

Abstract
In the last step of glycolysis Pyruvate kinase catalyzes the irreversible conversion of ADP and phosphoenolpyruvate to ATP and pyruvic acid, both crucial for cellular metabolism. Thus pyruvate kinase plays a key role in controlling the metabolic flux and ATP production. The hallmark of the activity of different pyruvate kinases is their tight modulation by a variety of mechanisms including the use of a large number of physiological allosteric effectors in addition to their homotropic regulation by phosphoenolpyruvate. Binding of effectors signals precise and orchestrated movements in selected areas of the protein structure that alter the catalytic action of these evolutionarily conserved enzymes with remarkably conserved architecture and sequences. While the diverse nature of the allosteric effectors has been discussed in the literature, the structural basis of their regulatory effects is still not well understood because of the lack of data representing conformations in various activation states. Results of recent studies on pyruvate kinases of different families suggest that members of evolutionarily related families follow somewhat conserved allosteric strategies but evolutionarily distant members adopt different strategies. Here we review the structure and allosteric properties of pyruvate kinases of different families for which structural data are available.
Keywords: allosteric enzyme, cryptosporidium, crystal structure, glycolysis, holo‐enzyme, protein structure, pyruvate kinase
Abbreviations
- CpPyK
Cryptosporidium parvum PyK
- F6P
Fructose 6‐phosphate
- FBP
Fructose 1,6‐bisphosphate
- FDP
Fructose 2,6‐disphosphate
- G6P
Glucose 6‐phosphate
- HK
Hexokinase
- LA
Lactic acid
- PEP
Phosphoenolpyruvate
- PFK
Phosphofructokinase
- PyK
Pyruvate kinase
- PYR
Pyruvic acid
1. INTRODUCTION
Glycolysis is the central pathway for the utilization of glucose in most organisms. In this oxygen‐independent pathway, glucose is metabolized into the highly versatile metabolite, pyruvic acid (PYR), while producing ATP and NADH (Figure 1).1, 2 Under aerobic conditions, PYR is decarboxylated to acetyl‐CoA, which can be utilized for energy production by cellular respiration in mitochondria3 or for fatty acid biosynthesis.1, 4 In oxygen‐limited environments, PYR can be anaerobically oxidized to lactic acid (LA). Moreover, PYR can be converted to glucose (via gluconeogenesis), alanine (by alanine transaminase), or oxaloacetate (by pyruvate carboxylase). To meet cellular needs, the rate of conversion of glucose into PYR is tightly regulated by three glycolytic enzymes, hexokinase (HK), phosphofructokinase (PFK), and pyruvate kinase (PyK).5
Figure 1.
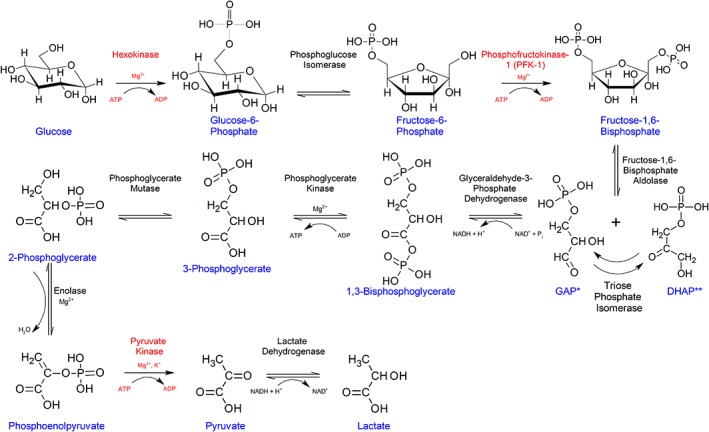
Flowchart showing different reactions in the glycolytic pathway. Reactions in the glycolytic pathway are shown with chemical structures of the substrates, products and cofactors including metal ions at each step. Enzymes responsible for catalyzing the irreversible regulatory steps are indicated in red. *GAP and **DHAP are abbreviations for glyceraldehyde 3‐phosphate and dihydroxyacetone phosphate, respectively. In some hyperthermophilic archaea, GAP is irreversibly oxidized to 3‐phosphoglycerate (3PG) by glyceraldehyde 3‐phosphate ferrodoxin oxidoreductase or nonphosphorylating GAP dehydrogenase. Nevertheless, in the last reaction in the modified glycolytic pathway of these organisms PyK catalyzes the conversion of PEP to PYR. PyKs of the Thermoproteales archaea are activated by 3PG
In the last step of glycolysis, PyK (EC 2.7.1.40) catalyzes the irreversible transfer of phosphate from phosphoenolpyruvate (PEP) to ADP resulting in the production of PYR and ATP. PyK is a major regulator for controlling the metabolic flux and ATP production in glycolysis and is considered a potential drug target.6, 7, 8, 9 However, because of the conserved architecture of the PyK active site and the central role of glycolysis in all organisms, developing selective inhibitors targeting the active site is challenging. On the other hand, almost all PyKs are allosterically regulated by various physiological effectors.10, 11, 12, 13, 14, 15, 16 Therefore, the differences in the allosteric mechanisms of PyKs can be exploited for developing species‐ or cell‐type‐selective drugs.7, 8, 17, 18 Moreover, inhibition of PyK activity can be an effective therapeutic strategy against organisms that are dependent on glycolysis for energy production. One example is the globally important pathogen Cryptosporidium parvum, which because of the minimal metabolic capacity of its primitive mitochondria, relies exclusively on glycolysis for ATP production.19, 20, 21, 22, 23, 24, 25 Thus C. parvum PyK (CpPyK) may be an attractive therapeutic target for treatment of C. parvum infection. Recently, due to the discovery of the abundance of vertebrate PyK‐M2 isozyme in cancer cells this isoform has been the focus of considerable research.26, 27, 28 The strong interest in human PyK‐M2 isozyme is evident from the fact that one third of all PyK crystal structures deposited in the Protein Data Bank (PDB) describe human PyK‐M2 and its complexes (Table S1).
Analyses of PyKs of various organisms highlight the similarity of their overall structure and active site architecture as well as the remarkable versatility in regulation.11, 29, 30 However, our understanding of the regulatory mechanisms, especially the structural basis for transition among different activation states, is still evolving. The main bottleneck in characterizing the mechanisms of allosteric regulation is the lack of structural data for many PyKs in the ligand‐free state and in complex with various ligands. Of the 97 crystal structures of PyKs deposited in the PDB, structures representing all four states of the protein (apo, substrate‐complex, effector‐complex, and substrate‐effector complex) are available only for PyKs of Leishmania mexicana (LmPyK),10 Mycobacterium tuberculosis (MtbPyK),7 and human PyK‐M231 (Table S1).
Here we provide an overview of the current state of knowledge derived from structural and functional studies of PyKs. For general discussions and illustrations, we have mainly used LmPyK, which is one of the well‐studied members. We also discuss previously unpublished structural data on the CpPyK in the ADP‐bound state.
2. EVOLUTIONARY RELATIONSHIPS IN THE ALLOSTERIC REGULATION OF PYKS
A hallmark of PyK functions is the tight regulation of their activity by a variety of mechanisms including the use of many physiological allosteric modulators. To allow regulation of glycolytic flux in response to elevated PEP levels almost all tetrameric PyKs are homotropically activated by the substrate PEP.11 Moreover, PyKs are also regulated by heterotropic effectors with fructose 1,6‐bisphosphate (FBP) being the most widely known allosteric activator of bacterial, yeast, and mammalian enzymes.11, 32, 33, 34 On the other hand, trypanosomatid PyKs are modulated by fructose 2,6‐diphosphate (FDP).10 Other effectors alone or in combination modulate different PyKs based on the metabolic needs of the cells. A growing body of evidence suggests that evolution of PyKs is intimately connected with their allosteric properties.
PyKs exhibit an unusual phylogenetic tree structure that does not coincide with the universal tree topology of the three domains of life: Bacteria, Archaea, and Eukarya.35, 36 Phylogenetic analysis revealed clustering of PyKs in two groups. PyKs in cluster I, contain a Glu at position 117 (rabbit muscle PyK numbering, LmPyK residue 88) and those in cluster II contain a Lys at the corresponding position (Figure 2).37 PyKs in the two clusters differ in their dependence on monovalent cations.37 While PyKs containing Glu117 (cluster I) show an absolute requirement for K+ (or other monovalent cations), enzymes in cluster II are monovalent cation‐independent. The importance of the electrostatic charge of the residue at position 117 is underscored by the observation that K+ dependence can be removed by single amino acid substitution of Glu117Lys.38 Most PyKs with Lys117 also contain Leu113/Gln114 and a Val, Ile, or Leu residue at position 120, while in the majority of Glu117 PyKs, Thr, Lys, and Thr residues are present in positions 113, 114, and 120, respectively. It was proposed that K+‐dependent PyKs emerged from K+‐independent enzymes through single nucleotide substitutions.37
Figure 2.

