

Abstract
Stainless steels can become contaminated with radionuclides at nuclear sites. Their disposal as radioactive waste would be costly. If the nature of steel contamination could be understood, effective decontamination strategies could be designed and implemented during nuclear site decommissioning in an effort to release the steels from regulatory control. Here, batch uptake experiments have been used to understand Sr and Cs (fission product radionuclides) uptake onto AISI Type 304 stainless steel under conditions representative of spent nuclear fuel storage (alkaline ponds) and PUREX nuclear fuel reprocessing (HNO3). Solution (ICP-MS) and surface measurements (GD-OES depth profiling, TOF-SIMS, and XPS) and kinetic modeling of Sr and Cs removal from solution were used to characterize their uptake onto the steel and define the chemical composition and structure of the passive layer formed on the steel surfaces. Under passivating conditions (when the steel was exposed to solutions representative of alkaline ponds and 3 and 6 M HNO3), Sr and Cs were maintained at the steel surface by sorption/selective incorporation into the Cr-rich passive film. In 12 M HNO3, corrosion and severe intergranular attack led to Sr diffusion into the passive layer and steel bulk. In HNO3, Sr and Cs accumulation was also commensurate with corrosion product (Fe and Cr) readsorption, and in the 12 M HNO3 system, XPS documented the presence of Sr and Cs chromates.
1. Introduction
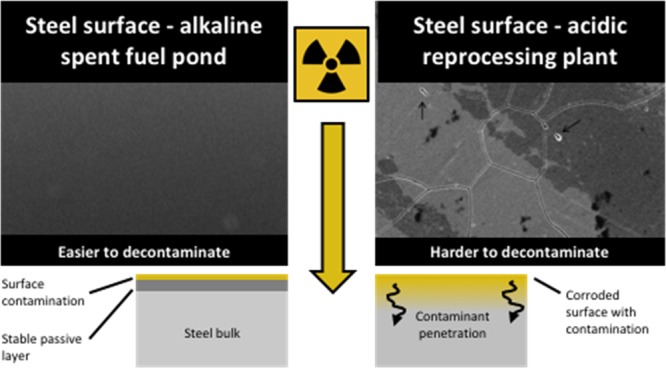
The development of a low carbon-based energy economy is a global priority, and nuclear power is an efficient, low-carbon energy source. However, the unavoidable generation of large amounts of solid radioactive waste and its costly management provide significant concerns.1,2 A major source of waste arises from the accumulation of radioactive species from aqueous solution onto the surfaces of nuclear plant materials.3 This “plate-out” process, if not controlled or mitigated, creates challenges for final material disposal as many of the materials are voluminous, can be hard to decontaminate, and are difficult to compress. The advancement of reliable and cost-effective decommissioning and decontamination strategies is consequently of great importance to minimize waste volumes, which, in turn, provides cost savings.
The widespread use of austenitic stainless steels, such as AISI Type 304L, as structural and storage materials4 at civil nuclear sites mean that these alloys become an important source of radioactive waste over a site’s lifetime. Reliable current estimates of the global amounts of nuclear industry radioactive stainless steels are not available, but they are a significant waste form (millions of tonnes).5 The selection of stainless steel in the nuclear industry is based on a combination of high radiation stability and excellent corrosion resistance, the latter afforded by the spontaneous formation of a nanometer-scale passivating Cr-oxide layer at the steel surface.6 It has been assumed that deposited radioactivity becomes bound to/impregnated within this passive layer during the accumulation process,7,8 although these phenomena have not been fully demonstrated. However, to make informed decisions regarding the design and optimization of effective decontamination treatments for these materials, an understanding of the principal chemical interactions driving radionuclide uptake onto or into the steel is critical.
The contamination phenomena witnessed during transport and storage of radioactive materials and liquors are not completely understood. It is known that, upon contact with aqueous solution, the passivating surface oxide layer of stainless steel, like many natural sorbents, is charged and interacts electrostatically with dissolved ions.9 On this basis, surface complexation models for radionuclide groundwater transport (e.g., Carrol et al.10) have been applied to describe the sorption properties of stainless steels.11,12 However, recent investigations on the contamination of pressurized water reactor (PWR) materials have shown sorption phenomena to be more complex, where complete characterization is not possible by utilization of charge principles alone.13 Here, the elemental composition and morphology of the passive layer were considered to provide chemical control of the accumulation process, which may facilitate sorption from boric acid coolant despite repulsive electrostatic interactions with the surface. The importance of the passive layer condition for the character of contamination strongly suggests that sorbent properties of stainless steels are inherited from this surface oxide film. It is clear that an assessment of the passivation state of the steel material is essential in understanding radionuclide sorption. However, the elemental composition, structure, and thickness of the surface oxide layer vary with the composition and potential of the passivating medium, complicating issues.14 Accordingly, the derived sorbent properties are expected to reflect both steel composition and environment chemistry, and therefore, “plate-out” phenomena must be investigated on an individual basis under the specific conditions of contamination.
Spent nuclear fuel storage and subsequent reprocessing to recover U and Pu are major activities at the back-end of a closed nuclear fuel cycle. Spent fuels are also stored for long periods prior to disposal in open fuel cycles. These activities have been implicated in stainless steel contaminant plate-out processes.15 Initially, spent fuels are stored in fuel ponds to facilitate the decay of short-lived fission products and to permit the dissipation of heat. The water in spent fuel ponds becomes contaminated with radionuclides, and in turn, the radionuclides can contaminate structural and storage materials (including steels) used in the ponds. After sufficient cooling, U and Pu recovery is then achieved in a closed cycle using solvent extraction in HNO3 via the PUREX process (or derivates of PUREX).16 The high acidity and strong oxidizing power of the reprocessing liquors likely modify the passivation state of steels used in the plant so that contamination processes here cannot be reasonably expected to be comparable with those described for PWR cooling circuit steel.13,17 However, this has not been investigated, and many uncertainties surrounding the mechanisms of fission product contamination for reprocessing plant materials also remain. To address these issues (contamination of spent fuel pond and reprocessing plant steels), it is necessary to characterize the surface chemistry of the steel materials in contact with the pond waters/HNO3 and identify the chemical processes that promote radionuclide uptake and accumulation on steel materials.
