

SUMMARY
HIV-1 uses the microtubule network to traffic the viral capsid core toward the nucleus. Viral nuclear trafficking and infectivity require the kinesin-1 adaptor protein FEZ1. Here, we demonstrate that FEZ1 directly interacts with the HIV-1 capsid and specifically binds capsid protein (CA) hexamers. FEZ1 contains multiple acidic, poly-glutamate stretches that interact with the positively charged central pore of CA hexamers. The FEZ1-capsid interaction directly competes with nucleotides and inositol hexaphosphate (IP6) that bind at the same location. In addition, all-atom molecular dynamic (MD) simulations establish the molecular details of FEZ1-capsid interactions. Functionally, mutation of the FEZ1 capsid-interacting residues significantly reduces trafficking of HIV-1 particles toward the nucleus and early infection. These findings support a model in which the central capsid hexamer pore is a general HIV-1 cofactor-binding hub and FEZ1 serves as a unique CA hexamer pattern sensor to recognize this site and promote capsid trafficking in the cell.
In Brief
In this paper, Huang et al. find that the viral cofactor FEZ1, a kinesin adaptor protein, uses multiple negatively charged amino-acid stretches to avidly interact with the positive center pores of the HIV-1 capsid protein hexamers, associating the virus particles to kinesin motors and thus promoting viral trafficking and infection.
Graphical Abstract
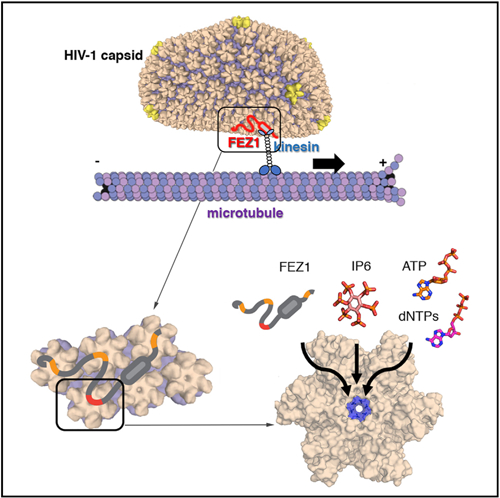
INTRODUCTION
The HIV-1 RNA genome is enclosed in the viral capsid, which is a protective protein shell and an interaction hub for cellular factors (Yamashita and Engelman, 2017). After entering a host cell, the capsid must reach the nuclear periphery, where the reverse transcribed viral DNA genome can be delivered into the nucleus for integration into the host genome. Because the capsid is too large to diffuse freely within the crowded cytoplasm environment, active transport along the microtubule network is required for capsid trafficking (Luby-Phelps, 2000). To accomplish the latter, HIV-1 and several other viruses hijack both kinesin and dynein motors to achieve net retrograde movement, even though kinesin motors move toward the cell periphery (Malikov et al., 2015). While counterintuitive, this bidirectional transport is a common mechanism in cellular cargo trafficking and enables efficient movement through the crowded cytoplasm (Müller et al., 2008; Welte, 2010). In addition, HIV-1 can stimulate the formation of a stable subset of microtubules that have long half-lives and higher affinities for kinesins, making them ideal for long-range cytoplasmic movement of cargos and viruses that exploit kinesins for retrograde movement (Fernandez et al., 2015; Malikov et al., 2015; Sabo et al., 2013).
The relationship between HIV-1 and microtubule trafficking appears to be more complex than simply getting the capsid to the nucleus. Microtubule stability and capsid transport are tightly connected with controlled capsid disassembly (termed uncoating). Uncoating is finely tuned to the viral life cycle, because the viral genome must initially remain protected by the capsid and later exit it for integration. Capsid uncoating is delayed and viral infectivity is reduced when interfering with microtubule networks, their motors or associated adaptors, or the regulators of their stability (Arhel et al., 2006; Bukrinskaya et al., 1998; Carnes et al., 2018; Dharan et al., 2017; Lukic et al., 2014; Malikov et al., 2015; McDonald et al., 2002; Pawlica and Berthoux, 2014). Although poorly understood, it has been suggested that the opposing forces applied by motor proteins and capsid-interacting factors at nuclear pores may contribute to capsid uncoating (Campbell and Hope, 2015; Gaudin et al., 2013).
Because the capsid is the exposed surface of the virus core during early infection, it is the most probable direct link between HIV-1 and the cytoskeleton. Capsid is known to be directly recognized by numerous cellular factors that either promote or inhibit infection (Delaney et al., 2017; Fritsche et al., 2016; Jia et al., 2015; Yamashita and Engelman, 2017), but little is known regarding whether or how it associates with the microtubule network. Although several microtubule-associated proteins cosediment with HIV-1 capsid assemblies, the dynein-adaptor protein BICD2 is one of the two microtubule-associated proteins demonstrated to date to directly bind capsid (Carnes et al., 2018; Dharan et al., 2017). As an adaptor protein, BICD2 can activate dynein-mediated transport while binding cargo, and HIV-1 may hijack this functionality for trafficking toward the nucleus. The other factor, a kinesin-1 adaptor protein, fasciculation and elongation protein zeta 1 (FEZ1), was identified as an important cofactor for early post-entry HIV-1 infection (Malikov et al., 2015; Malikov and Naghavi, 2017). FEZ1 promotes uncoating and trafficking of incoming viral cores to the nucleus in a kinesin-dependent manner, suggesting that HIV-1 coopts its adaptor function to facilitate early steps of viral infection. FEZ1 from mammalian cell lysates co-pellets with in vitro assembled capsid assemblies, indicating it may be a key link between capsid and the microtubule network (Malikov et al., 2015).
Molecular details by which microtubule-associated proteins recognize HIV-1 capsid are unknown. The fullerene cone morphology of the assembled capsid provides numerous unique and repetitive surface patterns that may be targeted by protein and small-molecule cofactors. Many capsid-binding host factors only weakly interact with the capsid protein (CA) monomer (Bhattacharya et al., 2014; Li et al., 2016; Liu et al., 2016). Instead, these host factors only recognize surfaces found in assembled capsids, which are built from 1,000 to 1,500 CA molecules. In the capsid, CA is arranged primarily as hexamer subunits, with 12 pentamer subunits necessary for capsid closure (Ganser et al., 1999; Li et al., 2000; Zhao et al., 2013). The 2- and 3-fold symmetric interfaces between hexamers, as well as intra-hexamer surfaces, provide unique binding sites for host factors (Ganser-Pornillos et al., 2008; Li et al., 2000). Because of the redundant nature of the capsid lattice, as well as numerous practical challenges when using assembled capsids (such as their heterogeneity and insolubility), it has been difficult to decipher the diverse capsid-binding modes of host factors.
Here, we use purified recombinant FEZ1 protein to demonstrate that it directly interacts with assembled capsid. Using soluble CA oligomers that capture various partial hexamer architectures (Summers et al., 2019), we show that FEZ1 specifically binds CA hexamers. We demonstrate that FEZ1 contains multiple hexamer-sensing motifs consisting of acidic, poly-glutamate stretches that bind at the highly conserved, positively charged hexamer central pore. Our all-atom molecular dynamic (MD) simulations provide details of this FEZ1-CA interaction. In addition, point mutations of the hexamer-binding FEZ1 sequence severely reduced retrograde viral trafficking and infectivity. The FEZ1-CA hexamer interaction directly competed with binding by deoxyribonucleotide triphosphates (dNTPs) and inositol hexaphosphate (IP6)—two small-molecule viral cofactors that bind in the same hexamer location (Dick et al., 2018; Jacques et al., 2016; Mallery et al., 2018). These findings support the notion that the central hexamer pore is a common cofactor-binding site for both proteins and small molecules. FEZ1 is a unique high-affinity CA hexamer pattern sensor, specifically targeting this site to form a bridge between capsid and kinesin motors to regulate HIV-1 intra-cellular trafficking and uncoating.
RESULTS
FEZ1 Directly Recognizes Assembled Capsid
We first sought to determine whether the FEZ1-capsid interaction observed previously using crude mammalian cell lysates is direct or whether additional cellular cofactors are involved. FEZ1 is predicted to be a natively unfolded protein with a putative coiled-coil region in its C-terminal half (Lanza et al., 2009) (Figure 1A). We recombinantly expressed full-length FEZ1 (FEZ11–392) and a series of FEZ1 truncation constructs in Escherichia coli and purified them to homogeneity (Figure S1A). We tested these FEZ1 constructs for their ability to copellet with in vitro assembled disulfide-crosslinked CA tubes, because these are well established to mimic the HIV-1 capsid lattice (Pornillos et al., 2009). The full-length protein, FEZ11–392, demonstrated strong CA tube copelleting under physiological ionic strength conditions (150 mM NaCl) (Figure 1B). Furthermore, we identified a truncated construct, FEZ192–198, which binds CA tubes equally well. In contrast, FEZ1 N- and C-terminal portions outside of this region did not show measurable copelleting under the same conditions (Figure 1B). These results demonstrate a direct interaction between FEZ1 and capsid that is not mediated by additional cellular factors. This lends support to a direct linkage between HIV-1 capsid and kinesin-mediated cytoskeleton trafficking.
Figure 1. FEZ1 Is a Specific CA Hexamer Sensor.

(A) Schematic diagram of the FEZ1 constructs (marked by the black dotted lines) used for capsid-binding assays. The yellow bar indicates the predicted coiled-coil region of FEZ1. The putative functional regions of FEZ1 are labeled with different colors.
(B) Copelleting assay of FEZ11–392, FEZ11–92, FEZ192–198, and FEZ1198–392 with 14C/45C crosslinked CA tubes in 150 mM NaCl. Full-length FEZ11–392 and FEZ192–198 copellet with CA tubes, while the N-terminal (FEZ11–92) and C-terminal (FEZ1198–392) regions do not. T, total load; S, supernatant; P, pellet.
(C–H) Size-exclusion chromatography (SEC)-binding assay of FEZ192–198 with different CA assemblies (schematics shown in cartoon insets). The concentrations of FEZ1 and CA monomer in each binding reaction were 33 and 98 μM, respectively. FEZ192–198 does not coelute with CA monomer (C), 1/3-hexamer (D), or 1/2-hexamerEE (E) in SEC assays. There is no elution shift of the mixture (solid purple) relative to the individual components (dotted lines). FEZ192–198 does coelute with the CA hexamer (G), and restoration of a hexamer from two 1/2-hexamers (cyan and pink cartoons) restores FEZ192–198 coelution (F), which coelutes at the same position as with CA hexamers. In contrast, FEZ192–198 does not coelute with hexamer-2foldon (purple cartoon), which also contains 6 CA subunits but arranged around the 3-fold capsid interface (H).
See also Figure S2.