Structure‐based sequence alignment of representative PyKs from various organisms. PROMALS3D (http://prodata.swmed.edu/promals3d/info/promals3d_help.html) was used for structure‐based alignment of amino acid sequences of PyKs of different organisms. Secondary structure assignment is based on consensus. Helices and strands are identified by domains A, B, and C. Each sequence is labeled with the PDBID for the PyK structure and the name of the organism it belongs to. Functionally important regions are labeled. Interacting residues identified in crystal structures are highlighted on the respective sequence. These residues were identified using PDBsum (http://www.ebi.ac.uk/pdbsum/). See also Table S4.*: K+‐binding residues [Note: Asn also interacts with ADP in CpPyK (6P0Y)]; #: Residues that interact with divalent cation; Yellow highlight: PEP‐binding residues; Light pink highlight: Residues that interact with ADP (CpPyK: 6P0Y) and ATP (LmPyK: 3HQP); Phosphate: Phosphate‐binding loop (6′‐phosphate of FBP and FDP and the phosphate of AMP interact with residues in this loop). Mobile: Mobile loop (in some PyK structure this loop is stabilized in effector‐bound form); Effector: Effector‐loop (usually stabilized in the presence of effectors); Orange highlight: Residues that interact with FDP in LmPyK complex (3HQP); Cyan highlight: Residues that interact with G6P (MtbPyK: 5WSB); Hinge: Bending residues that act as hinge for movement of the B‐domain upon substrate binding. The lengths of the hinge regions differ in different PyKs. Shown in italics are residues in these regions identified by DynDom using the crystal structures 3HQO (Apo) and 3HQN (substrate complex) of LmPyK (http://dyndom.cmp.uea.ac.uk/dyndom/)
Interestingly, enzymes of these two families use different allosteric activators. Cluster I PyKs are regulated by FBP11, 33 or FDP10, 39 while AMP or sugar monophosphates modulate the enzymes in cluster II.40, 41 PyKs belonging to both clusters are expressed in gamma‐proteobacteria,42 and in apicomplexan parasites in which cluster I and cluster II PyKs are found in cytosolic and apicoplast compartments, respectively.43, 44 Organisms carrying both families of PyKs include a number of clinically significant pathogens such as Escherichia coli,32, 40 Vibrio cholera,42 Salmonella typhimurium,45 Toxoplasma gondii,43, 46 and Plasmodium falciparum.44
PyKs of hyperthermophilic archaea Thermoproteales (but not thermophilic bacteria) are activated by 3‐phosphoglycerate (3PG) and not by FBP, FDP, or AMP.36, 47, 48 In the glycolytic pathway of these archaea, glyceraldehyde 3‐phosphate is irreversibly oxidized to 3PG.49, 50 A putative 3PG binding motif was identified in the sequence of PyKs of Thermoproteales, and based on phylogenetic and biochemical analyses it was concluded that these PyKs form an independent lineage.48 Their sequence contains a Ser residue in the position corresponding to Glu117 in rabbit muscle PyK and these enzymes are K+‐independent.35, 41
Thus members of different PyK families use different effectors, and the sequence and architecture of the effector binding sites and their allosteric strategies are linked with their evolution.8, 51 Nevertheless the amino acid sequence of different PyKs and the active site residues are very similar as expected for an evolutionarily conserved protein. The overall sequence identity between PyKs from evolutionarily distant organisms can be up to ~40% (Table S2).
3. ARCHITECTURE OF PYKS
3.1. Domain structure
Cytosolic PyKs are composed of 480–530 amino acids while the plastid and apicoplast‐specific PyKs carry an additional N‐terminal signal sequence.43, 44 The Plasmodium and Toxoplasma genome encodes two distinct proteins, the cytoplasmic form PyKI and the apicoplast form PyKII. Low sequence identity (~20%) between these forms suggests their distinct lineage. PyKII sequences include an N‐terminal apicoplast‐targeting signal of 130 residues and multiple insertions.52 PyKII of T. gondii is also localized in the mitochondria in addition to the apicoplast. PKI is similar to the glycolytic form of PyKs whereas PyKII is implicated in fatty acid biosynthesis.44 Notably, PyKII sequences possess a Lys residue in the position corresponding to Glu117 in rabbit muscle PyK, and therefore group with monovalent cation‐independent enzymes.
The majority of PyKs are homotetrameric proteins; however, other oligomeric forms have also been reported.53, 54, 55, 56 PyK monomers fold into three domains designated A, B, and C. In some eukaryotes an additional N‐terminal α‐helical domain of ~17 residues known as the N‐domain is present (Figure 3). Moreover, there is an extra C‐terminal domain in PyKs of some bacteria such as Bacillus,57, 58 Staphylococcus,6 and Cyanobacteria41 species. Generally, sequence identity among PyKs is highest in the A‐domains, followed by the B‐ and C‐domains (Table S2).
Figure 3.
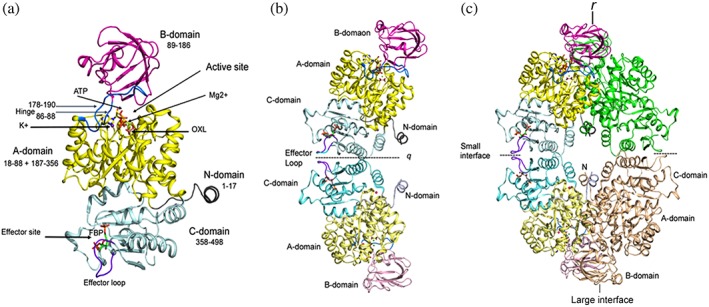
Domain organization and assembly of PyK. (a) Cartoon drawing of LmPyK monomer with domains colored as N (residues 1–17; gray), A (18–88 and 187–356; yellow), B (89–186; magenta), C (358–496; light cyan). Ligands ATP and oxalic acid (OXL) are shown as stick models. Metal ions (Mg2+ and K+) shown as green and purple spheres in the active site are indicated with an arrow. Hinges (86–88 and 178–190) are shown in marine blue. Effector loop (481–487) is shown in dark purple; FDP bound in the effector site is shown as stick model. The LmPyK structure (3HQP) containing ATP, OXL, FDP, Mg2+, and K+ was used in preparing this cartoon. (b) Cartoon drawing showing the LmPyK dimer and the C–C interface also known as the “small interface.” Domains in both subunits are colored similar to those in Figure 2a. Effector loops from each monomer are at the dimer interface. Dimer axis is designated q. (c) Cartoon drawing showing assembly of the LmPyK tetramer in 3HQP. The small and large interfaces are indicated. The large interface is formed by interactions of residues in the A‐domains of neighboring subunits across r axis
The A‐domain is the largest of the three domains and is composed of two segments spanning residues 18–88 and 187–356 (LmPyK numbering).10 It consists of a (β/α)8 barrel structure characterized by three helices located on top of the loops connecting the C‐terminal strands of the β‐sheet (Figures 3 and S1). These helices play a major role in catalysis and allosteric regulation. The PyK catalytic site is located on top of the barrel in a cleft between the A‐ and B‐domains (Figure 3a).
The B‐domain (residues 89–186) has a mixed β‐barrel topology and contains only one (or two) short α‐helix. The B‐domain is also known as the “lid domain” since upon binding the substrate it undergoes a bending motion which results in closing of the active site. The hinge residues for the bending are located at the intersections of A‐ and B‐domains, residues 86–88 and 178–190 (Figure 2). The bending angle is usually ~20°; however, comparison of the apo‐(3HQO) and substrate‐bound (3HQN) structures of LmPyK by using DynDom reveals a 40° rotation (Videos S1 and S2).59
Hinge residues 86–88 and several residues on both sides of the hinge play an important functional role in PyKs. As discussed above, the presence of Glu88 (equivalent to rabbit muscle PyK residue 117) determines the monovalent cation requirement of PyKs.37, 38 In LmPyK Asp83 and Thr84 are involved in coordination of the K+ ion. The sequence of residues, Asp83‐Thr‐Lys‐Gly‐Pro‐Glu‐Ile‐Arg‐Thr‐Gly92 (LmPyK numbering), at the intersection of the A‐ and B‐domain is generally conserved in FBP‐ or FDP‐activated PyKs of eukaryotes. Bacterial PyKs (such as E. coli PyK1 and Staphylococcus aureus PyK) that are activated by FBP, also have identical sequence in this region except at the C‐terminal position (Table S3). On the other hand, the sequence of this region differs in the AMP‐ and phosphosugar‐activated enzymes, such as E. coli PyK2 and MtbPyK, where Glu88 is replaced by Lys, both Thr residues are replaced by Leu or Val, and Lys85 is substituted by Gln. The hinge area is far from the effector binding sites, which are located within the C‐domain or between the A‐ and C‐domains (for G6P, discussed later). The C‐domain (residues 358–498) consists of five α‐helices and a five‐stranded mixed β‐sheet (Figures 3 and S2). The C‐domain of each PyK monomer is involved in interactions with the A‐domain of the same monomer. Moreover, residues in the C‐domains related by a two‐fold axis (referred to as the q‐axis) form the C–C dimer interface or small interface in the PyK tetramers (Figure S3). Interactions between residues in the A‐domains of neighboring subunits related by a two‐fold axis designated as the r‐axis form the large or A–A interface. The intermolecular contacts along the r‐interface are mostly contributed by residues belonging to helices Aα6, Aα7, and Aα8.60 Moreover, the N‐domain when present interacts with the A‐domain (Figure S4).