In this work, we seek to better understand 90Sr (t1/2 = 28.8 years) and 134+137Cs (t1/2 = ∼2 and 30.2 years, respectively) uptake onto and distribution in AISI Type 304 stainless steel surfaces across a range of solution chemistries representative of those found in the U.K. spent fuel storage and reprocessing streams. Strontium and cesium are high-yield fission products that are present in appreciable amounts in fuel pond waters and reprocessing stream liquors.18,19 Further, they comprise a significant fraction of the total activity deposited onto steels used for spent fuel storage and in reprocessing plants.3 Model contamination systems were used to simulate documented nuclear site conditions, and contamination was subsequently characterized using solution uptake data and analysis of the contaminated steel surface and near surface. A series of HNO3 matrices were used to model PUREX reprocessing streams, and 1 mM NaOH was used to simulate the conditions found in U.K. spent fuel ponds, where steel pond furniture is used to store spent fuel under alkaline conditions.19 The resulting data provide critical information to aid in decision-making regarding the cleaning and management of contaminated nuclear industry steels, where the ultimate goal is to remove these materials from radioactive waste sentencing and costly management.
2. Materials and Methods
2.1. Steel and Contamination Matrices
AISI Type 304 stainless steel was used in this study, and its composition is detailed in Table S1. This type of stainless steel is widely used in the nuclear industry (e.g., in reactors, reprocessing plants, storage facilities, etc.)4 and is considered to be the baseline stainless steel for research purposes. The procedure for achieving contamination of the stainless steel used in this study is fully described in our previous work.20 Briefly, solutions of 3, 6, and 12 M HNO3 and 1 mM NaOH (Aristar grade), which included 500 mg L–1 stable Sr and Cs, were used as the contaminant-bearing matrices to simulate reprocessing waste streams and alkaline fuel storage ponds at the Sellafield site. The stainless steel samples were reacted in these solutions for 30 days with the temperature of the solutions maintained at 60 °C.
2.2. Electron Microscopy
Steel surface morphology after immersion in the Sr- and Cs-bearing solutions was characterized by scanning electron microscopy (SEM) using a FEI Quanta 200 SEM at an accelerating voltage of 20 kV.
2.3. Glow Discharge Optical Emission Spectroscopy
Elemental depth analysis of steel coupons was performed with glow discharge optical emission spectroscopy (GD-OES) using a GD-Profiler 2 (Horiba Jobin Yvon). The area being sputtered during GD-OES analysis was 4 mm in diameter (see Section S2); thus, the elemental depth profiles created by this technique represent an averaged signal across an area of ∼0.13 cm2. The applied power used during analysis was 35 W, and the Ar gas pressure was 635 Pa. The elemental emission lines used for detection were 371.999 nm (Fe-I), 425.439 nm (Cr-I), 341.482 nm (Ni-I), 130.223 nm (O-II), 460.739 (Sr-II), 455.529 nm (Cs-I), and 396.157 nm (Al-I). The photon detector was calibrated using a 1 mm Al sheet prior to analysis and operated at a polychromator focal length of 500 mm and with 30 optical windows. To maintain the vacuum seal at the O-ring–sample interface, 304 stainless steel coupons of dimensions of 20 mm × 20 mm × 13 mm (l × w × t) were used for analysis.
2.4. Inductively Coupled Plasma Mass Spectrometry
Strontium and cesium uptake onto the steel surfaces was tracked by removing 1 mL aliquots of solution from exposure experiments at the following time intervals (0.17, 1, 6, 24, 168, 336, 504, and 672 h) followed by centrifugation (13,150g, 45 min). Thereafter, 500 μL of the resulting supernatant was withdrawn and diluted into 2% HNO3. Strontium, cesium, and alloying elements (Fe, Cr, and Ni) were analyzed using an Agilent 7500cx ICP-MS. Each uptake experiment was performed in triplicate, and the average values are provided. The amount sorbed to the steel at time t, qt (g m–2), was calculated as follows
where c0 and ct are the solution concentrations at time = 0 and t, respectively (g L–1), V is the volume of solution (L), and A is the surface area of the exposed steel surface (m2).
2.5. X-ray Photoelectron Spectroscopy
The composition of the surface oxide layer and the chemical environments of the contaminants were assessed using X-ray photoelectron spectroscopy (XPS). High-resolution scans were recorded using a Kratos AXIS Ultra DLD XPS with a monochromatic Al X-ray source (1486.69 eV). The relative fractions of Cr and Fe in the passive film were calculated as the relative mass ratio (Croxide/(Croxide + Feoxide)) using the Cr 2p peaks at ∼575 and 584 eV and the Fe 2p peaks located at ∼710 and 723 eV. Spectra were recorded in duplicate, and the resulting surface Fe/Cr ratios were averaged. Peak positions were charge-corrected by normalization with the C 1s ubiquitous hydrocarbon peak at 284.6 eV.
2.6. Time-of-Flight Secondary Ion Mass Spectrometry
The elemental distribution of the contaminants and the alloying elements on the steel surface were measured with an IONTOF TOF-SIMS5 of the reflectron type. Measurements were carried out using a pulsed Bi/Mn liquid metal ion gun (30 keV) operated at a 0.8 μA emission current. A 30 ns pulse width of the primary ion bunch at a 100 μs cycle time enabled a mass resolution of secondary ions in excess of 8000 amu. High-resolution imaging was performed on scanning areas of 50 μm × 50 μm. The most abundant isotopes were selected for characterization.
2.7. Laser Confocal Microscopy
GD-OES crater depths were quantified with a Keyence VK-X200K 3D laser confocal microscope at superfine resolution (2048 × 1536 pixels). Operating at a 200× magnification and a 0.5 μm step size, the area analyzed by the laser confocal microscope of the GD-OES crater was ∼6.0 mm × 6.5 mm.