FEZ1 Is a Specific CA Hexamer Pattern Sensor
To better define the specific capsid surface targeted by FEZ1, we used size-exclusion chromatography (SEC) coelution assays to test for interactions between FEZ192–198 and various soluble CA assemblies. We primarily used the FEZ192–198 construct because of its superior expression and solution behavior compared with full-length FEZ1. For CA assemblies, we first tested the established native CA monomers-dimers and disulfide-stabilized CA pentamers and hexamers (Pornillos et al., 2010; Pornillos et al., 2011). In addition, we developed an extensive library of stabilized CA oligomers that recapitulate the unique intra- and inter-hexamer surfaces found in the assembled capsid lattice (Summers et al., 2019). These new CA oligomers include a series of partial-hexamer assemblies, which subdivide the CA hexamer into smaller fragments, such as 1/3- and 1/2-hexamers. 1/3- and 1/2-hexamers contain two and three disulfide-linked CA monomers, respectively, arranged with 6-fold rotations just as they are in complete hexamers. 1/2-hexamer assemblies are discrete in solution but have the ability to dimerize into complete hexamers in certain conditions. This hexamer formation can be prevented by the addition of A42E/T54E mutations at exposed intra-hexamer surfaces (termed 1/2-hexamer-EE). Furthermore, we engineered a new CA hexamer that contains six CA molecules arranged as a trimer of dimers. This structure contains the 3-fold inter-hexamer interface and is stabilized by a fusion to the naturally trimeric bacteriophage T4 foldon domain (termed hexamer-2foldon).
Our comprehensive CA assembly library allowed us to unambiguously establish that FEZ1 specifically recognizes the CA hexamer, which is considered the repeating unit of the capsid lattice. We first tested for FEZ192–198 binding to native CA monomers-di-mers but observed no coelution between them (Figure 1C). Similarly, we observed no coelution between FEZ192–198 and either 1/3-hexamers or 1/2-hexamers that contained hexamer-preventing mutations (Figures 1D and 1E). In contrast, we consistently observed complete coelution between FEZ192–198 and CA hexamers (Figure 1G). A lack of interaction with CA dimers or partial hexamers, despite strong recognition of hexamers, demonstrates a novel FEZ1 capsid-lattice-sensing ability. We also detected strong FEZ192–198 coelution with the 1/2-hexamer assembly that is competent to dimerize into a hexamer. The observed elution profile closely resembles that of the hexamer-FEZ1 complex, suggesting that FEZ1 is able to bridge two 1/2-hexamers into a hexamer (Figure 1F). These data reinforce the idea that FEZ1 targets a complete CA hexamer. In addition, FEZ1 does not appear to require inter-hexamer surfaces for binding, because no coelution was observed with hexamer-2foldon, which contains native 2- and 3-fold inter-hexamer interfaces (Figure 1H). Despite their structural similarity to hexamers, we observed no FEZ1 coelution with CA pentamers (Figure S2). These data demonstrate that FEZ1 targets a highly specific surface pattern only found in complete hexamers. A binding mode of this nature has not been described for other capsid-binding proteins.
FEZ1 Uses Negatively Charged Poly-glutamate Regions to Bind CA Hexamers
The amino acid sequence of FEZ1 is notable in that it contains numerous negatively charged glutamate and aspartate residues (Figure 2A). A high concentration of negative charge is present within FEZ192–198, although many additional charged residues appear in the N-and C-terminal portions of the molecule. About one-third of the residues in FEZ1158–198 are negatively charged (underlined in Figure 2A). There is a stretch where nine of eleven residues are negatively charged (residues 181–191). We hypothesized that these acidic residues may interact with CA hexamers through electrostatic interactions.
Figure 2. FEZ1 Uses Negatively Charged Poly-glutamate Regions to Bind CA Hexamers.

(A) Protein sequence of the full-length FEZ11–392. Negatively charged residues (aspartate and glutamate) are in red. Poly-glutamate regions are marked with purple underlines, FEZ1178–188 is marked with a yellow box, and FEZ1158–198 is underlined in green.
(B–E) SEC-binding assays of different FEZ1 constructs with the CA hexamer. FEZ192–158 (B) and FEZ1158–182 (C) do not coelute with CA hexamer, while FEZ1158–198 (D) does. FEZ1158–198 182EA (E), with five consecutive glutamate residue 182EEEEE186 mutated to alanines, showed reduced interaction with CA hexamer. Schematics of the FEZ1 constructs are boxed in yellow and shown on top of the chromatograms. Red bars indicate the negatively charged residues of FEZ1.
(F) Summary table of the ITC data of the binding of FEZ1 92–198, FEZ1 178–188, and FEZ1 92–198 182EA with the CA hexamer. Data are represented as mean ± SD.
(G–I) Representative ITC curves of FEZ1 92–198 (G), FEZ1 92–198 182EA (H), and FEZ1178–188 (I) with the CA hexamer.
We performed molecular dissection of FEZ192–198 to identify its hexamer-binding regions. The first two-thirds of FEZ1, namely, FEZ192–158, did not show binding to CA hexamers in the SEC assay, while the highly negatively charged FEZ1158–198 demonstrated stable coelution. (Figures 2B and 2C). However, when residues in the C terminus of this interacting region were removed, the resulting FEZ1158–182 lost the ability to coelute with hexamers (Figure 2D). This suggests that residues in FEZ1 182–198 critically contribute to high-affinity CA hexamer binding. Consistent with this notion, when five sequential glutamates were mutated to alanines in the FEZ1158–198 stretch (FEZ1158–198 182EA), namely, 182EEEEE186 to 182AAAAA186, the mutation greatly reduced the FEZ1 CA-hexamer interaction (Figure 2E). Subsequent mapping identified the highly negatively charged FEZ1178–188 region (178SPDPEEEEEVL188) to be the minimal construct that retained strong interaction with CA hexamers (Figure S1B).
The SEC assay offers a quick, qualitative means to detect complex formation. However, it is only applicable to relatively stable complexes with binding affinity (dissociation constant [KD]) at the nanomolar or low micromolar range. To quantify the interaction, we used isothermal titration calorimetry (ITC) to directly measure the thermodynamic properties of the FEZ1 CA hexamer association. The binding of FEZ192–198 with CA hexamers is endothermic with a KD of 300 ± 60 nM (Figures 2F and 2G). This binding affinity is at least an order of magnitude higher than previously analyzed capsid-binding proteins such as CypA, TRIM5α, TRIMCyp, CPSF6, and Nup153 (Bhattacharya et al., 2014; Biris et al., 2012; Price et al., 2014; Yoo et al., 1997; Zhou et al., 2015), making FEZ1 the strongest CA hexamer binder known to date. The hexamer-binding affinity of the FEZ192–198 182EA mutant was reduced by approximately an order of magnitude (2.5 μM), highlighting the importance of the polyglutamate peptide sequence (Figures 2F and 2H). The binding stoichiometry was measured at slightly over one CA hexamer to FEZ1 (n = 1.3 ± 0.1 for FEZ192–198 and n = 1.6 ± 0.2 for FEZ192–198 182EA), partly because of the existence of multiple CA hexamer-binding sites within FEZ1 (one major and multiple additional weak sites, described later). When the minimal FEZ1178–188 construct containing the major CA hexamer-binding site is used, a comparable affinity was observed (KD = 180 ± 40 nM), with a 1:1 CA hexamer to FEZ1178–188 stoichiometry (n = 1.0 ± 0.2) (Figures 2F and 2I).
FEZ1 Specifically Recognizes the Positively Charged Center Pore of the CA Hexamer
Because FEZ1 uses a highly negatively charged region to interact with the CA hexamer, we examined the CA hexamer surface electrostatic potential distribution to identify the region bound by FEZ1. There is a ring of positively charged R18 residues lining the center pore of the capsid hexamer (Figure 3A). We investigated the importance of this highly positively charged region by mutating R18 to an oppositely charged residue (R18D) (Figures 3B and S3A). To verify that the R18D mutation does not affect the conformation of the CA hexamer, we determined the crystal structure of the R18D CA hexamer at 2.1Å resolution. Structural comparison between R18D and wild-type (WT) CA hexamers reveals minimal structural changes as measured by the root-mean-square deviation (RMSD) of ~0.5Å for all six CA molecules (Figures 3B and S3B; Table S1). We confirmed that the R18D mutation does not affect the morphology of CA tubes, as shown by negative-stain electron microscopy (EM) (Figure S4A).
Figure 3. FEZ1 Recognizes the Positively Charged Center Pore of CA Hexamers through Electrostatic Interactions.

(A) Comparison of the electrostatic potential surfaces of R18D (right) and WT (left) CA hexamers in the closed state (PDB: 3H47), as viewed from outside of the capsid (red, negative charge; blue, positive charge). The unit of the electrostatic potential map is kBT/e. Zoomed-in views of the hexamer center pores are shown in the middle inset, where the β-hairpin is not shown for clarity of the charges of the R18 (top) and R18D (bottom) pores.
(B) Left, overlay of the crystal structures of the WT CA hexamer (cyan) with the R18D CA hexamer (tan). The mutation does not change the hexamer structure. Right, electrostatic potential surface of the R18D CA hexamer shows the negatively charged center pore.
(C) R18D mutation substantially reduced the binding of FEZ192–227 with CA hexamer in SEC-binding assays (top) compared with the CA hexamer R18 (bottom).
(D) R18D mutation drastically reduced the copelleting of FEZ1 with CA tubes. T, total load; S, supernatant; P, pellet.
(E) Copelleting assays showing the binding of FEZ1 constructs to CA tubes at increasing ATP (left) or IP6 (right) concentrations. Only the FEZ1 bands are shown for clarity. FEZ11–392 is able to bind with CA tubes in the presence of physiological levels of ATP/IP6, and the increase of ATP/IP6 decreased binding. The binding of FEZ1178–188 to CA tubes is nearly abolished at 20 μM ATP/IP6, even though it has high affinity to individual CA hexamers. This difference is consistent with the avidity effect from multiple capsid-binding sites in full-length FEZ1. Center inset: structural illustrations of ATP (left, PDB: 6ERN) and IP6 (right, PDB: 6HBS) binding to the CA hexamer (middle) center R18 residues (blue).
See also Figures S3–S7.
Introduction of the R18D mutation drastically reduced the recognition of CA hexamers by FEZ1, while no binding reduction was observed by mutations in factor-binding sites at other known capsid-binding locations. In both SEC coelution assays and CA tube copelleting assays, the R18D mutation substantially reduced FEZ1 binding (Figures 3C and 3D). To confirm that the loss of FEZ1 binding does not result from destabilization of the R18D hexamer by negative charge repulsion at the R18D center pore, we tested FEZ1 binding to the neutral mutant R18A CA tubes. A similar reduction in FEZ1 binding was observed with the conservative R18A CA mutation (Figure S5), supporting direct binding at the R18 site rather than an indirect destabilization effect by R18D. To determine whether FEZ1 uses other established host factor-binding sites on capsid, we tested FEZ1-CA tube copelleting with various capsid mutations known to reduce capsid recognition by the host protein CypA (G89V and P90A) and the restriction factor MxB (P207S, G208R, and T210K) (Figure S4B) (Busnadiego et al., 2014; Gamble et al., 1996; Goujon et al., 2013; Kane et al., 2013; Matreyek et al., 2014). None of the aforementioned mutations reduced the FEZ1-CA tube interaction (Figure S4B). These results show that a specific electrostatic interaction at the hexamer center is necessary for FEZ1-capsid interactions.
