

SUMMARY
Non-homologous end joining (NHEJ) and homologous recombination (HR) are the two major pathways of DNA double-strand break (DSB) repair and both are highly conserved from yeast to mammals. Nej1 has a role in DNA end-tethering at a DSB, and the Mre11/Rad50/Xrs2 (MRX) complex is important for its recruitment to the break. Nej1 and Dna2-Sgs1 interact with the C-terminal end of Mre11, which also includes the region where Rad50 binds. By characterizing the functionality of Nej1 in two rad50 mutants, which alter the structural features of MRX, we demonstrate that Nej1 inhibits the binding of Dna2 to Mre11 and Sgs1. Nej1 interactions with Mre11 promote tethering and inhibit hyper-resection, and when these events are compromised, large deletions develop at a DSB. The work indicates that Nej1 provides a layer of regulation to repair pathway choice and is consistent with its role in NHEJ.
Graphical Abstract
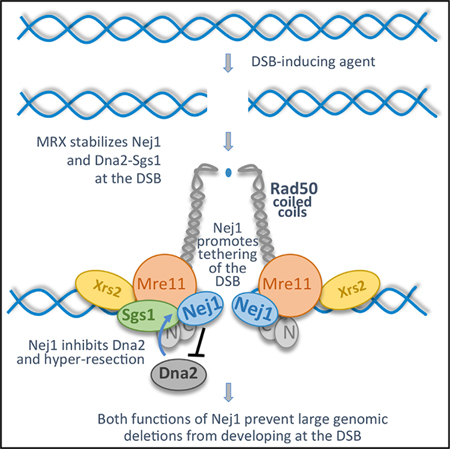
In Brief
Mojumdar et al. characterize the role of Nej1 during double-strand break repair. They show Nej1 promotes non-homologous end joining (NHEJ) by tethering the broken DNA ends and by inhibiting hyper-resection mediated by Dna2-Sgs1.
INTRODUCTION
Non-homologous end joining (NHEJ) and homologous recombination (HR) are the two central pathways of DNA double-strand break (DSB) repair. DNA end-tethering and 5′ resection are key processes at a DSB that impact repair pathway choice (Symington, 2016). NHEJ involves the direct ligation of broken ends with little or no processing, whereas HR requires 5′ DNA resection. If resection initiates, then NHEJ is no longer an option.
Repair factors that are recruited to the break are primarily categorized for their involvement in one of these two canonical pathways. For example, Nej1 is a core component of NHEJ, and its loss leads to end joining defects similar to those seen in ku70Δ and dnl4Δ mutant cells (Frank-Vaillant and Marcand, 2001; Valencia et al., 2001; Kegel et al., 2001; Chen et al., 2001). The initial recruitment of Nej1 to a DSB depends on yKu70/80 (Ku), and while Nej1 has no identified enzymatic activity, it has been shown to stimulate the ligase activity of Dnl4-Lif1 through its interactions with Lif1 (Chen and Tomkinson, 2011). Moreover, Nej1 promotes NHEJ indirectly by downregulating HR in a number of ways including stabilizing Ku, once it is recruited, which protects the DNA ends from nucleases (Chen and Tomkinson, 2011). Nej1 also helps in localizing Srs2 to the break, which inhibits Rad51 filament formation (Carter et al., 2009). Lastly, our previous work showed that Nej1 inhibits Dna2-Sgs1-dependent hyper-resection at the DSB; however, the mechanism behind this regulation remains unknown (Sorenson et al., 2017). The Nej1 human homolog, XLF, has been shown to facilitate NHEJ in the genome by aligning broken ends of a DSB in the genome prior to ligation (Reid et al., 2015; Graham et al., 2018). In vitro, Nej1 binds to DNA ends (Chen and Tomkinson, 2011); however, a role for Nej1 in end-tethering in vivo has not been determined.
The Mre11/Rad50/Xrs2 (MRX) complex is central to both NHEJ and HR. The structural features of MRX are important for end-tethering, and the endonuclease activity of Mre11 is important for HR by initiating resection at the break (Cannavo and Cejka, 2014). Ku is required for the efficient recruitment of MRX (Zhang et al., 2007); however, Ku and MRX function antagonistically in repair pathway choice (Clerici et al., 2008; Wu et al., 2008; Wasko et al., 2009; Balestrini et al., 2013). Both the CXXC hook motif and the extended coiled-coil region of Rad50 are important for DSB repair. Mutations in the extended regions of Rad50 transmit structural changes to the DNA binding globular head region of the complex where Rad50 interacts with Mre11 (Hohl et al., 2015; Park et al., 2017). The integrity of the hook is essential for tethering (Kaye et al., 2004; Lobachev et al., 2004); however, the impact of the coiled-coil region on end-tethering has not determined.
We show that MRX is essential for the recruitment of Nej1 to a DSB and its recruitment in turn regulates the level of Dna2 recovered at the break. Both Nej1 and Dna2 interact with the C-terminal end of Mre11, which is the same region in Mre11 where Rad50 binds to form the DNA binding globular head domain of the complex. Underscoring the importance of this Mre11 region in regulating interactions with repair factors, rad50 mutations in the distal coiled-coil region that transmit structural changes to the globular region reduce the levels of Nej1 and Dna2-Sgs1 recovered at a DSB. The association of Nej1 with MRX provides an important layer of regulation in DNA damage repair, which prevents genomic mutations from arising at a DSB. Nej1 has a role in two events that promote NHEJ. First, Nej1 functions in end-tethering, a process that directly affects NHEJ mediated repair. Second, Nej1 regulates the level of Dna2 nuclease that is recruited to the DSB, which indirectly promotes NHEJ through inhibiting resection.
RESULTS
Interactions between Nej1 and MRX at the DSB
We previously demonstrated that the loss of NEJ1 resulted in hyper-resection at a DSB (Sorenson et al., 2017). The MRX complex is one of the earliest factors recruited to the DSB and it is central to both NHEJ and HR in yeast; therefore, we determined whether Nej1 recruitment to a break depended on MRX (Wu et al., 2008, Mahaney et al., 2014, Sorenson et al.; 2017). Chromatin immunoprecipitation (ChIP) at the homothallic switching endonuclease (HO)-induced DSB showed that in rad50Δ mutant cells there was a significant reduction in the level of Nej1Myc recovered compared to wild-type (WT) cells (Figure 1A).
Figure 1. Nej1 Interact with MRX at DSB Site.

(A) Enrichment of Nej1Myc at DSB, at 0and 3-h time points, in WT (JC-1687), rad50Δ (JC-3311), rad50sc+h (JC-4526), rad50sc+h N873I (JC-4563), and no tag control (JC-727) were determined at 0.6 kb from DSB. The fold enrichment is normalized to recovery at the SMC2 locus.
(B) Y2H analysis between Nej1 fused to hemagglutinin tagged activation domain (HA-AD) and Mre11, Rad50, and Xrs2 fused to LexA-DBD was performed using a quantitative β-galactosidase assay as described previously in Bustard et al. (2012).
(C) Schematic representation of Mre11 and its functional domains. In green is the N terminus region (1–271 aa) of Mre11 that contains the four phosphodiesterase motifs, in orange is the region from 272–422 aa consisting of the DNA binding domain, and in gray is the C terminus region (423–692 aa) of Mre11 that contains the Rad50 binding site. Fragment generation is based on the sequence alignment shown in Hopfner et al. (2001).
(D) Y2H analysis was performed between Nej1 fused to HA-AD and Mre11 fragments (N-terminal, DBD, and C-terminal region) fused to LexA-DBD. All constructs are under galactose induction as shown in Figure S6, and quantitative β-galactosidase assays were performed as described in STAR Methods.
(E) Schematic showing Rad50 WT, sc+h, and sc+h N873I based on work described in Hohl et al. (2011, 2015).