3. Results and Discussion
3.1. Physical and Chemical Alteration of Steel Surfaces
To better understand the steel contamination mechanism, it is necessary to first study the physical state and elemental composition of the steel passive layer and its resulting corrosion state after exposure to the solution matrices.
SEM analysis (see Section S1 and Figure S1) revealed no discernible features of the surface morphology after immersion in the alkaline and less concentrated acid contaminating matrices (3 and 6 M HNO3) at 60 °C for 30 days. The uniform and low corrosion rate under moderately oxidizing solution conditions is associated with the formation of a protective Cr2O3 surface film that leaves the underlying steel material in a passive (unreactive/corrosion-resistant) state. In contrast, in the 12 M HNO3 matrix, the solution is sufficiently oxidizing to transform insoluble Cr(III) (present as Cr2O3) into soluble Cr(VI) species. The subsequent depletion of Cr in the passive layer deteriorates the film’s protective capabilities and may result in material failure.24 The breakdown of the passive layer under strongly oxidizing conditions is known as transpassive behavior and is characterized by the initiation of corrosion phenomena at the sites most susceptible to Cr depletion, for example, defects, interfaces, or grain boundaries.22,23 This was observed in the 12 M HNO3 matrix by the preferential corrosion attack at the grain boundaries (Figure S1C). Type 304 stainless steel also typically contains a small volume fraction of remaining δ-ferrite,21 and corrosion at these ferrite interface regions was also apparent. Depth analysis of the contaminated steel was then undertaken using GD-OES (Figure 1). The data are expressed as a function of “sputtering time” and estimated “sputtered depth” (see Section S2 for further details). Here, the O signal was initially intense, and the overlapping of the O and Cr maximum signals after a short sputtering time coincides with the steel passive layer. Further sputtering results in the complete loss of the O signal, thus corresponding to the bulk material. Of note, the O signal rapidly diminished after treatment in 12 M HNO3 (Figure 1C and Figure S3), which suggests that a stable surface oxide layer cannot be maintained under such strongly oxidizing conditions. The reduced signal of the protective passive film is expected to greatly increase the steel’s susceptibility to various forms of corrosive attack, a result confirmed by SEM analysis (Figure S1C).
Figure 1.

GD-OES elemental depth profiles of type 304 stainless steel after contamination in (A) 3 M (B) 6 M, and (C) 12 M HNO3 and (D) 1 mM NaOH aqueous solution at 60 °C for 30 days, with Sr and Cs as added contaminants. The O and Sr signals are scaled by factors of 10 and 100, respectively, for clarity. (A) and (D) have been redrawn from data presented by Lang et al.20 The x axis is expressed as sputtering time and estimated sputtered depth. For details on the conversion of time to depth, please see Section S2. Note that the x axis of (C) is different to the other panels. Data from (C) (12 M HNO3 sample) is also presented in Figure S3, with a redrawn x axis to allow better comparison of the oxygen data across all samples.
After reaction in the alkaline solution, the passive layer became substantially thicker than those grown under acidic conditions. This is shown by the increased persistence of the O signal in Figure 1D, which is accompanied by a significant Fe signal at this early sputtering stage. The growth of thicker films in alkaline media is related to the bilayer model that is generally accepted for the steel passive layer structure. The passive layer has been shown to be composed of an outer layer rich in various Fe oxides and an inner layer composed of Cr2O3.25,26 While this outermost oxide layer is relatively stable in neutral and alkaline solution, dissolution rapidly occurs in highly acidic solutions owing to Fe’s high solubility at acidic pH.27 Accordingly, in 12 M HNO3, the passive film will contract as the Fe oxide component is selectively leached into the solution. This behavior was confirmed by solution analysis (Figure 2A) that recorded a significant increase of Fe within 6 h of commencing Sr and Cs uptake studies. Thereafter, the solution Fe concentration decreased, which may be indicative of readsorption and/or reprecipitation of Fe corrosion products onto the steel surface. The dissolution of Cr into 12 M HNO3 may also be considered a characteristic of the transpassive behavior as it is known that effectively no Cr is released under passivating conditions.28 Chromium readsorption and/or reprecipitation was also observed (Figure 2A) during experimentation in 12 M HNO3. In the 3 and 6 M HNO3 experiments, Cr was not detectable, likely due to the formation of a stable Cr-rich passive layer at the steel surface in these solutions (Figure 1).
Figure 2.

(A) Time dependence of Cr and Fe solution concentrations in 3, 6, and 12 M HNO3 during the Sr and Cs uptake study onto type 304 stainless steel at 60 °C and (B) variation in the surface layer oxide content of type 304 stainless steel as a function of passivation treatment as determined by XPS (see the Supporting Information for details). Error bars are standard deviations expressed at 2σ.
Another important effect of selective Fe dissolution in acidic solution is the enrichment of Cr in the passive layer relative to the air (polished)- and alkaline-passivated specimens, as shown by XPS measurements (Figure 2B). On the other hand, the low solubility of Fe in alkaline solution yields a surface composition that is similar to the air (polished)-passivated material. According to the literature, the slight increase in surface Fe concentration at alkaline pH is due to the higher Cr solubility under basic conditions,26,29 although in this work, the presence of Cr in solution could not be detected (note that Cr could not be reliably detected below a concentration of several hundred μg L–1 in the alkaline samples due to the dilution required for analysis). An examination of the binding energy positions of the Fe and Cr 2p photoelectron peaks to identify the chemical forms of the passive layer components is provided in the Supporting Information.
3.2. Sr and Cs Kinetic Studies
In all solution matrices studied, Sr and Cs accumulation on the steel surface was reasonably rapid (Figure 3) with significant uptake of both elements occurring within 24 h. Uptake then continued but at a lower rate before an apparent steady state was reached at 30 days. Other radionuclide sorption studies have reported fast deposition rates onto austenitic stainless steel materials from acidic and alkaline pH solution matrices,13,15 suggesting that plate-out is an inherently fast process.
