = 0.004]. K–O are comparable to A–E, examining hM3Dq effects in control tissue. (O) Quantified ΔF/F0 (%) in artificial CSF and in 10 µM CNO [t(2) = 7.652, P = 0.017]. Statistical comparisons are two-tailed paired t-tests. Data are represented as mean ± SEM. Each circle represents an animal (each circle indicates the mean of two to three slices for a given animal). *P < 0.05, **P < 0.01.
DREADD-mediated DGC recruitment assessed by FosTRAP
The CamKIIα-hM3Dq slice imaging data bring to the forefront a potential confound: are excitatory DREADDs inducing focal seizure events that compromised SOR performance in control mice? To address this, all control mice injected with DREADDs or GFP in previous studies were FosTRAP animals so that we could observe in vivo DGC recruitment. Mice received CNO injections 1 h prior to the novel environment paradigm (Fig. 7A). The 0.3 mg/kg CNO dose used in the behavioural studies significantly increased the number of recruited DGCs in the novel environment (Fig. 7B and C). However, it took a 10-fold increase in CNO (3.0 mg/kg) to recruit a comparable number of DGCs to what we observed in epileptic animals (Figs 1E, 7B and C). We also examined mice that remained in their home cages, analysing any spontaneous activity (i.e. not behaviour-driven) CamKIIα-hM3Dq may evoke. No tested CNO dose increased DGC FosTRAPing, suggesting that DGC DREADD recruitment at these CNO doses is only modulatory and does not drive activity in the absence of relevant behavioural stimuli (Fig. 7E). Finally, we implanted CamKIIα-hM3Dq animals with hippocampal depth electrodes and generated a CNO dose-seizure response curve. Seizure-like events were not observed until 10–30 mg/kg CNO, up to 100-fold what we used in the behavioural studies (Supplementary Fig. 10). Consequently, we do not believe behavioural performance was compromised because of evoked seizure-like activity.
Figure 7.

FosTRAP confirms behavioural relevance of DREADDs and lack of seizures. (A) Schematic of novel environment procedure. (B) Confocal micrographs showing the FosTRAPed DGCs, viral expression, and the merge at each condition + CNO dose. (C–E) Cell density results, normalized to the GFP-expressing group in C. (C) Normalized cell density results quantifying the effect of different CNO doses on hM3Dq-mediated DGC recruitment [H(3, 17) = 9.816, P = 0.002, Kruskal-Wallis test with Dunn’s multiple comparisons correction]. (D) Normalized cell density results quantifying the effect of hM4Di on DGC recruitment [t(10) = 2.637, P = 0.025, Student’s t-test]. Note that the GFP group in D is the same data repeated from C. (E) Home cage normalized cell density analysis to examine spontaneous activity the DREADDs may recruit [F(3,13) = 0.2708, P = 0.845, one-way ANOVA]. Data are represented as mean ± SEM. Each circle represents an animal (number of animals indicated in parentheses) nsP > 0.05, *P < 0.05, **P < 0.01.
FosTRAP also confirmed that 1.5 mg/kg CNO successfully activated CamKIIα-hM4Di to reduce DGC recruitment. CamKIIα-hM4Di significantly decreased fosTRAPed DGCs compared to GFP controls in the novel environment [t(10) = 2.637, P = 0.025, Student’s t-test; Fig. 7B and D] such that FosTRAPed cell density was comparable to the minimal recruitment observed in the home cage (novel environment: 0.4317 ± 0.1, home cage: 0.4167 ± 0.1). Thus, the CNO doses in the behavioural studies successfully modulated DGC recruitment in vivo without inducing seizure-like events.
