

SUMMARY
Fragile X syndrome (FXS) is the leading heritable cause of intellectual disability and commonly co-occurs with autism spectrum disorder. Silencing of the Fmr1 gene leads to the absence of the protein product, fragile X mental retardation protein (FMRP), which represses translation of many target mRNAs. Excess translation of these targets is one cause of neuronal dysfunction in FXS. Utilizing the Drosophila model of FXS, we identified the mitogen-activated protein kinase kinase kinase (MAP3K) Wallenda/dual leucine zipper kinase (DLK) as a critical target of FMRP. dFMRP binds Wallenda mRNA and is required to limit Wallenda protein levels. In dFmr1 mutants, Wallenda signaling drives defects in synaptic development, neuronal morphology, and behavior. Pharmacological inhibition of Wallenda in larvae suppresses dFmr1 neurodevelopmental phenotypes, while adult administration prevents dFmr1 behavioral defects. We propose that in dFmr1 mutants chronic Wallenda/DLK signaling disrupts nervous system development and function and that inhibition of this kinase cascade might be a candidate therapeutic intervention for the treatment of FXS.
In Brief
Russo and DiAntonio identify a dysregulated MAPK signaling pathway in the fly model of fragile X syndrome. MAP3K Wnd/DLK drives dFmr1 mutant phenotypes, and pharmacological inhibition of Wnd/DLK prevents neural dysfunction in this model, thus highlighting a possible role for Wnd/DLK in the pathophysiology of fragile X syndrome.
Graphical Abstract
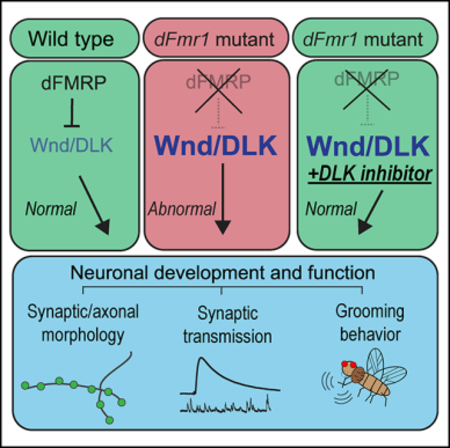
INTRODUCTION
Fragile X syndrome (FXS) is the most commonly inherited form of intellectual disability and is a leading cause of autism spectrum disorders (Bassell and Warren, 2008; Crawford et al., 2001). FXS is caused by a defect in the gene Fmr1, resulting in the absence of the Fmr1 gene product fragile X mental retardation protein (FMRP) (Pieretti et al., 1991; Warren and Nelson, 1994). The loss of FMRP drives a host of neurological symptoms of varying severity, including learning disabilities, behavioral deficits, sensory integration issues, and an increased propensity toward seizures (Bailey et al., 2000; Berry-Kravis et al., 2010; Contractor et al., 2015). One role of FMRP is to act as an RNA-binding protein (RNA-BP) that targets mRNAs and represses translation by stalling the ribosome and preventing the transcript from being translated into protein (Bassell and Warren, 2008). FMRP is a highly conserved protein, with orthologs in mice (FMRP) and Drosophila (dFMRP). The absence of FMRP in animal models causes abnormal neuronal morphology, physiology, and defects in behaviors such as hyperactivity, social assays, and sleep (Bardoni and Mandel, 2002; Bhogal and Jongens, 2010). Many mRNA targets of FMRP have been identified and are highly enriched for transcripts encoding proteins that govern synaptic development and function (Ascano et al., 2012; Darnell et al., 2011). For many target transcripts, the protein products are present at elevated levels in Fmr1-knockout neurons due to the loss of translational repression. Unfortunately, efforts to treat FXS by inhibiting or downregulating these target proteins have not succeeded (Erickson et al., 2017; Lee et al., 2018).
Defining the molecular factors underlying the neurobiological defects in FXS is critical to identify candidate therapies. The Drosophila model of FXS, in which the single ortholog of Fmr1, dFmr1, has been mutated and causes the absence of dFMRP, has been extremely fruitful in this context (Wan et al., 2000b). dFMRP is the lone ortholog of three human Fmr1 protein products—FMR1, FXR1, and FXR2—yet only expression of human FMR1 is sufficient to rescue neurological dysfunction in the Drosophila model of FXS (Coffee et al., 2010), highlighting the functional conservation between dFMRP and FMR1. dFmr1 mutants exhibit abnormal synaptic morphology and physiology at the larval neuromuscular junction (NMJ), which parallel observations of cortical and hippocampal neurons in the Fmr1KO mouse (Zhang et al., 2001; Comery et al., 1997; The Dutch-Belgian Fragile X Consortium, 1994). Adult flies also exhibit abnormal brain morphology and deficits in behaviors such as repetitive motions, learning and memory, and sleep (Dockendorff et al., 2002; Michel et al., 2004; Tauber et al., 2011). Hence, Drosophila is a powerful model to identify the molecular drivers of nervous system defects in FXS.
Studies of the larval NMJ have defined molecular pathways regulating neuronal development and function. We previously identified the conserved mitogen-activated protein kinase kinase kinase (MAP3K) Wallenda (Wnd) as a potent promoter of synaptic growth at the larval NMJ (Collins et al., 2006). Wnd and its mammalian ortholog dual leucine zipper kinase (DLK) have since been characterized as master regulators of neuronal stress pathways (Asghari Adib et al., 2018; Gerdts et al., 2016; Simon and Watkins, 2018). Activation of Wnd/DLK signaling triggers activation of downstream kinases and transcription factors that promote a stress response. Chronic activation of this stress response induces neuronal dysfunction in neurodegenerative disorders (Pichon et al., 2017). There is substantial evidence that the molecular pathways involved in the progression of neurodegenerative diseases are similarly dysregulated in neurodevelopmental disorders, including FXS, autism, and Down syndrome (Wang, 2015). Could dysregulated Wnd/DLK signaling, and subsequent upregulation of a conserved stress pathway, have a role in the pathophysiology of neurodevelopmental disorders? In seeking regulatory mechanisms controlling Wnd/DLK activity during synaptic development, we discovered that dFMRP regulates Wnd levels and function.
FMRP targets an estimated 4% of all neuronal mRNAs for translational repression (Ashley et al., 1993; Brown et al., 2001), so multiple targets likely account for the widespread and varied defects observed when FMRP-mediated translational control is lost. Nonetheless, there may be critical targets that are responsible for multiple phenotypes. Here, we identify the MAP3K Wnd/DLK as such a key target of dFMRP. dFMRP binds wnd mRNA and limits Wnd protein levels. Moreover, Wnd is required for defects in neuronal morphology, physiology, and behavior in the fly FXS model. Pharmacological inhibition of Wnd in dFmr1 mutant larvae corrects neurodevelopmental defects, while Wnd inhibition in the adult is sufficient to prevent behavioral defects in dFmr1 mutant flies. Taken together, these findings show that dysregulated Wnd signaling is a central driver of neural dysfunction in the fly model of FXS. We propose that the Wnd/DLK stress pathway may be an exciting candidate therapeutic target for FXS.
RESULTS
Overexpressing dFMRP Suppresses hiw Mutant Synaptic Overgrowth by Decreasing Levels of the Conserved MAP3K Wnd
We conducted an in vivo genetic screen in Drosophila to identify regulators of the conserved MAP3K Wnd. We leveraged Drosophila larvae mutant for the E3 ubiquitin ligase highwire (hiw). hiw mutant larvae exhibit dramatic overgrowth at the larval neuromuscular junction, defined by an increased branching of the synaptic terminal nerve and a substantial increase in the number of presynaptic boutons (Figure 1A; Collins et al., 2006). We previously identified Wnd as the Hiw target responsible for driving this extensive overgrowth, demonstrating that (1) Wnd protein levels are significantly increased in the hiw mutant nervous system, (2) the overgrowth phenotype is entirely suppressed in hiw;wnd double mutants, and (3) hiw-dependent NMJ overgrowth is a downstream readout of MAP kinase (MAPK) signaling through JNK and Fos (Collins et al., 2006). Thus, we reasoned that overexpressing proteins that normally limit Wnd protein levels or Wnd signaling would also suppress the hiw mutant overgrowth phenotype at the larval NMJ. We conducted a screen in hiw mutants to identify candidate genes with regulatory control over Wnd. We screened through transgenic constructs enriched for (1) genes with roles in nervous system development and (2) genes with orthologs for disease-related genes in mammals, and we examined NMJ morphology in these larvae to determine if overexpression suppressed hiw mutant overgrowth.
Figure 1. Overexpression of dFMRP Suppresses Synaptic Overgrowth and Decreases Wnd Protein Levels in hiw Mutant Larvae.

(A) Representative images of the NMJ synaptic terminal at muscle 4 in third-instar larvae. For all genotypes, the pan-neuronal driver Elav-GAL4 was used to express the given transgene, and the wild-type (WT) control is Elav-GAL4 in an otherwise wild-type background driving UAS-RFP. NMJs were stained for the presynaptic bouton marker DVGLUT (green) and nerve membrane marker HRP (red). Scale bar: 25 μm.
(B) Quantification of the mean (±SEM) number of DVGlut+ boutons per muscle 4 NMJ in each genotype. Overexpression of dFMRP completely suppresses NMJ overgrowth in hiw mutants.
(C) Representative images of larval ventral nerve cords (VNCs) from wild type (elav > RFP), hiw mutants (hiw elav > RFP), and hiw mutants overexpressing dFMRP (hiw elav > dFMRP). VNCs were stained for endogenous Wnd protein (green) and membrane marker HRP (red). Increased Wnd protein levels in hiw mutants is suppressed when dFMRP is overexpressed. Scale bar: 50 μm.
(D) Quantification of the mean (±SEM) intensity of Wnd staining in the VNCs for each genotype.
(E) Representative images of larval VNCs from wild type (elav > UAS-WndKDGFP), hiw (hiw,elav > UAS-WndKDGFP), and hiw overexpressing dFMRP (hiw,elav > UAS-WndKDGFP,UAS-dFMRP). VNCs were stained for the transgenic Wnd protein using anti-GFP (green) and membrane marker HRP (red). Scale bar: 50 μm.
(F) Quantification of the mean (±SEM) intensity of GFP staining in the VNCs for each genotype.
Statistical tests and exact p values reported in Table S1. *p = 0.05; **p = 0.01; ***p = 0.001; ****p = 0.0001, NS, not significant; p > 0.05.
See also Figure S1.