3.2. Active site
Amino acids from both A‐ and B‐domains contribute to the formation of the active site consisting of highly conserved residues that bind to the substrates and metal ions, which also interact with the substrates. The PyK active site includes three positively charged residues, two Arg and a Lys; and four negatively charged residues, two Asp and two Glu residues. In addition to a monovalent cation, most PyKs require a divalent (Mg2+or Mn2+) cation, which has been implicated in enzyme activation and in the formation of the substrate‐complex with ADP.61 Cations have also been suggested to induce domain movement.62 Amino acids responsible for binding to the divalent cation are within the A‐domain.63, 64, 65 In LmPyK10 the Mg2+ ion (or Mn2+) interacts with the carboxylate side chains of Glu241 and Asp265. Additionally, the divalent cation also coordinates to the oxygen atoms of PYR or other ligands.66 A second divalent cation forms coordination with ADP or ATP in the holo‐enzyme structures (3HQP, 6P0Y) but does not directly bind to the protein. The K+ ion coordinates to Asn51, Ser53, Asp83, and Thr84 in the A‐domain (Table S4).
PEP and ADP interact mainly with residues located in the A‐domain. Moreover, an Arg from the B‐domain also binds to ADP. Currently, crystal structures of PyK in the ADP‐bound state have been reported for human PyK‐M2 (3GR4) and CpPyK (6P0Y) only. In 3GR4, PyK‐M2 is also bound to FBP and tartaric acid but lacks any metal ion in the active site, while the structure of holo‐CpPyK contains ADP, Mg2+, and K+. In both structures two conserved Arg residues and a conserved Asn residue form hydrogen bonds with ADP (Figure 4). Additionally, in holo‐CpPyK, a conserved His79 (55) is also involved in hydrogen bonding to ADP. In the LmPyK/ATP complex, the phosphate oxygen atoms of the γ‐phosphate group of ATP form additional interactions with side chains of Asn51 and Lys 238. Moreover, Mg2+ and K+ ions also coordinate to the terminal phosphate (Figure S5).
Figure 4.
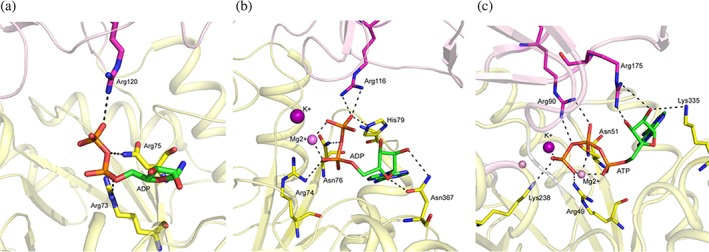
ADP binding site in PyK. Interactions between PyK residues and ADP are shown in stick model. Residues from both the A‐ and B‐domains are involved in binding to the substrate. (a) Cartoon drawing showing human PyK‐M2 (3GR4) active site. The A‐ and B‐domains are shown in light yellow and light magenta color. The C‐atoms of the residues in the A‐ and B‐domains, which interact with ADP, are shown in magenta and yellow, respectively. No metal ion was present in the PyK‐M2 complex. (b) Cartoon drawing showing holo‐CpPyK (6P0Y) active site. The A‐ and B‐domains are shown in light yellow and light magenta color. The C atoms of the residues in the A‐ and B‐domains, which interact with ADP, are shown in magenta and yellow, respectively. K+ and Mg2+ ions are shown as purple and pink spheres. (c) Cartoon drawing showing LmPyK (3HQP) active site with ATP bound. The A‐ and B‐domains are shown in light yellow and light magenta color. The C‐atoms of the residues in the A‐ and B‐domains, which interact with ADP, are shown in magenta and yellow, respectively. K+ and Mg2+ ions are shown as purple and pink spheres
So far, a structure of PyK in complex with PEP has been reported only for the Trypanosoma brucei enzyme (TbPyK; 4HYV). In this complex, PEP engages in coordination with the bound Mg2+ and K+ ions in the active site.66 Five conserved residues participate in hydrogen bonding with PEP: side chain atoms of Arg50, Lys239, and Thr297, and the backbone nitrogen atoms of Gly264 and Asp265 (Figure 5a). Structures of PYR complexes have been determined for TbPyK and mammalian M1 and M2 isozymes (Table S1). In these complexes PYR is involved in coordination with the Mg2+ and forms hydrogen bonds with all residues as observed in the PEP complex except Arg50 (which is hydrogen bonded to the phosphate moiety of PEP in 4HYV). In the TbPyK PYR complex (4KCT), the Lys239 sidechain moves closer to the product and forms a hydrogen bond with O3 atom of PYR [Figure 5b].
Figure 5.
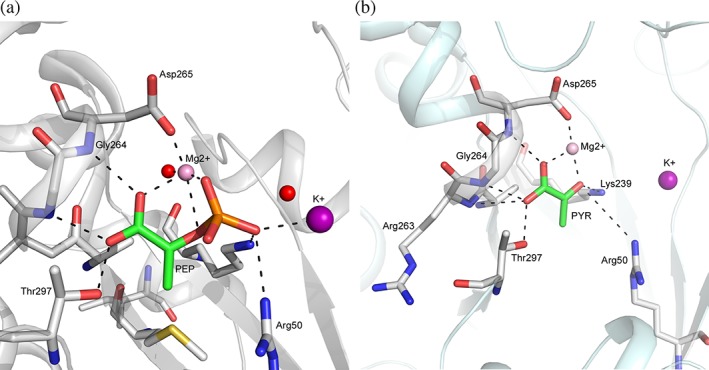
PEP and PYR binding sites in PyK. Interactions of PyK residues and metal ions with PEP and PYR in TbPyK. (a) Cartoon drawing of TbPyK structure (4KCT) showing PYR in stick model (C: green) and residues (C: white) forming interactions with PEP. K+ and Mg2+ and ions are shown as purple and pink spheres. Two water molecules in close proximity are shown as red spheres. (b) Cartoon drawing of TbPyK structure (4HYV) showing PEP in stick model (C: green) and residues (C: white) forming interactions with PEP. Mg2+ and K+ ions are shown in rose and purple spheres
3.3. Effector binding site
The canonical allosteric effector binding sites in PyKs are located within the C‐domain.33 This site, first identified in the crystal structure of yeast PyK1 bound with FBP, is approximately 40 å away from the catalytic site (1A3W).33 More recently, FDP has been found to bind in the same site in LmPyK (3HQP, 3HQQ)10 and Trypanosoma cruzi PyK (TcPyK, 4KS0).51
The effector binding site is lined by three loops: a “phosphate‐binding loop” (LmPyK residues 400–404), a “mobile loop” (448–453), and an “effector loop” (481–487). The effector remains buried in a pocket formed by the phosphate‐binding loop (connecting the Cβ1 strand and Cα3 helix) and the first two turns of the Cα5 helix (Figure S2). The 6′‐phosphate group of FBP and FDP occupies overlapping positions in their complexes and engages in interactions with the residues in the “phosphate‐binding loop” (Figure 6).10, 33 Additionally, Arg and a Ser immediately C‐terminal to this loop also form hydrogen bonds with the 6′‐phosphate. The sequence of the phosphate‐binding loop usually contains multiple threonine and serine residues.
Figure 6.
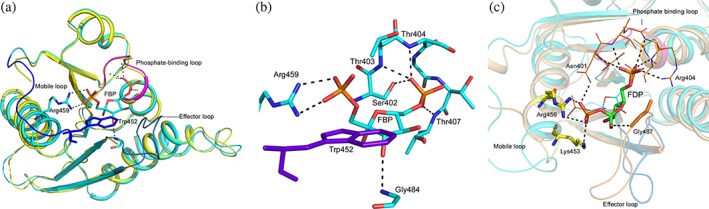
FBP and FDP binding sites in PyKs. (a) Superposition of yeast PyK1 without (1A3X; yellow) and with (1A3W; cyan) bound FBP molecule. Residues in the phosphate‐ binding loop (magenta) are involved in binding to the 6′‐phosphate group. Effector loop (slate) remains disordered in FBP‐free state. The mobile loop (purple) is also disordered in the absence of FBP. Arg459 (stick model, C: cyan) on the Cα5 helix engages in binding to the 1′‐phosphate and Trp452 (stick model, C: dark purple) forms hydrophobic interaction with the sugar moiety. (b) Close‐up view showing interactions in the FBP binding site of yeast PyK. (c) Superposition of the C‐domains of yeast PyK1 (1A3W) in cyan and LmPyK (3HQP) light orange shows the overlapping of the FBP (shown in line) and FDP (shown in stick) binding sites. The 2′‐phosphate of FDP interacts with side chains of Arg456 and Lys453 (immediately before the Cα5 helix) and the hydroxyl oxygen of the sugar forms a hydrogen bond with the backbone nitrogen atom of Gly487 on the LmPyK effector loop. Residues in the phosphate binding loop (residues 401–406; shown as line; C: deep orange) interact with the 6′‐phosphate. Backbone nitrogen atoms of Asn401, Ser402, Thr403, and Arg404 form hydrogen bonds with the 6′‐phosphate. Moreover, LmPyK Asn401 and Arg404 sidechains interact with the 2′‐phosphate and 6′‐phosphate, respectively
In yeast PyK1 the 1′‐phosphate of FBP forms a salt bridge with the side chain of Arg459 located on the Cα5 helix33 (Figure 6a,b). This Arg residue is present in PyKs of yeast, vertebrates and trypanosomatids. In PyK‐FDP complexes, the 2′‐phosphate of FDP is hydrogen‐bonded to the Arg residue in the corresponding position on the Cα5 helix (Arg456 and Arg457 in LmPyK and TcPyK, respectively) and the side chain of a Lys residue (Lys453 and Lys454 in LmPyK and TcPyK, respectively) that is conserved in trypanosomatids (Figure 6c).
