

Abstract
Activation of the Fas death receptor leads to the death of motoneurons in culture. To investigate the role of Fas in programmed cell death and pathological situations, we used several mutant mice deficient for Fas signaling and made a novel transgenic FADD-DN (FAS-associated death domain-dominant-negative) strain. In vitro, motoneurons from all of these mice were found to be resistant to Fas activation and to show a delay in trophic deprivation-induced death. During normal development in vivo, no changes in motoneuron survival were observed. However, the number of surviving motoneurons was twofold higher in animals deficient for Fas signaling after facial nerve transection in neonatal mice. These results reveal a novel role for Fas as a trigger of axotomy-induced death and suggest that the Fas pathway may be activated in pathological degeneration of motoneurons.
Keywords: Fas, motoneuron, programmed cell death, FADD, facial nerve, axotomy
Introduction
The involvement of death receptor signaling in programmed, as well as in pathological neuronal death, has been the subject of many studies. One example is the triggering of neuronal death in the developing chick retina by binding of NGF to its low-affinity receptor p75 (Frade et al., 1996), a member of the tumor necrosis factor receptor (TNFR) family of transmembrane receptors. Although other death receptors in this family such as TNFR1 and Fas have been studied primarily in the immune system (Ashkenazi and Dixit, 1998), recent evidence has accumulated, suggesting that they may also control neuronal death (Raoul et al., 2000). TNFα binding to TNFR1 contributes to the death of NGF-dependent peripheral neurons during development (Barker et al., 2001), and we showed that activation of Fas could trigger the death of motoneurons in vitro (Raoul et al., 1999). These results raised the possibility that an active killing mechanism through Fas may be relevant in vivo.
To analyze the role of Fas in developmental and pathological motoneuron death, we now used different mice impaired for Fas signaling. We developed and studied a transgenic mouse overexpressing a dominant-negative (DN) form of FAS-associated death domain (FADD), a Fas adaptor molecule (Chinnaiyan et al., 1995) that is also required for signaling by TNFR1 (Hsu et al., 1996; Yeh et al., 1998). In these mice, the interaction between the death receptor and the FADD death domains does not occur because the death domain of FADD is deleted, meaning that recruitment of procaspase-8 to the death-inducing signaling complex (DISC) does not occur. We also used two other mutants: a spontaneous loss-of-function mutation at the Fas locus (lpr/lpr) (Adachi et al., 1993) and a Fas-/- mouse (Adachi et al., 1995), both of which develop lymphoadenopathy and autoimmune diseases.
We investigated the in vivo impact of Fas signaling impairment on developmental and lesion-induced motoneuron death in these three mutants. Our results provide the first evidence for a role of Fas in mediating motoneuron death after axotomy of the facial nerve and suggest that alternative pathways can be used for execution of programmed cell death (PCD) during motoneuron development.
Materials and Methods
Animals. Fas-/- mice were kindly supplied by Dr. S. Nagata (Osaka University Medical School, Osaka, Japan). lpr/lpr and control mice were purchased from (The Jackson Laboratory, Bar Harbor, ME) All mutants were maintained on a C57BL/6 genetic background. Fas-/- and Fas+/- animals were genotyped by PCR according to Adachi et al. (1995).
Generation ofβ-actin-FADD-DN transgenic mice. A cDNA encoding for a dominant-negative form of FADD fused to an amino terminal AU1 epitope tag (AU1/FADD-DN) was cloned into the HindIII-cloning site of the expression vector pBAP (a gift from William L. Perry and Nancy Jenkins, National Institutes of Health, Bethesda, MD). Transgenic founders were identified by Southern blot analysis using an FADD-DN probe to detect the transgenic construct. Heterozygous founder mice were back-crossed for >10 generations with C57BL/6 mice to establish founder lines on a pure C57BL/6 genetic background. Transgenic lines had no evident phenotype by observation and were maintained as heterozygotes. Wild-type littermates were used as controls. The following primers were used for genotyping: human β-actin promoter sense primer (5′-ATTCTCGCAGGATCAGTCGA-3′) and human FADD-DN construct antisense primer (5′-TGGTGGATCCGACGCTTCGGAGGTAG-3′).
Whole-mount terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) experiments were performed on whole embryonic day (E) 12.5 spinal cords as described previously (Yamamoto and Henderson, 1999).
Western immunoblot analysis. Tissues were lysed in 125 mm Tris-HCl, pH 8.0, 2 mm EDTA, pH 8.0, 2.5% SDS, and 10% β-mercaptoethanol. Fifteen micrograms of protein lysate (determined by Bio-Rad Bradford assay; Bio-Rad, Hercules, CA) were fractionated by SDS gel electrophoresis and transferred to a polyvinylidene difluoride membrane (Millipore, Bedford, MA). Blots were probed with either a monoclonal antibody against human FADD (Alexis Biochemicals, San Diego, CA) or a polyclonal antibody that recognizes both endogenous FADD and the human FADD-DN construct (Calbiochem, La Jolla, CA).