The β-hairpin region of R18D CA adopted the closed conformation (Jacques et al., 2016) under our crystallization condition. A comparison of the R18D CA hexamer structure with known CA hexamer structures in the open or closed state is shown in Figure S6. It has been reported that the β-hairpin region is highly dynamic, with a predominantly open conformation with pH < 7 and a closed conformation with pH > 7 (Jacques et al., 2016). We tested whether the pH-dependent open and closed β-hairpin conformations affect FEZ1 binding by carrying out binding assays at pH 6, 7, and 8. The results showed that in all three conditions, FEZ1 was able to interact with the CA tubes at a similar level (Figure S7). This is likely because of the dynamic nature of the CA β-hairpin and the conformational flexibility of FEZ1, which is predicted to be unstructured in its unbound form. It is conceivable that the binding energy of FEZ1 enables a shift in the equilibrium toward the open conformation of the CA β-hairpin so that FEZ1 is able to reach into the R18 pore for a stable interaction. This hypothesis is supported by our MD simulation shown later, demonstrating FEZ1’s ability to access the hexamer center pore.
FEZ1 Interacts with a Conserved Cofactor-Binding Site on Capsid
To date, no other host proteins have been described to bind at the CA hexamer center using the R18 residue. However, R18 has been shown to be critical in the binding and translocation of nucleotides into the capsid for reverse transcription (Jacques et al., 2016) and in the binding of the small-molecule cofactor IP6 for maturation and stability of capsid (Dick et al., 2018; Márquez et al., 2018). To support our hypothesis that FEZ1 binds in this region of the CA hexamer, we performed competition assays using ATP, dNTPs, or IP6 to compete with FEZ1 for CA tube copelleting (Figures 3E and S4C). As expected, the addition of nucleotides or IP6 reduced FEZ1-CA tube interaction in a concentration-dependent manner. These data substantiate FEZ1 as the first protein known to bind to the same conserved, highly positively charged hexamer center targeted by important small-molecule cofactors.
The nucleotide and IP6 titration results also demonstrate that FEZ1 has the ability to bind capsid under physiological conditions. Full-length FEZ1 retained CA tube binding in the presence of up to 5 mM ATP and 200 μM IP6 (Figure 3E), which are the cellular concentrations of these molecules. The binding was diminished or abolished at higher cofactor concentrations. The construct containing a minimal binding motif (FEZ1178–188), which interacts with CA hexamers at a high affinity (Figures 2F and 2I), lost its binding to CA tubes at 20 μM ATP or 20 μM IP6 (Figure 3E). This effect at a low ATP/IP6 concentration supports that the loss of binding is a consequence of direct competition at the hexamer center where these cofactors interact, rather than an indirect charge screening effect of these charged molecules at high concentrations.
FEZ1 Uses Multiple Negatively Charged Regions to Avidly Bind Capsid
It may appear intriguing that full-length FEZ1 required higher nucleotide triphosphate (NTP) or IP6 concentrations to abolish binding to CA tubes than that needed for FEZ1178–188; the latter showed high-affinity binding to CA hexamers similar to longer FEZ1 constructs (Figures 2F, 2G, and 2I). These data suggest that FEZ1 contains multiple CA hexamer-interacting regions. In our SEC and ITC assays using the CA hexamer as the binding partner, which has a single targeting site, the minimal FEZ1178–188 interacted as effectively as the longer constructs. In contrast, when using CA tubes that contain many hexamer-targeting sites, the simultaneous binding by multiple regions in full-length FEZ1 allows a stronger interaction through the avidity effect. The FEZ1 sequence contains numerous highly negatively charged patches throughout the molecule (Figure 2A). Our data demonstrated efficient CA tube binding by FEZ192–198 (Figure 1B), which likely contains multiple interaction sites, in addition to the strong binding region in FEZ1178–188. Although the N- and C-terminal regions (FEZ11–92 and FEZ198–198) did not bind CA tubes under the conditions tested (150 mM NaCl) (Figure 1B), the abundance of negatively charged segments in these regions indicates that they potentially contain weaker auxiliary binding sites.
To test the hypothesis that FEZ1 contains multiple capsid-binding sites, we performed binding assays at low ionic strength conditions under which the low-affinity electrostatic interactions may sustain. Even at a low-salt condition of 50 mM NaCl, FEZ11–92 and FEZ1198–392 did not coelute with CA hexamers in SEC-binding assays (Figures 4A and 4B). However, they efficiently copelleted with CA tubes under this condition (Figure 4C). These data show that the individual interactions in these auxiliary regions are weak but multiple interactions allowed avid binding to CA tubes. We confirmed that these sites also interact with CA hexamers at the R18 location, because the CA R18D mutation abolished or severely abrogated binding by all FEZ1 constructs (Figure 4C). A binding mode of this nature is supported by the salt dependence of CA tube binding by FEZ1 constructs (Figures 4E and 4F). A longer construct with more capsid-interacting stretches, FEZ192–227, was able to retain binding in the presence of higher salt concentrations compared with a shorter construct with fewer binding stretches, FEZ192–158. These results support a model in which primary hexamer-targeting motifs in FEZ1 (FEZ1158–198) drive FEZ1-capsid interaction through avidity, with auxiliary binding provided by additional FEZ1 acidic stretches (Figure 4D).
Figure 4. FEZ1 Has Multiple Negatively Charged Regions for Avid Binding to the Capsid.

(A andB) SEC-binding assay showing that FEZ11–92 (A) or FEZ1189–392 (B) does not coelute with CA hexamer. There is no elution shift of the mixture (solid purple) relative to the individual components (dotted lines).
(C) Copelleting assays show that capsid-binding sites in both FEZ11–92 and FEZ1189–392 can be detected in low salt (50 mM NaCl); the binding is abolished when using R18D CA tubes (bottom row).
(D) Cartoon schematic showing the interaction of Fez1 with multiple hexamers.
(E and F) Binding of FEZ1 constructs to CA tubes is reduced and abolished at different salt concentrations. FEZ192–227 interacts with CA tubes at physiological salt concentrations (150 mM) (E), while FEZ192–158 only binds at lower salt concentrations (F), consistent with multiple binding sites in FEZ1.
All-Atom MD Simulation Shows a FEZ1-Hexamer Interaction Mode Driven by the CA R18 Residue
MD simulation has been shown to be a tool in understanding virus capsids, and, in particular, the HIV-1 capsid (Freddolino et al., 2006; Kotecha et al., 2015; Zhao et al., 2013). To investigate the interaction of FEZ1 with the CA hexamer, we built an all-atom model and set up a canonical 2-μs-long MD simulation. To prevent model bias by the initial simulation setup, the FEZ1178–188 peptide was initially placed outside of the CA hexameric pore more than 24Å from R18 (Figure 5A). The dynamic β-hairpin structure (Jacques et al., 2016) and the flexibility of the FEZ1 peptide allow FEZ1 to reach down into the center of the R18 ring. During the simulation, the FEZ1178–188 segment rapidly translocated into the pore at the beginning of the simulation and stably remained inside the hexamer pore. The stability of binding was conferred by interactions between FEZ1 and the R18 residue for the remainder of the simulation (Figure 5B; Video S1). This rapid conformational change, followed by a stable conformation, demonstrates strong interactions between FEZ1178–188 and the CA hexamer. In addition, FEZ1178–188 was recruited from the exterior of the capsid toward the interior cavity, which shows weak electrostatic shielding provided by solvent, water, and ions.
Figure 5. All-Atom Molecular Dynamic Simulations Identify FEZ1 Interactions at the CA Hexamer Center.

(A) All-atom model of FEZ1178–188 is placed at the center of the simulation box containing a CA hexamer, water, and 150 mM NaCl. Before the simulation, the initial location of the FEZ1178–188 segment is 24Å from R18. N-terminal domain (NTD) of CA (CANTD; gold), C-terminal domain (CTD) of CA (CACTD; blue), and FEZ1178–188 (red) are shown in a ribbon representation.
(B) Binding of FEZ1 to the CA hexamer after 2 μs of MD simulation. Surface representation of the internal hexamer center pore showing that FEZ1 interacts with the R18 residues of the CA hexamer (cyan and blue sticks) through negatively charged acidic residues (magenta and red sticks).
(C) List of the top 10 residues in the CA hexamer that are interacting with FEZ1178–188, showing that FEZ1178–188 is stably bound to R18 of the CA hexamer. The occupancy is calculated throughout the length of the simulation.
(D) Dynamics of FEZ1178–188 near a CA hexamer. FEZ1 quickly travels from the exterior of the hexamer to the interior (500 ns). After 500 ns, the interaction between FEZ1 and CA plateaus. The root-mean-square deviation (RMSD) plot of the FEZ1178–188 Cα atoms (in gray) and the electrostatic interactions with the CA hexamer (in red) during the simulation are shown.
To identify the detailed molecular contacts between FEZ1 and the CA hexamer, we performed statistical analysis on the contacts observed during the simulation. The contact analysis reveals that FEZ1 binds to the CA hexamer driven by the electrostatic interactions between the poly-glutamate stretch in FEZ1178–188 and the R18 residue in CA hexamers (Figures 5C and 5D). Moreover, the stability of these salt bridges was demonstrated by the high-contact occupancies exhibited during the simulation. Specifically, the salt bridges between E182-R18 and E183-R18 contacts both showed occupancy larger than 99.5%. Results from our MD simulations suggest that FEZ1178–188 negatively charged residues strongly bind to the positively charged R18 residue inside the CA hexamer center pore through salt bridges. An important detail revealed by the simulations is the ability of FEZ1 to rapidly access the CA-hexamer cavity and stably bind to it.
Mutations in FEZ1 Negatively Charged Regions Reduce HIV-1 Early Trafficking and Infectivity
Our preceding data show that FEZ1 makes specific contacts with the HIV-1 capsid through highly negatively charged patches and that the primary capsid-targeting regions are in FEZ1158–198, which contains a strong CA hexamer-interacting site in FEZ1178–188. To determine the importance of the charged regions during early HIV-1 infection in natural target cells, we first tested the effect of mutations in the hexamer-targeting region FEZ1178–188. Human microglia CHME3 stably expressing control FLAG alone, FLAG-tagged WT, or FLAG-tagged FEZ1178–188E/A (full-length FEZ1 that has all glutamates in the region 178–188 mutated to alanine) were selectively depleted for endogenous FEZ1, followed by infection with HIV-1 pseudotyped with the WT envelope and carrying a luciferase reporter gene (Malikov and Naghavi, 2017). Compared with CHME3 cells expressing exogenous WT FEZ1, HIV-1 infection was moderately impaired in cells expressing FEZ11–392 178–188EA (Figure 6A). Given the function of FEZ1 as a kinesin-1 adaptor protein that facilitates microtubule-based movement of incoming HIV-1 capsid cores (Malikov et al., 2015; Malikov and Naghavi, 2017), we then tested whether these effects on early infection reflected defects in the transport of incoming viral particles to the nucleus. To do so, CHME3 cells depleted for endogenous FEZ1 and expressing exogenous forms of WT or mutant FEZ1, as described earlier, were infected with HIV-1 pseudotyped with WT envelope and whose core was labeled using GFP-tagged Vpr. These cells were then imaged by live cell microscopy. Rapid, long-range, bi-directional movement of HIV-1 particles was observed in cells expressing exogenous WT FEZ1, and the motility of HIV-1 particles was not significantly affected in CHME3 cells expressing FEZ11–392 178–188EA (Figure 6C; Video S2). It is somewhat surprising that mutation of this major CA hexamer-targeting region of FEZ1 did not show a detectable trafficking defect. This is possibly because the many secondary binding sites in FEZ11–392 178–188EA collectively still allow avid binding of HIV-1 capsid and the reduction (not major loss) in binding was below the threshold of detection in our trafficking measurements.