The strongest hit from this screen was overexpression of Drosophila FMRP (dFMRP), which is encoded by the dFmr1 gene. hiw mutants exhibit increased branching and bouton number at the larval NMJ compared to wild type (WT), and driving a Wnd-RNAi in the nervous system of hiw mutants fully suppresses the overgrowth phenotype (Figures 1A and 1B). Overexpressing dFMRP in the hiw mutant phenocopied Wnd knockdown by completely suppressing NMJ overgrowth (Figures 1A and 1B), restoring the number of synaptic boutons to wild-type levels (Figure 1B). Overexpression of dFMRP suppresses synaptic overgrowth in a second hiw mutant allele (Figures S1A and S1B), demonstrating that suppression is not an allele-specific effect. Overexpression of dFMRP in an otherwise wild-type background does not significantly alter the bouton number at the NMJ (Figures S1A and S1B), consistent with dFMRP blocking hiw-dependent signaling rather than inhibiting a parallel synaptic growth pathway. These results are consistent with a prior study that demonstrated overexpression of dFMRP can suppress morphological defects in hiw mutant class IV dendritic arborization (C4da) sensory neurons (Kim et al. 2013). Kim et al. demonstrated that FMRP represses translation of Dscam1 and that Wnd signaling enhances Dscam1 translation, and so they proposed a model in which FMRP and Wnd act antagonistically to tune Dscam1 levels. However, we found that while expression of FMRP fully rescues the hiw synaptic growth defect, loss of Dscam1 does not rescue (data not shown). Instead, expression of FMRP phenocopies a loss of Wnd, leading us to test the hypothesis that the Wnd protein levels might be downregulated by dFMRP.
To test if dFMRP decreases Wnd protein levels in the hiw mutant nervous system, we used the pan-neuronal driver Elav-GAL4 to overexpress either a control upstream activating sequence (UAS) transgene (RFP) or dFMRP in a hiw null background (hiwΔN) and measured endogenous Wnd protein levels in the ventral nerve cord (VNC) of third instar larvae. Examination of the VNCs reveals a substantial increase in endogenous Wnd levels in the hiwΔN background over wild-type controls, in which Wnd protein is just barely detectable (Figures 1C and 1D). Overexpression of dFMRP in the hiw mutant nervous system completely suppresses this increase in Wnd protein in the larval VNC (Figures 1C and 1D), showing that excess dFMRP decreases Wnd protein levels. We next tested whether dFMRP can regulate a transgenic Wnd construct under the control of the exogenous UAS-promoter in hiw mutants. Similar to endogenous Wnd, levels of the UAS-WndKDGFP transgene are increased in hiw mutant VNCs relative to WT—this is completely suppressed by overexpression of dFMRP (Figures 1E and 1F). Hence, dFMRP can regulate Wnd protein levels regardless of whether wnd is expressed from the endogenous or exogenous locus and so is unlikely to regulate Wnd levels via a transcriptional mechanism. We conclude that overexpressing dFMRP suppresses hiw mutant overgrowth by downregulating Wnd protein levels.
dFMRP Is Required to Limit Wnd Levels in the Nervous System and Interacts with Wnd mRNA
Given that overexpressing dFMRP can negatively regulate Wnd protein levels, we hypothesized that endogenous dFMRP normally regulates Wnd protein levels. To test this, we compared endogenous Wnd protein levels in wild-type VNCs to Wnd levels in two different transheterozygous dFmr1 mutant backgrounds: the null allele dFmr1Δ50M (Zhang et al., 2001) over a deficiency line in which dFmr1 locus is excised (Df(3R)by62 (labeled in Figure 2 as dFmr1(1)) and dFmr1Δ50M over a second null allele, dFmr12 (labeled in Figure 2 as dFmr1(2); Dockendorff et al., 2002). Western blotting for dFMRP protein confirms that these transheterozygotes lack dFMRP in the nervous system (Figure 2A). Wnd protein levels are significantly increased in the VNCs of dFmr1 mutants relative to wild type (Figures 2A and 2B). Neuronal expression of a wild-type dFmr1 rescue transgene (Dockendorff et al., 2002) reverts Wnd protein levels to those of wild-type flies (Figures 2A and 2B). Thus, dFMRP negatively regulates Wnd protein expression in the nervous system, and the absence of dFMRP results in increased Wnd protein.
Figure 2. dFMRP Is Required to Limit Wnd Levels in the Nervous System and Binds Wnd mRNA, Suggesting Translational Repression.

(A) Representative western blots of Wnd, dFMRP, and Tubulin from larval VNCs taken from wild type, two different dFmr1-transheterozygous mutants, and a dFmr1 transheterozygous mutant with a wild-type dFmr1 genomic rescue construct inserted on a different chromosome.
(B) Quantification of mean (±SEM) Wnd levels in each genotype. Wnd levels were increased in both dFmr1 mutants, and the genomic rescue of dFmr1 abolished the increase.
(C) qPCR quantification of wnd mRNA levels in wild-type versus dFmr1 mutants, presented as fold change relative to wild type. The wnd mRNA levels did not differ between wild-type and dFmr1 mutants.
(D) Representative western blots of wild-type and dFmr1 mutant larval VNCs after ex vivo incubation in cycloheximide (CHX) for the indicated times. Wnd levels are higher at baseline in dFmr1 mutants, but protein degradation is not inhibited.
(E) Quantification of mean (±SEM) Wnd levels in each genotype at each time point of CHX treatment. Wnd degradation after CHX treatment did not differ between wild-type and dFmr1 mutants.
(F) Representative immunoblot from wild-type and dFmr1 mutants. INPUT (left side of blot): 10% of the lysate prior to immunoprecipitation was collected for input controls, and blotting for dFMRP confirms the absence of dFMRP protein in dFmr1 mutants. IP (right side of the blot): dFMRP protein was immunoprecipitated from Drosophila heads, and as expected, no dFMRP was detected from dFmr1 mutants.
(G) RT-PCR products from input and immunoprecipitated samples from wild-type and dFmr1 flies. All mRNAs probed (listed on left of image) were present in the RNA input samples extracted from both genotypes. Futsch mRNA was used as a positive control and immunoprecipitated with dFMRP. rp49 mRNA was used as a negative control and did not immunoprecipitate with dFMRP. wnd mRNA immunoprecipitated with dFMRP in wild-type flies. No wnd mRNA is detected in dFmr1 mutant immunoprecipitation samples.
(H) qPCR quantification of wnd mRNA product detected from immunoprecipitated samples, normalized to wnd mRNA from input, and presented as relative mRNA levels compared to wild-type immunoprecipitated wnd mRNA. For dFmr1 mutants, all CT values were >38 or undetectable. All undetectable CT values were set to 40 for the purposes of quantification.
Statistical tests and exact p values reported in Table S1. For all quantifications: *p = 0.05; **p = 0.01; ***p = 0.001; ****p = 0.0001; NS, not significant; p > 0.05.
dFMRP is an RNA binding protein that can stall translation from its target mRNAs. While this is a likely mechanism by which dFMRP is regulating Wnd levels, we sought to test other possible regulatory mechanisms. First, the absence of dFMRP can trigger transcriptional changes (Korb et al., 2017), so we conducted qPCR in wild-type and dFmr1 larval brains to determine if wnd mRNA levels are increased in dFmr1 mutants. We observed no significant difference in wnd mRNA in the dFmr1 nervous system relative to wild type (Figure 2C). Taken with our data that excess dFMRP does not require the endogenous locus to decrease Wnd protein (Figures 1E and 1F), we conclude that dFMRP does not regulate wnd at the level of transcription. Second, Wnd protein stability could be increased in dFmr1 mutants, which could also drive increased Wnd protein levels. We used the protein synthesis inhibitor cycloheximide (CHX) to assess turnover of Wnd protein. Wild-type and dFmr1 nervous systems were exposed to CHX for 0, 6, 12, and 18 h, and we probed for Wnd protein levels at each time point. As expected, dFmr1 mutants have higher Wnd protein at baseline (Figure 2D); however, the relative rate of Wnd degradation over the time course did not vary between wild type and dFmr1 (Figures 2D and 2E), demonstrating that Wnd protein stability is not increased in dFmr1 mutants.
We then employed RNA immunoprecipitation (RNA-IP) to test the hypothesis that wnd is an mRNA target of dFMRP in vivo in the fly, which would suggest regulation at the level of translation. We collected lysates from the heads of both wild-type and dFmr1 null flies and confirmed that dFMRP could be pulled down from wild-type flies, while no dFMRP was present in dFmr1 mutants (Figure 2F). The pulldowns and subsequent western blots were performed in non-denaturing buffers to preserve any protein-protein interactions between dFMRP and Wnd. We did not detect any Wnd coimmunoprecipitating with dFMRP protein (Figure 2F). We next conducted RT-PCR to assess whether wnd mRNA immunoprecipitated with dFMRP. The mRNA for Futsch, a known target of dFMRP in the fly nervous system, was successfully detected in the immunoprecipitated samples from wild-type flies (Figure 2G). Further, the mRNA for rp49, a housekeeping gene that is commonly used as a negative control in dFMRP studies (Bienkowski et al., 2017), was not detected in immunoprecipitated samples (Figure 2G). wnd mRNA coimmunoprecipitated with dFMRP and can be observed after RT-PCR of the RNA isolated from wild-type samples immunoprecipitated with dFMRP (Figure 2G). The qPCR of immunoprecipitated RNA samples confirmed that wnd mRNA is present in wild-type samples and virtually absent in dFmr1 mutant samples (Figure 2H). Thus, we conclude that dFMRP binds wnd mRNA in vivo, and Wnd protein is negatively regulated by dFMRP in wild-type animals likely through translational control. These findings are consistent with high-throughput studies conducted in flies and mice that identified both Wnd and DLK, respectively, among FMRP bound transcripts (Darnell et al., 2011; McMahon et al., 2016).
Excess Wnd in the dFmr1 Mutant Nervous System Drives Defects in Synaptic Morphology
Wnd levels are increased in the dFmr1 mutant nervous system, and excess Wnd can trigger numerous neuronal phenotypes, including defects in NMJ morphology, axon development, and synaptic physiology (Collins et al., 2006; Goel and Dickman, 2018; Li et al., 2017; Shin and DiAntonio, 2011; Xiong et al., 2010), so we hypothesized that Wnd might be a functionally relevant target required for defects observed in dFmr1 mutant neurons. dFmr1 mutant NMJs exhibit well-established morphological phenotypes, with an increase in the number of synaptic boutons that is largely driven by a profusion of satellite boutons, which surround larger boutons rather than occurring individually along the nerve terminal and may represent an arrested state of bouton development (Siller and Broadie, 2011; Zhang et al., 2017; Gatto and Broadie, 2008; Zhang et al., 2001). We first tested if NMJ morphological defects in dFmr1 mutant larvae are Wnd dependent. We generated double-mutant flies for dFmr1 and wnd to conduct epistasis analysis of dFmr1 mutant NMJs. We recombined the dFmr1Δ50M and df(dFmr1) alleles with two different, previously validated alleles of wnd—wnd-1 and wnd-3 (Collins et al., 2006)—to generate four unique double-mutant lines in which both dFMRP and Wnd protein are absent. dFmr1 larvae exhibit synaptic overgrowth with abundant satellite boutons, while dFmr1,Wnd double mutants do not exhibit increases in total bouton number (Figures 3A and 3B) or in satellite bouton number (Figures 3A and 3C). Larvae mutant for wnd alone show no substantial differences in NMJ morphology relative to wild type (Figures 3A–3C). Hence, Wnd is necessary for morphological defects at the dFmr1 mutant NMJ.