Motoneuron culture. Motoneuron cultures were prepared from E12.5 mouse spinal cords essentially as described previously (Arce et al., 1999) but with minor modifications. Metrizamide gradient purification was replaced by Optiprep (Nycomed Pharma, Oslo, Norway) gradient centrifugation. Optiprep solution (5.2% v/v) was freshly prepared in 10 mm Tricine, pH 7.8, 4.4% (w/v) glucose. In most experiments, the magnetic column step was omitted, and motoneurons in the enriched Optiprep fraction were identified by morphological criteria. Culture medium was Neurobasal medium (Invitrogen, San Diego, CA) supplemented as described previously (Arce et al., 1999).
Assay of motoneuron survival after Fas treatment. For cell survival measurement, motoneurons were plated at a density of 1500–2000 cells per well in four-well plates in the presence of a mixture of neurotrophic factors (NTFs) (1 ng/ml human BDNF, R & D Systems, Minneapolis, MN; 100 pg/ml rat glial-derived neurotrophic factor (GDNF), Sigma, St. Louis, MO; 10 ng/ml rat ciliary neurotrophic factor (CNTF), R & D Systems) added at the time of cell seeding. Motoneurons were grown for 16 hr at 37°C before being incubated in the presence of NTFs with purified hamster anti-mouse Fas antibody Jo2 (PharMingen, San Diego, CA) at the indicated concentrations diluted in Neurobasal medium. Surviving motoneurons were counted 2 d after treatment under phase-contrast. Results were expressed relative to the value for NTFs. All assays were performed in duplicate or triplicate wells in two to four independent experiments.
Assay of motoneuron survival after trophic deprivation. Mouse motoneurons were seeded in 3.5 cm diameter Petri dishes at a density of 2000 cells per dish. Surviving motoneurons were counted at 1, 2, 3, and 4d in vitro (DIV) under phase contrast. Motoneuron survival was calculated as a ratio between the number of cells cultured in the absence versus in the presence of NTFs. At least three experiments with triplicate dishes were performed for each mouse genotype examined. Statistical analysis of results of each experiment was performed (Student's t test; p < 0.05).
Facial nerve transection and facial motoneuron counting. Postnatal day (P) 3 lpr/lpr and FADD-DN pups (as well as control pups; n = 5 for each genotype) were anesthetized using pentobarbital, and the skin behind the left ear was cut open. The facial nerve was transected and the wound sutured with Ethicon (New Brunswick, NJ) (6/0; 0.7 metric; C1) before the pups were warmed up and returned to the mother. One week after surgery, the pups were killed for counting of facial motoneurons on both sides (ipsilateral to axotomy and contralateral) in a double-blind manner before genotyping of the corresponding animals. After perfusion with 4% paraformaldehyde in PBS, brains were removed, postfixed in the same fixative, dehydrated, and embedded in paraffin. Sections of 8 μm were stained with cresyl violet, and facial motoneurons were counted in every fifth section.
For each brain, the total number of surviving motoneurons on the axotomized side was evaluated, and a rescue index was defined as the ratio between the number of ipsilateral on contralateral facial motoneurons. The difference between mutant samples (n = 5 for each genotype) and wild-type counterparts (n = 5 for each genotype) was statistically analyzed (one-way ANOVA; all pairwise multiple comparison procedures: Tukey test; p < 0.001; p < 0.05).
Results
Generation and preliminary characterization of β-actin-FADD-DN transgenic mice
We generated transgenic mice overexpressing a FADD-DN construct lacking the death effector domain (DED) responsible for the interaction of FADD with caspase-8. This construct blocks apoptosis of mouse thymocytes and T cells in vivo when expressed under the lck promoter in another transgenic model (Newton et al., 1998; Zornig et al., 1998). The FADD-DN cDNA-coding sequence and an AU1 epitope tag were subcloned into the pBAP vector containing the ubiquitous human β-actin promoter (Fig. 1A). Transgenic lines were maintained as heterozygotes and showed no evident phenotype. Western blot analysis using an antibody specific for the human FADD protein revealed the presence of 18 kDa protein in lysates from the brain, heart, and to a much lesser extent, liver from adult transgenic mice, whereas no clear signal was detected in lysates from a control littermate (Fig. 1B). Many tissues from transgenic animals displayed no detectable expression (e.g., kidney, lymph nodes, and thymus; data not shown).
Figure 1.
