
Extended Data Figure 7. Ibogaine docking and molecular dynamics simulations.
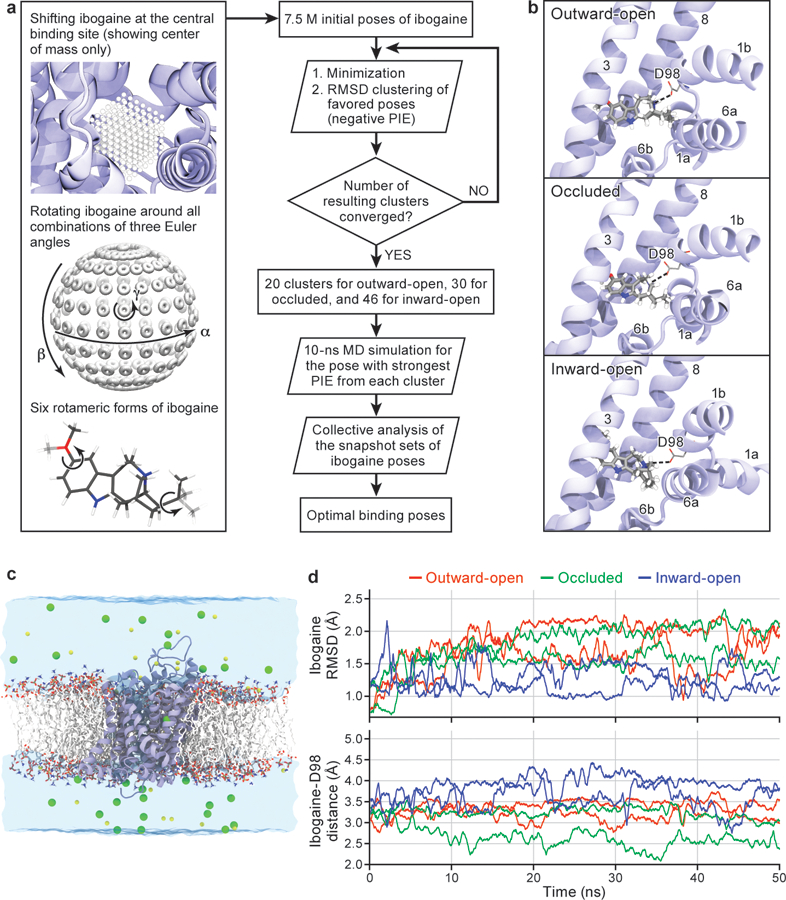
a, Workflow of ligand docking. b, Optimal binding poses of ibogaine in the central binding site of the outward-open, occluded, and inward-open conformations. For clarity, only TM helices surrounding the central binding site, i.e., TM1, TM3, TM6, and TM8, are shown. The interaction between ibogaine and Asp98 of SERT, both shown in sticks, is highlighted. c, Simulation system used to study the structural stability and ibogaine binding of different conformations of SERT (two independent 50 ns simulations for each conformation), showing the transporter in cartoon, with 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine lipids drawn in sticks, bulk water in a transparent surface, and solute ions (100 mM NaCl for the outward-open simulation) in yellow (Na+) and green (Cl−) spheres. d, Structural stability of bound ibogaine measured as the mass-weighted root-mean-square deviation (RMSD, including hydrogen atoms) of the ligand, as well as the Asp98-ibogaine (O-N) distance. The trajectories of outward-open, occluded, and inward-open SERT are plotted in red, green, and blue respectively, and shown for two independent simulations.