Figure 3. Wnd Is Required for Synaptic Morphology Defects in dFmr1 Mutants.

(A) Representative images of the NMJ synaptic terminal and boutons at muscle 4 in third-instar wild type, Wnd mutant, dFmr1 mutant, and dFmr1,Wnd double mutant larvae. NMJs were stained for the presynaptic bouton marker DVGLUT (green) and nerve membrane marker HRP (red). Representative bouton images are shown in the inset panels to the right of the respective genotype.
(B) Quantification of the mean (±SEM) number of boutons per muscle 4 NMJ in each genotype. Total bouton number is increased in dFmr1 mutants relative to wild type, which is suppressed in dFmr1,Wnd double mutants.
(C) Quantification of the mean (±SEM) number of satellite boutons per muscle 4 NMJ in each genotype. Satellite bouton number is increased in dFmr1 mutants relative to wild type, which is suppressed in dFmr1,Wnd double mutants.
(D) Representative images of the NMJ synaptic terminal and boutons at muscle 4 in third-instar larvae. The Elav-GAL4 driver was used in either a wild-type or dFmr1 mutant background to drive the respective transgenes. NMJs were stained for the presynaptic bouton marker DVGLUT (green) and nerve membrane marker HRP (red). Representative bouton images are shown in the inset panels to the right of the respective genotype.
(E) Quantification of the mean (±SEM) number of total boutons per muscle 4 NMJ in each genotype.
(F) Quantification of the mean (±SEM) number of satellite boutons per muscle 4 NMJ in each genotype. Suppression by Wnd-RNAi, JNK-RNAi, and Fos-DN for both total and satellite bouton number demonstrates that presynaptic Wnd signaling drives aberrant NMJ morphology in dFmr1 mutants.
Statistical tests and exact p values reported in Table S1. Scale bar: 25 μm. *p = 0.05; **p = 0.01; ***p = 0.001; ****p = 0.0001; NS, not significant; p > 0.05.
See also Figure S2.
We next asked if Wnd can drive other neuronal morphological defects in dFmr1 mutants. We have previously demonstrated that hiw mutants exhibit Wnd-dependent defects in fly mushroom body (MB) morphology (Shin and DiAntonio, 2011), and MB morphology is also disrupted in dFmr1 mutants (Michel et al., 2004; Siller and Broadie, 2011). dFmr1 phenotypes include thin or misprojected α and/or β lobes (Michel et al., 2004). Thus, we tested if dysregulated Wnd also drives MB developmental defects in dFmr1 mutants. We examined the MBs of wild-type, dFmr1 mutants, and double dFmr1,Wnd mutants immediately after eclosion. MB phenotypes are highly sensitive to genetic background (Michel et al., 2004), and in our dFmr1Δ50M/df(dFmr1) adults, the most penetrant phenotype was malformation of the α lobe. The α lobe is substantially thinner than the β lobe, while in wild-type MBs, the width of each lobe is roughly the same (Figures S2A–S2D). We quantified the width of each fly’s α and β lobe and present these data as “α/β lobe width ratio” (Figure S2B for schematic; see Method Details for quantification details). The ratio for dFmr1 MBs is significantly decreased relative to WT (Figure S2C) due to abnormally thin α lobes. After categorizing all the α lobes across genotypes, we found that thin a lobes are a highly penetrant phenotype in mutants, affecting about 80% of the dFmr1 brains analyzed (Figure S2D). In contrast, there was no significant decrease in the MB lobe width ratio relative to wild type in dFmr1,Wnd double mutants (Figures S2C and S2D). Hence, Wnd is required for MB lobe malformations in dFmr1 mutants, demonstrating that the effects of dysregulated Wnd signaling can impact neuronal morphology beyond the larval NMJ.
We returned to the NMJ to assess whether Wnd functions in the neuron to disrupt synaptic development in dFmr1 mutants. Many important FMRP targets are regulated postsynaptically (Darnell and Klann, 2013); however, the previously described role for Wnd in promoting synaptic growth at the NMJ is presynaptic (Collins et al., 2006). To test this, we employed the panneuronal elav-GAL4 driver in a dFmr1 mutant background to drive an RNAi against Wnd exclusively in neurons. dFmr1 mutants expressing a control transgene have an increase in the number of synaptic and satellite boutons, compared to a wild-type control (Figures 3D–3F). Presynaptic knockdown of Wnd fully rescued these bouton phenotypes at the larval NMJ (Figures 3D–3F). We next tested if Wnd functions through its canonical MAP kinase cascade, in which the downstream MAP kinase JNK activates the transcription factor Fos (Collins et al., 2006; Xiong et al., 2010), by (1) RNAi knockdown of JNK and (2) overexpression of a dominant-negative transgene for the transcription factor Fos (Fos-DN) in the dFmr1 mutant background. We observed a complete suppression of the increases in bouton number and the formation of satellite boutons in the dFmr1 mutant background with either JNK or Fos loss-of-function (Figures 3D–3F). While other MAP3Ks can also signal through JNK and Fos, the full suppression from inhibition of Wnd, JNK, or Fos is consistent with the model that Wnd signaling through a JNK-/Fos-dependent transcriptional program is required for defects in dFmr1 NMJ synapse morphology.
Excess Wnd in the dFmr1 Mutant Nervous System Drives Defects in Synaptic Physiology
Having determined that Wnd is necessary in dFmr1 mutant larval NMJs to drive defects in synaptic terminal morphology, we next sought to determine whether Wnd alters synaptic transmission at the NMJ. Both the fly and mouse models of FXS exhibit significant changes in synaptic transmission (Contractor et al., 2015; Drozd et al., 2018; Repicky and Broadie, 2009). At the larval NMJ in Drosophila, previously reported phenotypes include increased evoked release (Zhang et al., 2001) and decreased quantal size (Gatto and Broadie, 2008). We tested if increased Wnd in dFmr1 mutants leads to changes in synaptic transmission at the larval NMJ. We performed electrophysiological recordings at the larval NMJ of dFmr1 mutants and observed that relative to wild type, there is an increase in quantal content (Figures 4A and 4D). Notably, and consistent with previous studies, evoked excitatory postsynaptic potential (EPSP) amplitudes are slightly but not significantly increased (Figures 4A and 4B; Repicky and Broadie, 2009). However, the amplitudes of miniature, spontaneous excitatory events (mEPSPs) are significantly decreased in dFmr1 mutants (Figures 4A and 4C), indicating that increased quantal content is driven by a decrease in postsynaptic response to the neurotransmitter in a single vesicle; thus, the nerve releases more vesicles to compensate for the decreased postsynaptic response. Such homeostatic compensation is well described at the Drosophila NMJ (Davis, 2013; Petersen et al., 1997). In dFmr1,Wnd double mutants, the decrease in mEPSP amplitude is abolished (Figure 4C), and quantal content is restored to wild-type levels (Figure 4D). Neuronal depletion of Wnd via RNAi also suppressed the decrease in mEPSP amplitude and increase in quantal content in dFmr1 mutants (Figures S3A–S3D). Hence, neuronal Wnd is required for the decrease in quantal size and the increase in quantal content during neurotransmission at the dFmr1 mutant NMJ.
Figure 4. Abnormal Synaptic Transmission at the dFmr1 Mutant Larval NMJ Requires Wnd.

(A) Representative EPSP and mEPSP electrophysiological traces from larval NMJs at Muscle 6 from wild type, dFmr1 mutant, and double dFmr1,Wnd mutants.
(B) Quantification of mean (±SEM) EPSP amplitudes.
(C) Quantification of mean (±SEM) mEPSP amplitudes. Reduced mEPSP amplitude in dFmr1 mutants is suppressed in dFmr1,Wnd double mutants.
(D) Quantification of quantal content, which was calculated individually per cell by dividing the mean EPSP amplitude by the mean mEPSP amplitude, and then averaged per genotype. Increased quantal content in dFmr1 mutants is abolished in dFmr1,Wnd double mutants.
(E) Representative images of wild type, dFmr1, and dFmr1,Wnd NMJs stained for the glutamate receptor subunits DGluRIIA (red) and DGluRIIB (green). Terminal membrane is stained with HRP (blue). In dFmr1 larvae, there is a Wnd-dependent decrease in DGluRIIA levels while DGluRIIB levels remained roughly the same.
(F) Quantification of the ratio of DGluRIIA levels to DGluRIIB levels, which was derived from analyzing the fluorescent intensities of each subunit across the entire terminal. dFmr1 NMJs have a lower DGluRIIA/DGluRIIB ratio, which is suppressed in dFmr1, Wnd. Statistical tests and exact p values reported in Table S1. Scale bar: 10 μm. *p = 0.05; **p = 0.01; ***p = 0.001; ****p = 0.0001; NS, not significant; p > 0.05.
See also Figure S3.
What are the Wnd-dependent mechanisms driving decreased quantal size and increased quantal content at the dFmr1 mutant synaptic terminal? We first assessed active zones and postsynaptic densities via staining for the active zone protein Bruchpilot (Figures S3E–S3G) and the common glutamate receptor subunit DGluRIII (also known as DGluRIIC; Figures S3E–S3G) and detected no obvious change in levels or localization, suggesting that synapse formation is largely normal in dFmr1 mutants. We next tested if postsynaptic mechanisms governing sensitivity to neurotransmitters are altered by Wnd. Increased Wnd signaling in the neuron reduces the sensitivity of the postsynaptic muscle via downregulation of glutamate receptors (Goel and Dickman, 2018). Further, dFMRP regulates the ratio of different subclasses of ionotropic glutamate receptor subunits at the larval NMJ (Pan and Broadie, 2007; Tessier and Broadie, 2012), and we have previously shown that the ratio of the two alternative subunits, DGluRIIA and DGluRIIB, regulates quantal size (DiAntonio et al., 1999). Thus, we tested if excess Wnd is driving changes in glutamate receptor subunit expression in dFmr1 mutants. We observed that the ratio of DGluRIIA to DGluRIIB expression is decreased in dFmr1 mutants in a Wnd-dependent manner (Figures 4E and 4F). Less DGluRIIA decreases quantal size (DiAntonio et al., 1999), so this would explain the decreased mEPSP amplitudes in dFmr1 mutants. Taken together, these findings indicate that changes in synaptic transmission at the NMJ in dFmr1 mutants are due to increased neuronal Wnd signaling altering postsynaptic glutamate receptor subunit composition.
dFmr1 Mutant Repetitive Grooming Behaviors are Wnd Dependent
Both Drosophila and mammalian models of FXS display behavioral abnormalities (Drozd et al., 2018). Given that Wnd is a critical downstream target of dFMRP in neuronal development and synaptic physiology, we sought to determine if this regulatory relationship extends beyond nervous system development and is required for normal behavior in Drosophila adults. One known dFmr1 mutant behavioral phenotype is excessive, repetitive grooming (Tauber et al., 2011). Adult flies execute stereotyped exploratory behavior when introduced into a novel chamber, alternating between bouts of walking, stopping, and grooming (Phillis et al., 1993; Soibam et al., 2013). dFmr1 mutants groom excessively, spending significantly more total time grooming and more time in each bout of grooming than do wild-type flies. This grooming phenotype emerges as flies age (Tauber et al., 2011). Adapting a previously published assay (Figure 5A; Tauber et al., 2011), we tested if dFmr1,Wnd double mutants retain this defect in grooming. We observed that wild-type and dFmr1 mutant flies groom the same amount at 10 days of age, but by day 25, grooming in wild-type flies remains stable while dFmr1 mutants groom themselves excessively (Figures 5B and 5C; Videos S1, S2, and S3). Excitingly, dFmr1,Wnd double mutants did not display any excessive grooming activity and were statistically indistinguishable from wild type at both ages tested (Figures 5B and 5C; Videos S1, S2, and S3). wnd mutants alone did not show any repetitive grooming behaviors (Figures 5B and 5C). Thus, Wnd is necessary for the excessive grooming behavior of dFmr1 mutant flies.