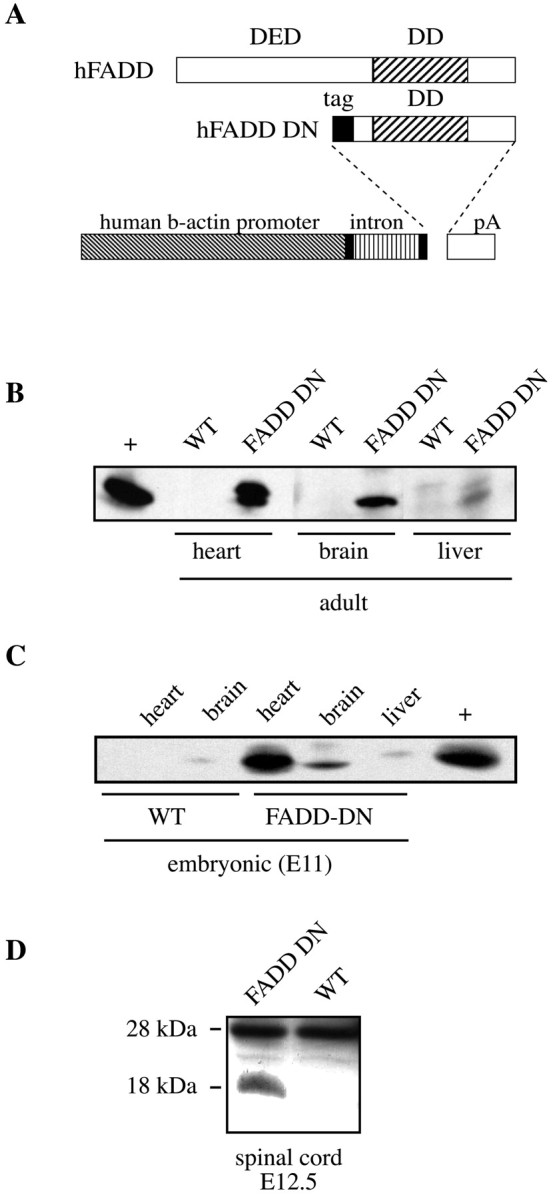
Generation and characterization of FADD-DN mice. A, Scheme of the FADD-DN construct: the DED of human (h) FADD was deleted and replaced by an N-terminal AU-1 tag, whereas the death domain (DD) was kept. The gene is under the control of a human β-actin promoter. B, C, Expression of FADD-DN in adult (B) and E11 (C) tissues was shown by Western blot analysis, using a mouse monoclonal anti-human FADD antibody. In B and C, a positive control (+) corresponding to thymus from lck promoter-FADD-DN transgenic mouse is shown. Expression under theβ-actin promoter is surprisingly restricted, primarily to the heart and the nervous system. D, Expression of FADD-DN and endogenous FADD in E12.5 spinal cord shown by Western blot analysis using a rabbit polyclonal anti-FADD antibody recognizing both human and murine forms. WT, Wild-type.
Expression at E11 showed the same profile as in the adults (Fig. 1C), and by reverse transcriptase-PCR, we detected the presence of transgenic mRNA in lysates of spinal cords from E12.5 FADD-DN mice (data not shown). This was confirmed (Fig. 1D) using a polyclonal antibody recognizing both human FADD-DN (18 kDa) and murine FADD (28 kDa). These studies show that (1) the amount of FADD-DN protein is compatible with a dominant-negative function and (2) any effect should be relatively tissue-specific and exclude immune tissues, the usual sites of action of the Fas/FasL system.
Death of cultured motoneurons induced by trophic deprivation is slowed down but not abolished by mutations in the Fas pathway
We showed previously that trophic deprivation-induced death of embryonic motoneurons involves the Fas/Fas-L pathway (Raoul et al., 1999). To evaluate the impact of mutations affecting Fas (lpr/lpr, Fas-/-) and the Fas/TNFR1 pathway (FADD-DN) on this process, embryonic motoneurons from mutant and control mice were cultured in the presence or absence of a mixture of trophic factors (BDNF, CNTF, and GDNF). Survival was evaluated as a percentage of the value in the presence of trophic factors at different times (1–4 d in culture). When deprived of trophic support, motoneurons from mutant mice showed a significant increase in survival at both 1 and 2 d in culture compared with their control counterparts (Fig. 2A--C). This provides three lines of genetic evidence for the role of the Fas system in trophic deprivation-induced death, previously deduced from the use of soluble Fas extracellular domain (Raoul et al., 1999). Nevertheless, this initial delay did not result in long-term protection, because at day 4, motoneurons from mutant mice and controls displayed similar survival values (Fig. 2A--C). To demonstrate that Fas signaling was affected in these mutants, embryonic motoneurons were cultured in the continuous presence of the optimal mixture of trophic factors (BDNF, CNTF, and GDNF) and treated 16 hr after seeding with an agonistic anti-Fas antibody at various concentrations (1, 10, and 100 ng/ml). At days 3 and 5, surviving motoneurons were counted by phase-contrast microscopy. Up to 50% of C57BL/6 control and Fas+/- motoneurons were killed by the anti-Fas antibody with a typical dose–response curve (Raoul et al., 1999), whereas lpr/lpr, Fas-/-, and FADD-DN motoneurons were not sensitive to the anti-Fas treatment after 3 d (Fig. 2D--F) or 5 d (data not shown). In accordance with the role of FADD as adaptor molecule downstream of both Fas and TNFα, we tested whether FADD-DN motoneurons were also resistant to TNFα-induced death. Although a small fraction of control cells was found to be sensitive to soluble TNFα (100 ng/ml), FADD-DN motoneurons displayed complete resistance to TNFα (Fig. 2G). Interestingly, Fas and TNFα did not show any additive effect in killing control motoneurons (Fig. 2H).