The furanose ring of FBP is involved in hydrophobic interactions with the aromatic side chain of Trp452 located at the C‐terminal end of a flexible loop linking Cβ3 strand to Cα5 helix. This Trp residue is conserved in PyKs of yeast and vertebrate PyKs but not in other species. In some PyK structures this “mobile loop” is disordered in the absence of a bound activator (yeast PyK, 1A3X). The length and the amino acid sequence of the mobile loop vary in PyKs of different organisms.67
In general, the effector loop, which joins Cβ4 and Cβ5, remains disordered in the absence of a bound effector and becomes ordered upon binding the effector in the pocket. In yeast PyK, the loop is stabilized upon FBP‐binding via formation of a hydrogen bond between the 3′‐hydroxyl oxygen of the sugar moiety and the backbone nitrogen atom of Gly484. Three additional residues, Gly490, His491, and Ser492, from this loop are also involved in nonbonded interactions with FBP. In TcPyK the effector loop transitions from disordered to an ordered state (4KRZ vs. 4KS0) upon binding FDP. However, in the structure of TcPyK with Ponceau‐S dye bound at the ADP binding site (3QV9) the effector loop is ordered even in the absence of a bound effector (Figure S6a).8 A similar ordering of the effector has been noticed in the structure of CpPyK bound to ADP (6P0Y) (Figure S6b,c). The sequence and the length of the effector loop show some similarities in PyKs of evolutionarily related organisms. It should be noted that trypanosomatid PyKs can also bind FBP; however, as compared to FDP at least a 1,000‐fold higher concentration of FBP is required to produce a weaker allosteric effect.51
AMP binds in the same pocket as FBP and FDP. In the MtbPyK AMP complex (5WS9), the AMP phosphate group forms hydrogen bonds with Thr374(402), Gln375(403), Ser376(404), and Thr379(407) in the phosphate‐binding loop, and with a conserved Gly457(497) in the effector loop (Figure 7). The adenine ring is in a hydrophobic area and forms stacking interactions with Phe373, Trp398, and Met425. While Phe373 and Met425 are present in some AMP‐activated PyKs, Trp398 is unique in MtbPyK. Interaction of Trp398 with AMP requires flipping of the side chain. In the absence of the effector, this effector loop is highly flexible (5WS9).
Figure 7.
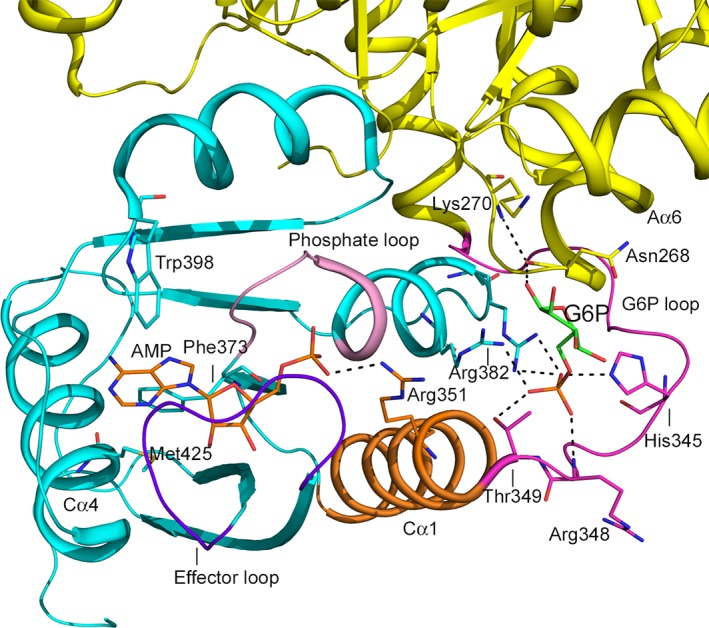
Interactions between the allosteric effector sites in MtbPyK. Cartoon drawing of MtbPyK (5WSB) showing stick models of AMP (C: orange) and G6P (C: green) in MtbPyK. AMP is bound in the canonical effector binding site in the C‐domain (cyan) and G6P is bound at a site between the C‐ and the A‐domain (yellow). The AMP phosphate group is hydrogen bonded to several residues in the phosphate‐binding loop (light magenta). The effector loop (residues 451–458; purple blue) wraps around AMP. The adenine ring of AMP is in a hydrophobic environment contributed by Phe373, Trp398, and Met425. The AMP phosphate group also interacts with Arg351 (stick, C: orange) on the Cα1 helix (orange), which acts as a link between the site for binding of AMP and G6P, and is connected to the G6P loop (residues 338–349). Phosphate group of G6P forms hydrogen bonds with His345, Arg348, and Thr349, which is at the N‐terminal end of the Cα1 helix. The backbone nitrogen atom of Lys270 from the A‐domain forms a hydrogen bond with the sugar moiety of G6P. Moreover, the main chain of Asn268 forms a hydrogen bond to the sugar, thus stabilizing the interactions between the A‐ and C‐domain
3.4. Noncanonical binding site for G6P
Recently, a binding site for G6P was identified in the crystal structures of MtbPyK.7 This site, located at the interface of the A‐ and C‐domains near the canonical effector binding pocket, is formed by three helices Aα6, Cα1, Cα2, and the G6P‐loop at the N‐terminal end of Cα1. Arg382 and Arg385 located on Cα2, and His345, Arg348, and Thr349 on the G6‐loop are involved in binding G6P. Asn268's main chain nitrogen atom also forms a hydrogen bond with the sugar. Sequence of the G6P loop varies in PyKs of different organisms but is identical in different species of Mycobacterium.7 Remarkably, G6P and AMP exhibit synergistic allosteric activity.7 In the crystal structure of MtbPyK complexed with both AMP and G6P (5WSB), the Cα1 helix acts as a bridge between the two allosteric sites; Arg351 in Cα1 interacts with AMP and Thr349 is hydrogen bonded to G6P, thus forming a hydrogen‐bonded network (Figure 7).
4. MECHANISMS OF ALLOSTERIC REGULATION
According to the classical Monod‐Wyman‐Changeux model of allostery, oligomeric enzymes undergo symmetrical transitions between the ligand‐free T‐ and ligand‐bound R‐states. The enzyme structure is stabilized in the R‐state by ligand binding.68, 69, 70, 71 However, structural details in all states are rarely known. For example, although allosteric activation of E. coli PyK1 (EcPyK1) by FBP has been long known, the crystal structure in the R‐state remains elusive. From comparison of the T‐state structure of EcPyK1 with that of mammalian PyK‐M1 R‐state (FBP‐bound) structure, a concerted rotation of all three domains of each PyK monomer during allosteric transition was identified. Specifically, these structures revealed a combination of two types of rigid body movements, which included a rotation of the B‐ and C‐domains of each subunit and a rotation of every subunit in the tetramer.14 The overall structure of individual domains does not change but the domains connected by flexible hinges undergo rigid body motion across the pivot. Currently, five mechanistic variations for allosteric regulations of PyKs are proposed. These mechanisms describe how the binding of effector(s) at the allosteric site activates the enzyme by promoting conformational changes within each subunit and by influencing interactions between neighboring subunits. In most of these cases the allosteric effect enhances the stability and rigidity of the tetramer in an active R‐state. These effects are induced mainly via alterations of the inter‐subunit interactions at the A–A and C–C interfaces in the tetramer. These five mechanisms are as follows:
A “rock and lock” model has been proposed for allosteric activation of LmPyK wherein transition between the inactive T‐state and active R‐state is accompanied by a symmetrical rigid body rocking motion of the A‐ and C‐domains of each monomer in the tetramer. The R‐state is stabilized by eight salt bridges between the monomers in each dimer across the C–C interface which lock the tetramer in a rigid state. The increased rigidity of the tetramer upon ligand binding results in a seven‐fold higher reaction rate.10 Structures of TcPyK with and without bound FDP (4KS0 and 4KRZ) demonstrated that this enzyme also follows the “rock and lock” model and adheres to a similar transition between the T‐ and R‐state, which involves a rigid body rocking motion of the A‐ and C‐domain cores.51 Although a true T‐state crystal structure for TbPyK has not been determined, comparison with the corresponding LmPyK structure revealed a similar rigid body rotation of the A‐ and C‐domain cores. The R‐state for TcPyK and TbPyK is stabilized via a conserved salt‐bridge between Asp483 (LmPyK residue 482) and Arg494(493) across the C–C interface (Figure S7).66 Asp483 is located within the effector loop, and thus the conformation of this loop is critical to the allosteric control of these enzymes.51, 66 In the effector‐free state this loop is usually flexible.
