

Abstract
Drug distribution is a necessary component of models to predict human pharmacokinetics. A new membrane-based tissue-plasma partition coefficient (Kp) method (Kp,mem) to predict unbound tissue to plasma partition coefficients (Kpu) was developed using in vitro membrane partitioning [fraction unbound in microsomes (fum)], plasma protein binding, and log P. The resulting Kp values were used in a physiologically based pharmacokinetic (PBPK) model to predict the steady-state volume of distribution (Vss) and concentration-time (C-t) profiles for 19 drugs. These results were compared with Kp predictions using a standard method [the differential phospholipid Kp prediction method (Kp,dPL)], which differentiates between acidic and neutral phospholipids. The Kp,mem method was parameterized using published rat Kpu data and tissue lipid composition. The Kpu values were well predicted with R2 = 0.8. When used in a PBPK model, the Vss predictions were within 2-fold error for 12 of 19 drugs for Kp,mem versus 11 of 19 forKp,dPL. With one outlier removed for Kp,mem and two for Kp,dPL, the Vss predictions for R2 were 0.80 and 0.79 for the Kp,mem and Kp,dPL methods, respectively. The C-t profiles were also predicted and compared. Overall, the Kp,mem method predicted the Vss and C-t profiles equally or better than the Kp,dPL method. An advantage of using fum to parameterize membrane partitioning is that fum data are used for clearance prediction and are, therefore, generated early in the discovery/development process. Also, the method provides a mechanistically sound basis for membrane partitioning and permeability for further improving PBPK models.
SIGNIFICANCE STATEMENT
A new method to predict tissue-plasma partition coefficients was developed. The method provides a more mechanistic basis to model membrane partitioning.
Introduction
The volume of distribution and clearance equally determine the half-life of a drug. The steady-state volume of distribution (Vss) can be predicted using empirical methods (Obach et al., 1997), computational approaches (Ghafourian et al., 2004; Lombardo et al., 2006; Zhivkova and Doytchinova, 2012), physiologic equations (Oie and Tozer, 1979; Lombardo et al., 2004; Korzekwa and Nagar, 2017a), and tissue:plasma partition coefficients (Kp). The Kp prediction methods are widely used since they describe the distribution in physiologically based pharmacokinetic (PBPK) models. While some methods require an in vivo component (Arundel, 1997; Björkman, 2002; Jansson et al., 2008; Poulin and Theil, 2009), others use more readily available in vitro inputs.
Several factors influence drug distribution, including partitioning into membranes and other lipids, binding to proteins (primarily plasma proteins), pH partitioning (e.g., lysosomes), transporters, and membrane permeability. Most models represent tissue interactions with in vitro surrogates. The Poulin and Krishnan (1995) model originally described phospholipid partitioning with the octanol:water partition coefficient (P) and assumed phospholipid composition to be represented by 30% octanol and 70% water. They developed Kp prediction equations that included an additional surrogate for neutral lipid partitioning in adipose tissue (Poulin and Theil, 2000; Poulin et al., 2001), which was modified by Berezhkovskiy (2004). Rodgers and Rowland (2006) developed two equations for prediction of unbound Kp (Kpu): one for acids, neutrals, and weak bases, and another for moderate-to-strong bases (Rodgers et al., 2005). Drug partitioning into erythrocytes was used to parameterize the interaction of bases with acidic phospholipids (APs). It was assumed that ionized bases interact only with APs, while uncharged molecules interact only with neutral phospholipids (Rodgers et al., 2005; Rodgers and Rowland, 2006).
In most currently used composition-based models, log P is used to model the phospholipid partitioning (0.3P). A shortcoming of using log P to represent phospholipid partitioning is the lack of orientation-specific interactions with phospholipid membranes (Balaz, 2009; Nagar and Korzekwa, 2012, 2017). Additionally, both neutral and ionized bases are known to interact with all phospholipids, and not just net-neutral and net-acidic phospholipids, respectively. Therefore, current methods to calculate Kp appear to be based on mechanistically unsound assumptions. Previously, we used microsomal partitioning [fraction unbound in microsomes (fum)] instead of log P to parameterize phospholipid partitioning in a Vss model (Korzekwa and Nagar, 2017a). Partitioning into microsomes (unsorted phospholipid vesicles) is used extensively in clearance predictions and can be determined experimentally or predicted (Austin et al., 2002; Hallifax and Houston, 2006; Poulin and Haddad, 2011; Nagar and Korzekwa, 2017). A benefit of using fum to represent phospholipid partitioning is that it measures interactions with all phospholipids for both charged and uncharged species.
Previous studies have compared different Kp prediction methods and their ability to predict both tissue Kp and/or Vss (De Buck et al., 2007; Poulin and Theil, 2009; Jones et al., 2011; Graham et al., 2012; Zou et al., 2012; Chan et al., 2018). These studies came to different conclusions on the most accurate Kp model, which was primarily dependent on the drug data set used (De Buck et al., 2007; Graham et al., 2012). Graham et al. (2012) showed that the Rodgers et al. (2005) method was able to better predict Kp and Vss for different classes of drugs than other composition-based models. The Poulin and Theil (2009) method led to good Vss predictions, but required in vivo data (Kpu muscle) (Graham et al., 2012). More recently, Chan et al. (2018) compared the ability of composition-based Kp models and preclinical extrapolation to predict Vss. Composition-based models predicted Vss with accuracy similar to preclinical extrapolation. They noted that the Rodgers method was able to predict Vss well for drugs with log P values less than 3, and that many drugs with large errors in Vss for composition-based models also had errors in preclinical extrapolation (Chan et al., 2018).
This report evaluates a model to predict Kp using fum to represent membrane partitioning. Plasma protein binding and microsomal partitioning values were determined experimentally for 19 drugs. Tissue Kp values were calculated for each compound using a differential phospholipid Kp prediction method (Kp,dPL) (Rodgers et al., 2005; Rodgers and Rowland, 2006), as well as a method that uses fum to parameterize membrane partitioning [the membrane-based Kp prediction method (Kp,mem)]. Simulations were run and their ability to predict Vss and concentration-time (C-t) profiles was determined. More mechanistically sound assumptions for Kpu will be required when expanding current perfusion-limited PBPK models to include explicit membrane partitioning and permeability. Models to predict Kpu bases on experimental partitioning into membranes may allow a facile transition to models that limit permeability with explicit membrane compartments (Nagar et al., 2014).
Materials and Methods
Materials.
A Harvard Apparatus (Holliston, MA) 96-well equilibrium dialyzer and single-plate Harvard Apparatus plate rotator were used for equilibrium dialysis experiments. Human plasma was obtained from US Biologic (Salem, MA) and Innovative Research Inc. (Novi, MI). Rat liver microsomes were obtained from BD Biosciences (San Jose, CA) and Corning Life Sciences (Tewksbury, MA). Warfarin, fluconazole, glyburide (glybenclamide), ketoprofen, fenofibrate, +/cis-diltiazem hydrochloride, +/− verapamil hydrochloride, caffeine, betaxolol hydrochloride, DMSO, nicardipine hydrochloride, metoprolol tartrate, felodipine, and nafcillin sodium were obtained from Sigma Aldrich (St. Louis, MO). Quinidine gluconate, formic acid, acetonitrile, and diphenhydramine hydrochloride were obtained from Fisher Scientific (Norristown, PA). Mibefradil hydrochloride was obtained from Cayman Chemical Company (Ann Arbor, MI). Diclofenac sodium was obtained from Calbiochem (Burlington, MA). Fenofibric acid was obtained from Kano Laboratories (Nashville, TN). One milligram per milliliter solutions of phenytoin, diazepam, and midazolam in methanol were obtained from Cerilliant (a Sigma Aldrich company). The 100 mM PBS and 0.3 mM MgCl2 dialysis buffer was composed of magnesium hydrochloride hexahydrate (Fisher Scientific), potassium phosphate monobasic (Sigma Aldrich), and potassium phosphate dibasic (Fisher Scientific). An Agilent 1100 HPLC and API 4000 mass spectrometer and Agilent 1100 HPLC and API 4000 Q-Trip mass spectrometer were used to determine the concentrations for equilibrium dialysis. Mathematica version 11.0 (Wolfram, Champaign, IL) was used for all compartmental modeling and simulations. Literature data from plots were digitized using Engauge Plot Digitizer version 10.4 (GitHub, San Francisco, CA).
Probe Drug Selection and Data Collection.