Selectively increasing DGC activity compromises spatial object recognition performance
While DGCs are the dentate gyrus’s principal signalling population, mossy cells are also dentate glutamatergic neurons that powerfully affect circuit activity. Mossy cell numbers are selectively decreased in the pilocarpine model, but are present in control animals and can express CamKIIα-DREADDs. Recent work demonstrates that mossy cells have opposing effects on dentate gyrus activity (increasing mossy cell activity induces DGC quiescence, suppressing mossy cells increases DGC activity; Bui et al., 2018). Hence, we wanted to ensure DREADD manipulation restricted to DGCs reproduced our findings. Proopiomelanocortin (POMC) is expressed in DGCs and a subpopulation of hypothalamic neurons (McHugh et al., 2007). We used a POMC-Cre recombinase line to direct FLEX-switched DREADD viral expression (AAV5-hsyn-DIO-hM3Dq-mcherry) to DGCs specifically. POMC-hM3Dq activation recreated the SOR performance deficit compared to POMC-Cre animals expressing Cre-GFP [t(22) = 2.196, P = 0.039, Student’s t-test; Fig. 8A]. POMC-hM3Dq did not affect novel object recognition performance [t(22) = 0.3256, P = 0.748, Student’s t-test; Fig. 8B], demonstrating hippocampal-specificity. Interestingly, POMC-hM3Dq mice were more anxious on the open field test [t(20) = 3.857, P = 0.001, Student’s t-test; Fig. 8C]. Viral expression (Supplementary Fig. 2) revealed the FLEX-switch virus travelled farther than the CamKIIα virus and affected ventral DGCs in addition to dorsal DGCs (CamKIIα: 950 ± 150 µm of tissue, FLEX-switch: 2100 ± 200 µm). We examined ambulation during the SOR test trial to see if POMC-hM3Dq mice displayed anxiogenic-like behaviour; we found no difference in thigmotaxis between POMC-hM3Dq mice and POMC-GFP mice [t(21) = 0.8083, P = 0.428, Student’s t-test; Fig. 8D], nor in the time the groups spent exploring objects (Supplementary Fig. 9E), suggesting that the SOR deficit was not a function of induced anxiety. Finally, voltage sensitive dye imaging showed CNO bath application (10 µM) significantly increased responses [t(3) = 4.302, P = 0.023, two-tailed paired t-test; Fig. 8E–I], confirming that Cre-driven hM3Dq enhanced DGC activation as we expected. Thus, increasing DGC activity is sufficient to compromise cognitive abilities independently of other circuit phenotypes associated with epilepsy.
Figure 8.

Selectively activating DGCs compromises SOR. POMC-Cre mice were injected with either GFP or hM3Dq and received 0.3 mg/kg CNO 1 h prior to test trials throughout behavioural testing. (A) SOR performance [t(22) = 2.196, P = 0.039]. (B) Novel object recognition performance [t(22) = 0.3256, P = 0.748]. (C) Open field anxiety analysis [t(20) = 3.857, P = 0.001]. (D) SOR test trial anxiety analysis [t(20) = 0.8083, P = 0.428]. (E) Representative micrograph of voltage sensitive dye recording configuration in an hM3Dq-expressing slice. Scale bar = 425 µm. (F and G) ΔF/F0 (%) max response in artificial CSF (aCSF) (F) and 10 µM CNO (G). (H) Representative traces of ΔF/F0 in artificial CSF (top) and CNO (bottom). (I) Quantification of response to stimulation in the DGC+molecular layer [t(3) = 4.302, P = 0.023]; each circle represents the average of two to three slices per animal. Statistical comparisons were Student’s t-tests (A–D) or two-tailed paired t-tests (I). Data are represented as mean ± SEM. Each circle represents an animal (number of animals indicated in parentheses). nsP > 0.05, *P < 0.05, **P < 0.01.
Discussion
In these studies, we sought to manipulate net circuit activity by bidirectional DREADD modulation to effect behavioural outcomes, particularly exploring if circuit-based therapeutic approaches could correct epilepsy-associated cognitive dysfunction. Remarkably, acutely reducing DGC hyperactivity in chronically epileptic animals rescued their behavioural performance on the dentate-mediated SOR task (Figs 2C and 3B). This restoration of function was surprising given that the epileptic dentate gyrus exhibits many mechanistic changes, including (i) increased birth and aberrant circuit integration of adult-born DGCs (Parent et al., 1997); (ii) loss of GABAergic and glutamatergic hilar neurons (Blumcke et al., 2000; Kobayashi and Buckmaster, 2003) (Supplementary Fig. 1); (iii) changes in the DGC’s intrinsic properties (Bender et al., 2003); and (iv) recurrent mossy fibre sprouting (Tauck and Nadler, 1985) (Supplementary Fig. 1), which forms monosynaptic, autoexcitatory connections between DGCs (Scharfman et al., 2003), altering both DGC recruitment and their coding properties (Dengler et al., 2017).