Figure 3.

Effect of contact time on (A) Sr and (B) Cs uptake (qt) onto type 304 stainless steel during reaction in 3, 6, and 12 M HNO3 and 1 mM NaOH solution at 60 °C. Error bars are standard deviations expressed at 2σ.
In an effort to describe the Cs and Sr behavior over time, the solution data were analyzed with the Ho pseudo-second-order,30 Lagergren pseudo-first-order,31 and Elovich32 kinetic models. As these models have previously been used to describe the removal of low-valence metal ions from aqueous solution by solid substrates,33−35 it was postulated that the adsorption of Sr and Cs onto stainless steel may also be governed by similar kinetic descriptions. The pseudo-second-order kinetic model is based on the adsorption capacity of the sorbent and is expressed as follows30,36
and
where qe is the equilibrium sorption capacity (g m–2), k2 is the pseudo-second-order rate constant (m2 g–1 h–1), and h is the initial sorption rate (g m–2 h–1) as t → 0.
When the left-hand term is plotted against t, a linear plot of gradient 1/qe and a y-intercept of 1/k2qe2 are obtained. The adsorption pseudo-second-order kinetic plots from the Ho model are shown in Figure S5 for all for systems studied, where the values of qe and k2 can be calculated from the slope and intercept of the fits, respectively.
As per Figure S5, for all solution compositions investigated, consistently high correlations (R2 ≥ 0.99) were obtained for this pseudo-second-order kinetic model. In comparison, poorer correlations were obtained when the experimental data were analyzed with the other models (see Section S4 and Table S2). On the basis of these findings, it may be concluded that Sr and Cs contamination largely follows pseudo-second-order kinetics over the entire reaction period. In addition, the theoretical qe values (Table S3), obtained from the gradients of the pseudo-second-order kinetic plots (Figure S5), were found in all instances to be in excellent agreement with the experimental values, further supporting the model selection. It should be noted that experimental equilibrium sorption capacities were determined by taking an average of the qt values measured at the last three time intervals (336, 504, and 672 h). In all instances, low standard deviations (≤10 %) were calculated, supporting our assumption that equilibrium uptake had been reached within 336 h.
A comparison of the theoretical equilibrium sorption values showed a marked influence of contaminating solution composition on the extent of Sr contamination (Table S3), where uptake increased with HNO3 molarity and was greatest under alkaline conditions. In contrast, the amount of Cs reacting with the steel surface was found to be largely independent of solution composition and was only marginally higher at alkaline pH (Table S3). The effect of solution pH on metal ion uptake onto austenitic stainless steel surfaces has previously been rationalized by electrostatic surface complexation models based on the acido-basic properties of the surface hydroxyl groups of the passive layer.11,12 The pH point of zero charge for austenitic stainless steels is known to be acidic, reportedly within the pH range of 3–4.37 Accordingly, in the alkaline matrix, the steel surface will acquire an overall negative charge that will interact favorably with accumulating metal ions. As might be expected from the difference in charge density, the adsorption behavior of Cs+ is less sensitive to electrical forces, and hence, the increased affinity for sorption at alkaline pH is more apparent for Sr2+. In the case of the HNO3 solutions, the steel surface now exerts a positive potential that acts to inhibit cationic sorption. The steel’s higher affinity for Sr uptake under increasingly acidic conditions (3 to 12 M HNO3) cannot be accounted for by charge principles and suggests that additional processes are controlling sorption behavior at acidic pH. This result is consistent with one of the principal assumptions of the pseudo-second-order kinetic model, which states that the sorption mechanism includes a chemical sorption process.30,36 The inconsistent sorption capacities may suggest that Sr uptake is more sensitive to surface speciation changes derived from variations in the exact steel passivation condition or to other chemical effects, for example, coprecipitation of Sr with the products of steel corrosion. Finally, there was a marked increase in the rate of Cs uptake onto the steel surface in 12 M HNO3 (Table S3). This too may reflect changes in the steel passivation state or other chemical effects. Such reaction pathways for Sr and Cs were further tested using surface-sensitive techniques (see below).
3.3. Contaminant Depth Distribution
In addition to alloy depth profiling analysis, the depth distribution of Sr and Cs within the steel matrix was also evaluated with GD-OES. Under passivating conditions (alkaline and 3 and 6 M HNO3), Sr was contained at the steel near the surface (upper 10–20 nm; Figure 1). Alignment of the Sr maximum signal with the corresponding O and Cr signals (Figure 1) demonstrated enrichment of this contaminant within the Cr2O3 passive layer, irrespective of the composition of the passive film (in the alkaline system, the passive layer had an outer layer rich in Fe that then transitioned to a Cr-rich layer). These results collectively suggest that, under passivating conditions, the depth of Sr contamination is not significantly impacted by the composition of the passive layer or the contaminating solution.
The Cr2O3 component, which is traditionally considered the source of corrosion resistance, was observed to also afford protection of the underlying material from contamination phenomena. On this basis, it is not surprising that Sr bulk material diffusion was prominent after contamination in 12 M HNO3 (Figure 1C), where breakdown of the Cr2O3 passive layer was a characteristic feature for this specimen. It was determined by measurement of the GD-OES crater depth (Figure S2) that the Sr penetrating depth in 12 M HNO3 was ∼150 nm (Figure 1C).
For Cs depth characterization, no signal could be detected for any of the acidic systems investigated. This is likely due to the small amounts of Cs accumulating on the steel surface and the insensitivity of optical techniques for Cs detection.38 In contrast, when the steel was reacted under alkaline conditions in the presence of Cs, Cs uptake was higher, and this resulted in Cs concentrations that permitted GD-OES analysis. As shown in Figure 4, this showed that Cs enrichment was only observed to ∼15 nm, within the Cr-enriched oxide layer of this sample, in a similar fashion to Sr (Figure 1). Collectively, these depth profiling results demonstrate that a stable Cr2O3 film is an adequate barrier against the mobilization of contaminants into the bulk material.