Figure 2.
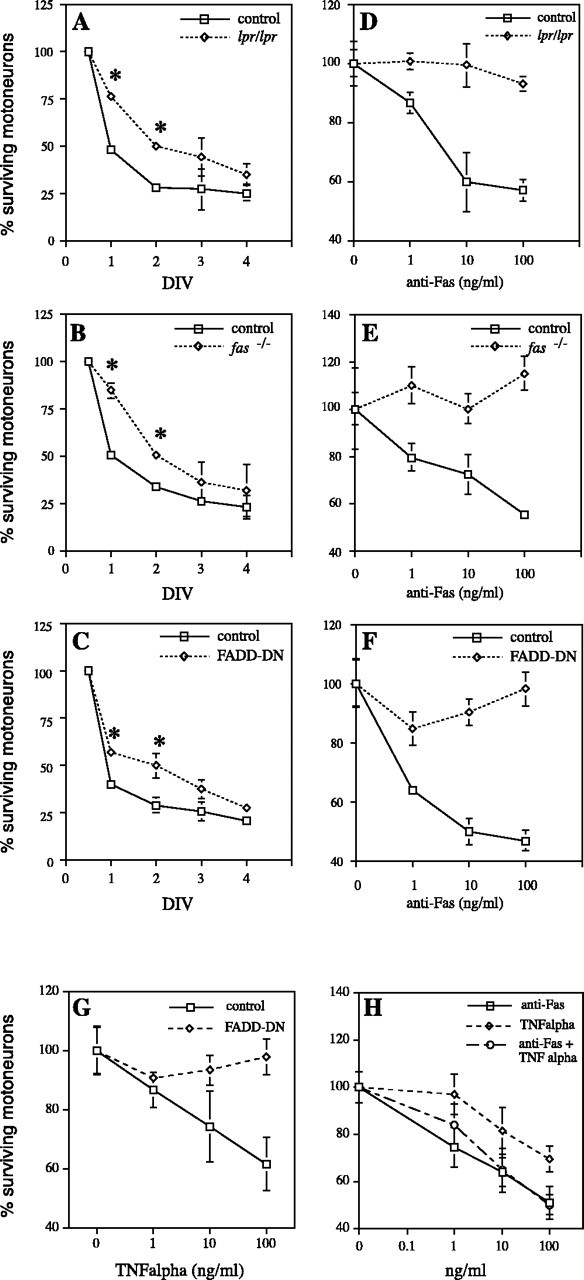
Survival properties of mutant Fas mice motoneurons in vitro. Trophic deprivation-induced death of cultured motoneurons is delayed but not abolished by mutations affecting Fas. lpr/lprmice(A),Fas-/-mice(B), and FADD-DNmice(C)are shown. The percentage of motoneuron survival was calculated as a ratio between the number of cell scultured in the absence versus presence of NTFs (DIV). The number of living motoneurons at 12 hr was defined as 100%. (asterisk indicates Student'sttest,p<0.05). Motoneurons from mutant Fas mice are resistant to exogenous activation and combined treatment with anti-Fas antibody, and TNFα shows no additive effect on motoneuron death induction. lpr/lpr (D), Fas-/- (E), FADD-DN (F, G), and wild-type (H) mice are shown. Treatment with anti-Fas antibody (D–F) and TNFα (G, H) was performed 16 hr after cell plating in the continued presence of NTFs, and surviving motoneurons were counted 2d after. Counts are expressed as a percentage of the surviving motoneurons with NTFs alone.
Mutations affecting the Fas pathway do not modify the pattern or the outcome of spinal motoneuron developmental death
The delay in the death of Fas mutant motoneurons observed in vitro prompted a study of the potential influence of Fas during developmental death in vivo. Motoneuron death during PCD was quantified using whole-mount TUNEL on E12.5 spinal cords from lpr/lpr, Fas-/-, and FADD-DN. The pattern of death profiles (data not shown) and the number of TUNEL+ cells (Table 1) did not differ between mutant (lpr/lpr or Fas-/-) and control (C57BL/6 or Fas+/-) spinal cords. Because the TUNEL technique may not have allowed the detection of small differences spread over the period of PCD, the final outcome of motoneuron PCD was also analyzed in FADD-DN and control mice after the end of PCD (at P10) by counting lumbar motoneurons. No significant difference was observed between mutant mice (1233 ± 68; n = 7) and controls (1265 ± 71; n = 7).