Human PyK‐M2 uses a novel mechanism based on transition between different oligomeric states during allosteric activation.31 This isozyme transitions between an inactive monomer and an active tetrameric form when bound to the activator.72, 73 In the absence of FBP, salt bridges and hydrogen bonds between residues across the C–C interface result in a stable inactive T‐state tetramer. Moreover, stacking interactions between two Trp residues across the interface further stabilize the T‐state. Upon binding of FBP, the monomer undergoes a rigid body rotation.31 The effector loop wraps around FBP forcing the side chains of Trp515 and Arg516 to orient inward. As a result the hydrogen bonding network that stabilizes the T‐state is broken (Figure S8a). The stacking interactions are also disrupted since the Trp482 sidechain forms a hydrogen bond with the 1′‐phosphate group of FBP. Finally, the R‐state is stabilized by insertion of a disordered residue Lys421 into the hole formed by residues 390–420 (Figure S8b). A somewhat similar but much tighter “peg in hole” arrangement is observed in the constitutively active PyK‐M1 tetramer. The concerted rocking motions of the monomers appropriately position Arg342 near the active site for stabilization of the short glycine‐rich Aα6′ helix of an adjacent monomer (Figure S8c). On the other hand, the allosteric inhibitor Phe binds in a pocket located between active site and effector site near the pivot point for rigid body rotation. In the Phe complex of PyK‐M2, a rigid body rotation of the A‐ and C‐domains forces Lys421 out of the binding pocket that is responsible for the stabilization of the active R‐state tetramer.
-
AMP and G6P were found to synergistically activate MtbPyK. A “rock‐shape‐lock” mechanism based on a concerted rocking motion has been proposed to explain the activation mechanism.7 In MtbPyK AMP‐binding is accompanied by an interplay of two loops: the canonical effector loop (AMP‐loop) spanning residues 451–458, which remains flexible in the effector‐free state, and a C‐terminal tail loop comprising residues 467–472. During the allosteric transition, the A‐ and C‐domains of each monomer simultaneously undergo a rigid body rotation disrupting the C–C interface interactions between the AMP loop and the tail loop, as well as a hydrogen bond between two adjacent C‐domains. The relocation of the AMP loop and reorganization of the tail loop disrupt interface hydrogen bonds and form new interactions within each monomer enhancing interactions between allosteric ligands and each monomer. Thermal stability analysis and enzyme kinetic data suggest enhanced rigidity of the R‐state MtbPyK in the presence of AMP and G6P.7
Simultaneously during T‐ to R‐state conversion, the Aα6′–Aα6 helix bundle located at the A–A interface also undergoes a rotation around the hinge (Asn268); the concerted rocking motions form the R‐state structure and the active architecture of the catalytic site. Moreover, movement of the Aα7′–Aα7 helix pair generates new hydrogen bonds across the A–A interface with Aα6′–Aα6 through reorientation of the sidechain of Arg290.
Transition from T‐ to fully ligated R‐state (both AMP and G6P bound) is accompanied by rotation and shift of both Cα1 and Cα4‐helices that bind G6P and AMP, respectively. Binding of one activator induces a similar rotation of its binding helix and a similar, though smaller, movement of the other helix. Displacement of these helices gives rise to the synergistic allosteric effect where reshaping of the allosteric sites by binding of either activator favors the binding of the other (Figure 7).7 In the presence of both AMP and G6P, a network of hydrogen bonds links the Cα1 helix with AMP and G6P.
The crystal structure of EcPyK1 in the activator‐bound state has not been reported. Two early models were proposed for allosteric activation of EcPyK1 by FBP. The “domain rotation” model proposed reorientation of individual domains within each monomer as well as reorientation of each monomer within the tetramer. On the other hand, according to the “rigid body reorientation” model only monomers within the tetramer reorient with respect to each other and this increases the rigidity of the active site. A combination of time‐resolved electrospray ionization mass spectrometry coupled to hydrogen‐deuterium exchange studies and mutagenesis were employed to study the mechanism of allosteric activation by FBP on EcPyK1.34 These studies demonstrated enhanced conformational flexibility upon FBP binding at the allosteric site. Specifically, FBP binding causes destabilization of an α‐helix bridging the allosteric site and the active site in the same monomer. This results in a loss of stability in the assembly of the (β/α)8‐barrel domain and the active site loops.34
Finally, activation of PyK of Geobacillus stearothermophilus and several other bacteria (such as S. aureus) is thought to involve an additional C‐terminal domain (C′) found in these proteins. In these PyKs the effector loop is located within this domain.74 Presence of the allosteric activator ribose‐5‐phosphate (R5P) enhances thermal stability of the G. stearothermophilus PyK tetramer.75 The C′‐domains of these bacterial PyKs are predicted to be involved in interactions with adjacent monomers across the C–C interface resulting in the stabilization of the tetramer.51 Currently, no structural data is available for the activator‐bound state of any PyK of this category.
5. STRUCTURALLY AND FUNCTIONALLY DISTINCT PYKS
5.1. Vertebrate PyK isozymes
Vertebrates demonstrate exquisite regulation of PyK activities by a combination of strategies including an elaborate distribution of four isozymes (L, R, M1, and M2) in different tissues. The L and R isozymes are dominant in the liver and erythrocytes respectively, and both forms are allosterically regulated by FBP. Recently, in human liver PyK switching of the allosteric loop from an open to closed conformation in the presence of FBP has been attributed to the change in interactions at the C–C interface which leads to the altered substrate affinity of the enzyme.76 Muscle specific isoforms, PyK‐M1 and PyK‐M2, are expressed from the same gene but differ in 22 residues located within a 56 residue region situated at the C–C interface close to the allosteric effector‐binding site. PyK‐M1 is a highly active nonallosteric form found in tissues, such as heart, brain, and skeletal muscle that need to rapidly generate large amounts of ATP. However, in muscle and brain, M1 is allosterically inhibited by phenylalanine.77, 78, 79 On the other hand, PyK‐M2 is expressed in all proliferative cells, leukocytes, lung, spleen, kidney, and adipose tissue, and is the dominant isoform in fetuses. The activity of the M2 isozyme is modulated by FBP and amino acids. In the absence of FBP, PyK‐M2 exists as a mixture of monomer (80%), dimer (10%), and tetramer (10%). Binding of allosteric activators to PyK‐M2 induces active tetramer formation while inhibitors, oncoproteins and post‐transcriptional modifications promote the inactive monomer/dimer state. Transition of PyK‐M2 between these on and off modes provides the cells with nutrient sensing and growth signaling mechanisms.31, 80 Elevated levels of PyK‐M2 (compared to other isoforms) in nearly all cancer cells suggest its potential association with cancer. Recently, an observed correlation between high PyK‐M2 and low survival rate has been reported in patients with hepatocellular carcinoma.81 Additional mechanisms for regulation of PyK‐M2, such as the nitrosylation of specific Cys residues have also been proposed.80 However, PyK‐M2 is also present in normal cells,27, 82 and currently there is no evidence of a metabolic shift to this isozyme during tumorigenesis.83 Therefore, additional studies are needed to establish how regulation of PyK‐M2 might support the altered glucose metabolism in cancer cells and the metabolic re‐routing of glucose from cellular respiration to its increased utilization in the biosynthesis of macromolecules in proliferating cells.26, 31, 84, 85
5.2. C. parvum PyK (CpPyK)
The unusual and extremely reduced mitochondria of Cryptosporidium lack the classical respiratory chain19, 20 and therefore, cannot carry out most of the biochemical functions usually performed by this organelle. As an intestinal pathogen, C. parvum has adapted to the microaerophilic environment and relies exclusively on glycolysis for ATP production.86 The C. parvum genome contains a single pyk gene encoding the glycolytic form of the enzyme, which demonstrates distinct functional and structural properties as compared to other PyKs.87, 88 Crystal structures of CpPyK in the apo‐form (4DRS)88 and in the holo‐form in complex with ADP, Mg2+, and K+ have been determined (6P0Y) (Table S5). Notably, in these structures, a unique covalently cross‐linked dimer is observed wherein Cys26 of each monomer forms a disulfide bridge with Cys312 of the other at the A–A interface.88 Notably, both Cys26 and Cys312 are unique in the CpPyK sequence, and no other disulfide‐linked PyK dimer has been reported. These disulfide bonds may contribute to the increased structural stability of CpPyK dimers as suggested from the calculated ΔG diss values for the dimers in apo‐ and holo‐CpPyK crystal structures (Table S6). However, additional studies are needed to investigate the role of these intramolecular crosslinks. The ADP, Mg2+, and K+ binding sites in CpPyK overlap with positions occupied by substrate and metal ions in other PyKs (Figure S5). The holo‐CpPyK structure also exhibits domain closure as seen in other PyKs (Video S2). A Cl− ion was modeled in the same position between the A‐ and C‐domain where a sulfate ion was found in the apo‐enzyme structure. However, the functional significance of this anion‐binding is not known.