Figure 4.
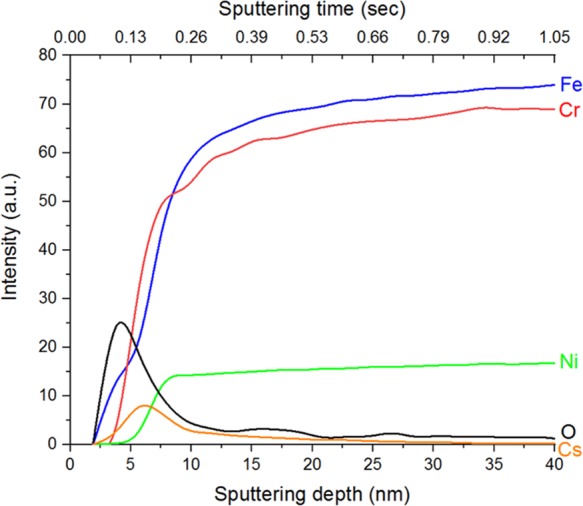
GD-OES elemental depth profiles of type 304 stainless steel contaminated in 1 mM NaOH aqueous solution at 60 °C for 30 days, with Sr and Cs added as contaminants. The O and Cs signals are scaled by factors of 10 and 100, respectively, for clarity. The x axis is expressed as sputtering time and estimated sputtered depth. For details on the conversion of time to depth, please see Section S2.
3.4. Spatial Distribution
Time-of-flight secondary ion mass spectrometry (TOF-SIMS) data from type 304 stainless steel after contamination in 12 M HNO3 (Figure 5) clearly show the presence of numerous Fe and Cr hotspots of ∼2 μm in diameter on the steel surface. These features were not found at the steel grain boundaries and may be formed by readsorption/reprecipitation of once dissolved Fe and Cr (Figure 2A) onto the steel surface. The remarkably similar morphology and locations of Sr enrichment with Fe and Cr hotspots are noteworthy and suggest that Sr contamination may occur due to coadsorption and/or coprecipitation with Fe/Cr species. Similar findings have been reported in a previous radionuclide/stainless steel sorption study.39 The possibility that coadsorption/coprecipitation reactions control Sr uptake here is also consistent with the observed time dependence of Fe and Sr sorption as both solution profiles exhibit a significant decrease after 6 h to reach an equilibrium state within 30 days. Further, after the first day of reaction, Cr was also readsorbed onto the steel commensurate with Sr (Figures 2A and 3A). On this basis, the increasing Sr uptake under more acidic conditions may, in part, be rationalized by the higher Fe and Cr solution concentrations that are available to participate in a coadsorption/coprecipitation process. This inference is further supported by Sr XPS measurements (see Section 3.5 for further details), although the XPS data implicate Cr in Sr uptake and not Fe. Finally, due to the spatial limitations of the GD-OES analysis (which sputters ∼0.13 cm2 of the sample), the impact that the micrometer-sized hotspots could have on Sr depth penetration into the steel (Figure 1C) cannot be assessed. Their role thus warrants further investigation.
Figure 5.

(A) Secondary electron image of the sputtered area and (B–H) the corresponding TOF-SIMS maps. The type 304 stainless steel specimen was contaminated in 12 M HNO3 for 30 days at 60 °C. Arrows show the positions of the grain boundaries.
TOF-SIMS data from the 12 M HNO3 Cs system showed that Cs accumulation on the steel surface was more homogeneous, with no obvious correlation evident with Fe and Cr (Figure 5) or accumulation at the grain boundaries. As such, sorption may be the dominant control facilitating Cs reaction with the steel surface at acidic pH; however, XPS analysis (see Section 3.5 for details) of this sample indicated that Cs reaction with Cr could also be a factor governing Cs uptake.
Under the passivating conditions of the alkaline pH system, as expected, the decreased Fe and Cr solubility resulted in a marked reduction in the extent of alloying element readsorption to the steel surface (Figure 6). Nevertheless, correlation of Fe, Cr, and Sr hotspots was still apparent (although not as pronounced as for the 12 M HNO3 sample (Figure 5)). Cesium was also correlated with hotpots of Fe and, to a lesser extent, Cr. The exact role of readsorption of Fe and Cr species in facilitating Sr and Cs adsorption/coprecipitation in the alkaline system is not clear, but XPS analysis again implicated Cr as a factor governing Sr uptake to the steel. Unfortunately, Cs XPS analysis was not possible for this sample (see Section 3.5 for further details).
Figure 6.

(A) Secondary electron image of the sputtered area and (B–H) the corresponding TOF-SIMS maps. The type 304 stainless steel specimen was contaminated in 1 mM NaOH for 30 days at 60 °C. Arrows show the positions of the grain boundaries.
For the 12 M HNO3 and alkaline samples, it is also interesting to note that there was no preferential accumulation of either Sr or Cs along the grain boundaries (Figures 5 and 6). These findings (from stainless steel reacted at 60 °C) contradict the proposed Cs contamination mechanisms for stainless steel at the elevated temperatures found in nuclear reactors (∼650 °C).40 Here, Cs preferentially accumulates at the steel grain boundaries and then penetrates into the steel subsurface. The contrast between the two studies suggests that temperature plays an important role in steel contamination mechanisms at nuclear sites and, for our study, further implicates the hotspots as possible vectors allowing contaminant penetration into the steel subsurface. Depth analysis collected at a micrometer spatial scale is required to confirm this.
Finally, the solution, GD-OES, and TOF-SIMS data indicate that minimization of corrosion product solution concentrations by frequent renewal of the reprocessing streams and storage pond solutions may prove to be an effective method to limit the extent of Cs and Sr contamination of stainless steel.