Table 1.
Cell death in embryonic spinal cords of mice deficient for the Fas pathway
|
Mouse genotype |
TUNEL+ profiles (E12.5 thoracic spinal cord) |
|---|---|
| C57BL/6 | 598 ± 33 |
| (n = 3) | |
| lpr/lpr | 616 ± 26 |
| (n = 3) | |
| Fas+/- | 602 ± 64 |
| (n = 3) | |
| Fas-/- | 594 ± 52 |
|
|
(n = 3)
|
Number of TUNEL + motoneurons in the thoracic part of E12.5 spinal cords of lpr/lpr and Fas-/- mice with their respective controls. Values correspond to mean ± SD.
Fas signaling is involved in axotomy-induced death of facial motoneurons
Because Fas and FADD signaling did not seem to be an absolute requirement for developmental motoneuron death, we tested the hypothesis of their involvement in lesion-induced motoneuron death and chose facial nerve axotomy as a paradigm. We first confirmed that facial motoneurons were, like spinal motoneurons, sensitive to Fas-induced death. Indeed, agonistic anti-Fas antibody triggered the death of ∼50% of cultured facial motoneurons 48 hr after treatment (Fig. 3A). Moreover, facial motoneurons from FADD-DN mice were Fas-resistant (Fig. 3A). As with spinal motoneurons, counting cresyl violet-stained facial motoneurons after the period of naturally occurring PCD showed no significant differences between FADD-DN (2685 ± 92; n = 4) and wild-type (2825 ± 138; n = 4) littermates. Similar results were obtained comparing lpr/lpr (2410 ± 110; n = 4) to C57BL/6 control mice (2356 ± 155; n = 4).
Figure 3.
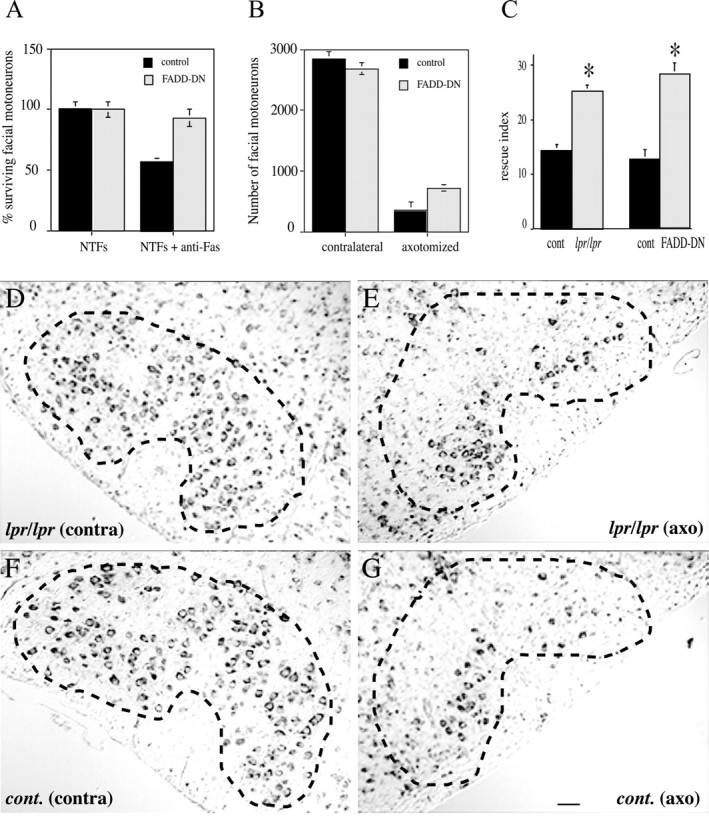
The Fas death pathway is activated after early postnatal (P3) facial nerve transection. A, Facial motoneurons are sensitive to Fas-induced death when isolated from E13.5 wild-type mice and are resistant when isolated from Fas mutant mice. Facial nuclei were localized and dissected after a protocol developed by Jean Livet, Vilma Arce, and Odile Delapeyrière (personal communication). Motoneurons were purified, cultured, treated, and counted as in Figure 2. B–G, Facial motoneurons survive better from axotomy in Fas mutant mice. Motoneurons were counted in cresyl violet-stained sections throughout the facial nucleus of P10 mice whether axotomized or not at P3 (mean ± SEM; *p < 0.001; n = 5 for each genotype). D–G, Cresyl violet staining of comparable serial sections from contralateral (contra) and axotomized (axo) facial nuclei of lpr/lpr (D, E) and C57BL/6 control (F, G) mice 1 week after lesion (P10). B, G, Total number of facial motoneurons (B) and rescue index (G) defined as the ratio between the number of motoneurons in the axotomized nucleus and the number of motoneurons in the contralateral one of the same animal inlpr/lprcompared with C57BL/6 control mice, and in FADD-DN mice compared with wild-type littermates (mean± SEM; *p < 0.001; n = 5 for each genotype). Dashed lines in D–G indicate the border of the facial nucleus. Scale bar: (in G) D–G, 100 μm.