Figure 6. FEZ1 Capsid-Binding Regions Contribute to Early HIV-1 Infection.

(A–D) CHME3 cells stably expressing control FLAG (control), WT (FEZ1), or mutant FEZ1 (FEZ11–392 178–188EA or FEZ11–392 158–198EA, alanine substitution mutants of glutamates in the region 178–188 or 158–198 of full-length FEZ1, respectively) were treated with an siRNA targeting the FEZ1 3′UTR (FEZ1nc) to deplete endogenous FEZ1. Cells were then infected with HIV-1 luciferase reporter virus carrying the WT envelope (HIV-1-WT-Luc) followed by measurement of luciferase activity (A and B). The error bars represent SD from at least three independent experiments. Knockdown levels of endogenous FEZ1 in each sample from (A) and (B) are shown in the lower panels, which were analyzed by western blotting using anti-FEZ1 antibodies. GAPDH served as loading control. (C and D) CHME3 cells depleted for endogenous FEZ1 and expressing control FLAG or exogenous forms of WT or mutant FEZ1 as in (A) and (B), respectively, were infected with HIV-1-WT-Luc containing GFP-Vpr (HIV-1-WT-Luc-GFP-Vpr) and imaged live at 3 frames per min for 2 h, followed by quantification of the percentage of viral particles within 2 μm of the nucleus at the indicated times after infection. The error bars represent SD from at least three independent experiments.
(E) Quantification of the average distance (micrometers per 2.5 min) traveled by viral particles toward the nucleus (retrograde motility shown as black symbols) or away from the nucleus (anterograde motility, shown as white symbols) in CHME3 cells as in (D).
We then looked into the capsid-targeting regions in FEZ1158–188, which allows avid capsid binding through many negatively charged residues dispersed in multiple patches. We constructed the FLAG-tagged FEZ1158–198E/A, which is full-length FEZ1 that has all glutamates in the region 158–198 mutated to alanine. We tested the effect of FEZ1158–198E/A expression on the early infection and transport of incoming HIV-1 particles in cells depleted for endogenous FEZ1. HIV-1 infection was significantly reduced in CHME3 cells expressing FEZ1158–198E/A (Figure 6B). Moreover, FEZ1158–198E/A expression in CHME3 cells failed to rescue HIV-1 transport to the nucleus, indicating that the motility of incoming HIV-1 particles was significantly reduced (Figure6D; Video S2). Imaging was conducted after spinoculation-based infection, and after time taken to set acquisition locations, the times shown in the figure indicate the period of imaging beginning 20 min post-infection. Therefore, in control and FEZ1 cells, but not FEZ11–392 158–198EA cells, a significant number of particles are already trafficking in the cytosol and approaching the nucleus. Moreover, as infection progresses, the number of particles in control and FEZ1 cells decreases because of movement out of the focal plane and/or uncoating. To confirm that the defect in early transport reflected impairment in microtubule motor activity, cells were treated and infected as described earlier but imaged at a higher frame rate of 1 frame per second (fps) over a 300 s period, followed by measurements of distances traveled by viral particles in either retrograde or anterograde directions (Malikov et al., 2015). In the control small interfering RNA (siRNA)-treated cells and WT FEZ1-expressing cells, particles on average traveled longer retrograde than anterograde distances, resulting in a net forward movement toward the nucleus (Figure 6E). In contrast, in cells expressing FEZ11–392 158–198EA, viral particles did not exhibit differences in retrograde versus anterograde movement, resulting in no net forward movement toward the nucleus, which is in line with findings that overall transport and early infection are impaired in the presence of this mutant. These results support the notion that multiple negatively charged regions of FEZ1contribute to early HIV-1 infection through avid interactions with the viral capsid and that the negative charges within the 158–198 motif are specifically required for FEZ1 to control HIV-1 transport to the nucleus.
DISCUSSION
Many viruses use the microtubule network and their associated motor proteins for transportation inside the cell. However, unlike several other viruses, evidence for direct engagement of HIV-1 with microtubule motors is lacking. From studies of FEZ1 and BICD2, it is becoming evident that HIV-1 engages motors indirectly through these kinesin-1 and dynein adaptors, respectively. Outside the context of infection, FEZ1 is known to act as a kinesin-1 adaptor protein that regulates kinesin-1 activity and links it to cargos to transport vesicles and organelles along microtubules in neurons (Blasius et al., 2007; Bloom and Horvitz, 1997; Chua et al., 2012; Kuroda et al., 1999; Toda et al., 2008). It is reported that FEZ1 interacts with the tail of kinesin-1 heavy chain using its C-terminal coiled-coil region 231–308 (Blasius et al., 2007). However, although it was shown that FEZ1 binds in vitro assembled CA-NC complexes and promotes early HIV-1 trafficking to the nucleus (Malikov and Naghavi, 2017; Malikov et al., 2015), the structural basis by which HIV-1 capsids engage motor adaptors remains poorly defined. It was not known whether FEZ1 directly interacts with HIV-1 capsid and, if it does so, which molecular determinants drive the interaction.
The work presented here demonstrates that FEZ1 is a unique HIV-1 capsid pattern sensor that uses highly negatively charged acidic stretches to interact with the positively charged center pore of the CA hexamer. Although the interaction is driven by electrostatic contacts with multiple charged stretches on FEZ1, the recognition of CA hexamers appears to be highly specific, because no binding was detected with CA pentamers that contain similar charge characters (Figure S2). Consistent with FEZ1’s role in early viral transport, we found that FEZ1 mutations that abolish its interaction with CA tubes greatly reduced both accumulation of virus particles around the nucleus and HIV-1 infectivity. These results establish a solid link between FEZ1 and intracellular trafficking of HIV-1, substantiating its roles as an adaptor bridging viral capsid and kinesin-1 (Figure 7A).
Figure 7. Schematic Models of HIV-1 Capsid Trafficking on Microtubules and a Conserved Targeting Site at the CA Hexamer Center.

(A) Schematic of FEZ1 (red) bridging HIV-1 capsid to the kinesin motor (blue) and microtubule for trafficking (right). Other potential HIV-1 tracking machinery (BICD2/dynein/dynactin) is shown on the left. The arrows indicate the direction of trafficking.
(B) Center of the CA hexamer is a conserved common interacting site for proteins (FEZ1), nucleotides (NTPs and dNTPs), and other cellular cofactors (IP6).
The interaction between FEZ1 and HIV-1 capsid follows a common theme found in the cases of many other capsid-interacting proteins, in which simultaneous binding at multiple, otherwise weak individual contact sites creates an avidity effect for a stable interaction (Li and Sodroski, 2008; Yang et al., 2012; Goldstone et al., 2014; Buffone et al., 2015; Li et al., 2016). Although FEZ1 contains a reasonably high-affinity CA hexamer-interacting site (FEZ1178–188), the electrostatic nature of the interaction does not sustain the ionic strength conditions and the direct competition from high concentrations of dNTPs/NTPs and IP6 molecules in cells. HIV-1 solves this problem by using the repeating nature of the capsid lattice to interact simultaneously with numerous negative patches within FEZ1. This highlights the advantageous capsid-targeting mode employed by many cellular protein factors, in which only relatively weak individual interaction sites need to be developed and the avidity effect increases the overall binding affinity by orders of magnitude (Rudnick and Adams, 2009; Vauquelin and Charlton, 2013). Furthermore, targeting multiple sites enables a capsid pattern-sensing ability that ensures productive binding to the assembled capsid that contains the viral genome.
An important discovery from our work is that the center pore of the CA hexamer is a conserved interaction hub for both small-molecule and protein cofactors in the cell (Figure 7B). The positively charged center of the CA hexamer, generated by residue R18, serves as an ideal interaction site with the highly negatively charged FEZ1 protein. The critical role of the R18 residue has been well documented. When mutated, there are aberrant and fewer capsids, resulting in strikingly reduced HIV-1 infectivity (Forshey et al., 2002; von Schwedler et al., 2003). This ring of R18 has been indicated to form a pore for the recruitment of nucleotides for reverse transcription inside the capsid (Jacques et al., 2016) and for coordinating the polyanion IP6 to stabilize the capsid (Mallery et al., 2018). We demonstrate here that this CA hexamer pore can also be targeted by protein cofactors (Figure 7B). Our MD simulations show that FEZ1 is able to reach down into this arginine ring in the center of the CA hexamer, possibly because of its inherently flexible and unstructured nature. It is conceivable that other cellular proteins with flexible, negatively charged regions can also bind HIV-1 capsid through this interaction mode.
Many HIV-1 early post-entry infection events remain to be discovered. Here we find that FEZ1 acts as a kinesin-1 adaptor protein that interacts with the HIV-1 capsid directly via avid electrostatic interactions, thereby linking HIV-1 particles to the host transport system. Overall, our study demonstrates FEZ1 as a novel HIV-1 capsid pattern sensor, which targets the conserved cellular cofactor-binding site at the CA hexamer center and supports the requirement of this virus-host factor interaction for efficient transport and early infection of HIV-1.
STAR⋆METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
This study did not generate new unique reagents. Plasmids generated in this study are available upon request. Further information and requests for plasmids, resources, and reagents should be directed to and will be fulfilled by the Lead Contact, Yong Xiong (yong.xiong@yale.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial Strains
All molecular cloning was carried out in E. coli XL10-Gold Ultracompetent cells. All recombinant proteins were expressed and purified from the E. coli strain BL21(DE3). Both cell lines were routinely cultured at 37°C while shaking at > 220 RPM. XL10-Gold cells were grown in Luria Broth and BL21(DE3) cells were grown in either Luria Broth (starter culture) or Terrific Broth (for protein expression).
Human Cell Lines
Human microglia CHME3 cells (from undetermined sex) were previously described (Malikov et al., 2015).