A diverse set of drugs made up of acids, bases, and neutrals was selected to compare the prediction methods (Table 1). Drugs were considered neutral when primarily uncharged at physiologic pH (7.4). Unless noted otherwise, the pKa values of acids (pKa,a) and bases (pKa,b) for neutrals were set at 14 and 1, respectively. Any significant ionized and neutral fraction was considered by both methods. The probe drugs were selected based on the availability of literature intravenous pharmacokinetics data, as well as drug-specific parameters. Average experimental intravenous bolus and/or infusion C-t profiles for 19 drug studies were collected from the literature. If the data were represented as graphical C-t profiles, the plots were digitized. When average subject weight was available, simulations were conducted to reproduce the observed Vss for that average weight. The observed clearance and the steady-state volume of distribution were determined by compartmental analysis using standard equations with Mathematica. Specifically, one- to three-compartment models were evaluated to generate the C-t profiles from the experimental data. Experimental clearance and Vss values were determined from these compartmental models. The best model for the experimental data was determined by the corrected Akaike information criterion values (Akaike, 1974) and residual plots. For all drugs, microrate constants were well defined and use of noncompartmental analysis was not required. Experimental clearance values were assumed for all further modeling efforts and experimental Vss values were compared with predicted values.
TABLE 1.
Pharmacokinetic parameters of test drugs
| Test Drug | Class | Type | Number of Subjects | Weight | Dose | Duration | Number of Points | Vss | CL | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| kg | mg | min | l | l/h | ||||||
| Betaxolol | B | Infusion | n = 10 | 73.6 | 8.94 | 30 | 17 | 360 | 11 | Ludden et al., 1988 |
| Diltiazem | B | Infusion | n = 12 | 63 | 15 | 30 | 13 | 306 | 97 | Hermann et al., 1983 |
| Diphenhydramine | B | Bolus | n = 8 | 98.0 | 56 | N/A | 12 | 788 | 43 | Scavone et al., 1990 |
| Metoprolol | B | Infusion | n = 5 | 66 | 3.9 | 10 | 20 | 274 | 59 | Regårdh et al., 1974 |
| Mibefradil | B | Infusion | n = 6 | 70a | 20 | 30 | 16 | 187 | 17 | Clozel et al., 1991 |
| Nicardipine | B | Bolus | n = 6b | 67.0 | 10 | N/A | 12 | 62 | 76 | Campbell et al., 1985 |
| Quinidine | B | Infusion | n = 12b | 65.3 | 244 | 22 | 12 | 227 | 18.5 | Ueda et al., 1976 |
| Verapamil | B | Infusion | n = 20 | 70a | 10 | 5 | 15 | 266 | 49 | McAllister and Kirsten, 1982 |
| Caffeine | N | Infusion | n = 10 | 79.5 | 350 | 30 | 17 | 42.8 | 5.2 | Blanchard and Sawers, 1983 |
| Diazepam | N | Infusion | n = 24 | 78.1 | 5 | 1 | 21 | 89.5 | 1.33 | Agarwal et al., 2013 |
| Felodipine | N | Infusion | n = 10 | 74 | 2.5 | 30 | 22 | 320 | 41.7 | Edgar et al., 1985 |
| Fluconazole | N | Infusion | n = 6 | 70a | 50 | N/A | 13 | 59.3 | 1.25 | Ripa et al., 1993 |
| Midazolam | N | Bolus | n = 6 | 67.6 | 10 | N/A | 15 | 51.2 | 18 | Heizmann et al., 1983 |
| Phenytoin | N | Infusion | n = 6 | 78.1 | 275 | 6 | 15 | 38.8 | 1.76 | Gugler et al., 1976 |
| Diclofenac | A | Infusion | n = 6 | 65 | 46.5 | 2 | 15 | 9.23 | 17.6 | Willis et al., 1980 |
| Glyburide | A | Infusion | n = 10 | 77.8 | 2 | 60 | 19 | 11.78 | 4.88 | Debruyne et al., 1987 |
| Ketoprofen | A | Bolus | n = 7 | 70a | 100 | N/A | 12 | 9.9 | 5.02 | Debruyne et al., 1987 |
| Nafcillin | A | Infusion | n = 6 | 70a | 475 | 7 | 9 | 20.4 | 33.9 | Waller et al., 1982 |
| Warfarin | A | Bolus | n = 6 | 66.8 | 100 | N/A | 8 | 7.66 | 0.179 | O’Reilly et al., 1971 |
A, acids; B, bases; N, neutral; CL, clearance.
Individual weights not provided and 70 kg assumed.
Individual C-t not provided. C-t profile simulated from average parameters.
Literature physiologic data were used for Kp predictions and PBPK modeling (Brown et al., 1997; Poulin and Theil, 2002; Fenneteau et al., 2010; Ye et al., 2016). For drug-specific parameters [log P, pKa, and blood to plasma (BP)] (Table 2), experimental values from the literature were preferred over calculated/predicted values, and if more than one experimental value was found, then the experimental values were averaged. Human BP values could not be found for betaxolol and nafcillin. For betaxolol, the values of the fraction unbound in plasma (fup) were similar for rat and human; therefore, the rat BP value of 2.0 was used. For nafcillin, a value of 0.55 (1-hematocrit) was used, which is the BP ratio of similar compounds in humans (Greene et al., 1978). Also, this will not affect Vss predictions since BP is not included in the Kp equations for acids. Protein binding was experimentally determined for all compounds (Table 2), with the exception of caffeine due to caffeine contamination in all plasma samples. A caffeine fup value of 0.72 was determined by averaging values found in the literature.
TABLE 2.
Drug-specific parameters
| Compound | Class | fup (n = 4)a | fum (n = 4)a | Log P | pKa,a | pKa,b | BP | Log Dvo | CL | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| l/h | ||||||||||
| Betaxolol | B | 0.50 (12%) | 0.77 (3%) | 2.81 | 14 | 9.4 | 2b | 1.78 | 10.2 | Riddell et al., 1987; Recanatini, 1992; Rodgers and Rowland, 2007 |
| Diltiazem | B | 0.26 (8%) | 0.48 (2%) | 2.7 | 14 | 7.7 | 1 | 1.88 | 97.5 | Rekker and Mannhold, 1992; Obach, 1999; Ishihama et al., 2002 |
| Diphenhydramine | B | 0.44 (4%) | 0.84 (4%) | 3.27 | 14 | 8.98 | 0.74 | 2.30 | 43 | Albert et al., 1975; Sangster, 1994; Hansch et al., 1995; Obach, 1999 |
| Metoprolol | B | 0.87 (17%) | 0.80 (3%) | 1.88 | 14 | 9.7 | 1.14 | 0.746 | 58.8 | Hansch et al., 1995; Rodgers and Rowland, 2007 |
| Mibefradil | B | 0.031 (11%) | 0.034 (15%) | 3.07 | 14 | 10.2 | 0.64 | 2.07 | 15.5 | Welker et al., 1998; Nagar and Korzekwa, 2017 |
| Nicardipine | B | 0.0024 (7%) | 0.039 (12%) | 3.82 | 14 | 8.6 | 0.71 | 2.90 | 31.2 | Sangster, 1993; Rodgers and Rowland, 2007 |
| Quinidine | B | 0.15 (8%) | 0.815 (10%) | 3.52 | 14 | 8.94 | 0.92 | 2.07 | 14 | Sangster, 1994; Obach, 1999; Nagar and Korzekwa, 2017 |
| Verapamil | B | 0.088 (20%) | 0.37 (19%) | 3.79 | 14 | 8.92 | 0.74 | 2.88 | 49 | Sangster, 1994; Hansch et al., 1995; Robinson and Mehvar, 1996; Obach, 1999 |
| Caffeine | N | 0.72c | 0.98 (6%) | −0.07 | 14 | 1.04 | 1.01 | −1.43 | 5.2 | Hansch et al., 1995; Rodgers and Rowland, 2007 |
| Diazepam | N | 0.012 (9%) | 0.74 (4%) | 2.82 | 14 | 3.4 | 0.64 | 1.79 | 1.33 | Maguire et al., 1980; Sangster, 1993; O’Neil, 2006 |
| Felodipine | N | 0.0017 (12%) | 0.023 (27%) | 3.86 | 14 | 5.07 | 0.7 | 2.95 | 41.7 | Diez et al., 1991; Uchimura et al., 2010; Pandey et al., 2013 |
| Fluconazole | N | 0.93 (14%) | 0.94 (14%) | 0.8 | 14 | 1.77 | 1 | −0.79 | 1.25 | Debruyne et al., 1987; Debruyne, 1997; Rodgers and Rowland, 2007 |
| Midazolam | N | 0.033 (4%) | 0.71 (4%) | 3.15 | 14 | 6.01 | 0.53 | 2.16 | 18 | Heizmann et al., 1983; Rodgers and Rowland, 2007 |
| Phenytoin | N | 0.18 (7%) | 0.83 (3%) | 2.21 | 8.32 | 1 | 0.61 | 1.11 | 1.61 | Stella et al., 1998; Brittain, 2007; Uchimura et al., 2010 |
| Diclofenac | A | 0.0014 (18%) | 0.78 (4%) | 4.51 | 4.15 | 1 | 0.55 | 3.68 | 17.6 | Sangster, 1994; Obach, 1999; Avdeef, 2003 |
| Glyburide | A | 0.0012 (16%) | 0.72 (9%) | 4.29 | 5.38 | 1 | 0.57 | 2.59 | 4.81 | Austin et al., 2002; Li et al., 2017 |
| Ketoprofen | A | 0.0041 (9%) | 0.95 (4%) | 3.12 | 4.45 | 1 | 0.56 | 2.06 | 5.02 | Sangster, 1993, 1994; Rodgers and Rowland, 2007; Ye et al., 2016 |
| Nafcillin | A | 0.123 (6%) | 0.94 (14%) | 2.7 | 2.6 | 1 | 0.55d | 1.66 | 33.9 | Wishart et al., 2018 |
| Warfarin | A | 0.0076 (13%) | 0.98 (16%) | 2.7 | 5.05 | 1 | 0.55 | 1.66 | 0.179 | Hiskey et al., 1962; Hansch et al., 1995; Obach, 1999 |
A, acids; B, bases; N, neutral; CL, clearance; vo, vegetable oil.