Despite this ensemble of disruptive, epilepsy-associated circuit changes, our results demonstrate that the chronically epileptic dentate gyrus retains the underlying capability to perform its inherent function: to code information. Ordinarily the dentate gyrus is sparsely active, which allows the circuit to differentiate incoming inputs. Outside of seizure events, the epileptic dentate gyrus recruits 3.5-fold more DGCs in behavioural contexts than the normal dentate (Fig. 1D). Incoming inputs may overwhelm DGCs, masking the coding-permissive integrated output they propagate downstream in noise. One could conceptualize appropriate dentate gyrus activity as a bell curve or tuning curve, with epilepsy pushing DGCs too far along the activity spectrum, resulting in poor coding and concomitant behavioural deficits. Imposing constraint on DGC hyperactivity, thus, allows the dentate gyrus to operate within a functional window such that the circuit salvages some of its coding and orthogonalization capabilities (Supplementary Fig. 5B).
Our findings are consistent with the hypothesis that brain circuits are tuned optimally to generate behaviour, and that neurological disease can disrupt this tuning to effect measurable behavioural deficits. Using varying KORD recruitment, we were able to create our hypothesized network excitability-behaviour curve (Supplementary Fig. 5B) within a single epileptic cohort (Fig. 3B). We found similar evidence for this network excitability-behaviour tuning curve when we modulated DGC activity in control mice, which presumably already have properly tuned circuits; increasing or decreasing DGC recruitment compromised performance (Fig. 3C and Supplementary Fig. 5C). These data collectively provide multiple lines of evidence demonstrating the necessity to tune the dentate gyrus to an appropriate activity level to affect successful behavioural outcomes.
Appropriate circuit tuning is not a novel concept for network function; homeostatic plasticity is a ubiquitous mechanism maintaining suitable activity levels in the healthy brain. Furthermore, an ever-growing body of literature demonstrates that modulating specific neural targets can improve cognitive outcomes via optogenetics (Roy et al., 2016; Smith et al., 2016; Yang et al., 2018), chemogenetics (Fortress et al., 2015; Lopez et al., 2016; Wang et al., 2018), or deep brain stimulation (Lee et al., 2013; Hao et al., 2015). However, this is among the first studies in which a targeted circuit manipulation—particularly such an acute manipulation—can rescue cognitive function in fully developed chronic epilepsy, a disease in which circuit properties are dramatically and permanently altered from their previously functional state. Izadi et al. (2019) recently demonstrated a similar cognitive recovery in chronic TLE rats using medial septum stimulation. These data along with our findings are promising for patients who suffer with cognitive deficits: therapeutically targeting net circuit excitability, a downstream network outcome, can restore cognitive function independently of correcting the tonic pathological alterations in the epileptic dentate gyrus.
Several lines of evidence (discussed below) suggest it is unlikely that DREADD activation improved SOR performance due to seizure modulation. In the systemic pilocarpine TLE mouse model, seizures occur in repeated 4–8-day clusters with 5–20 seizures per day; clusters are separated by more than a week during which there are few to no seizures (Supplementary Fig. 3). Overall, average aggregate seizure frequencies are approximately 1–3/day (Supplementary Fig. 3). In the present studies, we were unlikely to significantly affect seizure severity as we activated the DREADDs focally and acutely (over a period of hours). The DREADDs were expressed only in the dorsal dentate gyrus (Supplementary Fig. 2), and the ventral hippocampus may be involved more implicitly in seizure generation (discussed below). Furthermore, we took steps to minimize the probability of seizure occurrence during behavioural testing periods. We attempted to select mice that were in an inter-seizure cluster period; mice evidencing seizures during preliminary handling, habituation, or testing periods were not included in analyses as they likely were mid-seizure cluster. Additionally, mice continued to display seizures even when we recruited dorsally-expressed inhibitory DREADDs acutely (during behavioural training/testing, Supplementary Fig. 6) or over several days (Supplementary Fig. 7).