METHOD DETAILS
Protein expression and purification
N-terminal 6 × His-tagged, GST-tagged, or MBP-tagged FEZ1 constructs were cloned into pET-28a (Novagen) with mutants created by QuickChange mutagenesis. They were transformed and expressed in Escherichia coli BL21 (DE3) (Lucigen) grown in TB media to an OD600 of 0.6 and induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) overnight at 16°C. The cells were harvested by centrifugation (5000 rpm, 10 min, 4°C) and resuspended in lysis buffer containing 50 mM Tris (pH 7.5), 400mM NaCl, and 0.1mM Tris(2-carboxyethyl)phosphine (TCEP) and followed by lysis with a microfluidizer. The lysate was centrifuged at 13,000 rpm for 40 min, 4°C and proteins were purified by passing through a nickel affinity column (QIAGEN), a HiTrapQ anion exchange column (GE Heathcare) in 20 mM Tris (pH 8.0) using a 0–1000 mM NaCl (with 0.1mM TCEP) gradient elution, followed by a Superdex-200 gel-filtration column (GE Heathcare) in buffer 25 mM Tris (pH 7.0), 50 mM NaCl and 0.1 mM TCEP. The protein purity was examined by SDS-PAGE. All CA constructs were cloned into pET-11a (Novagen). CA proteins were overexpressed in E. coli BL21(DE3) cells at 25°C for 12 h by induction with 0.5 mM IPTG at OD600 0.6–0.8. CA proteins were purified by 25% w/v ammonium sulfate precipitation, dialysis into low-salt buffer (25 mM HEPES pH 7, 0.1 mM TCEP), and cation exchange chromatography. A list of the FEZ1 constructs used in this paper is summarized in Table S2.
Analytical size-exclusion chromatography (SEC)
The interaction between FEZ1 and cross-linked CA (A14C/E45C) hexamers was examined by size exclusion chromatography. CA hexamers were assembled as described previously (Pornillos et al., 2009). Purified samples of FEZ1 (80 μM) were mixed with CA hexamer (20 μM) and applied to a Superdex 200 10/300 GL column (GE Healthcare) pre-equilibrated in 25 mM Tris, pH 8.0, 50 mM NaCl buffer. FEZ1 binding tests with small capsid assemblies (Figure 1) used 200 μL reaction volumes with 33 μM FEZ1 and 98 μM CA monomer (of the appropriate assembly). The UV absorbance at 280 nm was recorded to monitor the elution of FEZ1 and CA hexamer. For a typical SEC run, the eluted peak has a total volume of ~2 mL, resulting in a dilution factor of ~10.
CA Tube Pelleting Assay with purified FEZ1 proteins
Cross-linked CA (A14C/E45C) R18 (WT) and R18D tubes were dialyzed overnight at 4°C into assembly buffer (1 M NaCl, 25 mM Tris pH 8.0), followed by dialysis into binding buffer (50 mM NaCl, 25 mM Tris pH 8.0) (Pornillos et al., 2009). FEZ1 proteins were spun at 20,000 3 g for 30 min at 4°C. FEZ1 (5 μM) proteins were added to CA tubes and incubated at room temperature for 30 min. Subsequently, 7 μL aliquots were withdrawn and labeled as total. The remaining was pelleted at 20,000 3 g for 30 min at 4°C. Total, supernatant, and pellet samples were analyzed by SDS-PAGE. Pelleting experiments with ATP and dNTPs were performed as described with ATP concentrations as described and 20 mM dNTPs.
Isothermal Titration Calorimetry
The affinity of the FEZ1 protein for CA hexamer was determined using isothermal titration calorimetry (Nano ITC; TA Instruments). FEZ1 and the CA hexamer were dialyzed overnight in the buffer 25 mM Tris (pH 8), 50 mM NaCl. The CA hexamer (120 μM concentration in the syringe) was added in 30 injections of 1.6 μL into a 15 μM solution of purified FEZ1 protein at 25°C in the cell. Data were analyzed using the NanoAnalyze software from TA Instruments. Results are from 3 independent experiments, and is represented as mean ± standard deviation.
Crystallization and data collection
Crystallization was performed using the microbatch under-oil method (Chayen et al., 1992). Protein sample at 2 mg/mL was mixed with precipitant at a 1:1 ratio (1 μL protein:1 μL precipitant) at room temperature. R18D CA in 50 mM NaCl, 25mM Tris pH 8 was crystallized in the condition containing 0.1M Tris-HCl pH 8.5, 8% PEG 8000. Crystals were cryoprotected with precipitant containing 25% (Vol/Vol) glycerol before freezing in liquid nitrogen. All diffraction data were collected at the Advanced Photon Source NE-CAT beam-line 24-IDE. Data statistics are summarized in Table S1.
Structure determination and refinement
The structure was solved by molecular replacement using PHASER (McCoy et al., 2007) using the previously published CA structure (PDB: 3H47) as a search model. The model was refined with iterative rounds of TLS and restrained refinement using Refmac5 (Vagin et al., 2004) followed by rebuilding the model to the 2Fo–Fc and the Fo–Fc electron density maps using Coot (Emsley and Cowtan, 2004). Refinement statistics are summarized in Table S1. PyMol (Schrodinger, 2015) with the APBS (Adaptive Poisson-Boltzmann Solver) (Baker et al., 2001) plug-in was used to calculate the surface electrostatic potential.
Negative stain electron microscopy
To prepare samples for negative stain, the CA tubes were assembled as described above. Aliquots (3 μl) were adsorbed to a glow-discharged, 400-mesh, carbon-coated copper grid and stained with fresh uranyl formate (2%). Images were recorded on a TF20 electron microscope (FEI) equipped with a field-emission gun at the indicated magnification on a 4k × 4k Gatan UltraScan charge-coupled device camera (Gatan).
Atomic model building for molecular dynamic (MD) simulations
Initial coordinates of the HIV-1 CA hexamer were generated from a native full-length HIV-1 capsid protein (PDB: 4XFX) (Gres et al., 2015), applying a six-fold symmetry. The two missing loops, residue 5 to 9 and residue 222 to 231, in the original 4XFX model were built using the model builder algorithm Modeler (Eswar et al., 2006). In addition, the protonation states of the titratable residues, particularly histidine, asparagine, lysine and cysteine, at pH 7.4, were determined and assigned by PDB: 2PQR (Dolinsky et al., 2007). There was no available atomic model of human FEZ1 protein when conducting this study. The initial model of the FEZ1178–188 peptide was built by using the ab initio modeling protocol in Rosetta (Leaver-Fay et al., 2011). The glutamate and aspartate residues were all in deprotonated form, which resulted in −6 charges for this peptide. After being equilibrated in explicit water for 0.5 μs, this FEZ1 peptide was combined with the CA hexamer model and placed at the entrance of the CA hexamer center channel. This complex was then solvated in a 110Å × 120Å × 90 Å TIP3P (Jorgensen, 1998) water box and neutralized by 150 mM NaCl. The total atom count of this resulting FEZ1-CA hexamer model was ~122 K.
System preparation for MD simulation
After model building, the prepared model underwent a two-step energy minimization. The CA and FEZ1 protein were fixed at the first 10,000 steps of minimization, while the water molecules and ions were free to move. In the second minimizing stage, the fixed atoms were relieved but the heavy atoms of the proteins were restrained by a force constant of 10 Kcal/molÅ2 for another 10,000 steps. The minimized model was then thermalized to 310 K with a rate of 1,000 steps/K while the harmonic restraints on protein heavy atoms were maintained. Next, these harmonic restraints on the heavy atoms were gradually released during a 100,000 equilibration run under 310 K and 1 bar. Minimization, thermalization and equilibration runs were carried out in NAMD 2.12 (Phillips et al., 2005) at a time step of 2 fs. All bonds to hydrogen were constrained with the SHAKE (solute) or SETTLE (solvent) algorithms. Temperatures were regulated by the Langevin thermostat algorithm employing a damping coefficient of 1 ps−1. The pressure was maintained by the Nose-Hoover Langevin piston pressure control, allowing isotropic cell scaling.
MD simulation production runs on Anton2
The production runs of Fez1-CA hexamer system were performed for 2 ms on the special purpose supercomputer Anton2 (Shaw et al., 2014) at the Pittsburgh Supercomputer Center (PSC). The CHARMM 36 m (Huang et al., 2017) force field for protein was employed in the MD simulations. During the simulation, the temperature (310 K) and pressure (1 bar) were maintained by employing the Multigrator integrator (Lippert et al., 2013) and the simulation time-step was set to 2.5 fs/step, with short-range forces evaluated at every time step, and long-range electrostatics evaluated at every second time step. Short-range non-bonded interactions were cut off at 17Å; long range electrostatics were calculated using the k-Gaussian Split Ewald method (Shan et al., 2005). The Cα atoms involved in 2- and 3-fold inter-hexamer interfaces, namely helices 9, 10 and 11, were restrained with a force constant 1.0 Kcal mol−1Å−2, to mimic the interactions with neighboring CA-hexamers in the P6 periodic cell.
Viruses
To generate HIV-1 luciferase reporter virus pseudotyped with WT envelope (HIV-1-WT-Luc), 293T cells were co-transfected with a plasmid encoding WT envelope along with the plasmid, pNL4.3.luc.R-.E-(AIDS Reagent Repository no 3418). A GFP-tagged Vpr expressing vector was included in these co-transfections to generate a fluorescent version of this virus, HIV-1-WT-Luc-GFP-Vpr (Delaney et al., 2017).
Generation of stable pools
Retroviruses encoding either control Flag (Control), or C’ terminally FLAG-tagged versions of full length human FEZ1 (FEZ1), or the FEZ1 mutants (FEZ11–392 178–188EA and FEZ11–392 158–198EA) were used to infect human microglia CHME3 cells followed by selection to generate stable expressing pools, as previously described (Malikov et al., 2015).
RNAi and measurement of early HIV-1 infection
For transient knockdowns, CHME3 cells were transfected with 60 pmol of a custom-made siRNA duplex targeting the FEZ1 3′-UTR (FEZ1nc), obtained from Ambion (siRNA sense and antisense sequences are CUUAUACUCUUAAGACUAAtt and UUAGUCUUA AGAGUAUAAGct, respectively) using oligofectamine RNAiMAX transfection reagent (Invitrogen) as described previously (Malikov et al., 2015). siRNA-treated cells were infected with HIV-1-WT-Luc for 48 h. Cultures were then lysed and luciferase activity was measured using the Luciferase Assay System (Promega). Knockdown levels were confirmed by western blot analysis using anti-FEZ1 antibodies (Cat #4280 from Cell Signaling). GAPDH was detected using anti-GAPDH (Cat #sc-25778 from Santa Cruz). The results are representative of at least three independent experiments.