3.5. Speciation Analysis
Strontium accumulation on the steel surface was characterized by the corresponding Sr 3d3/2 and Sr3d5/2 XPS photoelectron peaks located at ∼135 and 133 eV, respectively. The surface concentrations obtained from contamination in 3 and 6 M HNO3 were too low to detect the Sr 3d lines. As a result, data are only presented for the 12 M HNO3 and 1 mM NaOH solution matrices (Figure 7), where Sr uptake was higher. For the 12 M HNO3 system, deconvolution of the Sr 3d core electron bands into the 3d3/2 and 3d5/2 components at 135.4 and 133.6 eV reveals the most probable bonding environment as SrCrO4.41,42 This assignment is based on the fact that other plausible chemical forms, such as Sr(NO3)2 and SrO, have significantly different 3d5/2 binding energy positions, reported at 134.0 and 132.6 eV, respectively.43 In combination with the GD-OES depth profiling and TOF-SIMS observations, this local bonding environment may be interpreted as the coadsorption and migration of Sr2+ into the Cr oxide passive layer. A comparison of the XPS spectrum obtained from the alkaline pH contamination reveals an additional component at 135.0 and 133.2 eV, corresponding to strontium carbonate.44 This result suggests that the coadsorption/coprecipitation of Sr with components of the contaminating solution occurs across a large pH range and may be an inherent feature to the Sr contamination process.
Figure 7.

High-resolution XPS spectra of the Sr 3d photoelectron lines for type 304 stainless steel contaminated in (A) 12 M HNO3 and (B) 1 mM NaOH at 60 °C for 30 days and (C) for the Cs 3d photoelectron lines for after contamination in 12 M HNO3 at 60 °C for 30 days.
With regard to Cs, after the reaction at alkaline pH, the Cs 3d photoelectron lines could not be detected from the steel due to spectral interference with the Fe 2p photoelectron peaks associated with the Fe-rich passive layer (see the Supporting Information for further information). Similarly, as previously described for GD-OES characterization, XPS analysis of Cs contamination under acidic conditions could not reliably be performed; as a result, data are only presented for the 12 M HNO3 matrix. Figure 7 shows the high-resolution XPS spectrum associated with the Cs 3d5/2 and 3d3/2 photoelectron peaks, which are observed at 723.9 and 737.8 eV, respectively. An examination of the literature suggests that the most probable chemical state is Cs2Cr2O7,45 which, analogous to the Sr assignment, may be interpreted as Cs complexation to the steel passive layer. It is not clear if coprecipitated species remain associated during Sr and Cs migration into the passive film since XPS cannot provide detailed information on the local arrangement of atoms around the central atom. Rather, the XPS technique can only distinguish different chemical environments by significant variation in the electron densities of the absorbing atomic species. On that basis, it is expected that the highly electron-withdrawing chromate and carbonate complexes observed in this study will dominate the Sr and Cs photoelectron line positions.
4. Conclusions
The results show that the extent and character of type 304 austenitic stainless steel contamination with the high-yield fission products Sr and Cs are intimately related to the corrosion state of the steel surface. Under alkaline conditions and in 3 and 6 M HNO3, the stainless steel is passivated, and contamination with Cs and Sr was identified as a surface-based phenomenon in which selective deposition in the Cr2O3 passive layer occurs. However, in 12 M HNO3 under transpassive conditions, the dissolving passive layer was found to be an ineffective barrier against Sr diffusion into the bulk material. Further, the partial dissolution of the steel surface directly led to an enhanced removal of Sr from the solution, with higher concentrations of Sr colocated with Fe and Cr hotspots, suggesting coprecipitation of the contaminants with Fe and Cr that had initially dissolved from the steel. Interestingly, the coincidence of Cs enrichment with Fe and Cr hotspots was also observed under basic conditions, but not for the HNO3 matrices. This would indicate that coprecipitation is not the dominant mode of Cs uptake at low pH and that alternative contamination pathways become available under acidic conditions. Future work should focus on whether other confounding factors present in spent fuel reprocessing and storage facilities (e.g., the presence of the other contaminant radionuclides, dissolved metals, and/or organics) would affect Cs and Sr contamination of steels.
This work has clear implications for the design and implementation of reprocessing schemes and subsequent decommissioning and post-operational clean-out (POCO) at nuclear licensed sites. If contaminant diffusion into the bulk material can be effectively inhibited during reprocessing or spent fuel storage, then removal of surface bound contaminants (e.g., via chemical washing) will likely prove to be a reliable decontamination technique. As such, these contaminated steels could be easily removed from radioactive waste sentencing. In contrast, chemical decontamination techniques may not be effective if the Sr/Cs penetrates into the steel passive layer or material bulk and/or coprecipitates with steel matrix elements. In these circumstances, more aggressive physical decontamination approaches may be required (e.g., shot blasting or ultrahigh pressure water jetting) to remove the upper layers of the material’s surface.
Acknowledgments
We gratefully acknowledge funding from the Sellafield Ltd. Centre of Expertise in Decontamination and Effluent Treatment and thank Ben Spencer (University of Manchester) for assistance with XPS measurements. G.T.W.L. and F.R.L. acknowledge funding from NE/M014088/1 and ST/N002474/1.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b01311.
Steel composition data, SEM data, additional GD-OES data interpretation, additional XPS data, and further details and results of solution modeling (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Brumfiel G. Fukushima set for epic clean-up. Nature 2011, 472, 146–147. 10.1038/472146a. [DOI] [PubMed] [Google Scholar]
- Rivasseau C.; Farhi E.; Atteia A.; Couté A.; Gromova M.; Saint Cyr D. d. G.; Boisson A.-M.; Féret A.-S.; Compagnon E.; Blignyabcd R. An extremely radioresistant green eukaryote for radionuclide bio-decontamination in the nuclear industry. Energy Environ. Sci. 2013, 6, 1230–1239. 10.1039/c2ee23129h. [DOI] [Google Scholar]
- International Atomic Energy Agency . Management of spent fuel from nuclear power reactors. In Proceedings of an international conference; STI/PUB/1661, International Atomic Energy Agency: Vienna, 2006.