To determine whether Fas is involved in axotomy-induced motoneuron death, facial nerve transection was performed on neonatal (P3) lpr/lpr mice and wild-type animals of the same genetic background (C57BL/6) as well as on P3 FADD-DN mice and their control littermates. Motoneurons were counted in both facial nuclei 1 week after lesion. A massive loss of motoneurons was seen in the facial nucleus ipsilateral to axotomy (Fig. 3E,G), whereas the contralateral nucleus was unmodified (Fig. 3D,E). Nevertheless, lpr/lpr and FADD-DN animals showed a higher number of surviving cells in the affected nucleus (Fig. 3B,E). To quantify this difference, five lpr/lpr and five control animals on one side and five FADD-DN mice with their control littermates on the other side were compared using a rescue index, defined as the ratio of the number of motoneurons in the axotomized nucleus to the number of motoneurons in the contralateral nucleus. The rescue index was higher by a factor of 1.8 in lpr/lpr and 2.1 in FADD-DN (p < 0.001), the percentage of surviving motoneurons increased from 14.3 to 25.2% and from 12.1 to 28.6%, respectively (Fig. 3C).
Discussion
The present results constitute the first demonstration of an in vivo role for Fas in controlling the death of motoneurons. After axotomy of the facial nerve in neonatal rodents, 90% of the motoneurons die by apoptosis (Sendtner et al., 1990; Li et al., 1994; Rossiter et al., 1996), and most of them synchronize their death between the fourth and seventh days after axotomy. The fraction of 25% that survives in the mutants therefore represents a significant but partial rescue compared with controls. Several studies on neonatal axotomy in mutant mice support the idea that the more upstream in a signaling cascade the gene of interest is, the more likely there will be redundant downstream effectors that will eventually bypass the blockade, leading to a partial and probably transient effect, as observed in our case. Transgenic or knock-out mice with gain or loss of function for major elements of the apoptotic pathways gave up to 100% saving, whereas transgenic mice overexpressing a soluble form of TNFR1 under a liver-specific promoter (Terrado et al., 2000) yielded the same rate of survival as in the present study. In this latter case, the effect on motoneuron survival was suggested to be peripheral, perhaps by affecting death mediated by immune system cells, because it is unlikely that a large molecule such as TNFR-fusion protein can cross the blood/brain barrier. In our study, the argument of an immune system-derived effect could stand for the lpr/lpr mice but not for the FADD-DN transgenic mice, which express the transgene essentially in brain and heart, very poorly in liver, and not at all in the thymus and lymph nodes. Together with our demonstration of a direct effect of Fas agonists on cultured facial motoneurons, this means that we favor a role for direct killing after axotomy. The source for Fas-L in the axotomized facial nucleus could be endogenous as shown previously for spinal motoneurons (Raoul et al., 1999), but it is also possible that FasL is supplied by invading lymphocytes, as is the case for TNFα after axotomy in adult animals (Raivich et al., 1998).
The absence of a complete rescue may also reflect a certain level of redundancy in the death receptors activated in vivo. In this respect, although FADD is downstream of both Fas and TNFR1, we did not observe a significantly higher level of rescue in FADD-DN than in lpr/lpr mice, in which only Fas is impaired. This result indicates that TNFR1, which also uses FADD signaling, does not by itself play an additional role, and that the population of motoneurons sensitive to TNFα most likely represents a subpopulation of the motoneurons killed after activation of Fas. Our results in vitro show that death in Fas mutant motoneurons is delayed but not blocked and suggest that after trying to activate the Fas pathway and failing, motoneurons can activate other pathways to accomplish the death process. One of these pathways may involve signaling through TNFR2 (which does not use FADD) because Raivich et al. (2002) showed that only the combined deletion of both TNFR1 and TNFR2 would prevent death of motoneurons after facial axotomy in the adult mouse.
This first report which shows the activation of the death receptor Fas as a trigger of axotomy-induced death is in accordance with the growing body of evidence supporting a role of the Fas pathway in neurodegenerative processes occurring in pathological conditions or as a consequence of injury. Fas expression was found to be associated with Alzheimer's disease (Nishimura et al., 1995), and a functional involvement of Fas activation was demonstrated after the induction of neuronal apoptosis by β-amyloid (Morishima et al., 2001). In that study, Fas-L was shown to be upregulated after activation of the c-Jun N-terminal kinase pathway induced by β-amyloid. Blocking the binding of Fas-L to Fas resulted in the partial rescue of neurons treated by β-amyloid. Increased levels of Fas, coincident with the cleavage of procaspase-8 to its active form and with the triggering of the mitochondrial apoptotic pathway, were also observed in neonatal rat thalamus during the early phase of apoptosis after hypoxia-ischemia (Northington et al., 2001).