Live imaging and analysis
CHME3 cells plated on 35-mm collagen-coated glass culture dishes (MatTeck, P35G-1.5–14C) were infected with HIV-1-WT-Luc-GFP-Vpr in CO2-independent media via spinoculation for 30 min. The media was changed and acquisition locations were set up within the first 20 min. Timestamps starting at 0 represent the start and period of live-cell video microscopy beginning at 20 min post-infection, performed at 3 frames per min for 2 h. Short interval acquisitions were performed at 1 fps for a period of 300 s as described previously (Malikov et al., 2015). Number of particles near nuclei were determined using MetaMorph imaging software (Molecular Devices). Up to 190 viral particles were counted per sample, and each experiment consisted of 2–4 cells. One-way ANOVA for correlated samples, with a confidence interval of 95% or greater was used to determine statistical significance in at least two independent experiments for each condition. Single virus particles were tracked in the TrackMate ImageJ plugin v2.8.1 (Tinevez et al., 2017) using estimated blob diameter 8.0 pixels, threshold 100 and frame to frame linking max distance 30 pixels. XML trace files were analyzed using Python 3.7.0 Jupyter notebooks (5.6.0) with Pandas (0.23.4) and Matplotlib (2.2.3) packages. In 3 independent experiments 1600–2800 tracks in 9–13 cells were analyzed per each condition. Equal-variances t test was used to determine statistical significances between datasets. The following representations of P values were used: *p < 0.05, **p < 0.01, ***p < 0.001.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed within published software as described in the Method Details section.
ITC experiments were repeated three times with different protein preparations. The mean and standard deviation values were calculated using Microsoft Excel. The number of mean ± standard deviation is shown in figure and figure legends of Figure 2.
Quantification of the measurement of luciferase activity is performed by using one-way ANOVA. Quantification of percentage of viral particles within 2 mm of the nucleus or average distance traveled by virus particles is done by the equal-variances t test. The number of experiments and the error bars representing standard deviation (SD) are given in the figures and figure legends of Figure 6.
DATA AND CODE AVAILABILITY
Data Deposition
The accession number for the R18D CA hexamer structure and diffraction data reported in this paper is PDB: 6OMT.
Software Availability
This study did not produce additional software. The software used in this paper has been published and is publicly available.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E. coli BL21(DE3) | Lucigen | Cat#60401 |
| E. coli XL10-Gold Ultracompetent Cells | Agilent | Cat#200315 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Terrific Broth | Research Products International | Cat#T5100–5000.0 |
| Luria Broth | Research Products International | Cat#L24400 |
| Isopropyl β-D-1-thiogalactopyranoside (IPTG) | American Bioanalytical | Cat#AB00841–00010 |
| complete EDTA-free protease inhibitor cocktail | Sigma | Cat#11873580001 |
| Tris (2-Carboxylethyl) phosphine Hydrochloride (TCEP) | Gold Bio | Cat#TCEP |
| phenylmethane sulfonyl fluoride (PMSF) | dot scientific | Cat#DSP20270–25 |
| 2-Mercaptoethanol (BME) | Alfa Aesar | Cat#J66742–0B |
| NuPAGE LDS Sample Buffer (4X) | Thermo Fisher | Cat#NP0008 |
| Dithiothreitol (DTT) | American Bioanalytical | Cat#AB00490–00100 |
| SimplyBlue SafeStain | Thermo Fisher | Cat#LC6060 |
| KOD Hot Start DNA polymerase | Novagen | Cat#710863 |
| Recombinant 6xHis-FEZ11–392 | This paper | N/A |
| Recombinant 6xHis-FEZ11–92 | This paper | N/A |
| Recombinant 6xHis-FEZ192–198 | This paper | N/A |
| Recombinant 6xHis-FEZ1198–392 | This paper | N/A |
| Recombinant 6xHis-FEZ192–158 | This paper | N/A |
| Recombinant 6xHis-FEZ192–227 | This paper | N/A |
| Recombinant 6xHis-FEZ192–198 182EA | This paper | N/A |
| Recombinant 6xHis-FEZ1178–188 | This paper | N/A |
| Recombinant 6xHis-FEZ1158–198 | This paper | N/A |
| Recombinant 6xHis-FEZ1158–198 182EA | This paper | N/A |
| Recombinant 6xHis-FEZ1158–182 | This paper | N/A |
| Recombinant CA 14C/45C | (Pornillos et al., 2009) | N/A |
| Recombinant CA 14C/45C/R18D | This paper | N/A |
| Recombinant CA 14C/45C/R18D/184A/185A | This paper | N/A |
| Recombinant CA 14C/45C/R18A | This paper | N/A |
| Recombinant CA 14C/45C/G89V | (Smaga et al., 2019) | N/A |
| Recombinant CA 14C/45C/P207S | (Smaga et al., 2019) | N/A |
| Recombinant CA 14C/45C/G208R | (Smaga et al., 2019) | N/A |
| Recombinant CA 14C/45C/T210K | (Smaga et al., 2019) | N/A |
| Recombinant CA 14C/184A/185A | (Summers et al., 2019) | N/A |
| Recombinant CA 45C/184A/185A | (Summers et al., 2019) | N/A |
| Recombinant CA 14C/42E/184A/185A | (Summers et al., 2019) | N/A |
| Recombinant CA 45C/54E/184A/185A | (Summers et al., 2019) | N/A |
| Recombinant CA 14C/42E-Mpro-184A/185A | (Summers et al., 2019) | N/A |
| Recombinant CA 45C/54C/184A/185A | (Summers et al., 2019) | N/A |
| Recombinant CA 42C/184A/185A | (Summers et al., 2019) | N/A |
| Recombinant CA 42C/54E/184A/185A | (Summers et al., 2019) | N/A |
| Recombinant CA 45C | (Summers et al., 2019) | N/A |
| Recombinant CA 14C | (Summers et al., 2019) | N/A |
| Recombinant CA 45C/54E | (Summers et al., 2019) | N/A |
| Recombinant CA 14C/42E | (Summers et al., 2019) | N/A |
| Recombinant CA 45C/54E(1–226)-foldon | (Summers et al., 2019) | N/A |
| Recombinant CA 14C/42E/204D/(1–221) | (Summers et al., 2019) | N/A |
| Recombinant CA 14C/45C/184A/185A | (Pornillos et al., 2009) | N/A |
| Recombinant CA 14C/45C-5L-SpyCatcher-6xHis | (Summers et al., 2019) | N/A |
| Recombinant CA 14C/45C-5L-SpyTag-MBP-6xHis | (Summers et al., 2019) | N/A |
| Recombinant CA 14C/45C/184A/185A-5L-Spy Tag-MBP-6xHis | (Summers et al., 2019) | N/A |
| Recombinant CA 14C/45C/184A/185A-5L-SpyCat-6xHis | (Summers et al., 2019) | N/A |
| Recombinant CA 14C/45C/P90A | (Summers et al., 2019) | N/A |
| Recombinant CA 45C/54C/184A/185A/P90A | (Summers et al., 2019) | N/A |
| Recombinant CA 14C/42E/184A/185A/P90A | (Summers et al., 2019) | N/A |
| Recombinant CA 42C/54E/184A/185A/P90A | (Summers et al., 2019) | N/A |
| Recombinant CA 14C/45C/P90A-5L-SpyCatcher-6xHis | (Summers et al., 2019) | N/A |
| Recombinant CA 14C/45C/P90A-5L-SpyTag-MBP-6xHis | (Summers et al., 2019) | N/A |
| Recombinant CA 14C/45C/184A/185A/P90A | (Summers et al., 2019) | N/A |
| Recombinant Mpro protease | (Yang et al., 2003) | N/A |
| Deposited Data | ||
| Crystal structure of CA R18D | This paper | PDB: 6EC2 |
| Oligonucleotides | ||
| All standard cloning primers for site-directed mutagenesis and Gibson assembly | Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| pET-11a | EMD Millipore | Cat#69436 |
| pRSFDUET-1 | EMD Millipore | Cat#71341 |
| pET28-a | EMD Millipore | Cat#69864 |
| pETDUET-1 | EMD Millipore | Cat#71146 |
| pMAL system | New England Biolabs | Cat#E8200S |
| Recombinant 6xHis-FEZ11–392 | This paper | N/A |
| Recombinant 6xHis-FEZ11–92 | This paper | N/A |
| Recombinant 6xHis-FEZ192–198 | This paper | N/A |
| Recombinant 6xHis-FEZ1198–392 | This paper | N/A |
| Recombinant 6xHis-FEZ192–158 | This paper | N/A |
| Recombinant 6xHis-FEZ192–227 | This paper | N/A |
| Recombinant 6xHis-FEZ192–198 182EA | This paper | N/A |
| Recombinant 6xHis-FEZ1178–188 | This paper | N/A |
| Recombinant 6xHis-FEZ1158–198 | This paper | N/A |
| Recombinant 6xHis-FEZ1158–198 182EA | This paper | N/A |
| Recombinant 6xHis-FEZ1158–182 | This paper | N/A |
| Plasmid: CA 14C/45C/R18D in pET-11a | This paper | N/A |
| Plasmid: CA 14C/45C/R18D/184A/185A in pET-11a | This paper | N/A |
| Plasmid: CA 14C/45C/R18A in pET-11a | This paper | N/A |
| Plasmid: CA 14C/45C/G89V in pET-11a | (Smaga et al., 2019) | N/A |
| Plasmid: CA 14C/45C/P207S in pET-11a | (Smaga et al., 2019) | N/A |
| Plasmid: CA 14C/45C/G208R in pET-11a | (Smaga et al., 2019) | N/A |
| Plasmid: CA 14C/45C/T210K in pET-11a | (Smaga et al., 2019) | N/A |
| Plasmid: CA 14C/184A/185A in pET-11 a | (Summers et al., 2019) | N/A |
| Plasmid: CA 45C/184A/185A in pET-11 a | (Summers et al., 2019) | N/A |
| Plasmid: CA 14C/42E/184A/185A in pET-11a | (Summers et al., 2019) | N/A |
| Plasmid: CA 45C/54E/184A/185A in pET-11 a | (Summers et al., 2019) | N/A |
| Plasmid: CA 14C/42E-Mpro-184A/185A in pET-11a | (Summers et al., 2019) | N/A |
| Plasmid: CA 45C/54C/184A/185A in pET-11 a | (Summers et al., 2019) | N/A |
| Plasmid: CA 42C/184A/185A in pET-11 a | (Summers et al., 2019) | N/A |
| Plasmid: CA 42C/54E/184A/185A in pET-11 a | (Summers et al., 2019) | N/A |
| Plasmid: CA 45C in pET-11a | (Summers et al., 2019) | N/A |
| Plasmid: CA 14C in pET-11a | (Summers et al., 2019) | N/A |
| Plasmid: CA 45C/54E in pET-11a | (Summers et al., 2019) | N/A |
| Plasmid: CA 14C/42E in pET-11a | (Summers et al., 2019) | N/A |
| Plasmid: CA 45C/54E(1–226)-foldon in pET-11a | (Summers et al., 2019) | N/A |
| Plasmid: CA 14C/42E/204D/(1–221) in pET-11a | (Summers et al., 2019) | N/A |
| Plasmid: CA 14C/45C/184A/185A in pET-11a | (Summers et al., 2019) | N/A |
| Plasmid: CA 14C/45C-5L-SpyCatcher-6xHis in pET-11a | (Summers et al., 2019) | N/A |
| Plasmid: CA 14C/45C-5L-SpyTag-MBP-6xHis in pET-11a | (Summers et al., 2019) | N/A |
| Plasmid: CA 14C/45C/184A/185A-5L-SpyTag-MBP-6xHis in pET-11a | (Summers et al., 2019) | N/A |
| Plasmid: CA 14C/45C/184A/185A-5L-SpyCat-6xHis in pET-11a | (Summers et al., 2019) | N/A |
| Plasmid: CA 14C/45C/P90A in pET-11a | (Summers et al., 2019) | N/A |
| Plasmid: CA 45C/54C/184A/185A/P90A in pET-11 a | (Summers et al., 2019) | N/A |
| Plasmid: CA 14C/42E/184A/185A/P90A in pET-11 a | (Summers et al., 2019) | N/A |
| Plasmid: CA 42C/54E/184A/185A/P90A in pET-11 a | (Summers et al., 2019) | N/A |
| Plasmid: CA 14C/45C/P90A-5L-SpyCatcher-6xHis in pET-11a | (Summers et al., 2019) | N/A |
| Plasmid: CA 14C/45C/P90A-5L-SpyTag-MBP-6xHis in pET-11a | (Summers et al., 2019) | N/A |
| Plasmid: CA 14C/45C/184A/185A/P90A in pET-11a | (Summers et al., 2019) | N/A |
| Plasmid: nanobody 37E7-6xHis in pET28-a | (Gray et al., 2017) | N/A |
| Software and Algorithms | ||
| NanoAnalyze v3.6.0 | TA Instruments | https://www.tainstruments.com/support/software-downloads-support/downloads/ |
| XDS | (Kabsch, 2010a, 2010b) | http://xds.mpimf-heidelberg.mpg.de/ |
| Phaser | (McCoy et al., 2007; Winn et al., 2011) | http://www.ccp4.ac.uk/html/phaser.html |