All 98 experimental values unless otherwise noted.
Rat BP was used.
An average of literature values was used.
0.55 was used (1-hematocrit).
The log Pvo (log of the vegetable oil:buffer partition coefficient) was calculated from log P using eq. 1 (Leo et al., 1971). This term is used to represent neutral lipid partitioning in adipose tissue in the Kp,dPL method; however, it is not used in the Kp,mem prediction method:
| (1) |
Microsomal Partitioning and Plasma Protein Binding.
Equilibrium dialysis was used to determine the fup and fum for the probe drugs using a protocol modified from prior studies (Kochansky et al., 2008; Curran et al., 2011; Di et al., 2017). Human plasma was adjusted to pH 7.4 by adding 1 M HCl. For fum determination, a 0.5 mg/ml rat liver microsomal solution was prepared from a 20 mg/ml pooled rat liver microsome stock solution. For highly bound compounds, a dilution method was used. Plasma was diluted using a 100 mM phosphate buffer and 3 mM MgCl2 solution to either 50% or 10% plasma. A 50% dilution of plasma was used for warfarin, while a 10% dilution was used for ketoprofen, nicardipine, glyburide, diclofenac, felodipine, and mibefradil. Drug solutions (2 μM) in either plasma or microsomes were added to wells on one side of the dialyzer, and blank 100 mM phosphate buffer with 3 mM of MgCl2 was added to the other side. The dialyzer plate was placed in the plate rotator, set to a speed of approximately 22 rotations per minute, and incubated for 22 hours at 37°C and 5% CO2. Liquid chromatography–tandem mass spectrometry was used to determine the concentration of drug in the buffer and the matrix.
The fraction unbound in a given matrix was determined by dividing the concentration of drug on the buffer side by the concentration of drug on the matrix side. For protein binding experiments using the dilution method, the fup value is calculated by eq. 2:
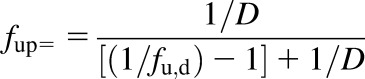 |
(2) |
where D is the dilution factor; fu,d is the fraction unbound in plasma measured in the diluted matrix; and fup is the fraction unbound in plasma.
Experimental fum values were measured at microsomal concentrations between 0.5 and 2 mg/ml and converted to values for 1 mg/ml (eq. 3) (Austin et al., 2002):
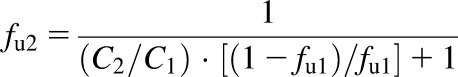 |
(3) |
where fu2 is the corrected unbound fraction; C2 is the 1 mg/ml microsomal protein concentration; C1 is the microsomal protein concentration used in assay; and fu1 is the fraction unbound in matrix measured during the assay. The average fraction unbound, S.D., and CV values were determined for each assay.
Simulations.
A generic PBPK model was used, with different tissues represented by 10 compartments (adipose, bone, brain, gut, kidney, liver, lungs, muscle, skin, and spleen), representing the major tissues in the body (Fig. 1). These compartments are linked via arterial and venous blood flows. The Kp values were predicted for each of the tissues using both methods (Kp,dPL and Kp,mem). The original Kp,dPL method uses two separate equations, one for acids, weak bases, and neutrals, and another for moderate-to-strong bases (eqs. 4–7) (Rodgers et al., 2005; Rodgers and Rowland, 2006).
Fig. 1.

Scheme for the generic PBPK model used in this study.
The Kp,dPL equation used for prediction of Kpu for strong-to-moderate bases is
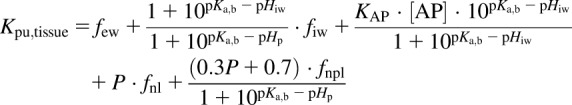 |
(4) |
where few is the fractional volume of extracellular water; fiw is the fractional volume of intracellular water; [AP] is the concentration of acidic phospholipids in the tissue; KAP is the association constant for acidic phospholipids in the tissue; fnl is the fractional volume of neutral lipids; fnpl is the fractional volume of neutral phospholipids; pKa,b is the basic ionization constant; and pKa,a is the acidic ionization constant.
The Rodgers equation used for prediction of Kpu for acids, neutrals, and weak bases is
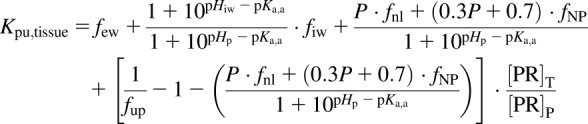 |
(5) |
where [PR]T/[PR]P is the plasma protein tissue (extracellular fluid) to plasma ratio, and NP denotes neutral phospholipid.
For eq. 4, the association constant for blood cells (BCs) is defined in eq. 6, and the tissue:plasma water partitioning coefficient for the BCs is defined in eq. 7:
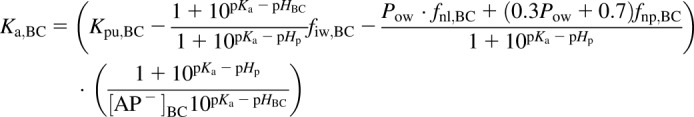 |
(6) |
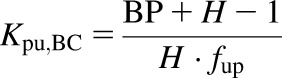 |
(7) |
The tissue:plasma partition coefficient (Kp) can be determined from Kpu by eq. 8:
| (8) |
For the Kp,mem method we use the previously reported equation (Korzekwa and Nagar, 2017a) that considers both phospholipid membrane partitioning with fum and neutral lipid partitioning with P (eq. 9):
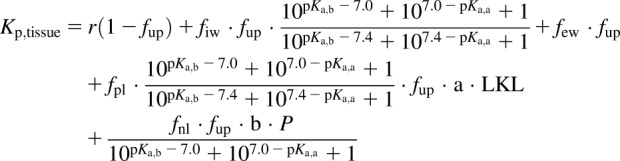 |
(9) |
where r is the protein ratio between the tissue and plasma; fpl is the fractional volume of phospholipid; LKL is the lipid binding constant; and a and b are parameterized coefficients. Plasma and tissue pH values were assumed to be 7.4 and 7.0, respectively. As described previously for Vss predictions (Korzekwa and Nagar, 2017a), the tissue-specific r values for bases were decreased by 2.23-fold due to the lower amount of α-acid glycoprotein in the extracellular fluid relative to albumin (Rowland and Tozer, 2011).
Neutral lipid partitioning in adipose tissue is described by the vegetable oil:water partition coefficient in the Kp,dPL method, which is generally calculated from log P. In the Kp,mem prediction method log P is used directly. The a and b terms in eq. 9 were parameterized using the tissue Kp values and tissue composition data from Rodgers et al. (2005) and Rodgers and Rowland (2006). We excluded zwitterions and combined neutral and acidic phospholipids to obtain a fraction of the total phospholipids. Most fum values were calculated with our previously reported model (Nagar and Korzekwa, 2017) since experimental values are not available for this data set. The log Kpu values and the log of eq. 9 were used to fit a and b, with no additional weighting (log transformation results in 1/Y weighting). Outliers were identified using the BoxWhisker function (Frigge et al., 1989) in Mathematica with outliers defined as >1.5 times the interquartile range. Another model, which included an additional parameter for partitioning into adipose, was evaluated but did not improve predictions. The lipid concentrations multiplied by the lipid binding constant, L times KL, (LKL), was calculated from fum with eq. 10, using fum values normalized to 1 mg/ml microsomal protein:
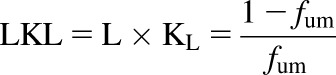 |
(10) |
Exposure overlap coefficients (EOCs) were used to quantify the ability to predict the shape of the C-t profile (eq. 11) (Nagar et al., 2017). They are calculated by determining the overlapping portion of the experimental and predicted C-t profile curves and dividing that area by the experimental area under the curve (AUC). Since the experimental clearance values were used for all predictions, both experimental and predicted C-t profiles will have the same AUC. This allows the EOC to be used as a direct comparison of curve shapes. Differences in the average EOCs were determined using the t test:
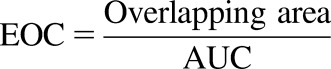 |
(11) |
Vss Predictions.