Concerning motoneurons more specifically, upregulation of Fas is associated with delayed motoneuron death after spinal cord ischemia in rabbits (Sakurai et al., 1998). In adult mice, transient spinal cord ischemia induces an increase in DISC formation as identified by coimmunoprecipitation using anti-Fas and anti-procaspase-8 antibodies, followed by an increase in activated caspase-8 (Matsushita et al., 2000).
All of these pieces of evidence suggest that the activation of the Fas pathway after injury might represent a widespread mechanism of neuronal death in pathological situations. Recently, we found that motoneurons derived from a model of amyotrophic lateral sclerosis (SOD1 transgenic mice) show increased susceptibility to Fas-induced death, outlining for Fas an additional potential projection in neurodegenerative diseases (Raoul et al., 2002). Thus, although Fas signaling may be growth-promoting in other contexts (Desbarats et al., 2003), our results indicate that the Fas-signaling pathway may be a new potential target for molecules aimed at preventing motoneuron death after injury and in motoneuron diseases.
Footnotes
This work was supported by Institut National de la Santè et de la Recherche Mèdicale, Centre National de la Recherche Scientifique, Association Française contre les Myopathies, European Commission Contract QLG3-CT-1999-00602, Amyotrophic Lateral Sclerosis Association, and GIP-Aventis. G.U. was supported by a Marie Curie European Economic Community fellowship. We are very grateful to Odile DeLapeyrière, Jean Livet, and Vilma Arce [Unitè Mixte de Recherche (UMR) 382] for help in developing facial motoneuron cultures. We are especially grateful to Gerard Evan for support and input at the initial phase of this work. We thank the animal units at Imperial Cancer Research Fund, Ian Rosewell for assistance with transgene microinjection, and Magali Chautan-Michaud (Trophos) for help in crossing and genotyping FADD-dominant-negative mice. We thank all members of UMR 382 for helpful comments on this manuscript and throughout this work.
Correspondence should be addressed to Dr. Brigitte Pettmann, Unitè Mixte de Recherche 382-IBDM, Campus de Luminy, Case 907, 13288 Marseille Cedex 09, France. E-mail: pettmann@ibdm.univ-mrs.fr.
G. Ugolini's present address: Lay Line Genomics Spa, c/o Parco Scientifico Biomedico San Raffaele, Via di Castel Romano 100, 00128 Roma, Italy.
C. Raoul's present address: Neurosciences Institute, Swiss Federal Institute of Technology, LENAAB-132, CH-1015 Lausanne, Switzerland.
Y. Yamamoto's present address: Department of Neurology D-4, Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan.
Copyright © 2003 Society for Neuroscience 0270-6474/03/238526-06$15.00/0
B.P. and A.-O.H. contributed equally to this work.
References
- Adachi M, Watanabe-Fukunaga R, Nagata S ( 1993) Aberrant transcription caused by the insertion of an early transposable element in an intron of the Fas antigen gene of lpr mice. Proc Natl Acad Sci USA 90: 1756-1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi M, Suematsu S, Kondo T, Ogasawara J, Tanaka T, Yoshida N, Nagata S ( 1995) Targeted mutation in the Fas gene causes hyperplasia in peripheral lymphoid organs and liver. Nat Genet 11: 294-300. [DOI] [PubMed] [Google Scholar]
- Arce V, Garces A, de Bovis B, Filippi P, Henderson C, Pettmann B, deLapeyriere O ( 1999) Cardiotrophin-1 requires LIFRbeta to promote survival of mouse motoneurons purified by a novel technique. J Neurosci Res 55: 119-126. [DOI] [PubMed] [Google Scholar]
- Ashkenazi A, Dixit VM ( 1998) Death receptors: signaling and modulation. Science 281: 1305-1308. [DOI] [PubMed] [Google Scholar]
- Barker V, Middleton G, Davey F, Davies AM ( 2001) TNFalpha contributes to the death of NGF-dependent neurons during development. Nat Neurosci 4: 1194-1198. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, O'Rourke K, Tewari M, Dixit VM ( 1995) FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 81: 505-512. [DOI] [PubMed] [Google Scholar]
- Desbarats J, Birge RB, Mimouni-Rongy M, Weinstein DE, Palerme JS, Newell MK ( 2003) Fas engagement induces neurite growth through ERK activation and p35 upregulation. Nat Cell Biol 5: 118-125. [DOI] [PubMed] [Google Scholar]
- Frade JM, Rodriguez-Tebar A, Barde YA ( 1996) Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature 383: 166-168. [DOI] [PubMed] [Google Scholar]