Figure 5. dFmr1 Mutant Repetitive Grooming Behaviors Are Wnd Dependent.

(A) Illustration depicting experimental setup and timeline to assay adult Drosophila grooming behavior. For all genotypes, flies were tested at 10 and 25 days to assess grooming duration and grooming bout length. All analysis was performed blinded to genotype and in random order.
(B) Quantification of percent of time spent grooming during a 3-min trial. There were no significant differences among wild-type, Wnd, or dFmr1,Wnd double mutants at any age.
(C) Quantification of average bout length within each 3-min trial at each age. There were no significant differences among wild-type, Wnd, or dFmr1,Wnd double mutants at any age.
*p = 0.05; **p = 0.01; ***p = 0.001; ****p = 0.0001; NS, not significant; p > 0.05. p values shown on figure represent statistical comparison to wild-type control within the same testing age.
Detailed statistical tests and additional comparisons available in Table S1.
A Chemical Inhibitor of Wnd Suppresses NMJ Defects in dFmr1 Mutant Larvae
The genetic data demonstrate that increased Wnd drives various neurological phenotypes in dFmr1 mutant larvae and flies. If excess Wnd signaling drives defects in dFmr1 mutants, then inhibiting the kinase activity of Wnd should also suppress dFmr1-dependent defects. Potent and selective kinase inhibitors of DLK, the mammalian ortholog of Wnd, block DLK-dependent neurodegenerative phenotypes (Patel et al., 2017; Pichon et al., 2017; Summers et al., 2018). We tested whether this DLK inhibitor (DLKi) can inhibit Wnd signaling. The most potent and best-described Wnd-dependent phenotype in Drosophila is the synaptic terminal overgrowth in hiw mutants, so we raised hiw mutant larvae on food dosed with the DLK inhibitor and assessed synaptic terminal morphology. In the hiw mutant, synaptic terminal overgrowth was fully suppressed when the larvae consumed DLK inhibitor-dosed food, while wild-type larvae raised on DLK inhibitor had no change in synaptic growth (Figures 6A and 6B). Thus, the DLK inhibitor suppresses the hiw NMJ phenotype, a downstream readout of increased Wnd MAP kinase signaling, as robustly as does genetic loss of wnd (Collins et al., 2006). Hence, we conclude that the DLK inhibitor is an effective inhibitor of the Wnd kinase in Drosophila. We next raised dFmr1 mutants on the DLK inhibitor and analyzed synaptic morphology. As with the hiw mutant, the DLK inhibitor suppresses aberrant synaptic morphology in dFmr1 mutant larvae (Figures 6C–6E). The dFmr1 mutants raised on the DLK inhibitor do not exhibit an increase in total or satellite boutons relative to wild type, while dFmr1 mutants raised on the vehicle retain these defects (Figures 6C–6E). Thus, pharmacological inhibition of Wnd kinase signaling can suppress synaptic development defects in the Drosophila FXS model.
Figure 6. An Inhibitor Against the Mammalian Ortholog of Wnd, DLK, Suppresses NMJ Morphology Defects in dFmr1 Mutant Larvae.

(A) Representative images of the NMJ synaptic terminal at muscle 4 in third-instar wild-type and hiw mutant larvae raised on food treated with either DMSO (vehicle control) or DLK inhibitor (DLKi). NMJs were stained for the presynaptic bouton marker DVGLUT (green) and nerve membrane marker HRP (red). Scale bar: 50 μm.
(B) Quantification of the mean (±SEM) number of boutons per muscle 4 NMJ in each genotype in each treatment condition. Feeding the DLK inhibitor to larvae is sufficient to suppress Wnd-dependent hiw synaptic overgrowth.
(C) Representative images of the NMJ synaptic terminal at muscle 4 in third-instar wild-type and dFmr1 mutant larvae raised on food treated with either DMSO (vehicle control) or DLK inhibitor. Representative bouton images are shown in the inset panels to the right of the respective genotype. Scale bar: 25 μm.
(D) Quantification of the mean (±SEM) number of total boutons per muscle 4 NMJ in each genotype in each treatment condition.
(E) Quantification of the mean (±SEM) number of satellite boutons per muscle 4 NMJ in each genotype. Feeding the DLK inhibitor to dFmr1 larvae is sufficient to suppress increases in total and satellite bouton number.
*p = 0.05; **p = 0.01; ***p = 0.001; ****p = 0.0001; NS, not significant; p > 0.05. p values shown on figure represent statistical comparison to wild type treated with DMSO.
Detailed statistical tests and additional comparisons available in Table S1.
Feeding Adult dFmr1 Mutant Flies the DLK Inhibitor Blocks Repetitive Grooming Behaviors
The ultimate goal in developing therapies for FXS is to ameliorate the symptoms associated with the disorder. Given that the DLK inhibitor treatment suppresses synaptic development abnormalities in dFmr1 mutant larvae, we tested if treatment with the DLK inhibitor could block excessive grooming behavior in dFmr1 mutant flies. Since the behavioral defects could result from prior developmental defects, we first asked if treating developing larvae with the DLK inhibitor would suppress behavioral defects in adults. Larvae remained on the DLK inhibitor from embryonic stage through pupation, and flies were moved to regular food within 8 h of eclosion. Grooming behavior was observed and quantified on days 10 and 25 post-eclosion. Developmental treatment with the DLK inhibitor did not reverse defects in grooming behavior. dFmr1 mutants raised as larvae on either the DMSO or DLK inhibitor displayed excessive grooming activities at day 25 relative to wild type (Figures 7A and 7B). We next asked if treating adult dFmr1 mutant flies with the DLK inhibitor, after larval and pupal development has occurred, is sufficient to block excessive grooming. Larvae were raised on regular food, and within 8 h of eclosion, adult flies to be tested for grooming were moved onto food dosed with the DMSO or DLK inhibitor and were examined for grooming behavior at days 10 and 25. Excitingly, feeding adult flies the DLK inhibitor is sufficient to prevent excessive grooming behavior in the dFmr1 mutants (Figures 7C and 7D). This suggests that aberrant Wnd signaling persists beyond development, and the deleterious effects of this signaling promotes excessive grooming behavior. Thus, Wnd signaling can be pharmacologically targeted to restore normal behavior to dFmr1 mutants, even after the bulk of neuronal development is complete, highlighting the possibility of targeting the Wnd/DLK signaling cascade for therapeutic intervention in FXS.
Figure 7. Feeding DLK Inhibitor to dFmr1 Mutant Adult Flies Prevents Repetitive Grooming Behavior.

(A) Wild-type and dFmr1 mutant larvae were raised from embryonic stages through pupation on food dosed with either vehicle (DMSO) or DLK inhibitor (50 uM). Adult flies were immediately moved to regular food after eclosion and consumed regular food throughout adulthood. Quantification of percent of time spent grooming during 3-min trials on days 10 and 25. dFmr1 mutants exhibited increased grooming in both conditions.
(B) Quantification of average bout length within each 3-min trial for wild-type and dFmr1 flies raised as larvae on either DMSO or DLK inhibitor. Average bout length was calculated by dividing the total number of bouts initiated by the total number of seconds spent grooming.
(C) Wild-type and dFmr1 mutant larvae were raised from embryonic stages through pupation on normal food, and adult flies were moved to food dosed with either DMSO or DLK inhibitor within 8 h of eclosion, limiting treatment to post-development. Quantification of percent of time spent grooming during a 3-min trial for wild-type and dFmr1 adult flies treated with either DMSO or DLK inhibitor post-eclosion.
(D) Quantification of average bout length within each 3-minute trial for wild-type and dFmr1 flies treated with either DMSO or DLK inhibitor post-eclosion. Feeding DLK inhibitor to dFmr1 mutant adults was sufficient to prevent repetitive grooming.
*p = 0.05; **p = 0.01; ***p = 0.001; ****p = 0.0001; NS, not significant; p > 0.05. p values shown on figure represent statistical comparison to wild-type control within the same testing age unless noted by an additional bar.
Detailed statistical tests and additional comparisons available in Table S1.
DISCUSSION
FXS is a common and pervasive neurodevelopmental disorder for which there are no effective therapeutic strategies. Here, we demonstrate that the highly conserved MAP3K Wnd is dysregulated in the Drosophila model of FXS. wnd is an mRNA target of dFMRP in the fly nervous system, and Wnd protein levels are elevated in dFmr1 mutants. Genetic depletion of wnd in larvae and adults lacking dFMRP suppresses aberrant neuronal morphology, physiology, and behavioral defects. Further, larvae and adults can be orally dosed with an inhibitor against the mammalian ortholog of Wnd to suppress NMJ development and adult behavioral defects. We conclude that aberrant Wnd signaling through its canonical MAP kinase cascade drives a wide variety of neurological defects imparted by the loss of dFMRP. This work highlights Wnd/DLK inhibition as a possible therapeutic strategy for FXS.
FMRP: One RNA Binding Protein, Many Targets
The search for a single, key target of FMRP that could be leveraged to alleviate the symptoms of FXS has been challenging and has yet to yield a viable therapeutic strategy (Erickson et al., 2017; Lee et al., 2018). This is perhaps not surprising, given that FMRP regulates a large set of target mRNAs, and the respective protein products have roles in virtually every aspect of neuronal function, including synaptic development and transmission (Ascano et al., 2012; Darnell et al., 2011). Moreover, loss of FMRP drives defects in many biological processes, including basal translation (Banerjee et al., 2018), epigenetic modification (Korb et al., 2017), metabolism (Weisz et al., 2018), and excitability (Contractor et al., 2015). As such, it was a surprising and exciting result that Wnd/DLK behaves as an essential, functionally relevant target for many of the neuronal phenotypes in the Drosophila model of FXS.