The Vss value was determined from the predicted Kp values and physiologic volumes (eq. 12):
| (12) |
where Vp is the plasma volume, and Vt represents the tissue volume.
Predicted Vss values were compared with observed values determined by compartmental modeling. To evaluate the predictive precision of the two methods, the absolute fold-error and absolute average fold error (AAFE) values were determined for all compounds and different subsets. Significance of differences in absolute fold errors were determined with a one-tailed t test. The AAFE determines the geometric mean of the absolute fold error (eq. 13) and is a measure of how precisely the two methods predict Vss:
| (13) |
where n is the number of drugs, and FE is the fold error.
Results
The parameters a and b in eq. 9 were fit to the experimental tissue reported Kpu values for the rat (Rodgers et al., 2005; Rodgers and Rowland, 2006). After sequential removal of 20 outliers (out of 401), the optimized parameters were a = 1383 ± 85 and b = 0.096 ± 0.029. The predicted versus observed Kpu values for 381 drugs are shown in Fig. 2. The R2 value for the fit was 0.80. There was no consistent characteristic for the removed outliers with the exception of the overprediction of phencyclidine in four tissues and the underprediction of basic compounds in the lung (five drugs).
Fig. 2.

Observed vs. predicted rat Kpu values from eq. 10: blue, adipose; red, bone; green, brain; purple, gut; light blue, heart; orange, kidney; magenta, liver; yellow, lung; light green, muscle; brown, skin; black, spleen.
Figure 3 and Table 3 show the observed versus predicted Vss values using either the Kp,dPL or Kp,mem prediction method. The accuracy of the Vss predictions was analyzed by determining the percentage of predictions within a range of absolute fold errors (Table 4) and AAFEs (Table 5). For the total data set of 19 drugs, the Kp,mem and Kp,dPL methods had comparable AAFE values (2.12 and 2.27, respectively). The Kp,mem and Kp,dPL methods had one (mibefradil) and two (diphenhydramine, felodipine) outliers, respectively. When these were excluded, the AAFEs were again comparable.
Fig. 3.

Observed Vss value vs. predicted Vss value for 19 drugs: blue, Kp,dPL; red, Kp,mem.
TABLE 3.
Observed and predicted Vss and EOC values for both methods
| Test Drug | Observed Vss | Predicted Vss | EOC | ||
|---|---|---|---|---|---|
| Kp,mem Method | Kp,dPL Method | Kp,mem Method | Kp,dPL Method | ||
| l | l | l | |||
| Betaxolol | 360 | 307 | 856 | 0.97 | 0.61 |
| Diltiazem | 306 | 320 | 309 | 0.76 | 0.80 |
| Diphenhydramine | 788 | 260 | 115 | 0.97 | 0.87 |
| Metoprolol | 274 | 429 | 217 | 0.76 | 0.87 |
| Mibefradil | 187 | 1470 | 53.2 | 0.81 | 0.82 |
| Nicardipine | 62 | 96.4 | 98.5 | 0.79 | 0.78 |
| Quinidine | 227 | 78.3 | 215 | 0.96 | 0.73 |
| Verapamil | 266 | 258 | 136 | 0.73 | 0.76 |
| Caffeine | 42.8 | 47.0 | 36.7 | 0.87 | 0.93 |
| Diazepam | 89.5 | 22.9 | 30.5 | 0.55 | 0.61 |
| Felodipine | 320 | 74.3 | 43.4 | 0.68 | 0.56 |
| Fluconazole | 59.3 | 83.4 | 49.1 | 0.85 | 0.99 |
| Midazolam | 51.2 | 64.1 | 113 | 0.82 | 0.71 |
| Phenytoin | 38.8 | 77.0 | 79.1 | 0.78 | 0.77 |
| Diclofenac | 9.23 | 7.98 | 7.89 | 0.84 | 0.84 |
| Glyburide | 11.78 | 10.2 | 10.0 | 0.86 | 0.87 |
| Ketoprofen | 9.9 | 8.54 | 8.47 | 0.89 | 0.89 |
| Nafcillin | 20.4 | 12.2 | 10.1 | 0.86 | 0.81 |
| Warfarin | 7.66 | 8.20 | 8.15 | 0.96 | 0.97 |
| Average | 0.82 ± 0.11 | 0.80 ± 0.12 | |||
TABLE 4.
Fraction of drugs in which the predictions had less than 1.5-, 2-, and 3-fold error
| Category | <1.5-Fold Error | <2-Fold Error | <3-Fold Error | |||
|---|---|---|---|---|---|---|
| Kp,mem | Kp,dPL | Kp,mem | Kp,dPL | Kp,mem | Kp,dPL | |
| All Compounds | 10/19 | 9/19 | 14/19 | 11/19 | 15/19 | 16/19 |
| Acids | 4/5 | 4/5 | 5/5 | 4/5 | 5/5 | 5/5 |
| Bases | 3/8 | 3/8 | 5/8 | 5/8 | 6/8 | 6/8 |
| Neutrals | 3/6 | 2/6 | 4/6 | 2/6 | 4/6 | 5/6 |
| Log P <3 | 5/9 | 5/9 | 8/9 | 5/9 | 8/9 | 9/9 |
| Log P >3 | 5/10 | 4/10 | 6/10 | 6/10 | 7/10 | 7/10 |
| fum < 0.8 | 6/11 | 4/11 | 8/11 | 6/11 | 8/11 | 9/11 |
| fum > 0.8 | 4/8 | 5/8 | 6/8 | 5/8 | 7/8 | 7/8 |
| fup < 0.1 | 6/10 | 4/10 | 7/10 | 6/10 | 7/10 | 8/10 |
| fup > 0.1 | 4/9 | 5/9 | 7/9 | 8/9 | 8/9 | 8/9 |
TABLE 5.
Absolute average fold error for Vss predictions using both the Kp,mem and Kp,dPL prediction methods
| Category | AAFE | AAFE (Excluding Outliers) | ||
|---|---|---|---|---|
| Kp,mem | Kp,dPL | Kp,mem | Kp,dPL | |
| All Compounds | 2.12 | 2.27 | 1.80 | 1.70 |
| Acids | 1.24 | 1.32 | 1.24 | 1.32 |
| Bases | 2.52 | 2.45 | 1.76 | 1.82 |
| Neutrals | 2.32 | 2.82 | 2.32 | 1.91 |
| Log P <3 | 1.66 | 1.68 | 1.66 | 1.68 |
| Log P >3 | 2.54 | 2.81 | 1.95 | 1.73 |
| fum < 80% | 2.36 | 2.41 | 1.81 | 1.92 |
| fum > 80% | 1.79 | 2.07 | 1.79 | 1.39 |
| fup < 10% | 2.45 | 2.42 | 1.84 | 1.86 |
| fup > 10% | 1.76 | 2.11 | 1.76 | 1.56 |
Ten different categories are compared in Tables 4 and 5. For predictions that were less than 1.5-fold error, Kp,mem scored higher in five categories, lower in two categories, and the same in three categories compared with Kp,dPL (Table 4); for predictions that were less than 3-fold error, Kp,mem scored lower in five categories and the same in five categories compared with Kp,dPL. When comparing all 19 drugs, the AAFEs with Kp,mem versus Kp,dPL were 2.12 versus 2.27, respectively (Table 5). Across the 10 categories, the AAFE was lower for Kp,mem than for Kp,dPL in eight categories. When outliers were excluded in each method, the AAFE was lower for Kp,mem than for Kp,dPL in five categories. For all categories listed in Table 5, there were no statistically significant differences in average fold error between Kp,mem and Kp,dPL.
Simulations.