| Refmac5 | (Adams et al., 2010) | http://www.ccp4.ac.uk/dist/html/refmac5.html |
| Phenix | (Emsley and Cowtan, 2004) | https://www.phenix-online.org/ |
| Coot | (Murshudov et al., 1997) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PyMOL(v1.6.0.0) | (Schrodinger, 2015) | https://pymol.org/2/ |
| Chimera (v13.0) | (Pettersen et al., 2004) | https://www.cgl.ucsf.edu/chimera/ |
| Other | ||
| Ni-NTA Agarose | QIAGEN | Cat#30230 |
| HiTrap Q HP 5mL | GE Healthcare | Cat#17115401 |
| HiTrap SP HP 5mL | GE Healthcare | Cat#17115201 |
| HiLoad 16/600 Superdex 200 PG | GE Healthcare | Cat#28990945 |
| Yarra SEC-3000 | Phenomenex | Cat#00F–4513–E0 |
| Superose6 10/300 GL | GE Healthcare | Cat#17517201 |
| Superdex 200 Increase 5/150 GL | GE Healthcare | Cat#28990945 |
| Pre-treated RC Tubing MWCO 10kD | Spectrum Labs | Cat#132120 |
| Slide-A-Lyzer Dialysis Cassettes, 10K MWCO, 0.5mL | Thermo Fisher | Cat#PI66383 |
| Amicon Ultra-15 Centrifugal Filter Units 10kDa | Millipore Sigma | Cat#UFC901024 |
| Amicon Ultra-15 Centrifugal Filter Units 30kDa | Millipore Sigma | Cat#UFC903024 |
| NuPAGE 4–12% Bis-Tris Midi gels | Invitrogen | Cat#WG1403B0X |
| Gen10 BT+ (Bis-Tris) Protein Mini Gel 4–12% | ConnSTEM | Cat#13A12 |
Highlights.
Kinesin adaptor protein FEZ1 directly interacts with HIV-1 capsid for trafficking
FEZ1 specifically targets the conserved center pore of capsid protein (CA) hexamers
FEZ1 uses electrostatic interactions to bind multiple CA hexamers in the capsid
FEZ1 capsid-binding residues are important for HIV-1 trafficking and infectivity
ACKNOWLEDGMENTS
We thank K. Zhou for assistance in performing negative-stain electron microscopy and the Xiong lab for discussion. We also thank the staff at the Advanced Photon Source beamlines 24ID-C and E and the National Synchrotron Light Source II beamline AMX. We are grateful to Dean Procter at North-western University for writing script for particle tracking analysis. This work was supported by NIH, NSF, and DOE grants P50GM082251 (to Y.X.), P30GM110758 (to J.R.P.), and R01GM101975 (to M.H.N.) and Collaboration Development Pilot Program awards from the Pittsburgh Center for HIV Protein Interactions (to Y.X. and J.R.P.), P30 GM124165 (NE-CAT beamlines), S10RR029205 (Pilatus detector), S10OD021527 (Eiger detector), and DE-AC02–06CH11357 (APS Synchrotron source). This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant ACI-1548562. Specifically, it used the Bridges system, which is supported by NSF award ACI-1445606, at the Pitts-burgh Supercomputing Center (PSC). Anton 2 computer time was provided by the PSC through grant R01GM116961 from the NIH. The Anton 2 machine at PSC was generously made available by D.E. Shaw Research. P.-T.H. was supported by the Taiwan Ministry of Education Scholarship to Study Abroad. B.J.S. was supported by the Predoctoral Program in Biophysics (NIH T32 GM008283).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.079.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arhel N, Genovesio A, Kim KA, Miko S, Perret E, Olivo-Marin JC, Shorte S, and Charneau P (2006). Quantitative four-dimensional tracking of cytoplasmic and nuclear HIV-1 complexes. Nat. Methods 3, 817–824. [DOI] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, and McCammon JA (2001). Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 98, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A, Alam SL, Fricke T, Zadrozny K, Sedzicki J, Taylor AB, Demeler B, Pornillos O, Ganser-Pornillos BK, Diaz-Griffero F, et al. (2014). Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc. Natl. Acad. Sci. USA 111, 18625–18630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biris N, Yang Y, Taylor AB, Tomashevski A, Guo M, Hart PJ, Diaz-Griffero F, and Ivanov DN (2012). Structure of the rhesus monkey TRIM5α PRYSPRY domain, the HIV capsid recognition module. Proc. Natl. Acad. Sci. USA 109, 13278–13283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasius TL, Cai D, Jih GT, Toret CP, and Verhey KJ (2007). Two binding partners cooperate to activate the molecular motor Kinesin-1. J. Cell Biol 176, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom L, and Horvitz HR (1997). The Caenorhabditis elegans gene unc-76 and its human homologs define a new gene family involved in axonal outgrowth and fasciculation. Proc. Natl. Acad. Sci. USA 94, 3414–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffone C, Schulte B, Opp S, and Diaz-Griffero F (2015). Contribution of MxB oligomerization to HIV-1 capsid binding and restriction. J Virol. 89, 3285–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukrinskaya A, Brichacek B, Mann A, and Stevenson M (1998). Establishment of a functional human immunodeficiency virus type 1 (HIV-1) reverse transcription complex involves the cytoskeleton. J. Exp. Med 188, 2113–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busnadiego I, Kane M, Rihn SJ, Preugschas HF, Hughes J, BlancoMelo D, Strouvelle VP, Zang TM, Willett BJ, Boutell C, et al. (2014). Host and viral determinants of Mx2 antiretroviral activity. J. Virol 88, 7738–7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell EM, and Hope TJ (2015). HIV-1 capsid: the multifaceted key player in HIV-1 infection. Nat. Rev. Microbiol 13, 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnes SK, Zhou J, and Aiken C (2018). HIV-1 Engages a Dynein-Dynactin-BICD2 Complex for Infection and Transport to the Nucleus. J Virol. 92, e00358–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chayen NE, Shaw Stewart PD, and Blow DM (1992). Microbatch crystallization under oil—a new technique allowing many small-volume crystallization trials. J. Cryst. Growth 122, 176–180. [Google Scholar]
- Chua JJ, Butkevich E, Worseck JM, Kittelmann M, Grønborg M, Behrmann E, Stelzl U, Pavlos NJ, Lalowski MM, Eimer S, et al. (2012). Phosphorylation-regulated axonal dependent transport of syntaxin 1 is mediated by a Kinesin-1 adapter. Proc. Natl. Acad. Sci. USA 109, 5862–5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney MK, Malikov V, Chai Q, Zhao G, and Naghavi MH (2017). Distinct functions of diaphanous-related formins regulate HIV-1 uncoating and transport. Proc. Natl. Acad. Sci. USA 114, E6932–E6941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharan A, Opp S, Abdel-Rahim O, Keceli SK, Imam S, Diaz-Griffero F, and Campbell EM (2017). Bicaudal D2 facilitates the cytoplasmic trafficking and nuclear import of HIV-1 genomes during infection. Proc. Natl. Acad. Sci. USA 114, E10707–E10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick RA, Mallery DL, Vogt VM, and James LC (2018). IP6 Regulation of HIV Capsid Assembly, Stability, and Uncoating. Viruses 10, E640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, and Baker NA (2007). PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 35, W522–W525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, and Sali A (2006). Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics Chapter 5, Unit-5.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez J, Portilho DM, Danckaert A, Munier S, Becker A, Roux P, Zambo A, Shorte S, Jacob Y, Vidalain PO, et al. (2015). Microtubule-associated proteins 1 (MAP1) promote human immunodeficiency virus type I (HIV-1) intracytoplasmic routing to the nucleus. J. Biol. Chem 290, 4631–4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forshey BM, von Schwedler U, Sundquist WI, and Aiken C (2002). Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol 76, 5667–5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freddolino PL, Arkhipov AS, Larson SB, McPherson A, and Schulten K (2006). Molecular dynamics simulations of the complete satellite tobacco mosaic virus. Structure 14, 437–449. [DOI] [PubMed] [Google Scholar]
- Fritsche LG, Igl W, Bailey JN, Grassmann F, Sengupta S, Bragg-Gresham JL, Burdon KP, Hebbring SJ, Wen C, Gorski M, et al. (2016). A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet 48, 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble TR, Vajdos FF, Yoo S, Worthylake DK, Houseweart M, Sundquist WI, and Hill CP (1996). Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 87, 1285–1294. [DOI] [PubMed] [Google Scholar]
- Ganser BK, Li S, Klishko VY, Finch JT, and Sundquist WI (1999). Assembly and analysis of conical models for the HIV-1 core. Science 283, 80–83. [DOI] [PubMed] [Google Scholar]
- Ganser-Pornillos BK, Yeager M, and Sundquist WI (2008). The structural biology of HIV assembly. Curr. Opin. Struct. Biol 18, 203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudin R, de Alencar BC, Arhel N, and Benaroch P (2013). HIV trafficking in host cells: motors wanted!. Trends Cell Biol. 23, 652–662. [DOI] [PubMed] [Google Scholar]
- Goldstone DC, Walker PA, Calder LJ, Coombs PJ, Kirkpatrick J, Ball NJ, Hilditch L, Yap MW, Rosenthal PB, Stoye JP, et al. (2014). Structural studies of postentry restriction factors reveal antiparallel dimers that enable avid binding to the HIV-1 capsid lattice. Proc Natl Acad Sci U S A 111, 9609–9614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goujon C, Moncorgé O, Bauby H, Doyle T, Ward CC, Schaller T, Hué S, Barclay WS, Schulz R, and Malim MH (2013). Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 502, 559–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray ER, Brookes JC, Caillat C, Turbé V, Webb BLJ, Granger LA, Miller BS, McCoy LE, El Khattabi M, Verrips CT, et al. (2017). Unravelling the Molecular Basis of High Affinity Nanobodies against HIV p24: In Vitro Functional, Structural, and in Silico Insights. ACS Infect. Dis 3, 479–491. [DOI] [PubMed] [Google Scholar]
- Gres AT, Kirby KA, KewalRamani VN, Tanner JJ, Pornillos O, and Sarafianos SG (2015). STRUCTURAL VIROLOGY. X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability. Science 349, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmüller H, and MacKerell AD Jr. (2017). CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacques DA, McEwan WA, Hilditch L, Price AJ, Towers GJ, and James LC (2016). HIV-1 uses dynamic capsid pores to import nucleotides and fuel encapsidated DNA synthesis. Nature 536, 349–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia X, Zhao Q, and Xiong Y (2015). HIV suppression by host restriction factors and viral immune evasion. Curr. Opin. Struct. Biol 31, 106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen WL (1998). Temperature dependence of TIP3P, SPC, and TIP4P water from NPT Monte Carlo simulations: Seeking temperatures of maximum density. J. Comput. Chem 19, 1179–1186. [Google Scholar]