In a previous study the enzymatic activity of CpPyK was shown to be not affected by known allosteric effectors of other PyKs including FBP, FDP, F6P, G6P, AMP, ATP, and R5P.87 Residues 508–517 corresponding to the canonical “effector loop” were missing from the apo‐CpPyK crystal structure (4DRS) but interestingly the loop becomes ordered in the holo‐enzyme structure despite the absence of any effector (Figure S9). The “effector loop” in each monomer swings out and interacts with the Cα5 helix of the subunit at the C–C interface. The Pro514 from the loop inserts into a hydrophobic pocket bordered by the C‐terminal residues Val523 and Pro526, and Glu481, Ile484, and Leu488 on Cα5 helix of the monomer across the interface (Figure S10). Moreover, in two of the four monomers Lys510 (which was disordered in the apo‐enzyme structure) in this loop forms a hydrogen bond with Glu433 of the same monomer thus enhancing their stability (Figure S10). A unique characteristic of this loop in the holo‐CpPyK structure is the formation of an intramolecular disulfide bond between Cys513 and Cys517 (Figure S9). Both Cys513 and Cys517 are unique in the CpPyK sequence, and this disulfide bonded effector loop is a distinct feature of CpPyK. Notably, the CpPyK sequence has an unusually high content of 21 Cys residues whereas most PyKs contain only 10–15. CpPyK enzyme remains active under reducing conditions.88 Additional structural and functional studies are necessary to characterize this distinct PyK.
6. CONCLUSION
While the early studies on pyruvate kinase promoted a universal design to one of the highly conserved enzymes in nature, adaptation and evolution have still provided unique avenues for differential regulation of PyKs in different organisms and different tissues. These adaptations demonstrate how microbes can compete for nutrients within their host environment and how various tissues can modulate their metabolic needs within a more complex body with competing demands. Altogether, this makes PyK a highly flexible and adaptable enzyme while still adhering to a mostly universal architecture. Having a clear understanding of these differences in regulations can eventually allow us to exploit these differences for developing novel therapeutic targets for a variety of diseases. However, to achieve this goal, there is a great need to increase our current library of structures that span multiple organismal isoforms in multiple states.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest with the contents of this article.
Supporting information
Video S1: Quicktime movie showing the morph between apo‐ and holo‐complex of LmPyK (3HQN and 3HQO, respectively)
Video S2: Quicktime movie showing the morph between apo‐ and holo‐complex of CpPyK (4DRS and 6P0Y, respectively)
Table S1 Crystal structures of PyKs (with references).
Table S2: Pairwise protein sequence comparisons of PyKs.
Table S3: Hinge region sequences.
Table S4: Site interactions in representative PyKs.
Table S5: Data collection and refinement statistics for holo‐CpPyK (6P0Y).
Table S6: Canonical dimers in L. mexicana, T. cruzi, M. tuberculosis, S. cerevisiae, E. coli PyKs versus covalent dimer in C. parvum PyK.
Figure S1. Close‐up view of the A‐domain.
Figure S2. Close‐up view of the C‐domain.
Figure S3. Inter‐subunit interactions involving effector loop residues at the C–C interface.
Figure S4. Interactions between monomers at the A–A interface.
Figure S5. Comparison of ADP and metal ion binding sites in holo‐CpPyK and LmPyK substrate activator complex.
Figure S6. Effector loop conformation in TcPyK in the effector‐free and effector‐bound states.
Figure S7. Rock and lock model for allosteric transition in Trypanosomatid PyKs.
Figure S8. Allosteric transitions in Human PyK‐M2.
Figure S9. Effector loop in holo‐CpPyK (6P0Y).
Figure S10. Ordering of the effector loop in holo‐CpPyK structure stabilizes the C–C dimer interface.
ACKNOWLEDGMENTS
A part of this work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Eiger 16M detector on 24‐ID‐E beam line is funded by a NIH‐ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, the U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract No. DE‐AC02‐06CH11357. A portion of the study on Cryptosporidium parvum pyruvate kinase was supported by a research grant #106493 35 RGGN from American Foundation for AIDS Research (amfAR) to DC.
Schormann N, Hayden KL, Lee P, Banerjee S, Chattopadhyay D. An overview of structure, function, and regulation of pyruvate kinases. Protein Science. 2019;28:1771–1784. 10.1002/pro.3691
Norbert Schormann and Katherine L. Hayden contributed equally to this study.
Funding information U.S. Department of Energy (DOE), Grant/Award Number: DE‐AC02‐06CH11357; NIH‐ORIP HEI, Grant/Award Number: S10OD021527; National Institutes of Health, Grant/Award Number: P30 GM124165; National Institute of General Medical Sciences; American Foundation for AIDS Research (amfAR), Grant/Award Number: #106493 35
REFERENCES
- 1. Nelson DL, Cox MM, Lehninger AL. Lehninger principles of biochemistry. 7th ed. New York, NY Houndmills, Basingstoke: W.H. Freeman and Company; Macmillan Higher Education, 2017; p. xxxiv, 1172, AS34, G20, I45. [Google Scholar]
- 2. Bar‐Even A, Flamholz A, Noor E, Milo R. Rethinking glycolysis: On the biochemical logic of metabolic pathways. Nat Chem Biol. 2012;8:509–517. [DOI] [PubMed] [Google Scholar]
- 3. Lehninger AL, Wadkins CL. Oxidative phosphorylation. Annu Rev Biochem. 1962;1962:31. [DOI] [PubMed] [Google Scholar]
- 4. Lehninger AL. Principles of biochemistry. 5th ed. New York: W.H. Freeman, 2008. [Google Scholar]
- 5. Tymoczko JL, Berg JM, Stryer L. Biochemistry: A short course. The glycolytic pathway. 2nd ed. New York: W. H. Freeman, 2012. [Google Scholar]
- 6. Zoraghi R, Worrall L, See RH, et al. Methicillin‐resistant Staphylococcus aureus (MRSA) pyruvate kinase as a target for bis‐indole alkaloids with antibacterial activities. J Biol Chem. 2011;286:44716–44725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhong W, Cui L, Goh BC, et al. Allosteric pyruvate kinase‐based “logic gate” synergistically senses energy and sugar levels in Mycobacterium tuberculosis . Nat Commun. 2017;8:1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Morgan HP, Mcnae IW, Nowicki MW, et al. The trypanocidal drug suramin and other trypan blue mimetics are inhibitors of pyruvate kinases and bind to the adenosine site. J Biol Chem. 2011;286:31232–312340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Adem S, Comakli V, Uzun N. Pyruvate kinase activators as a therapy target: A patent review 2011–2017. Expert Opin Ther Pat. 2018;28:61–68. [DOI] [PubMed] [Google Scholar]
- 10. Morgan HP, Mcnae IW, Nowicki MW, et al. Allosteric mechanism of pyruvate kinase from Leishmania mexicana uses a rock and lock model. J Biol Chem. 2010;285:12892–12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Enriqueta Muñoz M, Ponce E. Pyruvate kinase: Current status of regulatory and functional properties. Comp Biochem Physiol B Biochem Mol Biol. 2003;135:197–218. [DOI] [PubMed] [Google Scholar]
- 12. Mesecar A, Nowak T. Metal‐ion‐mediated allosteric triggering of yeast pyruvate kinase. 1. A multidimensional kinetic linked‐function analysis. Biochemistry. 1997;35:6792–6802. [DOI] [PubMed] [Google Scholar]
- 13. Fothergill‐Gilmore LA, Michels PA. Evolution of glycolysis. Prog Biophys Mol Biol. 1993;59:105–235. [DOI] [PubMed] [Google Scholar]
- 14. Mattevi A, Bolognesi M, Valentini G. The allosteric regulation of pyruvate kinase. FEBS Lett. 1996;389:15–19. [DOI] [PubMed] [Google Scholar]
- 15. Valentini G, Chiarelli L, Fortin R, Speranza ML, Galizzi A, Mattevi A. The allosteric regulation of pyruvate kinase. J Biol Chem. 2000;275:18145–18152. [DOI] [PubMed] [Google Scholar]
- 16. Carbonell JE, Feliu JE, Marco R, Sols A. Pyruvate kinase: Classes of regulatory isoenzymes in mammalian tissues. Eur J Biochem. 1973;37:148–156. [DOI] [PubMed] [Google Scholar]
- 17. Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine‐binding protein. Nature. 2008;452:181–186. [DOI] [PubMed] [Google Scholar]