(F) Enrichment of Xrs2HA at DSB, at 0- and 3-h time points, in WT (JC-4515), rad50Δ (JC-4516), rad50sc+h (JC-4518), rad50sc+h N873I (JC-4572), and no tag control (JC-727) were determined at 0.6 kb from DSB.
(G) Percentage cell survival upon chronic HO induction in WT (JC-727), rad50Δ (JC-3313), rad50sc+h (JC-4424), rad50sc+h N873I (JC-4561), nej1Δ (JC-1342), nej1Δ rad50Δ (JC-3314), nej1Δ rad50sc+h (JC-4476), and nej1Δ rad50sc+h N873I (JC-4597). Analysis was performed in triplicate from at least three biological replicate experiments. Statistical analysis is described in STAR Methods.
We next wanted to determine whether a physical interaction between Nej1 and MRX existed. Yeast two-hybrid (Y2H) was performed to obtain a quantitative measurement of Nej1 interaction(s) with each component of the MRX complex as previously described (Bustard et al., 2012; Mahaney et al., 2014). This approach was taken as coimmunoprecipitation (coIP) with Nej1 and is difficult because of its rapid degradation and short half-life (Carter et al., 2009; Deshpande and Wilson, 2007; Frank-Vaillant and Marcand, 2001; Mahaney et al., 2014). Nej1 was expressed as hemagglutinin (HA)-tagged prey and each subunit of MRX was expressed as LexA-tagged bait, and all constructs were under control of a galactose inducible promoter. Nej1 interacted with Mre11, but not Rad50 or Xrs2 (Figure 1B). To map the region in Mre11 where Nej1 binds, three fragments of Mre11 were expressed as bait. These included Mre11-N (1–271 aa), which contained the phosphodiesterase domain; Mre11-DBD (272–422 aa), which included the DNA binding domain; and Mre11-C (423–692 aa), which included the coiled-coil region of Mre11 where Rad50 binds and a DNA binding domain (Figure 1C). Nej1 interacted specifically with the C-terminal fragment of Mre11 (Figure 1D). Nej1 binding with Mre11-C was reduced relative to full-length Mre11 (Figures 1B and 1D). One explanation is that proper folding of the fragments could be compromised outside the context of the entire protein, underscoring the importance of full-length Mre11 for this interaction.
To understand the physiological importance of the Nej1-MRX interaction in DSB repair, we utilized two rad50 mutants, rad50sc+h and rad50sc+h N873I (Figure 1E). In both alleles, an internal deletion of 243 residues, including 105 and 129 residues flanking the hook, reduces the length of the coiled-coil from the distal end; however, the CXXC hook motif remains present (Hohl et al., 2011). In rad50sc+h, these changes were transmitted to the globular domain of the complex, where Rad50 interacts with Mre11; however, initial work showed that the expression of rad50sc+h did not affect Rad50-Mre11 interactions or MRX complex formation (Hohl et al., 2011). A suppressor mutation in rad50sc+h, resulting in the N→I change at position 873 between the distal and globular head of Rad50, restored some functionality of the MRX complex (Figure 1E; Hohl et al., 2015). The rationale for characterizing Nej1 in the context of these alleles was first, the sequence of Mre11 where Nej1 binds the complex remains intact, and second, NHEJ was markedly defective in rad50sc+h mutants (Hohl et al., 2011). These coiled-coil mutants had less of an impact on HR-mediated repair, indicating that the role of MRX in HR and NHEJ was distinct and separable (Hohl et al., 2011). To assess the localization of MRX to a DSB in these rad50 alleles, ChIP was performed with Xrs2HA. Xrs2HA enrichment at HO was detected in both rad50sc+h mutants, indicating MRX assembles in vivo at the break site (Figure 1F). Xrs2HA levels were reduced in rad50sc+h mutant cells, but not to rad50Δ levels, and recovery in rad50sc+h N873I mutants was similar to WT (Figure 1F). These data show that upon inclusion of N873I in rad50sc+h mutants, MRX association with the break is restored to levels indistinguishable from RAD50+ cells (Figure 1F).
We next measured cell survival during chronic expression of the HO endonuclease (Figure 1G). Survival rates are low under these conditions as cells survive only when the break is imprecisely repaired, which disrupts the HO recognition site and prevents further cleavage. The level of cell survival and the mating type of survivors provides information about how repair factors function at the break site. Consistent with initial work, the survival frequency for rad50sc+h was 25-fold lower than in WT cells (Figure 1G; Hohl et al., 2011). In contrast, the survival of rad50sc+h N873I was 3-fold lower than WT, thus the introduction of N873I into the rad50sc+h background resulted in an ~8-fold increase in survival. This increase might be attributed in part to the stable association of MRX at the DSB in rad50sc+h N873I compared to rad50sc+h, which could impact the recovery of other repair factors like Nej1. Indeed, reduced levels of Nej1Myc were observed in rad50sc+h mutant cells; however, recovery in rad50sc+h N873I was not different than WT (Figure 1A). To determine epistasis between NEJ1 and these rad50 alleles, nej1Δ was combined with rad50sc+h ± N873I. The survival rate dropped ~120-fold for both nej1Δ rad50sc+h and nej1Δ rad50sc+h N873I double mutants compared to rad50sc+h and rad50sc+h + N873I, respectively (Figure 1H). Interestingly, the survival rate of both double mutants was lower than in nej1Δ rad50Δ mutant cells. These findings raise the possibility that Nej1 promotes NHEJ by constraining an event that depends on MRX being at the DSB. The MRX complex has a central role in DNA end-tethering and 5′ resection. We reasoned that investigating Nej1 together with rad50sc+h ± N873I in these processes would be instructive for understanding the function of Nej1 in DSB repair.
Role of Nej1 in DNA End-Tethering at the DSB
The Nej1 homolog in humans, XLF, is critical for DNA end-tethering at a DSB, which is a critical step in repair (Reid et al., 2015; Graham et al., 2016, 2018; Mahaney et al., 2014); however, a role for Nej1 in this process has not been reported. To measure tethering in the HO-induced system, DNA regions proximal to the DSB were visualized by TetO integrated 3.2 kb upstream and the LacO array integrated 5.2 kb downstream of the cut site in cells expressing TetR-GFP and LacO-mCherry (Figures 2A and 2B). The level of end-to-end tethering upon DSB formation was determined by measuring the colocalization of GFP and mCherry foci. After HO induction to generate a DSB, ~20% of the DNA ends in WT cells were untethered (Figure 2C). In our system, the mean end-to-end distance in WT cells was 0.18 µm and this increased to 0.31 mm in nej1Δ. Moreover, the percentage of cells with untethered ends increased to 26% in cells lacking NEJ1 (Figure 2C). Lif1, which is the yeast equivalent of human XRCC4 and the binding partner of XLF, interacts with Xrs2 and stabilizes MRX at the break site (Chen et al., 2001; Palmbos et al., 2008; Zhang et al., 2007; Matsuzaki et al., 2008). Loss of LIF1, but not DNL4, resulted in a tethering defect that was similar to nej1Δ cells (Figure S1). Tethering defects were additive in nej1Δ lif1Δ doublemutant cells (Figure S1) and in line with earlier work that showed the loss of NEJ1 did not impact Lif1 localization to the DSB (Sorenson et al., 2017).
Figure 2. Nej1 Plays a Role in End-Tethering at DSB Site.

(A) Schematic representation of regions around the HO cut site on chromosome III. The ChIP probe used in this study is 0.6 kb from the DSB. The location of the RsaI site 0.15 kb from the DSB used in the qPCR resection assays is also indicated. Opposite sides of HO-induced DSB are tagged with GFP (3.2 kb from cut site) and mCherry (5.2 kb from cut site) repeats, which were used in end-tethering microscopy.
(B) Representative image of yeast cells with tethered (co-localized GFP and mCherry) and untethered (distant GFP and mCherry) ends.