- Hsu H, Shu HB, Pan MG, Goeddel DV ( 1996) TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 84: 299-308. [DOI] [PubMed] [Google Scholar]
- Li L, Oppenheim RW, Lei M, Houenou LJ ( 1994) Neurotrophic agents prevent motoneuron death following sciatic nerve section in the neonatal mouse. J Neurobiol 25: 759-766. [DOI] [PubMed] [Google Scholar]
- Matsushita K, Wu Y, Qiu J, Lang-Lazdunski L, Hirt L, Waeber C, Hyman BT, Yuan J, Moskowitz MA ( 2000) Fas receptor and neuronal cell death after spinal cord ischemia. J Neurosci 20: 6879-6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima Y, Gotoh Y, Zieg J, Barrett T, Takano H, Flavell R, Davis RJ, Shirasaki Y, Greenberg ME ( 2001) Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J Neurosci 21: 7551-7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Harris AW, Bath ML, Smith KG, Strasser A ( 1998) A dominant interfering mutant of FADD/MORT1 enhances deletion of autoreactive thymocytes and inhibits proliferation of mature T lymphocytes. EMBO J 17: 706-718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T, Akiyama H, Yonehara S, Kondo H, Ikeda K, Kato M, Iseki E, Kosaka K ( 1995) Fas antigen expression in brains of patients with Alzheimer-type dementia. Brain Res 695: 137-145. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Ferriero DM, Flock DL, Martin LJ ( 2001) Delayed neurodegeneration in neonatal rat thalamus after hypoxia-ischemia is apoptosis. J Neurosci 21: 1931-1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raivich G, Jones LL, Kloss CU, Werner A, Neumann H, Kreutzberg GW ( 1998) Immune surveillance in the injured nervous system: T-lymphocytes invade the axotomized mouse facial motor nucleus and aggregate around sites of neuronal degeneration. J Neurosci 18: 5804-5816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raivich G, Liu ZQ, Kloss CU, Labow M, Bluethmann H, Bohatschek M ( 2002) Cytotoxic potential of proinflammatory cytokines: combined deletion of TNF receptors TNFR1 and TNFR2 prevents motoneuron cell death after facial axotomy in adult mouse. Exp Neurol 178: 186-193. [DOI] [PubMed] [Google Scholar]
- Raoul C, Henderson CE, Pettmann B ( 1999) Programmed cell death of embryonic motoneurons triggered through the fas death receptor. J Cell Biol 147: 1049-1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoul C, Pettmann B, Henderson CE ( 2000) Active killing of neurons during development and following stress: a role for p75(NTR) and Fas? Curr Opin Neurobiol 10: 111-117. [DOI] [PubMed] [Google Scholar]
- Raoul C, Estevez A, Nishimune H, Cleveland D, deLapeyriere O, Henderson C, Haase G, Pettmann B ( 2002) Motoneuron death triggered by a specific pathway downstream of Fas. Potentiation by ALS-linked SOD1 mutations. Neuron 35: 1067-1083. [DOI] [PubMed] [Google Scholar]
- Rossiter JP, Riopelle RJ, Bisby MA ( 1996) Axotomy-induced apoptotic cell death of neonatal rat facial motoneurons: time course analysis and relation to NADPH-diaphorase activity. Exp Neurol 138: 33-44. [DOI] [PubMed] [Google Scholar]
- Sakurai M, Hayashi T, Abe K, Sadahiro M, Tabayashi K ( 1998) Delayed selective motor neuron death and fas antigen induction after spinal cord ischemia in rabbits. Brain Res 797: 23-28. [DOI] [PubMed] [Google Scholar]
- Sendtner M, Kreutzberg GW, Thoenen H ( 1990) Ciliary neurotrophic factor prevents the degeneration of motor neurons after axotomy. Nature 345: 440-441. [DOI] [PubMed] [Google Scholar]
- Terrado J, Monnier D, Perrelet D, Vesin D, Jemelin S, Buurman WA, Mattenberger L, King B, Kato AC, Garcia I ( 2000) Soluble TNF receptors partially protect injured motoneurons in the postnatal CNS. Eur J Neurosci 12: 3443-3447. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Henderson CE ( 1999) Patterns of programmed cell death in populations of developing spinal motoneurons in chicken, mouse, and rat. Dev Biol 214: 60-71. [DOI] [PubMed] [Google Scholar]
- Yeh WC, Pompa JL, McCurrach ME, Shu HB, Elia AJ, Shahinian A, Ng M, Wakeham A, Khoo W, Mitchell K, El-Deiry WS, Lowe SW, Goeddel DV, Mak TW ( 1998) FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 279: 1954-1958. [DOI] [PubMed] [Google Scholar]
- Zornig M, Hueber AO, Evan G ( 1998) p53-dependent impairment of T-cell proliferation in FADD dominant-negative transgenic mice. Curr Biol 8: 467-470. [DOI] [PubMed] [Google Scholar]