- Kabsch W (2010a). Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr 66, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010b). Xds. Acta Crystallogr. D Biol. Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane M, Yadav SS, Bitzegeio J, Kutluay SB, Zang T, Wilson SJ, Schoggins JW, Rice CM, Yamashita M, Hatziioannou T, and Bieniasz PD (2013). MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 502, 563–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha A, Seago J, Scott K, Burman A, Loureiro S, Ren J, Porta C, Ginn HM, Jackson T, Perez-Martin E, et al. (2015). Structure-based energetics of protein interfaces guides foot-and-mouth disease virus vaccine design. Nat. Struct. Mol. Biol 22, 788–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda S, Nakagawa N, Tokunaga C, Tatematsu K, and Tanizawa K (1999). Mammalian homologue of the Caenorhabditis elegans UNC-76 protein involved in axonal outgrowth is a protein kinase C zeta-interacting protein. J. Cell Biol 144, 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanza DC, Silva JC, Assmann EM, Quaresma AJ, Bressan GC, Torriani IL, and Kobarg J (2009). Human FEZ1 has characteristics of a natively unfolded protein and dimerizes in solution. Proteins 74, 104–121. [DOI] [PubMed] [Google Scholar]
- Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, et al. (2011). ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 487, 545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, and Sodroski J (2008). The TRIM5alpha B-box 2 domain promotes cooperative binding to the retroviral capsid by mediating higher-order self-association. J Virol. 82, 11495–11502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Hill CP, Sundquist WI, and Finch JT (2000). Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature 407, 409–413. [DOI] [PubMed] [Google Scholar]
- Li YL, Chandrasekaran V, Carter SD, Woodward CL, Christensen DE, Dryden KA, Pornillos O, Yeager M, Ganser-Pornillos BK, Jensen GJ, and Sundquist WI (2016). Primate TRIM5 proteins form hexagonal nets on HIV-1 capsids. eLife 5, e16269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippert RA, Predescu C, Ierardi DJ, Mackenzie KM, Eastwood MP, Dror RO, and Shaw DE (2013). Accurate and efficient integration for molecular dynamics simulations at constant temperature and pressure. J. Chem. Phys 139, 164106. [DOI] [PubMed] [Google Scholar]
- Liu C, Perilla JR, Ning J, Lu M, Hou G, Ramalho R, Himes BA, Zhao G, Bedwell GJ, Byeon IJ, et al. (2016). Cyclophilin A stabilizes the HIV-1 capsid through a novel non-canonical binding site. Nat. Commun 7, 10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luby-Phelps K (2000). Cytoarchitecture and physical properties of cytoplasm: volume, viscosity, diffusion, intracellular surface area. Int. Rev. Cytol 192, 189–221. [DOI] [PubMed] [Google Scholar]
- Lukic Z, Dharan A, Fricke T, Diaz-Griffero F, and Campbell EM (2014). HIV-1 uncoating is facilitated by dynein and kinesin 1. J. Virol 88, 13613–13625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malikov V, and Naghavi MH (2017). Localized Phosphorylation of a Kinesin-1 Adaptor by a Capsid-Associated Kinase Regulates HIV-1 Motility and Uncoating. Cell Rep. 20, 2792–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malikov V, da Silva ES, Jovasevic V, Bennett G, de Souza Aranha Vieira DA, Schulte B, Diaz-Griffero F, Walsh D, and Naghavi MH (2015). HIV-1 capsids bind and exploit the kinesin-1 adaptor FEZ1 for inward movement to the nucleus. Nat. Commun 6, 6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallery DL, Márquez CL, McEwan WA, Dickson CF, Jacques DA, Anandapadamanaban M, Bichel K, Towers GJ, Saiardi A, Böcking T, and James LC (2018). IP6 is an HIV pocket factor that prevents capsid collapse and promotes DNA synthesis. eLife 7, e35335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Márquez CL, Lau D, Walsh J, Shah V, McGuinness C, Wong A, Aggarwal A, Parker MW, Jacques DA, Turville S, and Böcking T (2018). Kinetics of HIV-1 capsid uncoating revealed by single-molecule analysis. eLife 7, e34772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matreyek KA, Wang W, Serrao E, Singh PK, Levin HL, and Engelman A (2014). Host and viral determinants for MxB restriction of HIV-1 infection. Retrovirology 11, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Cryst 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald D, Vodicka MA, Lucero G, Svitkina TM, Borisy GG, Emerman M, and Hope TJ (2002). Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 159, 441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller MJ, Klumpp S, and Lipowsky R (2008). Tug-of-war as a cooperative mechanism for bidirectional cargo transport by molecular motors. Proc. Natl. Acad. Sci. USA 105, 4609–4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, and Dodson EJ (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Pawlica P, and Berthoux L (2014). Cytoplasmic dynein promotes HIV-1 uncoating. Viruses 6, 4195–4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, and Schulten K (2005). Scalable molecular dynamics with NAMD. J. Comput. Chem 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, and Yeager M (2009). X-ray structures of the hexameric building block of the HIV capsid. Cell 137, 1282–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pornillos O, Ganser-Pornillos BK, Banumathi S, Hua Y, and Yeager M (2010). Disulfide bond stabilization of the hexameric capsomer of human immunodeficiency virus. J. Mol. Biol 401, 985–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pornillos O, Ganser-Pornillos BK, and Yeager M (2011). Atomic-level modelling of the HIV capsid. Nature 469, 424–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AJ, Jacques DA, McEwan WA, Fletcher AJ, Essig S, Chin JW, Halambage UD, Aiken C, and James LC (2014). Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog. 10, e1004459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnick SI, and Adams GP (2009). Affinity and avidity in antibody-based tumor targeting. Cancer Biother. Radiopharm 24, 155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo Y, Walsh D, Barry DS, Tinaztepe S, de Los Santos K, Goff SP, Gundersen GG, and Naghavi MH (2013). HIV-1 induces the formation of stable microtubules to enhance early infection. Cell Host Microbe 14, 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger. (2015). The PyMOL Molecular Graphics System, Version 1.8 (Schrodinger).
- Shan Y, Klepeis JL, Eastwood MP, Dror RO, and Shaw DE (2005). Gaussian split Ewald: A fast Ewald mesh method for molecular simulation. J. Chem. Phys 122, 54101. [DOI] [PubMed] [Google Scholar]
- Shaw DE, Grossman JP, Bank JA, Batson B, Butts JA, Chao JC, Deneroff MM, Dror RO, Even A, Fenton CH, et al. (2014). Anton 2: raising the bar for performance and programmability in a special-purpose molecular dynamics supercomputer. In Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis (IEEE Press), pp. 41–53. [Google Scholar]
- Smaga SS, Xu C, Summers BJ, Digianantonio KM, Perilla JR, and Xiong Y (2019). MxB restricts HIV-1 by targeting the tri-hexamer interface of the viral capsid. Structure, Published online May 10, 2019. 10.1016/j.str.2019.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers BJ, Digianantonio KM, Smaga SS, Huang P-T, Zhou K, Gerber EE, Wang W, and Xiong Y (2019). Modular HIV-1 Capsid Assemblies for Investigating Diverse Host-Capsid Recognition Mechanisms. Cell Host Microbe 26, 203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinevez JY, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL, and Eliceiri KW (2017). TrackMate: An open and extensible platform for single-particle tracking. Methods 115, 80–90. [DOI] [PubMed] [Google Scholar]
- Toda H, Mochizuki H, Flores R 3rd, Josowitz R, Krasieva TB, Lamorte VJ, Suzuki E, Gindhart JG, Furukubo-Tokunaga K, and Tomoda T (2008). UNC-51/ATG1 kinase regulates axonal transport by mediating motor-cargo assembly. Genes Dev. 22, 3292–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, and Murshudov GN (2004). REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D Biol. Crystallogr 60, 2184–2195. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, and Charlton SJ (2013). Exploring avidity: understanding the potential gains in functional affinity and target residence time of bivalent and heterobivalent ligands. Br. J. Pharmacol 168, 1771–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schwedler UK, Stray KM, Garrus JE, and Sundquist WI (2003). Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J. Virol 77, 5439–5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte MA (2010). Bidirectional transport: matchmaking for motors. Curr. Biol 20, R410–R413. [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, and Engelman AN (2017). Capsid-Dependent Host Factors in HIV-1 Infection. Trends Microbiol 25, 741–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Yang M, Ding Y, Liu Y, Lou Z, Zhou Z, Sun L, Mo L, Ye S, Pang H, et al. (2003). The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA 100, 13190–13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Ji X, Zhao G, Ning J, Zhao Q, Aiken C, Gronenborn AM, Zhang P, and Xiong Y (2012). Structural insight into HIV-1 capsid recognition by rhesus TRIM5alpha. Proc Natl Acad Sci U S A 109, 18372–18377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S, Myszka DG, Yeh C, McMurray M, Hill CP, and Sundquist WI (1997). Molecular recognition in the HIV-1 capsid/cyclophilin A complex. J. Mol. Biol 269, 780–795. [DOI] [PubMed] [Google Scholar]
- Zhao G, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning J, Ahn J, Gronenborn AM, Schulten K, Aiken C, and Zhang P (2013). Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 497, 643–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Price AJ, Halambage UD, James LC, and Aiken C (2015). HIV-1 Resistance to the Capsid-Targeting Inhibitor PF74 Results in Altered Dependence on Host Factors Required for Virus Nuclear Entry. J. Virol 89, 9068–9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data Deposition
The accession number for the R18D CA hexamer structure and diffraction data reported in this paper is PDB: 6OMT.
Software Availability
This study did not produce additional software. The software used in this paper has been published and is publicly available.