- Shaw R. D. Corrosion prevention and control at Sellafield nuclear fuel reprocessing plant. Br. Corros. J. 1990, 25, 97–107. 10.1179/bcj.1990.25.2.97. [DOI] [Google Scholar]
- Nieves L. A.; Chen S. Y.; Kohout E. J.; Nabelssi B.; Tilbrook R. W.; Wilson S. E. Analysis of disposition alternatives for radioactively contaminated scrap metal. J. Franklin Ins. 1998, 335, 1089–1103. 10.1016/S0016-0032(97)00055-0. [DOI] [Google Scholar]
- McGuire M. F.Stainless steels for design engineers; ASM International: Ohio, 2008. [Google Scholar]
- Castellani R.; Poulesquen A.; Goettmann F.; Marchal P.; Choplin L. Efficiency enhancement of decontamination gels by a super adsorbent polymer. Colloids Surf., A 2014, 454, 89–95. 10.1016/j.colsurfa.2014.04.022. [DOI] [Google Scholar]
- Wilbraham R. J.; Boxall C.; Goddard D. T.; Taylor R. J.; Woodbury S. E. The effect of hydrogen peroxide on uranium oxide films on 316L stainless steel. J. Nucl. Mater. 2015, 464, 86–96. 10.1016/j.jnucmat.2015.04.007. [DOI] [Google Scholar]
- McCafferty E.Introduction to corrosion science; Springer: Washington DC, 2006. [Google Scholar]
- Carrol S. A.; Roberts S. K.; Criscenti L. J.; O’Day P. A. Surface complexation model for strontium sorption to amorphous silica and goethite. Geochem. Trans. 2008, 9, 1–26. 10.1186/1467-4866-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouppert F., Rivoallan A., Largeron C.. An investigation of the chemical equilibria involving cesium, metal oxides and hydroxides on stainless steel. In-situ study of the contaminant sorption modes using fourier transform infrared spectrometry, transport and desorption of radio-contaminants In Waste Management ‘00 Conference Proceedings; Tucson, 2000.
- Fujii T.; Yamana H.; Takamiya K.; Watanabe M.; Moriyama H. Adsorption of fission products on a metal durface in nitric acid solutions: Radiochemical study using a multitracer. J. Radioanal. Nucl. Chem. 2002, 253, 199–204. 10.1023/A:1019629204861. [DOI] [Google Scholar]
- Kádár P.; Varga K.; Baja B.; Németh Z.; Vajda N.; Stefánka Z.; Kövér L.; Cserny I.; Tóth J.; Pintér T.; Schunk J. Accumulation of uranium, transuranium and fission products on stainless steel surfaces II. sorption studies in a laboratory model system. J. Radioanal. Nucl. Chem. 2011, 288, 943–954. 10.1007/s10967-011-1038-6. [DOI] [Google Scholar]
- Castle J. E. The composition of metal surfaces after atmospheric exposure: An historical perspective. J. Adhes. 2008, 84, 368–388. 10.1080/00218460802004477. [DOI] [Google Scholar]
- Adeleye S. A.; White D. A.; Taylor J. B. Ambient temperature contamination of process piping and the effects of pretreatment. Nucl. Technol. 1996, 113, 46–53. 10.13182/NT96-A35198. [DOI] [Google Scholar]
- Crossland I.Nuclear fuel cycle science and engineering; Woodhead Publishing: Cornwall, 2012. [Google Scholar]
- Hirschberg G.; Baradlai P.; Varga K.; Myburg G.; Schunk J.; Tilky P.; Stoddart P. Accumulation of radioactive corrosion products on steel surfaces of VVER type nuclear reactors. I .110mAg. J. Nucl. Mater. 1999, 265, 273–284. 10.1016/S0022-3115(98)00656-4. [DOI] [Google Scholar]
- Choppin G.R., Liljenzin J., Rydberg J.. Radiochemistry and nuclear chemistry; 3rd ed., Elsevier: Oxford, 2002. [Google Scholar]
- Wilson P. D.The nuclear fuel cycle: From ore to waste; Oxford University Press: Oxford, 1996. [Google Scholar]
- Lang A.; Engelberg D.; Smith N. T.; Trivedi D.; Horsfall O.; Banford A.; Martin P. A.; Coffey P.; Bower W. R.; Walther C.; Weiß M.; Bosco H.; Jenkins A.; Law G. T. W. Analysis of contaminated nuclear plant steel by laser-induced breakdown spectroscopy. J. Hazard. Mater. 2018, 345, 114–122. 10.1016/j.jhazmat.2017.10.064. [DOI] [PubMed] [Google Scholar]
- Mudali U. K.; Dayal R. K.; Gnanamoorthy J. B. Corrosion studies on materials of construction for spent nuclear fuel reprocessing plant equipment. J. Nucl. Mater. 1993, 203, 73–82. 10.1016/0022-3115(93)90432-X. [DOI] [Google Scholar]
- Priya R.; Mallika C.; Mudali U. K. Corrosion behavior of sensitized 304 SS in nitric acid medium containing oxidizing ions. Trans. Indian Inst. Met. 2014, 67, 459–467. 10.1007/s12666-013-0366-3. [DOI] [Google Scholar]
- Robin R.; Miserque F.; Spagnol V. Correlation between composition of passive layer and corrosion behavior of high Si-containing austenitic stainless steels in nitric acid. J. Nucl. Mater. 2008, 375, 65–71. 10.1016/j.jnucmat.2007.10.016. [DOI] [Google Scholar]
- Shankar A. R.; Niyanth S.; Vasudevan M.; Mudali U. K. Microstructural characterization and corrosion behavior of activated flux gas tungsten arc-welded and Multipass Gas Tungsten Arc-Welded stainless steel weld joints in nitric acid. Corrosion 2012, 68, 762–773. 10.5006/0515. [DOI] [Google Scholar]