Why might overactive Wnd/DLK signaling have such a profound impact on FXS? Activation of Wnd/DLK triggers wide-ranging effects throughout the neuron, including defects in the synapse, axon, and cell body. Asa MAP3K, Wnd phosphorylates downstream MAP kinases that both signal locally and activate a major transcriptional response governing critical cell stress pathways. We posit that reduction of Wnd/DLK signaling has such profound effects in suppressing dFmr1 mutant phenotypes because the consequences of Wnd activation are not limited to a single pathway or cellular compartment. Of course, other FMRP targets also must contribute to neuronal dysfunction, and overactive Wnd/DLK signaling likely interacts with these other dysregulated proteins. Indeed, increased Wnd/DLK signaling in dFmr1 mutants and hiw mutants yields very different synaptic phenotypes, suggesting that the impact of Wnd/DLK activation is influenced by other molecular abnormalities in the dFmr1 mutants. It will be critical in future studies to understand how downstream effectors of Wnd/DLK signaling interact with other dysregulated molecular pathways to govern the numerous neurological defects apparent in models of FXS.
DLK Signaling in Injury and Disease
Wnd/DLK signaling through its canonical MAP kinase cascade is a critical feature of the neuronal response to insult and injury. In acute axon injury paradigms, Wnd/DLK promotes distal axon degeneration by speeding up the turnover of axon survival factors (Summers et al., 2018; Walker et al., 2017). In the proximal axon, Wnd/DLK stimulates a retrograde signal to the neuronal cell body that can stimulate axon regeneration or neuronal cell death depending on the cell type (Shin et al., 2012; Watkins et al., 2013; Welsbie et al., 2013). This retrograde signal involves activation of the MAP kinase JNK and phosphorylation of the transcription factor Jun to mount the appropriate transcriptional response. In addition to responding to acute injury, there is emerging evidence that Wnd/DLK is chronically activated in neurodegenerative diseases. DLK and JNK signaling is active in both mouse models of and human patients with Alzheimer’s disease and amyotrophic lateral sclerosis (Pichon et al., 2017). The loss or inhibition of DLK protects against the progressive neuronal degeneration that occurs in these mouse models, paralleling our finding that progressive worsening of repetitive behaviors in the dFmr1 fly can be reversed by the DLK inhibitor. We hypothesize that low-level, chronic activation of Wnd/DLK signaling might be a shared feature of neurodevelopmental and neurodegenerative disorders. Indeed, it is estimated that 50%–70% of people with Down syndrome develop Alzheimer’s disease (Hartley et al., 2015; Head et al., 2012), and premutations in the Fmr1 gene can cause the neurodegenerative disorder fragile X-associated tremor/ataxia syndrome (Hagerman and Hagerman, 2013), so a critical link may exist between development and degeneration.
Wnd/DLK as a Candidate Therapeutic Target in FXS
FXS is characterized by neuroanatomical and functional defects that are established during development, leading to lifelong cognitive, sensory, and behavioral abnormalities. However, from our work in the fly model of FXS, we propose that the key factors underlying this neurodevelopmental disorder can be parsed into two stages: neuronal development is disrupted by increased Wnd/DLK signaling, and aberrant Wnd/DLK signaling persists throughout the lifetime of the organism, further impairing neuronal function. This model is consistent with prior studies of FMRP-regulated processes, including metabotropic glutamate receptor (mGluR)-dependent protein translation (Dölen et al., 2007; Hou et al., 2006; Huber et al., 2002). mGluR activation stimulates protein translation in dendritic spines, and normally FMRP inhibits translation to serve as a “brake”; the absence of FMRP leads to excessive translation of synaptic proteins and increased AMPA receptor internalization, driving synaptic dysfunction (Dölen and Bear, 2008; Muddashetty et al., 2007; Stoppel et al., 2017). Thus, not only is initial development impaired due to improper levels of specific synaptic proteins, but increased flux through mGluR-dependent translational mechanisms drive ongoing perturbations to synaptic plasticity and function. This concept, which has made downregulation of mGluR signaling an attractive treatment approach, is critical when considering Wnd/DLK as a therapeutic target in FXS—there are signaling abnormalities that can be alleviated through inhibition of Wnd/DLK’s kinase function even after development is complete.
The idea of targeting Wnd/DLK for treatment in FXS has other advantages. First, Wnd/DLK is a MAP3K with numerous downstream signaling effectors; if DLK inhibition does not lend itself well to therapeutic inhibition, further examination of the signaling cascade may identify alternative approaches and targets. Second, while DLK is essential for embryonic development, conditional knockout of DLK later in life does not impact viability (Miller et al., 2009), suggesting that DLK inhibition is a reasonable therapeutic strategy. Finally, highly selective inhibitors against DLK are currently being tested in patients as treatments for neurodegenerative disorders (Siu et al., 2018). Testing whether the FMRP-dependent regulation of DLK is conserved and functionally relevant in the mouse model of FXS is imperative, and positive results in such experiments may motivate additional translational studies.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Aaron DiAntonio (diantonio@wustl.edu). The double dFmr1,Wnd mutant fly strains generated in this study are available upon request. No other unique reagents were generated in this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Drosophila Rearing, Strains, and Genetics
Drosophila were cultured employing standard techniques, and all crosses were maintained at 25°C in 60% relative humidity. The following stocks were used in this study: hiwΔN (null allele of hiw; Wu et al., 2005), hiwND8 (strong hypomorphic allele of hiw; Wan et al., 2000a), wnd-1 and wnd-3 (Collins et al., 2006), DVGlut-Gal4 (Daniels et al., 2008), elav3E-Gal4 (Yao and White, 1994), dFmr12 (a gift from Thomas Jongens), WTR;dFmr13 (a gift from Thomas Jongens) (Dockendorff et al., 2002). Flies obtained from the Bloomington Stock Center included: UAS-RFP (#32218), UAS-wallenda-RNAi (TRiP Collection #25396), UAS-dFMRP (#6931), dFmr1Δ50M (#6930; Zhang et al., 2001), Df(3R)Exel6265 (#7732, deficiency line for dFmr1), UAS-JNK-RNAi (#57035), and UAS-FosDN (#7214).
In all experiments in which a GAL4 driver was employed, control larvae were generated by crossing virgins of the respective driver used to UAS-RFP males. Roughly equal numbers of male and female larvae and flies were tested (with the exception of hiw mutants, in which only males were assessed as hiw is on the X chromosome), and no sex-specific differences were observed in any experiment. Detailed genotypic information for each experiment is available in Table S1.
Standard techniques were used for recombination of the dFmr1 and wnd mutant alleles. All double mutants generated via recombination were tested for the presence of the respective mutant alleles via PCR, followed by western blots to verify the absence of protein product.
METHOD DETAILS
Immunocytochemistry
NMJs and VNCs: Third instar larvae were dissected in either cold PBS or Ca2+-free HL3 and fixed in either Bouin’s fixative for 10 min at RT or for 20 min in ice cold 4% paraformaldehyde. After fixing, larval preps were washed in PBS + 0.1% Triton X-100 (PBST), then blocked and stained in PBST + 5% goat serum. Larvae were incubated overnight in primary antibodies, including rabbit α-DVGLUT antibody (1:5000; Daniels et al., 2004), rabbit α-DGluRIII (1:1000; Marrus et al., 2004), mouse α-DGluRIIA (8B4D2, 1:100, Developmental Studies Hybridoma Bank), rabbit α-DGluRIIB (1:500; Marruset al., 2004), mouse α-Brp (NC82,1:100, Developmental Studies Hybridoma Bank) and rabbit α-Wallenda antibody (Collins et al., 2006) at 1:600 dilution. After incubation with primary, larvae were washed in PBST and incubated for 1 hour in the following secondary antibodies: Cy3and 649-conjugated goat α-HRP (1:1000, Jackson ImmunoResearch), AlexaFluor 488-conjugated α-rabbit (1:1000, Invitrogen), AlexaFluor 488-conjugated α-GFP (1:1000, Invitrogen), and Cy3 α-mouse (1:1000, Jackson ImmunoResearch). After secondary, larval preps were equilibrated in 70% glycerol in PBS, and mounted and imaged in Vectashield (Vector Laboratories).
Mushroom bodies: Within 12 hours of eclosion, adult flies were anesthetized on ice and decapitated with forceps. The brain was dissected and removed in cold PBS and fixed for 20 minutes in ice cold 4% paraformaldehyde. After fixing, brains were gently washed 3 times in PBS, and then washed in PBS + 0.1% Triton X-100 (PBST), then blocked overnight in PBST + 5% goat serum, rotating at 4°C. Brains were then incubated for at least 48 hours in primary antibody (mouse anti-FasII, Developmental Studies Hybridoma Bank, 1:5 dilution in block) rotating at 4°C. Brains were then washed three times in PBST for 10 minutes, and incubated in secondary antibody (Cy3 α-mouse) for 3 hours, rotating at room temperature. After secondary, brains were washed in PBS, and equilibrated directly in Vectashield for at least 2 hours at 4°C. Brains were then mounted and imaged.
Western Blots
To analyze protein levels in the third instar larval nervous system, ventral nerve cords (VNCs) were dissected and homogenized on ice in lysis buffer (100 mM NaCl, 100 mM Tris ph 8.0, 1 mM EDTA pH 8.0, 2 M urea, 1% v/v SDS, 20 mM Na3VO4, 1x Roche protease inhibitor) with a pestle. VNCs from 10 genetically identical flies were pooled into one lysate to achieve sufficient protein concentration, and the lysate was boiled for 10 minutes, and centrifuged at RT for 10 minutes at 13000 RPM. Gel electrophoresis and transfer were performed by standard procedures (Wu et al., 2005). Membranes were blocked for 1 hour and incubated overnight at 4°C in primary antibody solution consisting of 5% milk solution in Tris-buffered saline with 0.2% Tween-20 (TBST), and one of the following antibodies: rabbit α-Wallenda (1:800), mouse α-dFMRP (5B6, Developmental Studies Hybridoma Bank, 1:500), and mouse β-Tubulin (E7, Developmental Studies Hybridoma Bank, 1:100). Primary antibody solution was removed, membranes were washed 3 times with TBST, and membranes were then incubated with HRP conjugated secondary antibodies to either anti-mouse or anti-rabbit antibody (Jackson, 1:10,000). After 1 hr of incubation, membranes were washed 3 times for 10 minutes in TBST, 1 time for 10 minutes in TBS, and developed using Immobilon Western Chemiluminescent Substrate (EMD Millipore). Membranes were imaged on a G:Box Chemi-XX6 (Syngene) and quantified using ImageJ. Wnd band intensities were normalized to the intensity of their respective β-Tubulin loading control band, and intensities are represented as fold change over the control genotype.