This does not preclude the possibility that DREADDs were subtly affecting pathological-related activity during the behavioural studies; in addition to seizures, epileptic activity includes interictal spikes, which are known to disrupt cognitive performance (Shatskikh et al., 2006). It is possible that inhibitory DREADDs improved behavioural outcomes in part by blocking interictal burst activity. However, we do not believe the behavioural rescue was a function of seizure/interictal spike suppression. If such suppression were responsible for the improved performance, we would expect the ‘high’ KORD ligand dose to support successful behavioural outcomes, as it would be more effective in suppressing interictal activity. Instead, the animals, which performed well with the moderate dose, failed to discriminate with the higher dose (Fig. 3B), suggesting that modulating ongoing pathological activity was not the primary cause for the behavioural rescue.
However, the potential for both seizure and behavioural therapies suggests future studies: perhaps multifocal seizure models can be managed by discretely targeting multiple nodes within the propagation network. TLE patients and rodent models show damage in the substantia nigra (Turski et al., 1986), which is commonly recruited as seizures generalize. Other examples of affected regions include the amygdala (Turski et al., 1986; Bertram, 1997) and perirhinal cortex (Kelly and McIntyre, 1996; Vismer et al., 2015). Within the hippocampus, seizures in patients preferentially affect the anterior hippocampus, the region analogous with the ventral hippocampus in rodents. Several groups have found that TLE seizures more commonly start in the ventral hippocampus and propagate up the septotemporal axis to recruit dorsal regions (Racine et al., 1977; Toyoda et al., 2013); therefore, even within the hippocampus, one needs to target both dorsal and ventral DGCs to manage seizure propagation successfully. Encouragingly, chemogenetics have already proven successful in modulating seizure activity in focal epilepsy models (Kätzel et al., 2014) and in hippocampal interventions that span the septotemporal axis (Zhou et al., 2018). With modulatory techniques like chemogenetics, each circuit could be targeted and modified as needed, providing more personalized treatment and comprehensive behavioural and seizure management.
Several features make DREADDs an attractive and potentially translational therapeutic approach. For one, directed and deliberate DREADD expression coupled with the ligand’s specificity for the designer receptor minimize potential side-effects. Furthermore, these synthetic ligands possess no endogenous biological activity, but are highly bioavailable and can cross the blood–brain barrier. CNO has been found to back-metabolize to clozapine at higher rates than desired, making it a less selective ligand than originally expected (Gomez et al., 2017). However, multiple groups continue to engineer and to test alternative ligands that can interact with muscarinic DREADDs, to date validating Compound 21 and perlapine as CNO alternatives (Chen et al., 2015; Thompson et al., 2018).
The FosTRAP studies highlighted another important feature: how DREADDs modulate neural activity. Activating excitatory DREADDs with 10 times the CNO dose we used in behavioural tasks did not induce spontaneous activity when mice remained in familiar environments (Fig. 7E). Rather, the DREADDs only enhanced DGC activity when there were behaviourally relevant stimuli, i.e. exposure to a novel environment (Fig. 7C). This suggests that instead of forcing activity, DREADDs merely modulated the probability that DGCs would fire in response to endogenous afferent inputs. This proves interesting as effective, long-term treatment of cognitive symptoms in patients would require that the circuit can adapt to various contexts as needed, i.e. retain flexibility. Thus, interventions can not be overly deterministic. Rather than dictating neuronal responses, chemogenetics act to modulate the circuit to function within a window of activity, recruiting cell responses to endogenous inputs.