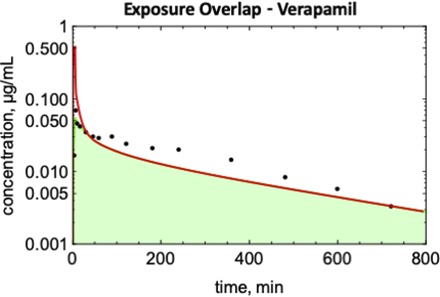
An example of EOC calculation is shown in Fig. 4 for verapamil. Concentration profiles were simulated for 19 drugs with both Kp prediction methods (Figs. 5–7). EOCs were determined for all 19 drugs (Table 3). Overall, there was no significant difference in the average EOC values for the Kp,mem or Kp,dPL method. However, there were some interesting deviations between the methods. The C-t profiles were poorly predicted by both methods for five drugs: mibefradil, diazepam, felodipine, diclofenac, and nafcillin. In addition, the Kp,dPL method poorly predicted the profiles for betaxolol and diphenhydramine. Some possible explanations for these discrepancies are discussed subsequently.
Fig. 4.

Determination of the exposure overlap coefficients for verapamil: red line, simulated C-t profile using the Kp,mem method; blue line, simulated C-t profile using the Kp,dPl method; green area, area of overlap.
Fig. 5.

Observed and predicted C-t profiles for bases: red lines, simulated C-t profile using the Kp,mem method; blue lines, simulated C-t profile using the Kp,dPl method; solid circles, experimental data.
Fig. 7.

Observed and predicted C-t profiles for acids: red lines, simulated C-t profile using the Kp,mem method; blue line, simulated C-t profile using the Kp,dPl method; solid circles, experimental data.
Fig. 6.

Observed and predicted C-t profiles for neutral molecules: red lines, simulated C-t profile using the Kp,mem method; blue lines, simulated C-t profile using the Kp,dPl method; solid circles, experimental data.
As discussed previously (Korzekwa and Nagar, 2017b), eqs. 5–7 indicate that the Kpu,tissue value, and ultimately the unbound Vss value, should be proportional to Kpu,BC (eq. 7), which was first experimentally observed by Hinderling (1997). Also, fup in the denominator of eq. 7 is ultimately multiplied by fup in the PBPK framework (eq. 8). Therefore, the predicted Vss values for bases should be relatively insensitive to fup when using eqs. 4–7. The impact on the predicted Vss after a 2-fold decrease in fup is shown in Table 6. As expected, changing fup for acids has little effect since plasma protein binding is high and Vss is low. For neutrals, a 2-fold decrease in fup results in an average 1.7-fold increase in Vss for both methods. For bases, a 2-fold decrease in fup results in an average 2-fold decrease in Vss for Kp,mem, but an average 1.05-fold increase with Kp,dPL.
TABLE 6.
Impact of errors in fup on Vss
| Compound | Fold Decrease in Vss upon a 2-Fold Decrease in fup | |
|---|---|---|
| Kp,mem | Kp,dPL | |
| Betaxolol | 1.97 | 0.95 |
| Diltiazem | 1.98 | 1.05 |
| Diphenhydramine | 1.94 | 0.82 |
| Metoprolol | 1.98 | 0.75 |
| Mibefradil | 2.00 | 0.95 |
| Nicardipine | 1.90 | 1.01 |
| Quinidine | 1.89 [average (bases)] | 1.00 [average (bases)] |
| Verapamil | 1.95 (1.95 ± 0.04) | 1.06 (0.95 ± 0.11) |
| Caffeine | 1.66 | 1.59 |
| Diazepam | 1.41 | 1.53 |
| Felodipine | 1.79 | 1.66 |
| Fluconazole | 1.82 | 1.70 |
| Midazolam | 1.78 [average (neutrals)] | 1.86 [average (neutrals)] |
| Phenytoin | 1.78 (1.71 ± 0.15) | 1.79 (1.69 ± 0.12) |
| Diclofenac | 1.01 | 1.01 |
| Glyburide | 1.05 | 1.03 |
| Ketoprofen | 1.01 | 1.00 |
| Nafcillin | 1.19 [average (acids)] | 1.09 [average (acids)] |
| Warfarin | 1.01 (1.05 ± 0.08) | 1.01 (1.03 ± 0.04) |
Discussion
Tissue partition coefficients, and ultimately Vss, are determined primarily by competition between plasma protein binding and lipid partitioning. The Kp,dPL method considers binding to neutral lipids such as triglycerides and neutral phospholipids in membranes, with acidic phospholipids considered separately (Rodgers et al., 2005; Rodgers and Rowland, 2006). The method assumes that neutral molecules only interact with neutral phospholipids, and only ionized bases interact with APs, an assumption questioned previously (Korzekwa and Nagar, 2017b). The major acidic phospholipid is phosphatidylserine and the major neutral phospholipid is phosphatidylcholine. Although phosphatidylcholine is net neutral and phosphatidylserine is net acidic, both molecules are zwitterions. Interactions between charged species in the polar head group region are dynamic processes with conformational changes occurring in a picosecond time frame (Tieleman et al., 1997). Balaz (2009) compiled experimental data evaluating the orientation of exogenous molecules in membranes. Hydrophilic molecules accumulate in the polar head group region, amphiphilic molecules accumulate at the interface, and hydrophobic molecules accumulate in the hydrophobic core. We have used this concept to develop quantitative models for membrane partitioning (Nagar and Korzekwa, 2017).
The Kp,mem model is based on previously reported Vss models (Korzekwa and Nagar, 2017a). The Kp model described herein uses the phospholipid component of tissues and fum to model membrane partitioning and log P for neutral lipid interactions. Equation 9 was parameterized using reported tissue composition data and tissue Kp values (Rodgers et al., 2005; Rodgers and Rowland, 2006). Only two constants were parameterized: the scaling factor for 1) membranes and 2) neutral lipids. Although the two methods use different mechanistic assumptions, the resulting fit for Kp,mem (Fig. 2) is similar to that reported by Rodgers et al. (2005) for Kpu parameterization.
The volume of distribution is generally low for acids due to high plasma protein binding and low partitioning into membranes and neutral lipids. At physiologic pH, most acids are negatively charged, and membranes have few hydrogen bond donors. Therefore, microsomal partitioning is low for acids, with fum values ranging from 0.72 to 0.98 (Table 2). Both the Kp,mem and Kp,dPL methods assume that only the neutral acids partitions into tissues, and both methods predict Vss with similar accuracy (AAFE = 2.12 and 2.27 for Kp,mem and Kp,dPL, respectively) (Table 5). This is expected since any model that restricts a compound with low fup to the plasma and extracellular space will predict a Vss value approximately equal to that of plasma proteins (∼7.5 l) (Rowland and Tozer, 2011). Underprediction of acids with Vss values >9, e.g., nafcillin, is frequently observed (Chan et al., 2018). Transporter activity (e.g., organic anion transporting polypeptides) could be one reason for the underprediction.
When two outliers for the Kp,dPL analysis (diphenhydramine and felodipine) are excluded, Vss predictions for neutrals using Kp,dPL improved from an AAFE value of 2.82 to 1.91 (Table 5). For diphenhydramine, the Kp,dPL method resulted in a 6.9-fold underprediction and Kp,mem gave a 3-fold underprediction (not an outlier). For felodipine, Kp,dPL resulted in a 7.4-fold underprediction and Kp,mem gave a 4.3-fold underprediction (not an outlier). The reason for these poor predictions is unknown but it may be difficult to predict Vss of a highly protein bound and partitioned neutral compound (felodipine).
The Kp,mem method uses a single equation for bases, neutrals, and acids and predicts Vss for bases with similar accuracy to Kp,dPL, which uses different equations for bases (eqs. 5–7). The Kp,dPL method assumes that ionized bases only interact with acidic phospholipids. This interaction is parameterized with BP, using the erythrocyte partition coefficient to parameterize binding of the ionized base to acidic phospholipids. Mechanistically, the assumption that bases bind only to acidic phospholipids is questionable. Hydrophobic bases bind to neutral phospholipids as well, with key interactions between the cation and negatively charged phosphate, and the hydrophobic region with the hydrophobic membrane core. From eqs. 5–7, it is clear that for moderate-to-strong bases with Vss values greater than the total body water, the Kp values are dominated by the acidic phospholipid binding terms. Therefore, although binding to only acidic phospholipids was assumed, a similar relationship is possible, assuming that ionized bases bind to all phospholipids. The ratio of neutral to acidic phospholipids is relatively constant across tissues (CV = 15%) (Rodgers et al., 2005), and total phospholipids can be substituted for neutral phospholipids. The KBC term in eq. 6 would be smaller, but the relevant phospholipid term in eq. 5 would be larger.