Cycloheximide Chase
To measure Wnd protein stability, an ex vivo preparation was employed to expose the intact nervous system to cycloheximide (CHX) for varying periods of time. Briefly, third instar larvae were dissected in ice-cold HL3.1. Upon exposure of the nervous system tissue and removal of gut tissue, dissected larvae were incubated in CHX (100 μg/mL) dissolved in HL3.1 for 6, 12, or 18 hours. After the indicated time, VNCs were dissected out and transferred to an Eppendorf tube, residual HL3.1 + CHX was removed with a pipette, and ice cold lysis buffer (described above) was added to VNC samples. 0hr time point samples were collected from VNCs after dissection in normal HL3.1. Samples were homogenized in lysis buffer, and a Micro BCA Assay was used to measure total protein and ensure equal loading across conditions and genotypes. Wnd, dFMRP, and Tub protein levels were measured and analyzed employing the western blot protocol described above. 8 to 10 VNCs were pooled per time point per genotype for each biological replicate of the CHX experiment, and 3 independent biological replicates were completed and are represented in the quantification. For each time point, Wnd protein intensities were normalized to levels of the loading control (Tub), and these values were normalized to the 0hr time point for the given genotype to present relative changes in Wnd levels over time.
qPCR
5 ventral nerve cords were dissected in ice cold PBS and incubated for 5 minutes followed by homogenization in Trizol, and the manufacturer’s instructions were followed to extract RNA. After nanodropping for concentration and DNase treatment (Promega) to eliminate genomic DNA, First-strand cDNA synthesis was performed using Quanta Biosciences qScript cDNA synthesis kit according to manufacturer’s instructions. cDNA was diluted for quantitative RT-PCR with Quanta Biosciences PerfeCTa SYBR Green FastMix with Rox. Data were analyzed using the ΔΔCT Method to calculate wnd mRNA fold change between WT and dFmr1 mutants. Quantification represents 5 biologically independent replicates. wnd mRNA levels were normalized to levels of the housekeeping gene rp49. The following primers were used to probe for rp49 (housekeeping control) and wnd:
rp49 forward TACAGGCCCAAGATCGTGAAG
rp49 reverse GACGCACTCTGTTGTCGATACC
wnd forward GGCAGGCTAAAGAACGAGACT
wnd reverse CCAAGCGGGACGGTAACAT
RNA Immunoprecipitation
Sample preparation and immunoprecipitation
~200 adult fly heads were collected and homogenized in 750 uL ice cold lysis buffer (50 mM Tris-HCL pH 7.4, 10 mM NaCl, 1% NP-40, 10mM Na3VO4, and 10mM NaF) with 1X protease inhibitors (Sigma-Aldrich, complete mini EDTA-free, #11836170001) and RNase inhibitor (RNAsin, 1U/μL, Thermo Fischer Scientific, #PRN2615). Homogenized lysate was spun at 4°C for 30 minutes and the pellet was discarded, and 500 uL of supernatant was collected. 50 uL of supernatant was collected and saved for Input blots. Protein G Dynabeads (Thermo Fischer Scientific, #10003D) were coupled to anti-dFMRP (Developmental Studies Hybridoma Bank, 25 μg antibody per 40 uL beads) by rotating end over end for 2 hours at RT. The remaining 450 uL of supernatant was precleared with blank beads for 1 hour rotating at RT. The supernatant was then transferred to tubes containing the Dynabeads conjugated to dFMRP antibody, and tubes were rotated overnight at 4°C. The following day, the beads were collected, washed 5 times for 1 minute each in a high-salt wash buffer (50 mM Tris-HCl pH 7.4,350 mM NaCl, 1% NP-40, and 1 U/μL RNAsin), and resuspended in 200 uL of lysis buffer. 50 uL was collected and processed for western blot to assess immunoprecipitation of dFMRP, and 150 uL was added to 100 uL of PBS and 750 uL Trizol to be processed for RNA extraction.
RNA extraction and qRT-PCR
Beads conjugated to the dFMRP protein were processed with Trizol following the manufacturer’s instructions (ThermoFisher Scientific, #15596026) to isolate mRNA species bound to dFMRP. Isolated RNA was Nanodropped to acquire RNA concentration, and then equal amounts of RNA from each genotype was treated with DNase (Sigma-Aldrich, DN25–1G) and cDNA was synthesized following the manufacturer’s instructions (qScript cDNA Synthesis Kit, Quantabio, VWR #733–1177). To probe for and amplify the presence of sequences of interest, qPCR was conducted on the cDNA purified from the dFMRP-bead complex, and the primer sequences for wnd and rp49 are the same as described above, and the primer sequences for Futsch (positive control for association with dFMRP) were:
Futsch forward ATCACCGCAAGTTTTGAAGG
Futsch reverse GCGAAGTCTTTTGGTGCTTC
To generate representative gel images, qPCR products were run on a 2% agarose gel and imaged using G:Box Chemi-XX6 (Syngene). Images are representative of 3 independently conducted RNA-immunoprecipitation experiments in which the results were indistinguishable.
Electrophysiology
Intracellular recordings were performed as described previously (Daniels et al., 2004). In brief, wandering 3rd instar larvae were dissected in Ca2+ free HL3.1 buffer (Feng et al., 2004) (70 mm NaCl, 5 mm KCl, 8 mm MgCl2, 10 mm NaHCO3, 5 mm trehalose, 5 mm HEPES, and 0 mm Ca2+, pH 7.2) and then washed with and recorded in HL3.1 buffer containing 0.35 mM Ca2+. Spontaneous mEPSPs and evoked EPSPs were recorded from muscle 6 in segments A2, A3, and A4 using borosilicate sharp electrodes (15–20 MΩ). Intracellular recordings were only used if a resting membrane potential between −60 and −80 mV could be maintained through the duration of the recording and if the muscle input resistance was > 5 MΩ. We did not observe significant differences in resting membrane potential or input resistance between genotypes in any experiment. 75 consecutive spontaneous events were measured per cell using MiniAnalysis Software (Synaptosoft, Decatur, GA) to determine mEPSP mean amplitude and frequency. Evoked EPSPs were recorded by sucking the end of a cut segmental nerve into a stimulating electrode and stimulating with a 1 ms pulse of sufficient amperage to recruit both axons innervating muscle 6. EPSP amplitude was calculated by averaging 75 consecutive evoked events at 2 Hz using pClamp9 software (Molecular Devices). Quantal content (QC) for each individual cell was estimated by dividing the average EPSP amplitude by the average mEPSP amplitude.
Imaging and Image Analysis
To assess synaptic growth, larvae stained for DVGlut and HRP were imaged using a 40x-oil or63x-oil immersion objective on a Leica TCS SPE confocal microscope. Images shown are maximal Z-projections of confocal stacks. Images for a given experiment (including experiments described below) were taken simultaneously using identical laser power, gain, and offset settings using the same step-size. Synaptic growth was quantified as previously described (Brace et al., 2014; Collins et al., 2006). Briefly, DVGlut-positive boutons were manually counted at larval muscle 4, segments A2 through A4, from at least 4 larvae per genotype. For Figures 3 and 6, both the total number of boutons and the number of satellite boutons were collected during counting. Satellite boutons were defined as boutons that were 1) smaller in size than immediately adjacent boutons and 2) emerging from an adjacent bouton rather than being spaced along a section of HRP positive nerve (Dickman et al., 2006; Lee and Wu, 2010).
To assess Wnd levels, larval ventral nerve cords were imaged using a 20x-oil immersion objective. Wnd levels were measured by analyzing confocal stacks for fluorescence intensity using ImageJ software (NIH; Schneider et al., 2012). A mask was created using the HRP channel to select the entire area encompassed by nervous system membrane. The mask was then applied to the stacks in which the Wnd channel had been isolated, and the mean intensity for the entire area was acquired. Mean Wnd/GFP intensities were normalized to the total area of the VNC compartment, and then presented as the fold change over the WT (control) intensity.
To assess levels of synaptic proteins (Brp, DGluRIII, DGluRIIA, DGluRIIB), NMJs were imaged using a63x-oil immersion objective, and confocal stacks were collected and analyzed for fluorescence intensity using ImageJ software (NIH). A mask was created using the HRP channel to select the entire synaptic terminal area, and the mask was applied to stacks in which the channel of interest had been isolated. The mean intensity for the entire area was acquired, normalized to the total area of the synaptic terminal, and presented as either 1) the ratio of expression of receptor subunits, found by dividing the mean DGluRIIA intensity by the mean DGluRIIB intensity (Figure 4F) or 2) the mean intensity of the given protein (Brp and DGluRIII) across the entire terminal in arbitrary units (Figures S3F and S3G).
To quantify mushroom body phenotypes in adult fly brains, mushroom body structures were imaged using a 20x-oil immersion objective. ImageJ software was used to measure the α and β lobe widths for each individual mushroom body. Using these data, an α/β lobe width ratio was calculated by dividing the α lobe width by the β lobe width for each individual mushroom body, and the average ratio for each genotype is presented. As the mushroom body is a symmetrical paired structure, each brain contains a set of two α lobes and two β lobes, both of which were quantified in each brain. Thus, each brain contributed an N of 2, with lobes exhibiting damage from the dissection being excluded from analyses. To calculate the percent penetrance of thin α lobe phenotypes, we defined “thin a lobe” as an α lobe with a width that was less than the WT mean width minus two-standard deviations. α lobes that met this criterion were classified as “thin,” and counted toward the percent of lobes exhibiting the thin phenotype.
DLK Inhibitor Experiments
GNE-3511 (MedChem Express HY-12947) referred to as DLKi, was resuspended from powder into 10 mM aliquots in DMSO. To dose larvae or flies with DLKi, the drug was diluted to either 35 uM (larvae) or 50 uM (adults) in the appropriate quantity of water to add to Formula 4–24 Instant Drosophila Medium Blue Food (Carolina, #173210) to make enough food to fill the bottom of an empty vial. For larval experiments, crosses were made directly on Blue Food such that egg-laying took place on food dosed with either control (DMSO in water) or DLKi, and larvae were exposed to drugged food for the entirety of development until dissection. For adults, flies were collected within 8 hours of eclosion and moved immediately to Blue Food with either control or DLKi. Adult flies were moved onto fresh Blue Food with the respective treatment every 5 days to ensure the drug remained active.
Grooming behavior
Grooming behaviors were observed and analyzed as previously described (Tauber et al., 2011). Briefly, male and female virgin flies of each genotype and/or drug condition were collected within 8 hours of eclosion and housed on either regular food (for genetic experiments in Figure 5 and larval dosing experiments in Figure 6) or Blue Food prepared and dosed with either drug vehicle (DMSO) or DLKi at 50 uM. 12 hours prior to testing, flies were lightly anesthetized on ice and housed individually and moved to the behavioral testing room. On the testing day, each individual fly was gently tapped into a testing chamber comprised of a small clear plastic Petri dish with an agar coated bottom. After one minute of acclimation to the chamber, grooming behaviors were observed for 3 minutes. A stopwatch was used to record the duration of grooming activities and count the number of grooming bouts initiated. After the trial, flies were rehoused on the appropriate medium. Flies were tested at days 10 and 25 post-eclosion. No significant differences in grooming were observed between male and female flies from the same genotype at any age or treatment, thus all data presented are from roughly equal numbers of male and female flies.
QUANTIFICATION AND STATISTICAL ANALYSIS
For all NMJ immunostaining and electrophysiological experiments, each NMJ terminal is considered an N of 1 since each presynaptic motor neuron is confined to its own muscle hemisegment. To control for variability between larvae within a genotype in these experiments, all data were collected from a minimum of 4 different larvae. With the exception of hiw experiments, in which only males were assessed because hiw is on the X chromosome, male and female larvae were used in equal numbers and it was confirmed that there are no statistical differences in NMJ morphology or physiology between males and females. For Wnd intensity experiments, each VNC contributed an N of 1.