Further studies are needed to determine if circuit-based therapies like chemogenetics are a viable translational therapy. One could explore how dentate gyrus tuning restoration affects coding in downstream structures, like CA1 or more downstream cortical regions, and if this circuit correction can positively affect more complex cognitive functions than the spatial task we used. Regarding chemogenetics specifically, it is important to note that we activated DREADDs acutely to improve behavioural deficits. DREADD-expressing neurons may develop compensatory changes in response to prolonged activation, which may decrease their effectiveness. However, to date, studies lasting several months have not shown such compensatory changes to sustained DREADD activation (Urban and Roth, 2015).
Many complex mechanistic alterations contribute to disease-related circuit disruptions and their corresponding behavioural phenotypes. This study demonstrates that the aggregate sum of these disruptions results in net changes in circuit excitability such that the disease-altered circuit moves away from a set activity point, which normally supports the successful coding of behavioural responses. Furthermore, our data highlight that restoring the circuit’s principal cell outputs to this appropriate activity point is a viable therapeutic option, as it restores circuit and behavioural functionality irrespective of the mechanisms underlying the disease-related deviation. In conclusion, this study demonstrates that restoring circuit tuning with circuit-based therapies can provide promising therapeutic results.
Supplementary Material
Acknowledgements
We thank Alicia White, Iraklis Petrof, and Shareen Nelson for technical assistance. We thank Marc Fuccillo, Frances Jensen, and Diego Contreras for their feedback on the manuscript.
Glossary
Abbreviations
- 4OHT
4-hydroxytamoxifen
- CNO
clozapine N-oxide
- DGC
dentate gyrus granule cell
- DREADDs
designer receptors exclusively activated by designer drugs
- FosTRAP
fos-targeted recombinase in active populations
- KORD
kappa opioid receptor DREADD
- SB
salvinorin B
- SOR
spatial object recognition
- TLE
temporal lobe epilepsy
Funding
This research was supported by NIH grants from the National Institute of Neurological Disorders and Stroke (NINDS R01s NS082046 and NS038572 to D.A.C), the National Institute for Mental Health (NIMH T32MH017168 to J.B.K., T32MH019112–27 to R.G.P.), and the Eunice Kennedy Shriver National Institute of Child Health and Human Development to the Intellectual and Developmental Disabilities Research Center at CHOP/PENN 1U54HD086984–01.
Competing interests
The authors report no competing interests.
References
- Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci 2007; 104: 5163–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender RA, Soleymani S V, Brewster AL, Nguyen ST, Beck H, Mathern GW et al. . Enhanced expression of a specific hyperpolarization-activated cyclic nucleotide-gated cation channel (HCN) in surviving dentate gyrus granule cells of human and experimental epileptic hippocampus. J Neurosci 2003; 23: 6826–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg AT, Zelko FA, Levy SR, Testa FM. Age at onset of epilepsy, pharmacoresistance, and cognitive outcomes: a prospective cohort study. Neurology 2012; 79: 1384–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram EH. Functional anatomy of spontaneous seizures in a rat model of limbic epilepsy. Epilepsia 1997; 38: 95–105. [DOI] [PubMed] [Google Scholar]
- Blumcke I, Suter B, Behle K, Kuhn R, Schramm J, Elger CE et al. . Loss of Hilar Mossy cells in ammon’s horn sclerosis. Epilepsia 2000; 41: S174–80. [DOI] [PubMed] [Google Scholar]
- Brooks-Kayal AR, Shumate MD, Jin H, Rikhter TY, Coulter DA. Selective changes in single cell GABA A receptor subunit expression and function in temporal lobe epilepsy. Nat Med 1998; 4: 1166–72. [DOI] [PubMed] [Google Scholar]