- 18. Stetak A, Veress R, Ovadi J, Csermely P, Keri G, Ullrich A. Nuclear translocation of the tumor marker pyruvate kinase M2 induces programmed cell death. Cancer Res. 2007;67:1602–1628. [DOI] [PubMed] [Google Scholar]
- 19. Putignani L, Tait A, Smith HV, et al. Characterization of a mitochondrion‐like organelle in Cryptosporidium parvum . Parasitology. 2004;129:1–18. [DOI] [PubMed] [Google Scholar]
- 20. Putignani L. The unusual architecture and predicted function of the mitochondrion organelle in Cryptosporidium parvum and hominis species: The strong paradigm of the structure‐function relationship. Parasitologia. 2005;47:217–225. [PubMed] [Google Scholar]
- 21. Henriquez FL, Richards TA, Roberts F, Mcleod R, Roberts CW. The unusual mitochondrial compartment of Cryptosporidium parvum . Trends Parasitol. 2005;21:68–74. [DOI] [PubMed] [Google Scholar]
- 22. Mather MW, Vaidya AB. Mitochondria in malaria and related parasites: Ancient, diverse and streamlined. J Bioenerg Biomembr. 2008;40:425–433. [DOI] [PubMed] [Google Scholar]
- 23. Vaidya AB, Mather MW. Mitochondrial evolution and functions in malaria parasites. Annu Rev Microbiol. 2009;63:249–267. [DOI] [PubMed] [Google Scholar]
- 24. Yu Y, Zhang H, Guo F, Sun M, Zhu G. A unique hexokinase in Cryptosporidium parvum, an apicomplexan pathogen lacking the Krebs cycle and oxidative phosphorylation. Protist. 2014;165:701–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. O'hara SP, Chen XM. The cell biology of cryptosporidium infection. Microbes Infect. 2011;13:721–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Christofk HR, Vander Heiden MG, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. [DOI] [PubMed] [Google Scholar]
- 27. Israelsen WJ, Vander Heiden MG. Pyruvate kinase: Function, regulation and role in cancer. Semin Cell Dev Biol. 2015;43:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li YH, Li XF, Liu JT, et al. PKM2, a potential target for regulating cancer. Gene. 2018;668:48–53. [DOI] [PubMed] [Google Scholar]
- 29. Miller G, Evans HJ. The influence of salts on pyruvate kinase from tissues of higher plants. Plant Physiol. 1957;32:346–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kayne FJ. Thallium (I) activation of pyruvate kinase. Arch Biochem Biophys. 1971;143:232–239. [DOI] [PubMed] [Google Scholar]
- 31. Morgan HP, O'Reilly FJ, Wear MA, et al. M2 pyruvate kinase provides a mechanism for nutrient sensing and regulation of cell proliferation. Proc Natl Acad Sci U S A. 2013;110:5881–5886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Waygood EB, Sanwal BD. The control of pyruvate kinases of Escherichia coli. I. Physicochemical and regulatory properties of the enzyme activated by fructose 1,6‐diphosphate. J Biol Chem. 1974;249:265–274. [PubMed] [Google Scholar]
- 33. Jurica MS, Mesecar A, Heath PJ, Shi W, Nowak T, Stoddard BL. The allosteric regulation of pyruvate kinase by fructose‐1,6‐bisphosphate. Structure. 1998;6:195–210. [DOI] [PubMed] [Google Scholar]
- 34. Donovan KA, Zhu S, Liuni P, et al. Conformational dynamics and allostery in pyruvate kinase. J Biol Chem. 2016;291:9244–9256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schramm A, Siebers B, Tjaden B, Brinkmann H, Hensel R. Pyruvate kinase of the hyperthermophilic crenarchaeote Thermoproteus tenax: Physiological role and phylogenetic aspects. J Bacteriol. 2000;182:2001–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Johnsen U, Hansen T, Schonheit P. Comparative analysis of pyruvate kinases from the hyperthermophilic archaea Archaeoglobus fulgidus, Aeropyrum pernix, and Pyrobaculum aerophilum and the hyperthermophilic bacterium Thermotoga maritima: Unusual regulatory properties in hyperthermophilic archaea. J Biol Chem. 2003;278:25417–25427. [DOI] [PubMed] [Google Scholar]
- 37. Oria‐Hernandez J, Riveros‐Rosas H, Ramirez‐Silva L. Dichotomic phylogenetic tree of the pyruvate kinase family: K+‐dependent and ‐independent enzymes. J Biol Chem. 2006;281:30717–30724. [DOI] [PubMed] [Google Scholar]
- 38. Laughlin LT, Reed GH. The monovalent cation requirement of rabbit muscle pyruvate kinase is eliminated by substitution of lysine for glutamate 117. Arch Biochem Biophys. 1997;348:262–267. [DOI] [PubMed] [Google Scholar]
- 39. Van Schaftingen E, Opperdoes FR, Hers HG. Stimulation of Trypanosoma brucei pyruvate kinase by fructose 2,6‐bisphosphate. Eur J Biochem. 1985;153:403–406. [DOI] [PubMed] [Google Scholar]
- 40. Waygood EB, Rayman MK, Sanwal BD. The control of pyruvate kinases of Escherichia coli. II. Effectors and regulatory properties of the enzyme activated by ribose 5‐phosphate. Can J Biochem. 1975;53:444–454. [DOI] [PubMed] [Google Scholar]
- 41. Knowles VL, Smith CS, Smith CR, Plaxton WC. Structural and regulatory properties of pyruvate kinase from the Cyanobacterium synechococcus PCC 6301. J Biol Chem. 2001;276:20966–20972. [DOI] [PubMed] [Google Scholar]
- 42. Guerrero‐Mendiola C, Garcia‐Trejo JJ, Encalada R, Saavedra E, Ramirez‐Silva L. The contribution of two isozymes to the pyruvate kinase activity of Vibrio cholerae: One K+‐dependent constitutively active and another K+‐independent with essential allosteric activation. PLoS One. 2017;12:e0178673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Saito T, Nishi M, Lim MI, et al. A novel GDP‐dependent pyruvate kinase isozyme from Toxoplasma gondii localizes to both the apicoplast and the mitochondrion. J Biol Chem. 2008;283:14041–14052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maeda T, Saito T, Harb OS, et al. Pyruvate kinase type‐II isozyme in Plasmodium falciparum localizes to the apicoplast. Parasitol Int. 2009;58:101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Garcia‐Olalla C, Garrido‐Pertierra A. Purification and kinetic properties of pyruvate kinase isoenzymes of Salmonella typhimurium . Biochem J. 1987;241:573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bakszt R, Wernimont A, Allali‐Hassani A, et al. The crystal structure of Toxoplasma gondii pyruvate kinase 1. PLoS One. 2010;5:e12736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. De La Vega‐Ruiz G, Dominguez‐Ramirez L, Riveros‐Rosas H, et al. New insights on the mechanism of the K(+‐) independent activity of crenarchaeota pyruvate kinases. PLoS One. 2015;10:e0119233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Johnsen U, Reinhardt A, Landan G, et al. New views on an old enzyme: Allosteric regulation and evolution of archaeal pyruvate kinases. FEBS J. 2019;286:2471–2489. [DOI] [PubMed] [Google Scholar]
- 49. Siebers B, Schonheit P. Unusual pathways and enzymes of central carbohydrate metabolism in Archaea. Curr Opin Microbiol. 2005;8:695–705. [DOI] [PubMed] [Google Scholar]
- 50. Brasen C, Esser D, Rauch B, Siebers B. Carbohydrate metabolism in Archaea: Current insights into unusual enzymes and pathways and their regulation. Microbiol Mol Biol Rev. 2014;78:89–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Morgan HP, Zhong W, Mcnae IW, Michels PA, Fothergill‐Gilmore LA, Walkinshaw MD. Structures of pyruvate kinases display evolutionarily divergent allosteric strategies. R Soc Open Sci. 2014;1:140120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chan M, Tan DS, Sim TS. Plasmodium falciparum pyruvate kinase as a novel target for antimalarial drug‐screening. Travel Med Infect Dis. 2007;5:125–131. [DOI] [PubMed] [Google Scholar]
- 53. Pawluk A, Scopes RK, Griffiths‐Smith K. Isolation and properties of the glycolytic enzymes from Zymomonas mobilis . Biochem J. 1986;238:275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nairn J, Duncan D, Gray LM, et al. Purification and characterization of pyruvate kinase from Schizosaccharomyces pombe: Evidence for an unusual quaternary structure. Protein Expr Purif. 1998;14:247–253. [DOI] [PubMed] [Google Scholar]
- 55. Plaxton WC, Smith CR, Knowles VL. Pyruvate kinase from Brassica napus (rapeseed) suspension cells. Arch Biochem Biophys. 2002;400:54–62. [DOI] [PubMed] [Google Scholar]
- 56. Lin M, Turpin DH, Plaxton WC. Pyruvate kinase isozymes from the green alga, Selenastrum minutum . Arch Biochem Biophys. 1989;269:219–227. [DOI] [PubMed] [Google Scholar]