(C) Scatter data plot showing the tethering of DSB ends, as measured by the distance between the GFP and mCherry foci in WT (JC4066), nej1Δ (JC-4364), rad50Δ (JC-4095), nej1Δ rad50Δ (JC-4355), rad50sc+h (JC-4466), nej1Δ rad50sc+h (JC-4533), rad50sc+h N873I (JC-4559), and nej1Δ rad50sc+h N873I (JC-4580). HO cutting in all strains was greater than 96% for all the experiments, and the analysis was performed on 100 cells from three biological replicate experiments.
Statistical analysis is described in STAR Methods.
Consistent with previous reports, rad50Δ mutants show a defect in tethering (Deshpande et al., 2014). Similar to rad50Δ mutants, cells carrying the rad50sc+h mutation showed an increase in the percentage of untethered ends to 44% and a >2-fold increase in the mean end-to-end distance to 0.43 mm (Figure 2C). The tethering defect in both rad50sc+h and rad50Δ mutants, as well as mre11Δ, was greater than that observed in nej1Δ mutants (Figures 2C and S1). This is independent of Mre11 nuclease activity, which was previously shown to have no effect on end-tethering (Kaye et al., 2004; Lobachev et al., 2004), and is consistent with the loss of tethering observed in cells lacking XRS2 (Oh et al., 2018).
In double-mutant cells, when nej1Δ was combined with rad50Δ or rad50sc+h, the tethering defect was epistatic, with no significant changes compared to the single mutants (Figure 2C). Cells harboring rad50sc+h N873I showed a deficiency in tethering too with a mean distance between foci that was 0.39 µm; however, the percentage of cells with untethered ends was significantly lower in rad50sc+h N873I (26.1%) compared to rad50sc+h (Figure 2C). Better tethering could be due directly to changes in the coiled-coil structure of Rad50 or indirectly to more MRX recruited to the DSB upon N873I inclusion (Figure 1F). Moreover, Nej1 is important for tethering, and in nej1Δ rad50sc+h N873I double mutants, the level of untethered ends increased to ~50%, a defect that was greater than that observed when nej1Δ was combined with the rad50sc+h or rad50Δ mutations (Figure 2C). The findings suggest Nej1 might function to inhibit a process distinct of tethering, which is compounded by a defect in MRX-dependent tethering and prompted our investigation of 5′ resection at the DSB.
Interplay of DNA End-Tethering and Resection in Maintaining Genomic Stability
Our previous work showed that Nej1 prevented hyper-resection and that this was dependent on MRX but not the nuclease activity of Mre11 (Sorenson et al., 2017). 5′ resection was measured by a qPCR-based approach 2 and 6 h after HO induction in G1-arrested cells. This method relies on an RsaI cut site located 0.15 kb from the DSB (Figure 3A). If resection progresses beyond the RsaI cut site, then the region would not be cleaved and would be amplified by PCR (Sorenson et al., 2017; Ferrari et al., 2015). A resection defect was observed in rad50sc+h mutant cells that was indistinguishable from the level of resection in rad50Δ mutants (Figure 3B). However, there was a different impact on resection in double mutants when nej1Δ was combined with rad50sc+h or rad50Δ. The resection defect in nej1Δ rad50Δ was similar to rad50Δ at 6 h; however, resection in nej1Δ rad50sc+h mutant cells was not statistically different from WT (Figure 3B). Interestingly, at an earlier 2-h time point, resection was observed in both nej1Δ rad50sc+h N873I and nej1Δ rasd50Δ, but not in nej1Δ rad50sc+h mutants (Figure 3B). Resection in nej1Δ rasd50Δ mutants defectively plateaued by 2 h; however, by 6 h in nej1Δ rad50sc+h N873I mutants, hyper-resection ensued similar to that in nej1Δ (Figure 3B).
Figure 3. Interplay of DNA End-Tethering and Resection in Maintaining Genomic Stability.

(A) Schematic representation of regions around HO cut site on chromosome III. The RsaI site used in the qPCR resection assays is 0.15 kb from the DSB.
(B and F) Resection of DNA 0.15 kb away from the HO DSB, as measured by percent single-stranded DNA (ssDNA), at 0, 2, and 6 h post DSB induction in G1 cells (B) or at 0, 30, 60, 90, and 120 min post DSB induction in cycling cells (F) in WT (JC-3585), nej1Δ (JC-3884), rad50Δ (JC-3882), nej1Δ rad50Δ (JC-3887), rad50sc+h (JC-4458), nej1Δ rad50sc+h (JC-4471), rad50sc+h N873I (JC-4567), and nej1Δ rad50sc+h N873I (JC-4569). Analysis was performed in triplicate from at least three biological replicate experiments. Statistical analysis is described in STAR Methods.
(C) Schematic representation of mating-type analysis of survivors from persistent DSB induction assays. Disruption of the MATa1 gene results in a sterile phenotype, and disruption of the MATα2 gene (~700 bp upstream of HO cut site) results in an a-like phenotype in the mating type assays. The mating phenotype is a readout for the type of repair: α survivors (mutated HO endonuclease in gray), sterile survivors (small insertions or deletions in yellow), and a-like survivors (>700 bp deletion in red).
(D and E) The mating phenotype of survivors (D) and mutagenic events (E) were determined by DNA sequencing in 20 sterile survivors (except in nej1Δ and nej1Δ rad50Δ because few survived) and 20 a-like survivors (except WT, rad50sc+h, and rad50sc+h N873I) and shown in Figure S2. Strains include WT (JC-727), nej1Δ (JC-1342), rad50Δ (JC-3313), nej1Δ rad50Δ (JC-3314), rad50sc+h (JC-4424), nej1Δ rad50sc+h (JC-4476), rad50sc+h N873I (JC-4561), and nej1Δ rad50sc+h N873I (JC-4597).
As mentioned, for a cell to survive continuous HO induction, the break must be repaired imprecisely. This can occur through pathways that introduce mutations, including NHEJ and MMEJ, or by single strand annealing (SSA), which requires long regions of homology and results in the loss of a large amount of genetic material during repair. The mating type of surviving cells is a proxy for the type of genomic alterations that develop during repair (Moore and Haber, 1996). Two genes that regulate mating type, MATα1 and MATα2, are located adjacent to the HOinduced DSB (Figure 3C). Large deletions (>700 bp) around the DSB, where both α1 and α2 have been disrupted, result in an a-like mating type (red, Figure 3C), whereas small deletions or insertions at the DSB lead to a sterile mating type (yellow, Figure 3C), The α-type survivors arise from disruptions in the HO endonuclease and are not a direct readout for repair at the DSB (gray, Figure 3C).
Repair events in rad50sc+h and rad50sc+h N873I survivors were similar to each other and to WT with small insertions and deletions (Figure 3D). Consistent with previous work, ~12% of nej1Δ mutant survivors incurred large deletions (Sorenson et al., 2017). We wanted to determine the genetic interaction between nej1Δ and rad50sc+h ± N873I in DNA end processing to assess the correlation between chromosomal deletions, tethering, and 5′ resection at the DSB.
Strikingly, > 50% of the nej1Δ rad50sc+h and nej1Δ rad50sc+h N873I survivors were a-like, which was a level exceeding that in nej1Δ rad50Δ double mutants at ~18% (Figure 3B; Sorenson et al., 2017). All sterile and a-like survivors, which are readouts for small deletions and/or insertions and large deletions, respectively, were verified by DNA sequencing (Figures 3E and S2). Importantly, resection was also determined in asynchronous cells. The rate of resection was faster at earlier time points (0–120 min) in asynchronous cells compared to G1-arrested cells; however, the same relative trend was observed (Figures 3B and 3F). This is relevant for correlating resection with the types of survivors that arise during persistent HO cutting.