- Bui AD, Nguyen TM, Limouse C, Kim HK, Szabo GG, Felong S et al. . Dentate gyrus mossy cells control spontaneous convulsive seizures and spatial memory. Science 2018; 359: 787–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla MK, Guzowski JF, Ramirez-Amaya V, Lipa P, Hoffman KL, Marriott LK et al. . Sparse, environmentally selective expression of arc RNA in the upper blade of the rodent fascia dentata by brief spatial experience. Hippocampus 2005; 15: 579–86. [DOI] [PubMed] [Google Scholar]
- Chen X, Choo H, Huang XP, Yang X, Stone O, Roth BL et al. . The first structure-activity relationship studies for designer receptors exclusively activated by designer drugs. ACS Chem Neurosci 2015; 6: 476–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho K-O, Lybrand ZR, Ito N, Brulet R, Tafacory F, Zhang L et al. . Aberrant hippocampal neurogenesis contributes to epilepsy and associated cognitive decline. Nat Commun 2015; 6: 6606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clelland CD, Choi M, Romberg C, Clemenson GD, Fragniere A, Tyers P et al. . A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science 2009; 325: 210–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter DA, Carlson GC. Functional regulation of the dentate gyrus by GABA-mediated inhibition. Prog Brain Res 2007; 163: 235–812. [DOI] [PubMed] [Google Scholar]
- de Lanerolle NC, Kim JH, Robbins RJ, Spencer DD. Hippocampal interneuron loss and plasticity in human temporal lobe epilepsy. Brain Res. 1989; 495: 387–95. [DOI] [PubMed] [Google Scholar]
- Dengler CG, Yue C, Takano H, Coulter DA. Massively augmented hippocampal dentate granule cell activation accompanies epilepsy development. Sci Rep 2017; 7: 42090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortress AM, Hamlett ED, Vazey EM, Aston-Jones G, Cass WA, Boger HA et al. . Designer receptors enhance memory in a mouse model of Down syndrome. J Neurosci 2015; 35: 1343–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner AR, Rowland DC, Hwang SY, Baumgaertel K, Roth BL, Kentros C et al. . Generation of a synthetic memory trace. Science 2012; 335: 1513–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert PE, Kesner RP, Lee I. Dissociating hippocampal subregions: a double dissociation between dentate gyrus and CA1. Hippocampus 2001; 11: 626–36. [DOI] [PubMed] [Google Scholar]
- Gomez JL, Bonaventura J, Lesniak W, Mathews WB, Sysa-Shah P, Rodriguez LA et al. . Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science 2017; 357: 503–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gröticke I, Hoffmann K, Löscher W. Behavioral alterations in the pilocarpine model of temporal lobe epilepsy in mice. Exp Neurol 2007; 207: 329–49. [DOI] [PubMed] [Google Scholar]
- Guenthner CJ, Miyamichi K, Yang HH, Heller HC, Luo L. Permanent genetic access to transiently active neurons via TRAP: targeted recombination in active populations. Neuron 2013; 78: 773–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao S, Tang B, Wu Z, Ure K, Sun Y, Tao H et al. . Forniceal deep brain stimulation rescues hippocampal memory in Rett syndrome mice. Nature 2015; 526: 430–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann U, Beck H, Dreier JPP, Ficker E, Stabel J, Zhang CLL. The dentate gyrus as a regulated gate for the propagation of epileptiform activity. Epilepsy Res Suppl 1992; 7: 273–80. [PubMed] [Google Scholar]
- Holmes GL. EEG abnormalities as a biomarker for cognitive comorbidities in pharmacoresistant epilepsy. Epilepsia 2013; 54: 60–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosford BE, Liska JP, Danzer SC. Ablation of newly generated hippocampal granule cells has disease-modifying effects in epilepsy. J Neurosci 2016; 36: 11013–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izadi A, Pevzner A, Lee DJ, Ekstrom AD, Shahlaie K, Gurkoff GG. Medial septal stimulation increases seizure threshold and improves cognition in epileptic rats. Brain Stimul 2019; 12: 735–42. [DOI] [PubMed] [Google Scholar]
- Jung MW, McNaughton BL. Spatial selectivity of unit activity in the hippocampal granular layer. Hippocampus 1993; 3: 165–82. [DOI] [PubMed] [Google Scholar]