Another implication of using BP to predict Kpu for bases is the insensitivity of Vss to measured fup. For acids, Vss is insensitive to fup since tissue partitioning is minimal. The Vss values for bases are expected to be proportional to fup since they partition heavily into tissues from the unbound concentration in cytosol (assumed to be equal to the unbound concentration in plasma). This is observed for Kp,mem but not for Kp,dPL (Table 6). This is a consequence of using BP (which includes fup) to calculate Kpu. The Vss predictions can be relatively accurate when BP is used to predict Kp for bases, since errors in fup are not manifest and unbound Vss for bases is proportional to erythrocyte partitioning (Hinderling, 1997). However, errors in fup can still result in many other inaccuracies, including in predictions of clearance and target activity.
Overall, the Kp,mem and Kp,dPL models give similarly accurate predictions, explaining 68% and 63% of the variance in Vss (80% and 79% without outliers), respectively. Several factors may explain the remaining variance. First, there can be significant variability in the Vss values measured across clinical studies. Not all pharmacokinetic data sets provide body weights. Also, experimental data from multiple sources are used, e.g., BP values; for example, Graham et al. (2012) observed a 7% decrease in accuracy when predicted instead of experimental log P values were used. For fup, differences between laboratories can be very large. Several recent publications discuss assay conditions for protein binding, including dilution and use of CO2 (Kochansky et al., 2008; Curran et al., 2011; Di et al., 2017). In this study, we measured a value of fup of 0.03 for mibefradil, whereas a value of <0.005 was reported previously (Clozel et al., 1991). Use of a smaller fup value would result in a better prediction with Kp,mem, but exclusion of our data is not justified.
Lysosomal partitioning affects tissue distribution for bases. For strong bases, partitioning into lysosomes due to pH differences results in lysosomal concentrations >200 times cytosolic concentrations. Assuming 5% lysosomes and 60% intracellular water in cells, partitioning of a strong base into lysosomes can increase the Kpu value by 6-fold. As discussed previously, while lysosomal partitioning certainly occurs it is likely to be highly correlated with phospholipid partitioning of bases (Korzekwa and Nagar, 2017a). Finally, although the vegetable oil:water partition constant would be a good surrogate for adiposomes if measured, this value is typically modeled using log P, which is not necessarily accurate (Korzekwa and Nagar, 2017b).
Transporter-mediated distribution can result in inaccurate predictions of Kp and Vss, particularly for some acids. Organic anion transporting polypeptide transporters can alter hepatic intracellular concentrations by two orders of magnitude (Kulkarni et al., 2016). Therefore, uptake into this organ alone can result in a 2-fold increase in Vss. Efflux transporters (e.g., P-glycoprotein and breast cancer resistance protein) will have a smaller impact. The decrease in Vss due to P-glycoprotein and breast cancer resistance protein at the blood-brain barrier would result in a 2% decrease in Vss. The impact from the liver would be even smaller since efflux transporters in the apical membrane would only decrease liver concentrations by 50% (Korzekwa and Nagar, 2014).
Since the AUC is determined by experimental clearance and dose in both methods, the AUC values for the simulations are normalized, and the EOC captures differences in the shapes of the C-t profile (Fig. 4). Several C-t profiles in Figs. 5–7 are not well predicted by either method. Although there are differences in the EOC for some drugs, the average EOCs for the Kp,mem and Kp,dPL prediction methods were not significantly different. Since clearance is constant, when the Vss value is overpredicted (e.g., betaxolol using Kp,dPL and mibefradil using Kp,mem) (Fig. 5), the terminal half-life is overpredicted. When the Vss value is underpredicted (e.g., diphenhydramine using Kp,dPL) (Fig. 5), the terminal half-life is underpredicted. Perhaps the most significant deficiency of the reported modeling approaches is the assumption of perfusion-limited distribution. As seen with diazepam, felodipine, diclofenac, and nafcillin, accurate C-t profiles are not predicted even when the Vss value is well predicted. For verapamil (Figs. 4 and 5), the distribution phase is not well predicted, presumably due to a combination of using a perfusion-limited model and experimental clearance. Clearly, multicompartmental distribution is not accurately modeled with perfusion-limited distribution.
In conclusion, Kp,mem can be used to predict Kpu with accuracy similar to Kp,dPL. An advantage of using fum to parameterize membrane partitioning is that fum is used for clearance prediction and is generated early in the discovery/development process. Also, differentiating between acidic and neutral phospholipids for bases and using 0.3P for neutral compounds is not mechanistically justified. Finally, since both the extent and rate of membrane partitioning and permeability are important, a mechanistically sound basis for membrane interactions is necessary for improved PBPK models.
Abbreviations
- AAFE
absolute average fold error
- AP
acidic phospholipid
- AUC
area under the curve
- BC
blood cell
- BP
blood to plasma
- C-t
concentration-time
- EOC
exposure overlap coefficient
- few
fractional volume of extracellular water
- fiw
fractional volume of intracellular water
- fnl
fractional volume of neutral lipids
- fum
fraction unbound in microsomes
- fup
fraction unbound in plasma
- Kp
tissue:plasma partition coefficient
- Kp,dPL
differential phospholipid tissue-plasma partition coefficient prediction method
- Kp,mem
membrane-based tissue-plasma partition coefficient prediction method
- Kpu
unbound tissue:plasma partition coefficient
- P
octanol:water partition coefficient
- PBPK
physiologically based pharmacokinetic
- pKa,a
ionization constant for acids
- pKa,b
ionization constant for bases
- Vss
steady-state volume of distribution
Authorship Contributions
Participated in research design: Holt, Nagar, Korzekwa.
Conducted experiments: Holt, Ye.
Performed data analysis: Holt, Nagar, Korzekwa.
Wrote or contributed to the writing of the manuscript: Holt, Nagar, Korzekwa.
Footnotes
This research was supported by the National Institutes of Health [Grants R01GM104178 and R01GM114369].
References
- Agarwal SK, Kriel RL, Brundage RC, Ivaturi VD, Cloyd JC. (2013) A pilot study assessing the bioavailability and pharmacokinetics of diazepam after intranasal and intravenous administration in healthy volunteers. Epilepsy Res 105:362–367. [DOI] [PubMed] [Google Scholar]
- Akaike T. (1974) A new look at the statistical model identification. IEEE Trans Autom Control 19:716–723. [Google Scholar]
- Albert KS, Hallmark MR, Sakmar E, Weidler DJ, Wagner JG. (1975) Pharmacokinetics of diphenhydramine in man. J Pharmacokinet Biopharm 3:159–170. [DOI] [PubMed] [Google Scholar]
- Arundel P. (1997) A multi-compartmental model generally applicable to physiologically-based pharmacokinetics. IFAC Proc Vol, 30:129–133. [Google Scholar]
- Austin RP, Barton P, Cockroft SL, Wenlock MC, Riley RJ. (2002) The influence of nonspecific microsomal binding on apparent intrinsic clearance, and its prediction from physicochemical properties. Drug Metab Dispos 30:1497–1503. [DOI] [PubMed] [Google Scholar]
- Avdeef A. (2003) Absorption and Drug Development: Solubility, Permeability, and Charge State, Wiley, Hoboken, NJ. [Google Scholar]
- Balaz S. (2009) Modeling kinetics of subcellular disposition of chemicals. Chem Rev 109:1793–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezhkovskiy LM. (2004) Volume of distribution at steady state for a linear pharmacokinetic system with peripheral elimination. J Pharm Sci 93:1628–1640. [DOI] [PubMed] [Google Scholar]
- Björkman S. (2002) Prediction of the volume of distribution of a drug: which tissue-plasma partition coefficients are needed? J Pharm Pharmacol 54:1237–1245. [DOI] [PubMed] [Google Scholar]