Mechanism of Resection Inhibition through Interactions between Nej1, Mre11, and Dna2-Sgs1
The rate of large deletions was similar in nej1Δ rad50sc+h and nej1Δ rad50sc+h N873I double mutants, yet hyper-resection was greater in rad50sc+h N873I than in rad50sc+h. This suggests that Nej1 could impact other factors at the DSB involved in DNA processing, a subtlety only revealed through comparing these two rad50 alleles. We previously showed that hyper-resection in nej1Δ mutants was dependent on Sgs1 and not Exo1 (Sorenson et al., 2017). Moreover, MRX is involved in the recruitment of Dna2-Sgs1 to the break (Chiolo et al., 2005; Mimitou and Symington, 2010), yet the physical interactions between these factors have not been characterized. We performed Y2H analysis with Dna2 or Sgs1 and each component of the MRX complex (Figures 4A and 4B; Figures S3A–S3D). The N-terminal domain of both Dna2 (Dna2-N) and Sgs1 (Sgs1-N) showed strong binding with Mre11 (Figures S3A and S3B). As Nej1 interacts with Mre11 through its C-terminal region (Figures 1B–1D), we were prompted to investigate whether there were also interactions between Nej1 and Dna2-Sgs1. Nej1 showed binding with the Sgs1-Hel fragment (Figure 4C), but there was no detectable interaction between Nej1 and the Dna2 fragments (Figure 4C). By Y2H we also determined the regulatory region of Dna2 (Dna2-N) interacted with the Sgs1-Hel fragment (Figure S3C) and the Mre11-C fragment (Figure S3D). Interestingly, in cells where NEJ1 was deleted, the interactions between Dna2-N and Mre11-C and between Dna2-N and Sgs1-Hel significantly increased (Figures 4E and S4A).
Figure 4. Nej1 Regulates Dna2 Recruitment to DSB.

(A) Schematic representation of Dna2 and its functional domains. In green is the N-terminal region, in red is the nuclease domain, and in yellow is in the C-terminal helicase domain.
(B) Schematic representation of Sgs1 and its functional domains. In gray is the N-terminal region, in blue is the helicase domain, and in green is in the C-terminal helicase and RNaseD C-terminal (HRDC) domain.
(C) Y2H analysis between Nej1 fused to HA-AD and either Dna2 or Sgs1 fragments fused to LexA-DBD. All constructs are under galactose induction (as shown in Figure S6), and quantitative β-galactosidase assays were performed as described in STAR Methods.
(D) Enrichment of Dna2HA at DSB, at 0- and 3-h time points, in WT (JC-4117), sgs1Δ (JC-4502), and no tag control (JC-727) were determined at 0.6 kb from DSB.
(E) Y2H analysis between Sgs1-Hel fused to HA-AD and Dna2-N fused to LexA-DBD was performed in WT cells (JC-1280) and in isogenic cells with nej1Δ (JC-4556) using a quantitative β-galactosidase assay and a drop assay on drop-out (-His, -Trp,-Leu) selective media plates.
(F) Enrichment of Nej1Myc at DSB, at 0and 3-h time points, in WT (JC-1687), nej1Δ (JC-4118), sgs1Δ (JC-4528), and no tag control (JC-727) were determined at 0.6 kb from DSB. Analysis was performed in triplicate from at least three biological replicate experiments. Statistical analysis is described in STAR Methods.
(G) Model showing Nej1 interacts with Mre11 and Sgs1. Nej1 inhibits the recovery of Dna2 at the DSB, which also interacts with both of these factors.
To assess whether Nej1 influenced the recruitment of Dna2Sgs1 to the DSB in vivo, we performed ChIP. The level of Dna2HA recovered at the break site doubled in nej1Δ, increasing significantly over WT (Figure 4D). In contrast, the level of Sgs1HA recovered in cells lacking NEJ1 was indistinguishable from WT cells (Figure S4B). When the converse experiment was performed, no significant difference was observed in level of Nej1 recovered at the DSB in cells where SGS1 was deleted (Figure 4F). In contrast, however, approximately half the level of Dna2HA was recovered in sgs1Δ mutant cells (Figure 4D). The loss of DNA2 is lethal, thus similar experiments could not be performed in cells carrying this deletion; however, our data show that when Dna2 is reduced in sgs1Δ mutants, Nej1 recovery does not change (Figure 4F). Interestingly, Nej1 recovery at the DSB significantly decreased in cells overexpressing Dna2 (Figures S4C and S4D), suggesting that Nej1 and Dna2 might compete for binding at the break, likely through the Mre11 C terminus where both interact. Taken together, these data suggest a model that is supported by Y2H and ChIP data, whereby Nej1 regulates resection by inhibiting the binding of Dna2 to Sgs1 and Mre11 at the DSB (Figure 4G).
Role of Nej1 in Regulation of HR Repair Pathway
The recruitment of Nej1 to a DSB was restored when the N873I mutation was combined with rad50sc+h (Figure 1F), thus we next determined whether the underlying resection phenotypes in the rad50sc+h ± N873I mutants (Figures 3B and 3F) stemmed from alteration in Dna2-Sgs1. We performed ChIP on Dna2 and Sgs1 in the rad50 alleles. We observed a reduction in Dna2 at the DSB in rad50sc+h mutants (Figure 5A). The level of Sgs1 was also reduced, approaching the level seen in rad50Δ mutants (Figure 5B). The enrichment level of Dna2 and Sgs1 in rad50sc+h mutants was above the non-tagged control (Figures 5A and 5B), indicating localization was reduced but not abolished. In contrast, the recruitment of both Dna2 and Sgs1 to the DSB in rad50sc+h N873I mutants was indistinguishable from WT (Figures 5A and 5B).
Figure 5. Role of Nej1 in Regulation of HR Repair Pathway.

(A) Enrichment of Dna2HA at DSB, at 0and 3-h time points, in WT (JC-4117), rad50Δ (JC-4503), rad50sc+h (JC-4531), rad50sc+h N873I (JC-4564), and no tag control (JC-727) were determined at 0.6 kb from DSB.
(B) Enrichment of Sgs1HA at DSB, at 0and 3-h time points, in WT (JC-4135), rad50Δ (JC-4138), rad50sc+h (JC-4457), rad50sc+h N873I (JC-4565), and no tag control (JC-727) were determined at 0.6 kb from DSB. The fold enrichment is normalized to recovery at the SMC2 locus. Analysis was performed in triplicate from at least three biological replicate experiments. Statistical analysis is described in STAR Methods.
(C) Model supported by microscopy data that determined tethering, qPCR that measured resection, and ChIP data assess the binding of Xrs2, Nej1, and Dna2Sgs1 at the DSB in (i) RAD50+, (ii) rad50sc+h, and (iii) rad50sc+h N873I.
(D and E) Drop assay with 5-fold serial dilutions on growth media YPAD, YPAD + 2% galactose, or YPAD + 0.02% MMS (D), and YPAD + 2.5 or 5 ug/ml phleomycin (E) using the following strains: WT (JC-727), rad50Δ (JC-3313), rad50sc+h (JC-4424), rad50sc+h N873I (JC-4561), nej1Δ (JC-1342), nej1Δ rad50Δ (JC-3314), nej1Δ rad50sc+h (JC-4476), and nej1Δ rad50sc+h N873I (JC-4597).
One model supported by our data is that changes in the globular head, resulting from the distal coiled-coil deletion in rad50sc+h, lead to a reduction in Dna2-Sgs1 recruitment. This could be attributed to changes in the interactions between Dna2-Sgs1 with MRX or could even reflect reduced MRX stability, as Xrs2 is also reduced in rad50sc+h (i and ii, Figures 1F and 5C). When N873I is introduced, structural changes in the distal end are suppressed and Nej1 and Dna2-Sgs1 binding to the globular head is restored. Thus, when the inhibitory influence of Nej1 is removed, hyper-resection follows (iii, Figure 5C). Resection also increases when NEJ1 is deleted in rad50sc+h mutants; however, levels do not reach that of rad50sc+h N873I because less Dna2-Sgs1 is localized (ii, Figure 5C).