In all immunostaining (larval and adult) experiments, animals were dissected and the data were analyzed blinded to genotype, and genotype was only decoded after the analysis was complete. For electrophysiology, the experimenter was blinded to genotype while recording and genotypes were decoded after traces were analyzed. For behavior, experimenter was blinded to genotype and condition, and individual flies were observed for grooming in random order, with each individual trial for each fly contributing an N of 1. Genotypes and conditions were decoded after the experiments were complete.
For all biochemistry experiments (Western blots and RNA-IP), each experiment was repeated at least three times with larvae and/or flies derived from independently made crosses. Each lane was loaded with protein from a pooled sample of 8–10 VNCs for a given genotype and experiment, and each experiment was independently conducted at least 3 times.
All data are presented as mean ± SEM. Detailed information on exact statistical tests used, N for each experiment, and exact p values can be found in Table S1, including additional statistical comparisons of interest that are not presented within the figures.
DATA AND CODE AVAILABILITY
This study did not generate or analyze datasets or code.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-DVGLUT | DiAntonio Lab | RRID: AB_2314346 |
| Rabbit anti-dGluRIII | DiAntonio Lab | N/A |
| Mouse anti-DGluRIIA | Developmental Studies Hybridoma Bank (DSHB) | DSHB Cat# 8B4D2 (MH2B); RRID: AB_528269 |
| Rabbit anti-DGluRIIB | DiAntonio Lab | N/A |
| Mouse anti-Bruchpilot | DSHB | DSHB Cat# NC82; RRID: AB_2314866 |
| Mouse anti-Fasicilin II | DSHB | Cat# 1D4; RRID: AB_528235 |
| Rabbit anti-Wallenda | DiAntonio Lab | N/A |
| Cy3 goat anti-HRP | Jackson ImmunoResearch | Cat# 123-165-021; RRID: AB_2338959 |
| 647 goat anti-HRP | Jackson ImmunoResearch | Cat# 123-605-021; RRID: AB_2338967 |
| AlexaFluor 488 goat anti-GFP | ThermoScientific - Invitrogen | Cat# A-21311; RRID: AB_221477 |
| AlexaFluor 488 goat anti-rabbit | ThermoScientific – Invitrogen | Cat# A-11034; RRID: AB_2576217 |
| Cy3 goat anti-mouse | Jackson ImmunoResearch | Cat# 115-165-146; RRID: AB_2338680 |
| Mouse anti-dFMRP | DSHB | Cat#5B6; RRID: AB_528253 |
| Mouse anti-beta tubulin | DSHB | Cat# E7; RRID: AB_2315513 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cycloheximide | Santa Cruz Biotechnology | Cat# Sc-3508 |
| Trizol | ThermoFisher | Cat# 15596018 |
| DNase | Sigma-Aldrich | Cat# DN25-1G |
| qScript cDNA synthesis kit | VWR, QuantaBio | Cat# 101414-098 |
| PerfeCTa SYBR Green FastMix - Rox | VWR, QuantaBio | Cat# 101414-278 |
| Protease inhibitor – cOmplete mini EDTA-free | SigmaAldrich | Cat# 11836170001 |
| RNase inhibitor - RNAsin | ThermoFischer Scientific | Cat# PRN2615 |
| DLK inhibitor – GNE-3511 | MedChem Express | Cat# HY-12947 |
| Formula 4-24 Instant Drosophila Medium Blue Food | Carolina Biological Supply | Cat# 173210 |
| Critical Commercial Assays | ||
| Pierce Protein Micro BCA Assay | ThermoFischer Scientific | Cat# 23227 |
| Protein G Dynabeads | ThermoFischer Scientific | Cat# 10003D |
| Experimental Models: Organisms/Strains | ||
| HiwΔN | DiAntonio Lab | RRID: BDSC_51637 |
| hiwND8 | DiAntonio Lab | RRID: BDSC_51638 |
| Wnd-1 | DiAntonio Lab | RRID: BDSC_51641 |
| Wnd-3 | DiAntonio Lab | RRID: BDSC_51999 |
| DVGlut-Gal4 | DiAntonio Lab | N/A |
| elav3E-Gal4 | DiAntonio Lab | N/A |
| dFmr12 | Jongens Lab | N/A |
| WTR;dFmr13 | Jongens Lab | N/A |
| dFmr1Δ50M,wnd-3 | This paper | N/A |
| dFmr1(df),wnd-1 | This paper | N/A |
| UAS-RFP | Bloomington Drosophila Stock Center (BDSC) | #32218 |
| Df(3R)Exel6265 (dFmr1(df)) | BDSC | #7732 |
| dFmr1Δ50M | BDSC | #6930 |
| UAS-dFMRP | BDSC | #6931 |
| UAS-JNK-RNAi | BDSC | #57035 |
| UAS-FosDN | BDSC | #7214 |
| UAS-wallenda-RNAi | BDSC | #25396 |
| Oligonucleotides | ||
| Primers for rp49, wnd, and futsch, see STAR Methods | This paper | N/A |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| MiniAnalysis | Synaptosoft | N/A |
| pClamp9 | Molecular Devices | N/A |
| Prism | GraphPad Software | N/A |
Highlights.
Drosophila dFMRP regulates MAP3K Wnd/DLK to promote nervous system function
Loss of dFMRP drives increased levels of Wnd/DLK protein in the nervous system
Increased Wnd in dFmr1 mutants causes defects in neuronal development and behavior
Wnd elimination or inhibition suppresses mutant phenotypes in the fly model of FXS
ACKNOWLEDGMENTS
This work was supported by funding from the NIH grants R01 NS065053 and R01 CA219866to A.D. and NIH F31 NS101827 to A.R. We thank Thomas Jongens for sharing dFmr1 flies. We thank Joe Dougherty and the Dougherty lab for technical advice and assistance. We also thank Jeff Milbrandt and the Milbrandt lab for helpful discussions. Lastly, we are grateful to all members of the DiAntonio Lab for fruitful scientific discussions and support.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.08.001.
REFERENCES
- Ascano M Jr., Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M, Dewell S, Hafner M, et al. (2012). FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 492, 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asghari Adib E, Smithson LJ, and Collins CA (2018). An axonal stress response pathway: degenerative and regenerative signaling by DLK. Curr. Opin. Neurobiol 53, 110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley C Jr., Wilkinson KD, Reines D, and Warren ST (1993). FMR1 protein: conserved RNP family domains and selective RNA binding. Science 262, 563–566. [DOI] [PubMed] [Google Scholar]
- Bailey DB Jr., Hatton DD, Mesibov G, Ament N, and Skinner M (2000). Early development, temperament, and functional impairment in autism and fragile X syndrome. J. Autism Dev. Disord 30, 49–59. [DOI] [PubMed] [Google Scholar]
- Banerjee A, Ifrim MF, Valdez AN, Raj N, and Bassell GJ (2018). Aberrant RNA translation in fragile X syndrome: From FMRP mechanisms to emerging therapeutic strategies. Brain Res. 1693 (Pt A), 24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardoni B, and Mandel J-L (2002). Advances in understanding of fragile X pathogenesis and FMRP function, and in identification of X linked mental retardation genes. Curr. Opin. Genet. Dev 12, 284–293. [DOI] [PubMed] [Google Scholar]
- Bassell GJ, and Warren ST (2008). Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60, 201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry-Kravis E, Raspa M, Loggin-Hester L, Bishop E, Holiday D, and Bailey DB (2010). Seizures in fragile X syndrome: characteristics and comorbid diagnoses. Am. J. Intellect. Dev. Disabil 115, 461–472. [DOI] [PubMed] [Google Scholar]
- Bhogal B, and Jongens TA (2010). Fragile X syndrome and model organisms: identifying potential routes of therapeutic intervention. Dis. Model. Mech 3, 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienkowski RS, Banerjee A, Rounds JC, Rha J, Omotade OF, Gross C, Morris KJ, Leung SW, Pak C, Jones SK, et al. (2017). The conserved, disease-associated RNA binding protein dNab2 interacts with the Fragile-X protein ortholog in Drosophila neurons. Cell Rep. 20, 1372–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brace EJ, Wu C, Valakh V, and DiAntonio A (2014). SkpA restrains synaptic terminal growth during development and promotes axonal degeneration following injury. J. Neurosci 34, 8398–8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown V, Jin P, Ceman S, Darnell JC, O’Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, et al. (2001). Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107, 477–487. [DOI] [PubMed] [Google Scholar]
- Coffee RL Jr., Tessier CR, Woodruff EA 3rd, and Broadie K (2010). Fragile X mental retardation protein has a unique, evolutionarily conserved neuronal function not shared with FXR1P or FXR2P. Dis. Model. Mech 3, 471–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CA, Wairkar YP, Johnson SL, and DiAntonio A (2006). Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron 51, 57–69. [DOI] [PubMed] [Google Scholar]
- Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, and Greenough WT (1997). Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc. Natl. Acad. Sci. USA 94, 5401–5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contractor A, Klyachko VA, and Portera-Cailliau C (2015). Altered Neuronal and Circuit Excitability in Fragile X Syndrome. Neuron 87, 699–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford DC, Acuña JM, and Sherman SL (2001). FMR1 and the fragile X syndrome: human genome epidemiology review. Genet. Med 3, 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels RW, Collins CA, Gelfand MV, Dant J, Brooks ES, Krantz DE, and DiAntonio A (2004). Increased expression of the Drosophila vesicular glutamate transporter leads to excess glutamate release and a compensatory decrease in quantal content. J. Neurosci 24, 10466–10474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels RW, Gelfand MV, Collins CA, and DiAntonio A (2008). Visualizing glutamatergic cell bodies and synapses in Drosophila larval and adult CNS. J. Comp. Neurol 508, 131–152. [DOI] [PubMed] [Google Scholar]
- Darnell JC, and Klann E (2013). The translation of translational control by FMRP: therapeutic targets for FXS. Nat. Neurosci 16, 1530–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KYS, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW (2013). Homeostatic signaling and the stabilization of neural function. Neuron 80, 718–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiAntonio A, Petersen SA, Heckmann M, and Goodman CS (1999). Glutamate receptor expression regulates quantal size and quantal content at the Drosophila neuromuscular junction. J. Neurosci 19, 3023–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickman DK, Lu Z, Meinertzhagen IA, and Schwarz TL (2006). Altered synaptic development and active zone spacing in endocytosis mutants. Curr. Biol 16, 591–598. [DOI] [PubMed] [Google Scholar]
- Dockendorff TC, Su HS, McBride SMJ, Yang Z, Choi CH, Siwicki KK, Sehgal A, and Jongens TA (2002). Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron 34, 973–984. [DOI] [PubMed] [Google Scholar]
- Dölen G, and Bear MF (2008). Role for metabotropic glutamate receptor 5 (mGluR5) in the pathogenesis of fragile X syndrome. J. Physiol 586, 1503–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dölen G, Osterweil E, Rao BSS, Smith GB, Auerbach BD, Chattarji S, and Bear MF (2007). Correction of fragile X syndrome in mice. Neuron 56, 955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drozd M, Bardoni B, and Capovilla M (2018). Modeling Fragile X Syndrome in Drosophila. Front. Mol. Neurosci 11, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson CA, Davenport MH, Schaefer TL, Wink LK, Pedapati EV, Sweeney JA, Fitzpatrick SE, Brown WT, Budimirovic D, Hagerman RJ, et al. (2017). Fragile X targeted pharmacotherapy: lessons learned and future directions. J. Neurodev. Disord 9, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Ueda A, and Wu C-F (2004). A modified minimal hemolymph-like solution, HL3.1, for physiological recordings at the neuromuscular junctions of normal and mutant Drosophila larvae. J. Neurogenet 18, 377–402. [DOI] [PubMed] [Google Scholar]