- Kätzel D, Nicholson E, Schorge S, Walker MC, Kullmann DM. Chemical–genetic attenuation of focal neocortical seizures. Nat Commun 2014; 5: 3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly ME, McIntyre DC. Perirhinal cortex involvement in limbic kindled seizures. Epilepsy Res 1996; 26: 233–43. [DOI] [PubMed] [Google Scholar]
- Kesner RP, Taylor JO, Hoge J, Andy F. Role of the dentate gyrus in mediating object-spatial configuration recognition. Neurobiol Learn Mem 2015; 118: 42–8. [DOI] [PubMed] [Google Scholar]
- Kheirbek MA, Drew LJ, Burghardt NS, Costantini DO, Tannenholz L, Ahmari SE et al. . Differential control of learning and anxiety along the dorsoventral axis of the dentate gyrus. Neuron 2013; 77: 955–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Buckmaster PS. Reduced inhibition of dentate granule cells in a model of temporal lobe epilepsy. J Neurosci 2003; 23: 2440–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS et al. . Rapid, reversible activation of AgRP neurons drdives feeding behavior in mice. J Clin Invest 2011; 121: 1424–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook-Magnuson E, Armstrong C, Bui A, Lew S, Oijala M, Soltesz I. In vivo evaluation of the dentate gate theory in epilepsy. J Physiol 2015; 593: 2379–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DJ, Gurkoff GG, Izadi A, Berman RF, Ekstrom AD, Muizelaar JP et al. . Medial septal nucleus theta frequency deep brain stimulation improves spatial working memory after traumatic brain injury. J Neurotrauma 2013; 30: 131–9. [DOI] [PubMed] [Google Scholar]
- Lee I, Hunsaker MR, Kesner RP. The role of hippocampal subregions in detecting spatial novelty. Behav Neurosci 2005; 119: 145–53. [DOI] [PubMed] [Google Scholar]
- Leutgeb JK, Leutgeb S, Moser M-B, Moser EI. Pattern separation in the dentate gyrus and CA3 of the hippocampus. Science 2007; 315: 961–6. [DOI] [PubMed] [Google Scholar]
- Lopez AJ, Kramar E, Matheos DP, White AO, Kwapis J, Vogel-Ciernia A et al. . Promoter-specific effects of DREADD modulation on hippocampal synaptic plasticity and memory formation. J Neurosci 2016; 36: 3588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lothman E, Stringer J. The dentate gyrus as a control point for seizures in the hippocampus and beyond. Epilepsy Res Suppl 1992; 7: 301–13. [PubMed] [Google Scholar]
- Marchant NJ, Whitaker LR, Bossert JM, Harvey BK, Hope BT, Kaganovsky K et al. . Behavioral and physiological effects of a novel kappa-opioid receptor-based DREADD in rats. Neuropsychopharmacology 2016; 41: 402–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh TJ, Jones MW, Quinn JJ, Balthasar N, Coppari R, Elmquist JK et al. . Dentate gyrus NMDA receptors mediate rapid pattern separation in the hippocampal network. Science 2007; 317: 94–9. [DOI] [PubMed] [Google Scholar]
- Nakashiba T, Cushman JD, Pelkey KA, Renaudineau S, Buhl DL, McHugh TJ et al. . Young dentate granule cells mediate pattern separation, whereas old granule cells facilitate pattern completion. Cell 2012; 149: 188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neunuebel JP, Knierim JJ. Spatial firing correlates of physiologically distinct cell types of the rat dentate gyrus. J Neurosci 2012; 32: 3848–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenaus A, Esclapez M, Houser CR. Loss of glutamate decarboxylase mRNA-containing neurons in the rat dentate gyrus following pilocarpine-induced seizures. J Neurosci 1993; 13: 4470–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira AMM, Hawk JD, Abel T, Havekes R. Post-training reversible inactivation of the hippocampus enhances novel object recognition memory. Learn Mem 2010; 17: 155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci 1997; 17: 3727–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine R, Rose PA, Burnham WM. Afterdischarge thresholds and kindling rates in dorsal and ventral hippocampus and dentate gyrus. Can J Neurol Sci 1977; 4: 273–8. [DOI] [PubMed] [Google Scholar]