The dynamic changes in these factors are informative for understanding the underlying alterations in resection. One prediction from this model is that when nej1Δ is combined with rad50sc+h and rad50sc+h N873I, the level of Dna2 recruited to the DSB would be restored to the level recovered in WT and nej1Δ, respectively. This proved difficult to measure directly because of the difference in growth rates and cut efficiency in the double-mutant combinations when Dna2 was tagged with an HA epitope. We did, however, determine the physiological impact of Nej1 on repair when resection is advantageous, such as when HR can be utilized. We determined methyl methanesulfonate (MMS) sensitivity in the various mutant backgrounds. Consistent with their initial characterization, the rad50sc+h allele grows better than rad50Δ mutants, and the addition of N873I partially suppressed the MMS sensitivity of rad50sc+h. These relative sensitivities paralleled growth on plates containing galactose, which can be repaired only by NHEJ (Figure 5D). In the double mutants where nej1Δ was also included, cell survival remained extremely low when NHEJ is the only option for repair (Figures 1G and 5D). In stark contrast, MMS resistance was fully restored in nej1Δ rad50sc+h ± N873I mutants but not in nej1Δ rad50Δ (Figure 5D). Similarly, sensitivity to phleomycin-induced DSBs was reversed in both double mutants, nej1Δ rad50sc+h and nej1Δ rad50sc+h N873I (Figure 5E). This rescue was largely dependent on Dna2-Sgs1 as both sgs1Δ nej1Δ rad50sc+h and sgs1Δ nej1Δ rad50sc+h N873I triple mutants showed decreased resection (Figures 6A and 6B) and extreme MMS sensitivity (Figure S5A).
Figure 6. Interplay of Nej1 and Sgs1 in End-Resection Step of HR Repair Pathway.

(A and B) Resection of DNA 0.15 kb away from the HO DSB, as measured by % ssDNA, at 0, 30, 60, 90, and 120 min post DSB induction in cycling cells. Strains used include WT (JC-727), nej1Δ (JC-1342), sgs1Δ (JC-3757), rad50sc+h (JC-4424), nej1Δ rad50sc+h (JC-4476), sgs1Δ rad50sc+h (JC-4478), nej1Δ sgs1Δ rad50sc+h (JC-4479) (A), and rad50sc+h N873I (JC-4561), nej1Δ rad50sc+h N873I (JC-4597), sgs1Δ rad50sc+h N873I (JC-4607), and nej1Δ sgs1Δ rad50sc+h N873I (JC-4605) (B). Analysis was performed in triplicate from at least three biological replicate experiments. Statistical analysis is described in STAR Methods.
DISCUSSION
The role of Nej1 in DSB repair has remained somewhat obscure compared to other components of the pathway including Ku, Lif1-Dnl4, and MRX. Here we demonstrate that Nej1 is important for end-tethering (Figure 2), a function not shared with either Ku or Dnl4 (Figure S1; Lobachev et al., 2004). Nej1 binds Lif1 (Carter et al., 2009; Deshpande and Wilson, 2007; Frank-Vaillant and Marcand, 2001; Mahaney et al., 2014; Sorenson et al., 2017), which is also important for tethering; however, surprisingly the defect in nej1Δ lif1Δ double mutants was additive, suggesting their role in tethering might be distinct of their physical interaction. In contrast, tethering in cells where nej1Δ is combined with the deletion of RAD50 or MRE11 was epistatic (Figures 2C and S1), which is consistent with Nej1 recruitment to the DSB being MRX dependent (Figure 1A).
Characterizing Nej1 in DSB repair was enabled by combining nej1Δ with two alleles of RAD50, rad50sc+h and rad50sc+h N873I. We find that the level of Nej1 recovered at the break in rad50sc+h N873I was indistinguishable from WT and that tethering in rad50sc+h N873I improved compared to rad50Δ and rad50sc+h. This improvement was dependent on Nej1 as the tethering defect in nej1Δ rad50sc+h N873I increased significantly, above all mutant combinations (Figure 2C). The mean distance between the broken DNA ends increased significantly in nej1Δ rad50sc+h N873I (0.55 µm) and was greater than nej1Δ rad50Δ (0.36 µm). The reason for this remains unclear; however, hyper-resection was also observed for nej1Δ rad50sc+h N873I double mutants, indicating that increased resection at the DSB could accentuate a tethering defect. Here we show that Nej1 and Dna2-Sgs1 physically interact with the C-terminal domain of Mre11 (amino acids 423–692), which includes the region in Mre11 where Rad50 binds (Lim et al., 2011) and the region in Mre11 recently shown to impact endtethering (Cassani et al., 2018).
In addition to its role in tethering, Nej1 inhibits hyper-resection mediated by Dna2-Sgs1 (Sorenson et al., 2017). The level of Nej1 and Dna2-Sgs1 recovered at the break was reduced in rad50sc+h mutants. These data indicate that the structural changes in MRX, caused by the sc+h mutation, were substantial enough to alter the recruitment of both HR and NHEJ repair factors to the DSB (Hohl et al., 2011, 2015). It has also been suggested that these structural changes impede the functionality of additional factors with compensatory nuclease activity, like Exo1 and Dna2-Sgs1; however, this had not been directly tested (Cejka et al., 2010; Cannavo et al., 2013; Hohl et al., 2015; Shim et al., 2010). When rad50sc+h was combined with the loss of SGS1, synergistic sensitivity to MMS was observed; however, when combined with exo1Δ, only minor sensitivity was observed (Figures S5A and S5B). These data indicate that in rad50sc+h mutants, the presence of Dna2-Sgs1 at the DSB, albeit at reduced levels, is critical for repair.
In rad50sc+h mutants, resection at early time points was defective in both asynchronously growing and G1-arrested cells (Figure 3F). However, the level of resection approaches WT by 4 h in asynchronous cultures (Figure S5C). One interpretation is that when repair can proceed via HR, the activation of nucleases in S phase results in more resection and partial suppression of MMS and phleomycin sensitivity (Ira et al., 2004). Growth of rad50sc+h compared to rad50Δ mutants on DNA damaging agents supports this model (Figures 5D and 5E). The presence of Nej1 prevented complete suppression because upon NEJ1 deletion, the double mutants became more resistant to MMS and phleomycin (Figures 5D and 5E). Moreover, we demonstrated that this suppression was completely dependent on Sgs1, which suggests that Nej1 might function outside of G1 to regulate Dna2-Sgs1 (Figure S5A).
Structural changes in the distal region of Rad50 also alter Mre11 nuclease function (Liu et al., 2016). 5′ resection was restored in rad50sc+h N873I mutants as was the recruitment of Dna2 and Sgs1 to the DSB. Similar to nej1Δ rad50sc+h double mutants, the deletion of NEJ1 in rad50sc+h N873I conferred some resistance to MMS and phleomycin. Moreover, sensitivity was greater when SGS1 was deleted in combination with rad50sc+h ± N873I compared to sgs1Δ (Figure 5A), suggesting that even when Rad50-Mre11 interactions improve with the addition on N873I, the Mre11 nuclease activity likely remained compromised. These data also indicate that Dna2-Sgs1, under Nej1 regulation, is critical for DNA damage repair in rad50sc+h N873I mutant cells (Hohl et al., 2015).
In all, our data support a model whereby the formation of large deletions stem from a decrease in end-tethering and an increase in 5′ resection. The relative contribution of each process to genomic loss at the break is difficult to determine in the various mutant backgrounds; however, Nej1 has a critical role in both. In the absence of NEJ1, resection increases and tethering decreases. Survivors harboring nej1Δ, rad50ΔD, and nej1Δ rad50Δ showed an increase in large deletions; however, the percentage of deletions is greater in nej1Δ compared to rad50Δ. In the absence of RAD50, tethering is defective, but in contrast to nej1Δ, resection decreases and fewer large deletions are seen. The level of large deletions in nej1Δ and nej1Δ rad50Δ mutants was similar; however, the reason for this might be somewhat different. The nej1Δ rad50Δ double mutants have a greater defect in tethering compared to nej1Δ, but the nej1Δ mutation leads to hyper-resection.