- Gatto CL, and Broadie K (2008). Temporal requirements of the fragile X mental retardation protein in the regulation of synaptic structure. Development 135, 2637–2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Summers DW, Milbrandt J, and DiAntonio A (2016). Axon self-destruction: new links among SARM1, MAPKs, and NAD+ metabolism. Neuron 89, 449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel P, and Dickman D (2018). Distinct homeostatic modulations stabilize reduced postsynaptic receptivity in response to presynaptic DLK signaling. Nat. Commun 9, 1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman R, and Hagerman P (2013). Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. Lancet Neurol. 12, 786–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley D, Blumenthal T, Carrillo M, DiPaolo G, Esralew L, Gardiner K, Granholm A-C, Iqbal K, Krams M, Lemere C, et al. (2015). Down syndrome and Alzheimer’s disease: Common pathways, common goals. Alzheimers Dement. 11, 700–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head E, Powell D, Gold BT, and Schmitt FA (2012). Alzheimer’s Disease in Down Syndrome. Eur. J. Neurodegener. Dis 1, 353–364. [PMC free article] [PubMed] [Google Scholar]
- Hou L, Antion MD, Hu D, Spencer CM, Paylor R, and Klann E (2006). Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron 51, 441–454. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, and Bear MF (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. USA 99, 7746–7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Wang X, Coolon R, and Ye B (2013). Dscam expression levels determine presynaptic arbor sizes in Drosophila sensory neurons. Neuron 78, 827–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korb E, Herre M, Zucker-Scharff I, Gresack J, Allis CD, and Darnell RB (2017). Excess Translation of Epigenetic Regulators Contributes to Fragile X Syndrome and Is Alleviated by Brd4 Inhibition. Cell 170, 1209–1223.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, and Wu C-F (2010). Orchestration of stepwise synaptic growth by K+ and Ca2+ channels in Drosophila. J. Neurosci 30, 15821–15833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AW, Ventola P, Budimirovic D, Berry-Kravis E, and Visootsak J (2018). Clinical Development of Targeted Fragile X Syndrome Treatments: An Industry Perspective. Brain Sci. 8, E214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhang YV, Adib EA, Stanchev DT, Xiong X, Klinedinst S, Sopina P, Jahn TR, Hume RI, Rasse TM, et al. (2017). Restraint of presynaptic protein levels by Wnd/DLK signaling mediates synaptic defects associated with the kinesin-3 motor Unc-104. eLife 6, e24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrus SB, Portman SL, Allen MJ, Moffat KG, and DiAntonio A (2004). Differential localization of glutamate receptor subunits at the Drosophila neuromuscular junction. J. Neurosci 24, 1406–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon AC, Rahman R, Jin H, Shen JL, Fieldsend A, Luo W, and Rosbash M (2016). TRIBE: Hijacking an RNA-Editing Enzyme to Identify Cell-Specific Targets of RNA-Binding Proteins. Cell 165, 742–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel CI, Kraft R, and Restifo LL (2004). Defective neuronal development in the mushroom bodies of Drosophila fragile X mental retardation 1 mutants. J. Neurosci 24, 5798–5809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BR, Press C, Daniels RW, Sasaki Y, Milbrandt J, and DiAntonio A (2009). Adual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat. Neurosci 12, 387–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muddashetty RS, Kelic S, Gross C, Xu M, and Bassell GJ (2007). Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J. Neurosci 27, 5338–5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L, and Broadie KS (2007). Drosophila fragile X mental retardation protein and metabotropic glutamate receptor A convergently regulate the synaptic ratio of ionotropic glutamate receptor subclasses. J. Neurosci 27, 12378–12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Meilandt WJ, Erickson RI, Chen J, Deshmukh G, Estrada AA, Fuji RN, Gibbons P, Gustafson A, Harris SF, et al. (2017). Selective Inhibitors of Dual Leucine Zipper Kinase (DLK, MAP3K12) with Activity in a Model of Alzheimer’s Disease. J. Med. Chem 60, 8083–8102. [DOI] [PubMed] [Google Scholar]
- Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, and DiAntonio A (1997). Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron 19, 1237–1248. [DOI] [PubMed] [Google Scholar]
- Phillis RW, Bramlage AT, Wotus C, Whittaker A, Gramates LS, Seppala D, Farahanchi F, Caruccio P, and Murphey RK (1993). Isolation of mutations affecting neural circuitry required for grooming behavior in Drosophila melanogaster. Genetics 133, 581–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichon CEL, Meilandt WJ, Dominguez S, Solanoy H, Lin H, Ngu H, Gogineni A, Ghosh AS, Jiang Z, Lee S-H, et al. (2017). Loss of dual leucine zipper kinase signaling is protective in animal models of neurodegenerative disease. Sci. Transl. Med 9, eaag0394. [DOI] [PubMed] [Google Scholar]
- Pieretti M, Zhang FP, Fu Y-H, Warren ST, Oostra BA, Caskey CT, and Nelson DL (1991). Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66, 817–822. [DOI] [PubMed] [Google Scholar]
- Repicky S, and Broadie K (2009). Metabotropic glutamate receptor-mediated use-dependent down-regulation of synaptic excitability involves the fragile X mental retardation protein. J. Neurophysiol 101, 672–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, and DiAntonio A (2011). Highwire regulates guidance of sister axons in the Drosophila mushroom body. J. Neurosci 31, 17689–17700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, Cho Y, Beirowski B, Milbrandt J, Cavalli V, and DiAntonio A (2012). Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron 74, 1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siller SS, and Broadie K (2011). Neural circuit architecture defects in a Drosophila model of Fragile X syndrome are alleviated by minocycline treatment and genetic removal of matrix metalloproteinase. Dis. Model. Mech 4, 673–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DJ, and Watkins TA (2018). Therapeutic opportunities and pitfalls in the treatment of axon degeneration. Curr. Opin. Neurol 31, 693–701. [DOI] [PubMed] [Google Scholar]
- Siu M, Sengupta Ghosh A, and Lewcock JW (2018). Dual Leucine Zipper Kinase Inhibitors for the Treatment of Neurodegeneration. J. Med. Chem 61, 8078–8087. [DOI] [PubMed] [Google Scholar]
- Soibam B, Shah S, Gunaratne GH, and Roman GW (2013). Modeling novelty habituation during exploratory activity in Drosophila. Behav. Processes 97, 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppel LJ, Auerbach BD, Senter RK, Preza AR, Lefkowitz RJ, and Bear MF (2017). β-arrestin2 couples metabotropic glutamate receptor 5 to neuronal protein synthesis and is a potential target to treat fragile X. Cell Rep. 18, 2807–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers DW, Milbrandt J, and DiAntonio A (2018). Palmitoylation enables MAPK-dependent proteostasis of axon survival factors. Proc. Natl. Acad. Sci. USA 115, E8746–E8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauber JM, Vanlandingham PA, and Zhang B (2011). Elevated levels of the vesicular monoamine transporter and a novel repetitive behavior in the Drosophila model of fragile X syndrome. PLoS ONE 6, e27100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier CR, and Broadie K (2012). Molecular and genetic analysis of the Drosophila model of fragile X syndrome. Results Probl. Cell Differ. 54, 119–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Dutch-Belgian Fragile X Consortium (1994). Fmr1 knockout mice: a model to study fragile X mental retardation. Cell 78, 23–33. [PubMed] [Google Scholar]
- Walker LJ, Summers DW, Sasaki Y, Brace EJ, Milbrandt J, and DiAntonio A (2017). MAPK signaling promotes axonal degeneration by speeding the turnover of the axonal maintenance factor NMNAT2. eLife 6, e22540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan HI, DiAntonio A, Fetter RD, Bergstrom K, Strauss R, and Goodman CS (2000a). Highwire regulates synaptic growth in Drosophila. Neuron 26, 313–329. [DOI] [PubMed] [Google Scholar]
- Wan L, Dockendorff TC, Jongens TA, and Dreyfuss G (2000b). Characterization of dFMR1, a Drosophila melanogaster homolog of the fragile X mental retardation protein. Mol. Cell. Biol 20, 8536–8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H (2015). Fragile X mental retardation protein: from autism to neurodegenerative disease. Front. Cell. Neurosci 9, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren ST, and Nelson DL (1994). Advances in molecular analysis of fragile X syndrome. JAMA 271, 536–542. [PubMed] [Google Scholar]
- Watkins TA, Wang B, Huntwork-Rodriguez S, Yang J, Jiang Z, Eastham-Anderson J, Modrusan Z, Kaminker JS, Tessier-Lavigne M, and Lewcock JW (2013). DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc. Natl. Acad. Sci. USA 110, 4039–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisz ED, Towheed A, Monyak RE, Toth MS, Wallace DC, and Jongens TA (2018). Loss of Drosophila FMRP leads to alterations in energy metabolism and mitochondrial function. Hum. Mol. Genet 27, 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsbie DS, Yang Z, Ge Y, Mitchell KL, Zhou X, Martin SE, Berlinicke CA, Hackler L Jr., Fuller J, Fu J, et al. (2013). Functional genomic screening identifies dual leucine zipper kinase as a key mediator of retinal ganglion cell death. Proc. Natl. Acad. Sci. USA 110, 4045–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Wairkar YP, Collins CA, and DiAntonio A (2005). Highwire function at the Drosophila neuromuscular junction: spatial, structural, and temporal requirements. J. Neurosci 25, 9557–9566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Wang X, Ewanek R, Bhat P, Diantonio A, and Collins CA (2010). Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J. Cell Biol 191, 211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao K-M, and White K (1994). Neural specificity of elav expression: defining a Drosophila promoter for directing expression to the nervous system. J. Neurochem 63, 41–51. [DOI] [PubMed] [Google Scholar]
- Zhang YQ, Bailey AM, Matthies HJG, Renden RB, Smith MA, Speese SD, Rubin GM, and Broadie K (2001). Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell 107, 591–603. [DOI] [PubMed] [Google Scholar]
- Zhang X-Y, Qi J, Shen Y-Q, Liu X, Liu A, Zhou Z, Han J, and Zhang ZC (2017). Mutations of PQBP1 in Renpenning syndrome promote ubiquitin-mediated degradation of FMRP and cause synaptic dysfunction. Hum. Mol. Genet 26, 955–968. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate or analyze datasets or code.