- Blanchard J, Sawers SJA. (1983) The absolute bioavailability of caffeine in man. Eur J Clin Pharmacol 24:93–98. [DOI] [PubMed] [Google Scholar]
- Brittain H. (2007) Profiles of Drug Substances, Excipients, and Related Methodology, Academic Press, Cambridge, MA. [Google Scholar]
- Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP. (1997) Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health 13:407–484. [DOI] [PubMed] [Google Scholar]
- Campbell BC, Kelman AW, Hillis WS. (1985) Noninvasive assessment of the haemodynamic effects of nicardipine in normotensive subjects. Br J Clin Pharmacol 20 (Suppl 1):55S–61S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan R, De Bruyn T, Wright M, Broccatelli F. (2018) Comparing mechanistic and preclinical predictions of volume of distribution on a large set of drugs. Pharm Res 35:87. [DOI] [PubMed] [Google Scholar]
- Clozel JP, Osterrieder W, Kleinbloesem CH, Welker HA, Schlappi B, Tudor R, Hefti F, Schmitt R, Eggers H. (1991) RO 40-5967: a new nonhydropyridine calcium antagonist. Cardiovasc Drug Rev 9:4–17. [Google Scholar]
- Curran RE, Claxton CRJ, Hutchison L, Harradine PJ, Martin IJ, Littlewood P. (2011) Control and measurement of plasma pH in equilibrium dialysis: influence on drug plasma protein binding. Drug Metab Dispos 39:551–557. [DOI] [PubMed] [Google Scholar]
- De Buck SS, Sinha VK, Fenu LA, Gilissen RA, Mackie CE, Nijsen MJ. (2007) The prediction of drug metabolism, tissue distribution, and bioavailability of 50 structurally diverse compounds in rat using mechanism-based absorption, distribution, and metabolism prediction tools. Drug Metab Dispos 35:649–659. [DOI] [PubMed] [Google Scholar]
- Debruyne D. (1997) Clinical pharmacokinetics of fluconazole in superficial and systemic mycoses. Clin Pharmacokinet 33:52–77. [DOI] [PubMed] [Google Scholar]
- Debruyne D, Hurault de Ligny B, Ryckelynck JP, Albessard F, Moulin M. (1987) Clinical pharmacokinetics of ketoprofen after single intravenous administration as a bolus or infusion. Clin Pharmacokinet 12:214–221. [DOI] [PubMed] [Google Scholar]
- Di L, Breen C, Chambers R, Eckley ST, Fricke R, Ghosh A, Harradine P, Kalvass JC, Ho S, Lee CA, et al. (2017) Industry perspective on contemporary protein-binding methodologies: considerations for regulatory drug-drug interaction and related guidelines on highly bound drugs. J Pharm Sci 106:3442–3452. [DOI] [PubMed] [Google Scholar]
- Diez I, Colom H, Moreno J, Obach R, Peraire C, Domenech J. (1991) A comparative in vitro study of transdermal absorption of a series of calcium channel antagonists. J Pharm Sci 80:931–934. [DOI] [PubMed] [Google Scholar]
- Edgar B, Regårdh CG, Johnsson G, Johansson L, Lundborg P, Löfberg I, Rönn O. (1985) Felodipine kinetics in healthy men. Clin Pharmacol Ther 38:205–211. [DOI] [PubMed] [Google Scholar]
- Fenneteau F, Poulin P, Nekka F. (2010) Physiologically based predictions of the impact of inhibition of intestinal and hepatic metabolism on human pharmacokinetics of CYP3A substrates. J Pharm Sci 99:486–514. [DOI] [PubMed] [Google Scholar]
- Frigge M, Hoaglin DC, Iglewicz B. (1989) Some implementations of the boxplot. Am Statistician 43:50–54. [Google Scholar]
- Ghafourian T, Barzegar-Jalali M, Hakimiha N, Cronin MTD. (2004) Quantitative structure-pharmacokinetic relationship modelling: apparent volume of distribution. J Pharm Pharmacol 56:339–350. [DOI] [PubMed] [Google Scholar]
- Graham H, Walker M, Jones O, Yates J, Galetin A, Aarons L. (2012) Comparison of in-vivo and in-silico methods used for prediction of tissue: plasma partition coefficients in rat. J Pharm Pharmacol 64:383–396. [DOI] [PubMed] [Google Scholar]
- Greene DS, Quintiliani R, Nightingale CH. (1978) Physiological perfusion model for cephalosporin antibiotics I: model selection based on blood drug concentrations. J Pharm Sci 67:191–196. [DOI] [PubMed] [Google Scholar]
- Gugler R, Manion CV, Azarnoff DL. (1976) Phenytoin: pharmacokinetics and bioavailability. Clin Pharmacol Ther 19:135–142. [DOI] [PubMed] [Google Scholar]
- Hallifax D, Houston JB. (2006) Binding of drugs to hepatic microsomes: comment and assessment of current prediction methodology with recommendation for improvement. Drug Metab Dispos 34:724–726, author reply 727. [DOI] [PubMed] [Google Scholar]
- Hansch C, Leo A, Hockman D. (1995) Exploring QSAR: Hydrophobic, Electronic, and Steric Constraints, ACS Publications, Washington, DC. [Google Scholar]
- Heizmann P, Eckert M, Ziegler WH. (1983) Pharmacokinetics and bioavailability of midazolam in man. Br J Clin Pharmacol 16 (Suppl 1):43S–49S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann P, Rodger SD, Remones G, Thenot JP, London DR, Morselli PL. (1983) Pharmacokinetics of diltiazem after intravenous and oral administration. Eur J Clin Pharmacol 24:349–352. [DOI] [PubMed] [Google Scholar]
- Hinderling PH. (1997) Red blood cells: a neglected compartment in pharmacokinetics and pharmacodynamics. Pharmacol Rev 49:279–295. [PubMed] [Google Scholar]
- Hiskey CF, Bullock E, Whitman G. (1962) Spectrophotometric study of aqueous solutions of warfarin sodium. J Pharm Sci 51:43–46. [DOI] [PubMed] [Google Scholar]
- Ishihama Y, Nakamura M, Miwa T, Kajima T, Asakawa N. (2002) A rapid method for pKa determination of drugs using pressure-assisted capillary electrophoresis with photodiode array detection in drug discovery. J Pharm Sci 91:933–942. [DOI] [PubMed] [Google Scholar]
- Jansson R, Bredberg U, Ashton M. (2008) Prediction of drug tissue to plasma concentration ratios using a measured volume of distribution in combination with lipophilicity. J Pharm Sci 97:2324–2339. [DOI] [PubMed] [Google Scholar]
- Jones RD, Jones HM, Rowland M, Gibson CR, Yates JWT, Chien JY, Ring BJ, Adkison KK, Ku MS, He H, et al. (2011) PhRMA CPCDC initiative on predictive models of human pharmacokinetics, part 2: comparative assessment of prediction methods of human volume of distribution. J Pharm Sci 100:4074–4089. [DOI] [PubMed] [Google Scholar]
- Kochansky CJ, McMasters DR, Lu P, Koeplinger KA, Kerr HH, Shou M, Korzekwa KR. (2008) Impact of pH on plasma protein binding in equilibrium dialysis. Mol Pharm 5:438–448. [DOI] [PubMed] [Google Scholar]
- Korzekwa K, Nagar S. (2014) Compartmental models for apical efflux by P-glycoprotein: part 2—a theoretical study on transporter kinetic parameters. Pharm Res 31:335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzekwa K, Nagar S. (2017a) Drug distribution part 2. Predicting volume of distribution from plasma protein binding and membrane partitioning. Pharm Res 34:544–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzekwa K, Nagar S. (2017b) On the nature of physiologically-based pharmacokinetic models—A priori or a posteriori? Mechanistic or empirical? Pharm Res 34:529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni P, Korzekwa K, Nagar S. (2016) Intracellular unbound atorvastatin concentrations in the presence of metabolism and transport. J Pharmacol Exp Ther 359:26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo A, Hansch C, Elkins D. (1971) Partition coefficients and their uses. Chem Rev 71:525–616. [Google Scholar]
- Li R, Bi YA, Vildhede A, Scialis RJ, Mathialagan S, Yang X, Marroquin LD, Lin J, Varma MVS. (2017) Transporter-mediated disposition, clinical pharmacokinetics and cholestatic potential of glyburide and its primary active metabolites. Drug Metab Dispos 45:737–747. [DOI] [PubMed] [Google Scholar]
- Lombardo F, Obach RS, Dicapua FM, Bakken GA, Lu J, Potter DM, Gao F, Miller MD, Zhang Y. (2006) A hybrid mixture discriminant analysis—random forest computational model for the prediction of volume of distribution of drugs in human. J Med Chem 49:2262–2267. [DOI] [PubMed] [Google Scholar]
- Lombardo F, Obach RS, Shalaeva MY, Gao F. (2004) Prediction of human volume of distribution values for neutral and basic drugs. 2. Extended data set and leave-class-out statistics. J Med Chem 47:1242–1250. [DOI] [PubMed] [Google Scholar]
- Ludden TM, Boyle DA, Gieseker D, Kennedy GT, Crawford MH, Ludden LK, Clementi WA. (1988) Absolute bioavailability and dose proportionality of betaxolol in normal healthy subjects. J Pharm Sci 77:779–783. [DOI] [PubMed] [Google Scholar]