The percentage of sterile survivors in rad50sc+h and rad50sc+h N873I was similar to WT; however, when these rad50 alleles were combined with the loss of NEJ1, the most common mutation was a 107-bp deletion adjacent to the HO cut site (Figures 3E and S2). The combined contribution of tethering and resection is somewhat confounded as the resection and tethering defect in nej1Δ rad50sc+h N873I was significantly higher than in nej1Δ rad50sc+h double mutants. Nevertheless, both of these double mutants showed a marked increase in the percentage of large deletions > 700 bp, which correlated with decreased tethering and increased resection, compared to rad50sc+h and rad50sc+h N873I single mutants.
Here, we show that Nej1 recruitment to the break depends on the MRX complex and identify functions of Nej1 that are important for DNA repair pathway choice, including end-tethering and 5′ resection at the DSB. Nej1 was recently shown to be important for joining incompatible DNA ends (Yang et al., 2015), and our data support a model whereby Nej1 not only promotes canonical NHEJ but also might promote imprecise NHEJ under certain conditions (Emerson et al., 2018; Yang et al., 2015). In all, our work underscores the importance of Nej1 and the structural features of Rad50 in NHEJ.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
As Lead Contact, Jennifer Cobb is responsible for all reagent and resource requests. Please contact Jennifer Cobb at jcobb@ucalgary.ca with requests and inquiries. There are no restrictions on the availability of strains or plasmids and this study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All the yeast strains used in this study are listed in Table S1 in supplemental information and were obtained by crosses. The strains were grown on various media for the experiments, and are described below. For experiments involving the induction of an HO DSB, YPLG media is used (1% yeast extract, 2% bactopeptone, 2% lactic acid, 3% glycerol and 0.05% glucose). For the continuous DSB assay, YPA plates are used (1% yeast extract, 2% bacto peptone, 0.0025% adenine) supplemented with either 2% glucose or 2% galactose. For the mating type assays, YPAD plates are used (1% yeast extract, 2% bacto peptone, 0.0025% adenine, 2% dextrose). For yeast 2-hybrid assays, standard amino acid drop out media lacking histidine, tryptophan and uracil is used and 2% raffinose is added as the carbon source for the cells.
Details of plasmids and primers used in this study are specified in Tables S2 and S3 in the Supplemental Information.
METHOD DETAILS
Tethering Microscopy
Cells derived from the parent strain JC-4066 were diluted and grown overnight in YPLG at 30°C to reach a concentration of 1×107 cells/ml. Cells were treated with 2% GAL for 3 hours and cell pellets were collected and washed 3 times with PBS. After the final wash, 50 µL PBS was left in the tube and 100 µL of 4% paraformaldehyde was added and the cells were incubated at room temperature for 5 min. Following paraformaldehyde treatment, the cells were washed 3 times with PBS (1ml per wash) and stored at 4°C until microscopy. Cells were placed on coverslips containing agarose pads. Z stack images were obtained in 0.25µm increments along the Z-plane to cover a total range of 3.75 µm using a LSM880 Carl Zeiss confocal microscope with a Plan Apochromat 63X/1.4 NA (oil immersion) objective and a camera Airyscan detector (Carl Zeiss). Genomic DNA was prepared from an aliquot taken after 3 hours of galactose induction to verify HO cutting. Acquistion and anaylsis software used was Zen Black (Carl Zeiss). ImageJ was used to measure the distance between the 2 foci representing either side of the DSB and the results were plotted.
Chromatin Immunoprecipitation
ChIP assay was performed as described previously (Mahaney et al., 2014). Cells were cultured overnight in YPLG at 25°C. Cells were then diluted to equal levels (5 3 106 cells/ml) and were cultured to one doubling (3–4 hr) at 30°C. 2% GAL was added to the YPLG and cells were harvested and crosslinked at various time points using 3.7% formaldehyde solution. Following crosslinking, the cells were washed with ice cold PBS and the pellet stored at —80°C. The pellet was re-suspended in lysis buffer (50mM HEPES pH 7.5, 1mM EDTA, 80mM NaCl, 1% Triton, 1mM PMSF and protease inhibitor cocktail) and cells were lysed using Zirconia beads and a bead beater. Chromatin fractionation was performed to enhance the chromatin bound nuclear fraction by spinning the cell lysate at 13,200rpm for 15 minutes and discarding the supernatant. The pellet was re-suspended in lysis buffer and sonicated to yield DNA fragments (~500bps in length). The sonicated lysate was then incubated in beads + anti-HA antibody or unconjugated beads (control) for 2 hr at 4°C. The beads were washed using wash buffer (100mM Tris pH 8, 250mM LiCl, 150mM (HA Ab) or 500mM (Myc Ab) NaCl, 0.5% NP-40, 1mM EDTA, 1mM PMSF and protease inhibitor cocktail) and protein-DNA complex was eluted by reverse crosslinking using 1% SDS in TE buffer, followed by proteinase K treatment and DNA isolation via phenol-chloroform-isoamylalcohol extraction. Quantitative PCR was performed using the Applied Biosystem QuantStudio 6 Flex machine. PerfeCTa qPCR SuperMix, ROX was used to visualize enrichment at HO2 (0.5kb from DSB) and HO1 (1.6kb from DSB) and SMC2 was used as an internal control.
qPCR based Resection Assay
Cells from each strain were grown overnight in 10ml YPLG to reach an exponentially growing culture of 1×107 cells/mL and were arrest cells in G1 with 15 µg/mL α-factor. 1 hr post α-factor addition to the media a second dose of 15 µg/mL α-factor was added for an additional 1 hr. Next, 2.5 mL of the cells were pelleted as t = 0 and 2% GAL, to induce a DSB, and 15 µg/mL α-factor, to maintain G1 arrest, was added to the remaining cells. 15 µg/mL α-factor was added every 1.5 hr post GAL addition to maintain cells in G1 for the duration of the experiment. 6 hours following GAL addition to the media 2.5 mL of the remaining cells were pelleted as the t = 6 hr time point. Genomic DNA was purified using standard genomic preparation methods and DNA was re-suspended in 100 mL ddH2O. Genomic DNA was treated with 0.005 µg/µL RNase A for 45 min at 37°C. 2 µL of DNA was added to tubes containing Cut Smart buffer with or without RsaI restriction enzyme and incubated at 37°C for 2 hr. Quantitative PCR was performed using the Applied Biosystem QuantStudio 6 Flex machine. PowerUp SYBR Green Master Mix was used to quantify resection at MAT1 (0.15kb from DSB) and MAT2 (4.8kb from DSB). Pre1 was used as a negative control. RsaI cut DNA was normalized to uncut DNA as previously described to quantify the % ssDNA / total DNA (Ferrari et al., 2015).
Continuous DSB assay and identification of mutations in survivors
Cells were grown overnight in YPLG media at 25°C to saturation. Cells were collected by centrifugation at 2500rpm for 3 minutes and pellets were washed 1x in ddH2O and re-suspended in ddH2O. Cells were counted and spread on YPA plates supplemented with either 2% GLU or 2% GAL. On the Glucose plates 1×103 total cells were added and on the galactose plates 1×105 total cells were added and 1×107 total cells were added for the other strains used. The cells were incubated for 3–4 days at room temperature and colonies counted on each plate. Survival was determined by normalizing the number of surviving colonies in the GAL plates to number of colonies in the GLU plates, and > 300 survivors were scored for each strain as previously described (Sorenson et al., 2017). Genomic DNA of sterile or a-type survivors was amplified with primers S1-S2 or A1-A2 (as deletions were too large to amplify with S1-S2), followed by DNA sequencing. Pre1 primers were used as a +ctrl for the PCR and all primer sequences are listed in Table S3. Survivors that were a type were verified to have large genomic deletions of ~800–1100bp by sequencing the PCR product amplified with A1-A2.