- Gui Y.; Zheng Z. J.; Gao Y. The bi-layer structure and the higher compactness of a passive film on nanocrystalline 304 stainless steel. Thin Solid Films 2016, 599, 64–71. 10.1016/j.tsf.2015.12.039. [DOI] [Google Scholar]
- Freire L.; Catarino M. A.; Godinhoc M. I.; Ferreira M. J.; Ferreira M. G. S.; Simões A. M. P.; Montemor M. F. Electrochemical and analytical investigation of passive films formed on stainless steels in alkaline media. Cem. Concr. Compos. 2012, 34, 1075–1081. 10.1016/j.cemconcomp.2012.06.002. [DOI] [Google Scholar]
- Liu J.; Zhang T.; Meng G.; Shao Y.; Wang F. Effect of pitting nucleation on critical pitting temperature of 316L stainless steel by nitric acid passivation. Corros. Sci. 2015, 91, 232–244. 10.1016/j.corsci.2014.11.018. [DOI] [Google Scholar]
- Haupt S.; Strehblow H.-H. The analysis of dissolved Cr3+ with the rotating ring-disc technique and its application to the corrosion of Cr in the passive state. J. Electroanal. Chem. Interfacial Electrochem. 1987, 216, 229–240. 10.1016/0022-0728(87)80209-7. [DOI] [Google Scholar]
- Freire L.; Carmezim M. J.; Ferreira M. G. S.; Montemor M. F. The passive behaviour of AISI 316 in alkaline media and the effect of pH: A combined electrochemical and analytical study. Electrochim. Acta 2010, 55, 6174–6181. 10.1016/j.electacta.2009.10.026. [DOI] [Google Scholar]
- Ho Y. S.; McKay G. Pseudo second order model for sorption processes. Process Biochem. 1999, 34, 451–465. 10.1016/S0032-9592(98)00112-5. [DOI] [Google Scholar]
- Lagergren S. K. About the theory of so-called adsorption of soluble substances. Sven. Vetenskapsakad. Handl. 1898, 24, 1–39. [Google Scholar]
- Taylor H. A.; Thon N. Kinetics of Chemisorption1. J. Am. Chem. Soc. 1952, 74, 4169–4173. 10.1021/ja01136a063. [DOI] [Google Scholar]
- Kamaraj R.; Vasudevan S. Evaluation of electrocoagulation process for the removal of strontium and cesium from aqueous solution. Chem. Eng. Res. Des. 2015, 93, 522–530. 10.1016/j.cherd.2014.03.021. [DOI] [Google Scholar]
- Mishra S. P. Removal behavior of hydrous manganese oxide and hydrous stannic oxide for Cs(I) ions from aqueous solutions. Sep. Purif. Technol. 2007, 54, 10–17. 10.1016/j.seppur.2006.08.018. [DOI] [Google Scholar]
- Juang R.-S.; Chen M.-L. Application of the Elovich equation to the kinetics of metal sorption with solvent-impregnated resins. Ind. Eng. Chem. Res. 1997, 36, 813–820. 10.1021/ie960351f. [DOI] [Google Scholar]
- Ho Y. S.; McKay G. Sorption of dye from aqueous solution by peat. Chem. Eng. J. 1998, 70, 115–124. 10.1016/S0923-0467(98)00076-1. [DOI] [Google Scholar]
- Hedberg Y.; Wang X.; Hedberg J.; Lundin M.; Blomberg E.; Wallinder I. O. Surface-protein interactions on different stainless steel grades: Effects of protein adsorption, surface changes and metal release. J. Mater. Sci.: Mater. Med. 2013, 24, 1015–1033. 10.1007/s10856-013-4859-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Z.; Allman S.; Brice D. J.; Martin R. C.; Andre N. O. Exploring laser-induced breakdown spectroscopy for nuclear materials analysis and in-situ applications. Spectrochim. Acta, Part B 2012, 74-75, 177–183. 10.1016/j.sab.2012.06.049. [DOI] [Google Scholar]
- Dombovári P.; Kádár P.; Kovács T.; Somlai J.; Radó K.; Krisztián R.; Varga I.; Buják R.; Varga K.; Halmos P.; Borszéki J.; Kónya J.; Nagy N. M.; Kövér L.; Varga D.; Cserny I.; Tóth J.; Fodor L.; Horváth A.; Pintér T.; Schunk J. Accumulation of uranium on austenitic stainless steel surfaces. Electrochim. Acta 2007, 52, 2542–2551. 10.1016/j.electacta.2006.09.007. [DOI] [Google Scholar]
- Sasaki K.; Tanigaki T.; Matsuyama M.; Fukumoto K.; Uno M. Cesium corrosion process in Fe-Cr steel. J. Nucl. Mater. 2013, 441, 315–323. 10.1016/j.jnucmat.2013.05.055. [DOI] [Google Scholar]
- Baghni I. M.; Lyon S. B.; Ding B. The effect of strontium and chromate ions on the inhibition of zinc. Surf. Coat. Technol. 2004, 185, 194–198. 10.1016/S0257-8972(03)01316-1. [DOI] [Google Scholar]
- Chen K.; Hyodo J.; Dodd A.; Ai N.; Ishihara T.; Jian L.; Jiang S. P. Chromium deposition and poisoning of La0.8Sr0.2MnO3 oxygen electrodes of solid oxide electrolysis cells. Faraday Discuss. 2015, 182, 457–476. 10.1039/C5FD00010F. [DOI] [PubMed] [Google Scholar]
- Vasquez R. P. X-ray photoelectron studies of Sr and Ba compounds. J. Electron Spectrosc. Relat. Phenom. 1991, 56, 217–240. 10.1016/0368-2048(91)85005-E. [DOI] [Google Scholar]
- Sosulnikov M. I.; Teterin Y. A. X-ray photoelectron studies of Ca, Sr and Ba and their oxides and carbonates. J. Electron Spectros. Relat. Phenom. 1992, 59, 111–126. 10.1016/0368-2048(92)85002-O. [DOI] [Google Scholar]
- Allen G. C.; Curtis M. T.; Hooper A. J.; Tucker P. M. X-ray photoelectron spectroscopy of chromium–oxygen systems. J Chem. Soc., Dalton Trans. 1973, 14, 1675–1683. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.