- 57. Sakai H. Possible structure and function of the extra C‐terminal sequence of pyruvate kinase from Bacillus stearothermophilus . J Biochem. 2004;136:471–476. [DOI] [PubMed] [Google Scholar]
- 58. Sakai H, Ohta T. Molecular cloning and nucleotide sequence of the gene for pyruvate kinase of Bacillus stearothermophilus and the production of the enzyme in Escherichia coli. Evidence that the genes for phosphofructokinase and pyruvate kinase constitute an operon. Eur J Biochem. 1993;211:851–859. [DOI] [PubMed] [Google Scholar]
- 59. Girdlestone C, Hayward S. The DynDom3D webserver for the analysis of domain movements in multimeric proteins. J Comput Biol. 2016;23:21–26. [DOI] [PubMed] [Google Scholar]
- 60. Mattevi A, Valentini G, Rizzi M, Speranza ML, Bolognesi M, Coda A. Crystal structure of Escherichia coli pyruvate kinase type I: Molecular basis of the allosteric transition. Structure. 1995;3:729–741. [DOI] [PubMed] [Google Scholar]
- 61. Baek Yh NT. Kinetic evidence for a dual cation role for muscle pyruvate kinase. Arch Biochem Biophys. 1982;217:491–497. [DOI] [PubMed] [Google Scholar]
- 62. Ou Y, Tao W, Zhang Y, Wu G, Yu S. The conformational change of rabbit muscle pyruvate kinase induced by activating cations and its substrates. Int J Biol Macromol. 2010;47:228–232. [DOI] [PubMed] [Google Scholar]
- 63. DA Muirhead HC, Barford D, Lorimerl CG, Fothergill‐Gilmore LA, Schiltz E, Schmitt W. The structure of cat muscle pyruvate kinase. EMBO J. 1986;5:475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Muirhead H, Clayden DA, Cuffe SP, Davies C. Crystallographic studies on the structure and catalytic activity of pyruvate kinase from skeletal muscle. Biochem Soc Trans. 1987;15:996–999. [DOI] [PubMed] [Google Scholar]
- 65. Larsen TM, Laughlin TL, Holden HM, Rayment I, Reed GH. Structure of rabbit muscle pyruvate kinase complexed with Mn2+, K+, and pyruvate. Biochemistry. 1994;33:6301–6309. [DOI] [PubMed] [Google Scholar]
- 66. Zhong WMH, Mcnae IW, Michels PA, Fothergill‐Gilmore LA, Walkinshaw MD. ‘In crystallo’ substrate binding triggers major domain movements and reveals magnesium as a co‐activator of Trypanosoma brucei pyruvate kinase. Acta Crystallogr. 2013;D69:1768–1779. [DOI] [PubMed] [Google Scholar]
- 67. Larkin MABG, Brown NP, Chenna R, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. [DOI] [PubMed] [Google Scholar]
- 68. Larsen TM, Benning MM, Wesenberg GE, Rayment I, Reed GH. Ligand‐induced domain movement in pyruvate kinase: Structure of the enzyme from rabbit muscle with Mg2+, K+, and L‐phospholactate at 2.7 a resolution. Arch Biochem Biophys. 1997;345:199–206. [DOI] [PubMed] [Google Scholar]
- 69. Monod JWJ, Changeux JP. On the nature of allosteric transitions: A plausible model. J Mol Biol. 1965;12:88–118. [DOI] [PubMed] [Google Scholar]
- 70. Perutz MF. Mechanisms of cooperativity and allosteric regulation in proteins. Q Rev Biophys. 1989;22:139–237. [DOI] [PubMed] [Google Scholar]
- 71. Changeux J‐P. The origins of allostery: From personal memories to material for the future. J Mol Biol. 2013;425:1396–1406. [DOI] [PubMed] [Google Scholar]
- 72. Ashizawa K, Mcphie P, Lin KH, Cheng SY. An in vitro novel mechanism of regulating the activity of pyruvate kinase M2 by thyroid hormone and fructose 1, 6‐bisphosphate. Biochemistry. 1991;30:7105–7111. [DOI] [PubMed] [Google Scholar]
- 73. Ashizawa K, Willingham MC, Liang CM, Cheng SY. In vivo regulation of monomer‐tetramer conversion of pyruvate kinase subtype M2 by glucose is mediated via fructose 1,6‐bisphosphate. J Biol Chem. 1991;266:16842–16846. [PubMed] [Google Scholar]
- 74. Suzuki K, Ito S, Shimizu‐Ibuka A, Sakai H. Crystal structure of pyruvate kinase from Geobacillus stearothermophilus . J Biochem. 2008;144:305–312. [DOI] [PubMed] [Google Scholar]
- 75. Lovell SC, Mullick AH, Muirhead H. Cooperativity in Bacillus stearothermophilus pyruvate kinase. J Mol Biol. 1998;276:839–851. [DOI] [PubMed] [Google Scholar]
- 76. McFarlane JS, Ronnebaum TA, Meneely KM, Chilton A, Fenton AW, Lamb AL. Changes in the allosteric site of human liver pyruvate kinase upon activator binding include the breakage of an intersubunit cation‐pi bond. Acta Crystallogr. 2019;F75:461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Feksa LR, Cornelio AR, Dutra‐Filho CS, De Souza Wyse AT, Wajner M, Wannmacher CM. Characterization of the inhibition of pyruvate kinase caused by phenylalanine and phenylpyruvate in rat brain cortex. Brain Res. 2003;968:199–205. [DOI] [PubMed] [Google Scholar]
- 78. Carminatti H, Jimenez De Asua L, Leiderman B, Rozengurt E. Allosteric properties of skeletal muscle pyruvate kinase. J Biol Chem. 1971;246:7284–7288. [PubMed] [Google Scholar]
- 79. Williams R, Holyoak T, McDonald G, Gui C, Fenton AW. Differentiating a ligand's chemical requirements for allosteric interactions from those for protein binding. Phenylalanine inhibition of pyruvate kinase. Biochemistry. 2006;45:5421–5429. [DOI] [PubMed] [Google Scholar]
- 80. Mitchell AR, Yuan M, Morgan HP, et al. Redox regulation of pyruvate kinase M2 by cysteine oxidation and S‐nitrosation. Biochem J. 2018;475:3275–3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhao R, Li L, Yang J, Niu Q, Wang H, Qin X, Zhu N, Shi A. Overexpression of pyruvate kinase M2 in tumor tissues is associated with poor prognosis in patients with hepatocellular carcinoma. Pathol Oncol Res. 2019;1–8 10.1007/s12253-019-00630-3. [DOI] [PubMed] [Google Scholar]
- 82. Harris RA, Fenton AW. A critical review of the role of M2PYK in the Warburg effect. Biochim Biophys Acta Rev Cancer. 2019;1871:225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bluemlein K, Gruning NM, Feichtinger RG, Lehrach H, Kofler B, Ralser M. No evidence for a shift in pyruvate kinase PKM1 to PKM2 expression during tumorigenesis. Oncotarget. 2011;2:393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Van Niekerk G, Engelbrecht AM. Role of PKM2 in directing the metabolic fate of glucose in cancer: A potential therapeutic target. Cell Oncol. 2018;41:343–351. [DOI] [PubMed] [Google Scholar]
- 85. Eigenbrodt E, Reinacher M, Scheefers‐Borchel U, Scheefers H, Friis R. Double role for pyruvate kinase type M2 in the expansion of phosphometabolite pools found in tumor cells. Crit Rev Oncog. 1992;3:91–115. [PubMed] [Google Scholar]
- 86. Mithieux G, Gautier‐Stein A. Intestinal glucose metabolism revisited. Diabetes Res Clin Pract. 2014;105:295–301. [DOI] [PubMed] [Google Scholar]
- 87. Denton H, Brown SM, Roberts CW, et al. Comparison of the phosphofructokinase and pyruvate kinase activities of Cryptosporidium parvum, Eimeria tenella and Toxoplasma gondii . Mol Biochem Parasitol. 1996;76:23–29. [DOI] [PubMed] [Google Scholar]
- 88. Cook WJ, Senkovich O, Aleem K, Chattopadhyay D. Crystal structure of Cryptosporidium parvum pyruvate kinase. PLoS One. 2012;7:e46875. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1: Quicktime movie showing the morph between apo‐ and holo‐complex of LmPyK (3HQN and 3HQO, respectively)
Video S2: Quicktime movie showing the morph between apo‐ and holo‐complex of CpPyK (4DRS and 6P0Y, respectively)
Table S1 Crystal structures of PyKs (with references).
Table S2: Pairwise protein sequence comparisons of PyKs.
Table S3: Hinge region sequences.
Table S4: Site interactions in representative PyKs.
Table S5: Data collection and refinement statistics for holo‐CpPyK (6P0Y).
Table S6: Canonical dimers in L. mexicana, T. cruzi, M. tuberculosis, S. cerevisiae, E. coli PyKs versus covalent dimer in C. parvum PyK.
Figure S1. Close‐up view of the A‐domain.
Figure S2. Close‐up view of the C‐domain.
Figure S3. Inter‐subunit interactions involving effector loop residues at the C–C interface.
Figure S4. Interactions between monomers at the A–A interface.
Figure S5. Comparison of ADP and metal ion binding sites in holo‐CpPyK and LmPyK substrate activator complex.
Figure S6. Effector loop conformation in TcPyK in the effector‐free and effector‐bound states.
Figure S7. Rock and lock model for allosteric transition in Trypanosomatid PyKs.
Figure S8. Allosteric transitions in Human PyK‐M2.
Figure S9. Effector loop in holo‐CpPyK (6P0Y).
Figure S10. Ordering of the effector loop in holo‐CpPyK structure stabilizes the C–C dimer interface.