Yeast 2-hybrid
The plasmids in Table S2 were constructed containing the gene encoding the region of the proteins – Sgs1, Dna2, Mre11, Nej1, Rad50 and Xrs2, using the primers listed in Table S3. The plasmids J-965 and J-1493 and the inserts were treated with BamHI and EcoRI and ligated using T4 DNA ligase. The plasmids were sequence verified. Reporter (J-359), bait (J-965) and prey (J-1493) plasmids, containing the gene encoding the desired protein under a galactose inducible promoter, were transformed into JC-1280 as previously described (Bustard et al., 2012). Cells were grown overnight in –URA –HIS –TRP media with 2% raffinose. Next day, cells were transferred into –URA –HIS –TRP media with either 2% GLU or 2% GAL and grown for 6 hr at 30°C. Cell pellets were resuspended and then permeabilized using 0.1% SDS followed by ONPG addition. β-galactosidase activity was estimated by measuring the OD at 420nm, relative β-galactosidase units were determined by normalizing to total cell density at OD600. Additionally, for drop assay cells were grown and spotted in ten-fold serial dilutions on plates containing 2% galactose lacking histidine and tryptophan (for plasmid selection) and leucine (for measuring expression from lexAop6-LEU2). Plates were photographed after 3 days of incubation at 30°C.
Western Blot
Cells were lysed by re-suspending them in lysis buffer (with PMSF and protease inhibitor cocktail tablets) followed by bead beating. The protein concentration of the whole cell extract was determined using the NanoDrop. Equal amounts of whole cell extract were added to wells. α-PGK was used as a loading control.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data in bar graphs represent the average of 3 biological replicates. Error bars represent the standard error of mean (SEM). Significance (p value) was determined using 1-tailed, unpaired Student’s t test p < 0.05*; p < 0.01**; p < 0.001***. Statistical analyses were performed in Prism7 (GraphPad).
DATA AND CODE AVAILABILITY
This study did not generate/analyze any code. Original data supporting the figures in the paper is available from the corresponding author on request.
Supplementary Material
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HA-probe mouse monoclonal antibody | Santa Cruz Biotechnology | F-7; RRID:AB_627809 |
| LexA-Probe mouse monoclonal antibody | Santa Cruz Biotechnology | 2–12; RRID:AB_627883 |
| Myc-Probe mouse monoclonal antibody | Abcam | ab32; RRID:AB_303599 |
| Peroxidase conjugated AffiniPure Goat α-Mouse antibody | Jackson ImmunoResearch | 115035174; RRID:AB_2338512 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| α-factor | EZBiolab | CP7206 |
| Adenine | Sigma | A8626 |
| Bacto™ Peptone | BD Biosciences | 211677 |
| Bacto™ Yeast extract | BD Biosciences | 212750 |
| Complete EDTA free protease inhibitor cocktail | Roche | 04693159001 |
| Dextrose | Sigma | D1912 |
| Difco™ Agar | BD Biosciences | 214530 |
| Dynabeads Sheep anti-Mouse IgG | Invitrogen | 20230531 |
| EDTA | VWR | 0322 |
| Ethanol | Commercial alcohols | P006EAAN |
| Formaldehyde | Sigma | F8775 |
| Galactose | Sigma | G0750 |
| Glycine | VWR | CA93291 |
| Glycogen | Roche | 10901393001 |
| Lactic acid | Sigma | 69785 |
| Lithium Chloride | EMD Millipore | LX03311 |
| Phenol-Chloroform-Isoamylalcohol | Invitrogen | 15593049 |
| PMSF | Sigma | 78830 |
| Potassium Chloride | EMD Chemicals | 1049360500 |
| Proteinase K | Invitrogen | 25530031 |
| Raffinose | US Biological | R1030 |
| RNase A | Sigma | R6513 |
| RsaI | New England Biolabs | R0167S |
| SDS | Avantor | 409502 |
| Sodium Acetate | VWR | BDH9278 |
| Sodium Chloride | Fisher Chemical | S64212 |
| Tris base | Fisher Chemical | BP1525 |
| Triton | Sigma | 9002931 |
| Zirconia Silica beads | BioSpec Products | 11079105z |
| Commercial Assays | ||
| PerfeCTa qPCR SuperMix, ROX | Quanta BioSciences Inc. | 89168–786 |
| PowerUp SYBR Green Master Mix | Applied Biosystems | A25743 |
| Western Lightning Plus-ECL | PerkinElmer | NEL105001EA |
| Experimental Models: Organisms/Strains | ||
| Yeast strains, see Table S1 | This study | N/A |
| Oligonucleotides | ||
| Primers, see Table S3 | This study | N/A |
| Recombinant DNA | ||
| Plasmids, see Table S2 | This study | N/A |
| Software | ||
| ImageJ | NIH | N/A |
| Prism7 | GraphPad | N/A |
Highlights.
The MRX complex stabilizes Nej1 at the break site
Nej1 inhibits Dna2 recruitment and HR
Nej1 and the coiled-coil region of Rad50 are important for tethering broken DNA ends
Defects in 5′ resection and end-tethering lead to large chromosome deletions at the DSB
ACKNOWLEDGMENTS
This work was supported by operating grants from CIHR MOP-82736, MOP137062, and NSERC 418122 awarded to J.A.C., CIHR operating grant MOP13639 to S.P.L.M., ERC grant (FP7/2007 2013/European Research Council grant agreement 281287) and funding from the Radiobiology program of the CEA Segment 4 to K.D., and NIH RO1 grant GM56888 and MSK Cancer Center Core Grant P30 CA008748 to J.H.J.P.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.018.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Balestrini A, Ristic D, Dionne I, Liu XZ, Wyman C, Wellinger RJ, and Petrini JH (2013). The Ku heterodimer and the metabolism of single-ended DNA double-strand breaks. Cell Rep 3, 2033–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustard DE, Menolfi D, Jeppsson K, Ball LG, Dewey SC, Shirahige K, SjÖgren C, Branzei D, and Cobb JA (2012). During replication stress, non-SMC element 5 (NSE5) is required for Smc5/6 protein complex functionality at stalled forks. J. Biol. Chem 287, 11374–11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo E, and Cejka P (2014). Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature 514, 122–125. [DOI] [PubMed] [Google Scholar]
- Cannavo E, Cejka P, and Kowalczykowski SC (2013). Relationship of DNA degradation by Saccharomyces cerevisiae exonuclease 1 and its stimulation by RPA and Mre11-Rad50-Xrs2 to DNA end resection. Proc. Natl. Acad. Sci. USA 110, E1661–E1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SD, Vigasová,,,,, Chen J, Chovanec M, and Aström SU (2009). Nej1 recruits the Srs2 helicase to DNA double-strand breaks and supports repair by a single-strand annealing-like mechanism. Proc. Natl. Acad. Sci. USA 106, 12037–12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassani C, Gobbini E, Vertemara J, Wang W, Marsella A, Sung P, Tisi R, Zampella G, and Longhese MP (2018). Structurally distinct Mre11 domains mediate MRX functions in resection, end-tethering and DNA damage resistance. Nucleic Acids Res 46, 2990–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka P, Cannavo E, Polaczek P, Masuda-Sasa T, Pokharel S, Campbell JL, and Kowalczykowski SC (2010). DNA end resection by Dna2Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature 467, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, and Tomkinson AE (2011). Yeast Nej1 is a key participant in the initial end binding and final ligation steps of nonhomologous end joining. J. Biol. Chem 286, 4931–4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Trujillo K, Ramos W, Sung P, and Tomkinson AE (2001). Promotion of Dnl4-catalyzed DNA end-joining by the Rad50/Mre11/Xrs2 and Hdf1/ Hdf2 complexes. Mol. Cell 8, 1105–1115. [DOI] [PubMed] [Google Scholar]