- Roy DS, Arons A, Mitchell TI, Pignatelli M, Ryan TJ, Tonegawa S. Memory retrieval by activating engram cells in mouse models of early Alzheimer’s disease. Nature 2016; 531: 508–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Sollas AL, Berger RE, Goodman JH. Electrophysiological evidence of monosynaptic excitatory transmission between granule cells after seizure-induced mossy fiber sprouting. J Neurophysiol 2003; 90: 2536–47. [DOI] [PubMed] [Google Scholar]
- Shatskikh TN, Raghavendra M, Zhao Q, Cui Z, Holmes GL. Electrical induction of spikes in the hippocampus impairs recognition capacity and spatial memory in rats. Epilepsy Behav 2006; 9: 549–56. [DOI] [PubMed] [Google Scholar]
- Simon P, Dupuis R, Costentin J. Thigmotaxis as an index of anxiety in mice. Influence of dopaminergic transmissions. Behav Brain Res 1994; 61: 59–64. [DOI] [PubMed] [Google Scholar]
- Smith AS, Williams Avram SK, Cymerblit-Sabba A, Song J, Young WS. Targeted activation of the hippocampal CA2 area strongly enhances social memory. Mol Psychiatry 2016; 21: 1137–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauck DL, Nadler J V. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci 1985; 5: 1016–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson KJ, Khajehali E, Bradley SJ, Navarrete JS, Huang XP, Slocum S et al. . DREADD agonist 21 is an effective agonist for muscarinic-based DREADDs in vitro and in vivo. ACS Pharmacol Transl Sci 2018; 1: 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda I, Bower MR, Leyva F, Buckmaster PS. Early activation of ventral hippocampus and subiculum during spontaneous seizures in a rat model of temporal lobe epilepsy. J Neurosci 2013; 33: 11100–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turski L, Cavalheiro EA, Sieklucka-Dziuba M, Ikonomidou-Turski C, Czuczwar SJ, Turski WA. Seizures produced by pilocarpine: neuropathological sequelae and activity of glutamate decarboxylase in the rat forebrain. Brain Res 1986; 398: 37–48. [DOI] [PubMed] [Google Scholar]
- Urban DJ, Roth BL. DREADDs (designer receptors exclusively activated by designer drugs): chemogenetic tools with therapeutic utility. Annu Rev Pharmacol Toxicol 2015; 55: 399–417. [DOI] [PubMed] [Google Scholar]
- Vardy E, Robinson JE, Li C, Olsen RHJ, DiBerto JF, Giguere PM et al. . A new DREADD facilitates the multiplexed chemogenetic interrogation of behavior. Neuron 2015; 86: 936–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vismer MS, Forcelli PA, Skopin MD, Gale K, Koubeissi MZ. The piriform, perirhinal, and entorhinal cortex in seizure generation. Front Neural Circuits 2015; 9: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Rein B, Zhang F, Tan T, Zhong P, Qin L et al. . Chemogenetic activation of prefrontal cortex rescues synaptic and behavioral deficits in a mouse model of 16p11.2 deletion syndrome. J Neurosci 2018; 38: 5939–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Yao C, Tian T, Li X, Yan H, Wu J et al. . A novel mechanism of memory loss in Alzheimer’s disease mice via the degeneration of entorhinal–CA1 synapses. Mol Psychiatry 2018; 23: 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Wei W, Mody I, Houser CR. Altered localization of GABAA receptor subunits on dentate granule cell dendrites influences tonic and phasic inhibition in a mouse model of epilepsy. J Neurosci 2007; 27: 7520–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Huguenard JR, Buckmaster PS. Increased excitatory synaptic input to granule cells from hilar and CA3 regions in a rat model of temporal lobe epilepsy. J Neurosci 2012; 32: 1183–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Nemes AD, Lee D, Ro EJ, Zhang J, Nowacki AS et al. . Chemogenetic silencing of hippocampal neurons suppresses epileptic neural circuits. J Clin Invest 2018; 129: 310–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting this study and the custom-written seizure MATLAB scripts are available from the corresponding author upon reasonable request. The MATLAB behavioural analysis code is available on Dr David Meaney’s website (https://www.seas.upenn.edu/∼molneuro/software.html).