- Maguire KP, Burrows GD, Norman TR, Scoggins BA. (1980) Blood/plasma distribution ratios of psychotropic drugs. Clin Chem 26:1624–1625. [PubMed] [Google Scholar]
- McAllister RG, Jr, Kirsten EB. (1982) The pharmacology of verapamil. IV. Kinetic and dynamic effects after single intravenous and oral doses. Clin Pharmacol Ther 31:418–426. [DOI] [PubMed] [Google Scholar]
- Nagar S, Korzekwa K. (2012) Commentary: nonspecific protein binding versus membrane partitioning: it is not just semantics. Drug Metab Dispos 40:1649–1652. [DOI] [PubMed] [Google Scholar]
- Nagar S, Korzekwa K. (2017) Drug distribution. Part 1. Models to predict membrane partitioning. Pharm Res 34:535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagar S, Korzekwa RC, Korzekwa K. (2017) Continuous intestinal absorption model based on the convection-diffusion equation. Mol Pharm 14:3069–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagar S, Tucker J, Weiskircher EA, Bhoopathy S, Hidalgo IJ, Korzekwa K. (2014) Compartmental models for apical efflux by P-glycoprotein—part 1: evaluation of model complexity. Pharm Res 31:347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obach RS. (1999) Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos 27:1350–1359. [PubMed] [Google Scholar]
- Obach RS, Baxter JG, Liston TE, Silber BM, Jones BC, MacIntyre F, Rance DJ, Wastall P. (1997) The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J Pharmacol Exp Ther 283:46–58. [PubMed] [Google Scholar]
- Oie S, Tozer TN. (1979) Effect of altered plasma protein binding on apparent volume of distribution. J Pharm Sci 68:1203–1205. [DOI] [PubMed] [Google Scholar]
- O’Neil M. (2006) The Merck Index–An Encyclopedia of Chemicals, Drugs, and Biologicals, Merck and Co., Inc., Whitehouse Station, NJ. [Google Scholar]
- O’Reilly RA, Welling PG, Wagner JG. (1971) Pharmacokinetics of warfarin following intravenous administration to man. Thromb Diath Haemorrh 25:178–186. [PubMed] [Google Scholar]
- Pandey MM, Jaipal A, Kumar A, Malik R, Charde SY. (2013) Determination of pKa of felodipine using UV-visible spectroscopy. Spectrochim Acta A Mol Biomol Spectrosc 115:887–890. [DOI] [PubMed] [Google Scholar]
- Poulin P, Haddad S. (2011) Microsome composition-based model as a mechanistic tool to predict nonspecific binding of drugs in liver microsomes. J Pharm Sci 100:4501–4517. [DOI] [PubMed] [Google Scholar]
- Poulin P, Krishnan K. (1995) A biologically-based algorithm for predicting human tissue: blood partition coefficients of organic chemicals. Hum Exp Toxicol 14:273–280. [DOI] [PubMed] [Google Scholar]
- Poulin P, Schoenlein K, Theil FP. (2001) Prediction of adipose tissue: plasma partition coefficients for structurally unrelated drugs. J Pharm Sci 90:436–447. [DOI] [PubMed] [Google Scholar]
- Poulin P, Theil FP. (2000) A priori prediction of tissue:plasma partition coefficients of drugs to facilitate the use of physiologically-based pharmacokinetic models in drug discovery. J Pharm Sci 89:16–35. [DOI] [PubMed] [Google Scholar]
- Poulin P, Theil FP. (2002) Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism-based prediction of volume of distribution. J Pharm Sci 91:129–156. [DOI] [PubMed] [Google Scholar]
- Poulin P, Theil FP. (2009) Development of a novel method for predicting human volume of distribution at steady-state of basic drugs and comparative assessment with existing methods. J Pharm Sci 98:4941–4961. [DOI] [PubMed] [Google Scholar]
- Recanatini M. (1992) Partition and distribution coefficients of aryloxypropanolamine β-adrenoceptor antagonists. J Pharm Pharmacol 44:68–70. [DOI] [PubMed] [Google Scholar]
- Regårdh CG, Borg KO, Johansson R, Johnsson G, Palmer L. (1974) Pharmacokinetic studies on the selective beta1-receptor antagonist metoprolol in man. J Pharmacokinet Biopharm 2:347–364. [DOI] [PubMed] [Google Scholar]
- Rekker R, Mannhold R. (1992) Calculation of Drug Lipophilicity: The Hydrophobic Fragmental Constant Approach, VCH. [Google Scholar]
- Riddell JG, Harron DWG, Shanks RG. (1987) Clinical pharmacokinetics of β-adrenoceptor antagonists. An update. Clin Pharmacokinet 12:305–320. [DOI] [PubMed] [Google Scholar]
- Ripa S, Ferrante L, Prenna M. (1993) Pharmacokinetics of fluconazole in normal volunteers. Chemotherapy 39:6–12. [DOI] [PubMed] [Google Scholar]
- Robinson MA, Mehvar R. (1996) Enantioselective distribution of verapamil and norverapamil into human and rat erythrocytes: the role of plasma protein binding. Biopharm Drug Dispos 17:577–587. [DOI] [PubMed] [Google Scholar]
- Rodgers T, Leahy D, Rowland M. (2005) Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate-to-strong bases. J Pharm Sci 94:1259–1276. [DOI] [PubMed] [Google Scholar]
- Rodgers T, Rowland M. (2006) Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci 95:1238–1257. [DOI] [PubMed] [Google Scholar]
- Rodgers T, Rowland M. (2007) Mechanistic approaches to volume of distribution predictions: understanding the processes. Pharm Res 24:918–933. [DOI] [PubMed] [Google Scholar]
- Rowland M, Tozer TN. (2011) Distribution of drugs extensively bound to plasma proteins, Clinical Pharmacokinetics and Pharmacodynamics: Concepts and Applications pp 695–701, Wolters Kluwer Health/Lippincott William & Wilkins, Philadelphia, PA. [Google Scholar]
- Sangster J. (1993) LOGKOW—A DataBank of Evaluated Octanol-Water Partition Coefficients, Sangster Research Laboratories, Montreal, Canada. [Google Scholar]
- Sangster J. (1994) Octanol-Water Partition Coefficients: Fundamentals and Physical Chemistry, Wiley, New York. [Google Scholar]
- Scavone JM, Luna BG, Harmatz JS, Von Moltke L, Greenblatt DJ. (1990) Diphenhydramine kinetics following intravenous, oral, and sublingual dimenhydrinate administration. Biopharm Drug Dispos 11:185–189. [DOI] [PubMed] [Google Scholar]
- Stella VJ, Martodihardjo S, Terada K, Rao VM. (1998) Some relationships between the physical properties of various 3-acyloxymethyl prodrugs of phenytoin to structure: potential in vivo performance implications. J Pharm Sci 87:1235–1241. [DOI] [PubMed] [Google Scholar]
- Tieleman DP, Marrink SJ, Berendsen HJC. (1997) A computer perspective of membranes: molecular dynamics studies of lipid bilayer systems. Biochim Biophys Acta 1331:235–270. [DOI] [PubMed] [Google Scholar]
- Uchimura T, Kato M, Saito T, Kinoshita H. (2010) Prediction of human blood-to-plasma drug concentration ratio. Biopharm Drug Dispos 31:286–297. [DOI] [PubMed] [Google Scholar]
- Ueda CT, Hirschfeld DS, Scheinman MM, Rowland M, Williamson BJ, Dzindzio BS. (1976) Disposition kinetics of quinidine. Clin Pharmacol Ther 19:30–36. [DOI] [PubMed] [Google Scholar]
- van De Waterbeemd H. (1993) Calculation of Drug Lipophilicity – The Hydrophobic Fragmental Constant Approach. Quantitative Structure-Activity Relationships 12:269-. [Google Scholar]
- Waller ES, Sharanevych MA, Yakatan GJ. (1982) The effect of probenecid on nafcillin disposition. J Clin Pharmacol 22:482–489. [DOI] [PubMed] [Google Scholar]
- Welker HA, Wiltshire H, Bullingham R. (1998) Clinical pharmacokinetics of mibefradil. Clin Pharmacokinet 35:405–423. [DOI] [PubMed] [Google Scholar]
- Willis JV, Kendall MJ, Jack DB. (1980) A study of the effect of aspirin on the pharmacokinetics of oral and intravenous diclofenac sodium. Eur J Clin Pharmacol 18:415–418. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z, et al. (2018) DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 46 (D1):D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye M, Nagar S, Korzekwa K. (2016) A physiologically based pharmacokinetic model to predict the pharmacokinetics of highly protein-bound drugs and the impact of errors in plasma protein binding. Biopharm Drug Dispos 37:123–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhivkova Z, Doytchinova I. (2012) Prediction of steady-state volume of distribution of acidic drugs by quantitative structure-pharmacokinetics relationships. J Pharm Sci 101:1253–1266. [DOI] [PubMed] [Google Scholar]
- Zou P, Zheng N, Yang Y, Yu LX, Sun D. (2012) Prediction of volume of distribution at steady state in humans: comparison of different approaches. Expert Opin Drug Metab Toxicol 8:855–872. [DOI] [PubMed] [Google Scholar]