- Chiolo I, Carotenuto W, Maffioletti G, Petrini JH, Foiani M, and Liberi G (2005). Srs2 and Sgs1 DNA helicases associate with Mre11 in different subcomplexes following checkpoint activation and CDK1-mediated Srs2 phosphorylation. Mol. Cell. Biol 25, 5738–5751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerici M, Mantiero D, Guerini I, Lucchini G, and Longhese MP (2008). The Yku70-Yku80 complex contributes to regulate double-strand break processing and checkpoint activation during the cell cycle. EMBO Rep 9, 810–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande RA, and Wilson TE (2007). Modes of interaction among yeast Nej1, Lif1 and Dnl4 proteins and comparison to human XLF, XRCC4 and Lig4. DNA Repair (Amst.) 6, 1507–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande RA, Williams GJ, Limbo O, Williams RS, Kuhnlein J, Lee JH, Classen S, Guenther G, Russell P, Tainer JA, et al. (2014). ATPdriven Rad50 conformations regulate DNA tethering, end resection, and ATM checkpoint signaling. EMBO J 33, 482–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson CH, Lopez CR, Ribes-Zamora A, Polleys EJ, Williams CL, Yeo L, Zaneveld JE, Chen R, and Bertuch AA (2018). Ku DNA End-Binding Activity Promotes Repair Fidelity and Influences End-Processing During Nonhomologous End-Joining in Saccharomyces cerevisiae. Genetics 209, 115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari M, Dibitetto D, De Gregorio G, Eapen VV, Rawal CC, Lazzaro F, Tsabar M, Marini F, Haber JE, and Pellicioli A (2015). Functional interplay between the 53BP1-ortholog Rad9 and the Mre11 complex regulates resection, end-tethering and repair of a double-strand break. PLoS Genet 11, e1004928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank-Vaillant M, and Marcand S (2001). NHEJ regulation by mating type is exercised through a novel protein, Lif2p, essential to the ligase IV pathway. Genes Dev 15, 3005–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham TG, Walter JC, and Loparo JJ (2016). Two-Stage Synapsis of DNA Ends during Non-homologous End Joining. Mol. Cell 61, 850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham TGW, Carney SM, Walter JC, and Loparo JJ (2018). A single XLF dimer bridges DNA ends during nonhomologous end joining. Nat. Struct. Mol. Biol 25, 877–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohl M, Kwon Y, Galván SM, Xue X, Tous C, Aguilera A, Sung P, and Petrini JH (2011). The Rad50 coiled-coil domain is indispensable for Mre11 complex functions. Nat. Struct. Mol. Biol 18, 1124–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohl M, Kochańczyk T, Tous C, Aguilera A, Krężel A, and Petrini JH (2015). Interdependence of the rad50 hook and globular domain functions. Mol. Cell 57, 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfner KP, Karcher A, Craig L, Woo TT, Carney JP, and Tainer JA (2001). Structural biochemistry and interaction architecture of the DNA doublestrand break repair Mre11 nuclease and Rad50-ATPase. Cell 105, 473–485. [DOI] [PubMed] [Google Scholar]
- Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth NM, et al. (2004). DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431, 1011–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye JA, Melo JA, Cheung SK, Vaze MB, Haber JE, and Toczyski DP (2004). DNA breaks promote genomic instability by impeding proper chromosome segregation. Curr. Biol 14, 2096–2106. [DOI] [PubMed] [Google Scholar]
- Kegel A, Sjöstrand JO, and Aström SU (2001). Nej1p, a cell type-specific regulator of nonhomologous end joining in yeast. Curr. Biol 11, 1611–1617. [DOI] [PubMed] [Google Scholar]
- Lim HS, Kim JS, Park YB, Gwon GH, and Cho Y (2011). Crystal structure of the Mre11-Rad50-ATPγS complex: understanding the interplay between Mre11 and Rad50. Genes Dev 25, 1091–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Sung S, Kim Y, Li F, Gwon G, Jo A, Kim AK, Kim T, Song OK, Lee SE, and Cho Y (2016). ATP-dependent DNA binding, unwinding, and resection by the Mre11/Rad50 complex. EMBO J 35, 743–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobachev K, Vitriol E, Stemple J, Resnick MA, and Bloom K (2004). Chromosome fragmentation after induction of a double-strand break is an active process prevented by the RMX repair complex. Curr. Biol 14, 2107–2112. [DOI] [PubMed] [Google Scholar]
- Mahaney BL, Lees-Miller SP, and Cobb JA (2014). The C-terminus of Nej1 is critical for nuclear localization and non-homologous end-joining. DNA Repair (Amst.) 14, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki K, Shinohara A, and Shinohara M (2008). Forkhead-associated domain of yeast Xrs2, a homolog of human Nbs1, promotes nonhomologous end joining through interaction with a ligase IV partner protein, Lif1. Genetics 179, 213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, and Symington LS (2010). Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J 29, 3358–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JK, and Haber JE (1996). Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol. Cell. Biol 16, 2164–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Lee SJ, Rothstein R, and Symington LS (2018). Xrs2 and Tel1 Independently Contribute to MR-Mediated DNA Tethering and Replisome Stability. Cell Rep 25, 1681–1692.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmbos PL, Wu D, Daley JM, and Wilson TE (2008). Recruitment of Saccharomyces cerevisiae Dnl4-Lif1 complex to a double-strand break requires interactions with Yku80 and the Xrs2 FHA domain. Genetics 180, 1809–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YB, Hohl M, Padjasek M, Jeong E, Jin KS, Krezel A, Petrini JH, and Cho Y (2017). Eukaryotic Rad50 functions as a rod-shaped dimer. Nat. Struct. Mol. Biol 24, 248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid DA, Keegan S, Leo-Macias A, Watanabe G, Strande NT, Chang HH, Oksuz BA, Fenyo D, Lieber MR, Ramsden DA, and Rothenberg E (2015). Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 112, E2575–E2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim EY, Chung WH, Nicolette ML, Zhang Y, Davis M, Zhu Z, Paull TT, Ira G, and Lee SE; S.E. (2010). Saccharomyces cerevisiae Mre11/ Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO J 29, 3370–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorenson KS, Mahaney BL, Lees-Miller SP, and Cobb JA (2017). The non-homologous end-joining factor Nej1 inhibits resection mediated by Dna2-Sgs1 nuclease-helicase at DNA double strand breaks. J. Biol. Chem 292, 14576–14586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS (2016). Mechanism and regulation of DNA end resection in eu-karyotes. Crit. Rev. Biochem. Mol. Biol 51, 195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia M, Bentele M, Vaze MB, Herrmann G, Kraus E, Lee SE, Schär P, and Haber JE (2001). NEJ1 controls non-homologous end joining in Saccharomyces cerevisiae. Nature 414, 666–669. [DOI] [PubMed] [Google Scholar]
- Wasko BM, Holland CL, Resnick MA, and Lewis LK (2009). Inhibition of DNA double-strand break repair by the Ku heterodimer in mrx mutants of Saccharomyces cerevisiae. DNA Repair (Amst.) 8, 162–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Topper LM, and Wilson TE (2008). Recruitment and dissociation of nonhomologous end joining proteins at a DNA double-strand break in Saccharomyces cerevisiae. Genetics 178, 1237–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Matsumoto Y, Trujillo KM, Lees-Miller SP, Osley MA, and Tomkinson AE (2015). Role of the yeast DNA repair protein Nej1 in end processing during the repair of DNA double strand breaks by non-homologous end joining. DNA Repair (Amst.) 31, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Hefferin ML, Chen L, Shim EY, Tseng HM, Kwon Y, Sung P, Lee SE, and Tomkinson AE (2007). Role of Dnl4-Lif1 in nonhomologous end-joining repair complex assembly and suppression of homologous recombination. Nat. Struct. Mol. Biol 14, 639–646. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze any code. Original data supporting the figures in the paper is available from the corresponding author on request.