

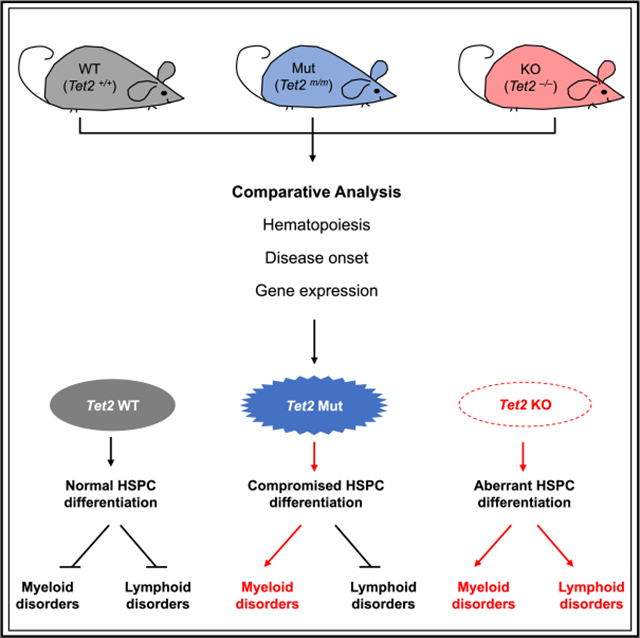
SUMMARY
The Ten-eleven translocation (TET) enzymes regulate gene expression by promoting DNA demethylation and partnering with chromatin modifiers. TET2, a member of this family, is frequently mutated in hematological disorders. The contributions of TET2 in hematopoiesis have been attributed to its DNA demethylase activity, and the significance of its nonenzymatic functions has remained undefined. To dissect the catalytic and non-catalytic requirements of Tet2, we engineered catalytically inactive Tet2 mutant mice and conducted comparative analyses of Tet2 mutant and Tet2 knockout animals. Tet2 knockout mice exhibited expansion of hematopoietic stem and progenitor cells (HSPCs) and developed myeloid and lymphoid disorders, while Tet2 mutant mice predominantly developed myeloid malignancies reminiscent of human myelodysplastic syndromes. HSPCs from Tet2 knockout mice exhibited distinct gene expression profiles, including downregulation of Gata2. Overexpression of Gata2 in Tet2 knockout bone marrow cells ameliorated disease phenotypes. Our results reveal the non-catalytic roles of TET2 in HSPC homeostasis.
In Brief
The DNA demethylase TET2 is commonly mutated in hematological disorders, but the significance of its enzymatic versus nonenzymatic roles in hematopoiesis remains undefined. Using Tet2 catalytic-mutant and knockout mice, Ito et al. find that Tet2 enzymatic activity is critical for myelopoiesis, while aberrant lymphopoiesis is mainly associated with complete loss of Tet2.
Graphical Abstract
INTRODUCTION
The Ten-eleven translocation (TET) family of dioxygenases (Tet1/2/3) are DNA-modifying enzymes expressed in various embryonic and adult cell types, including hematopoietic stem cells (HSCs) (Cimmino et al., 2011; Tahiliani et al., 2009). They regulate gene expression in part by promoting active and passive DNA demethylation through the conversion of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) and other oxidized derivatives (Ito et al., 2010, 2011; Tahiliani et al., 2009). TET2 is a unique member of this family that is highly expressed in the hematopoietic lineages. It is one of the most frequently mutated genes in myelodysplastic syndrome (MDS), an incurable clonal hematological disorder marked by pronounced reductions in blood cell production, significant dysplasia in the cells produced, or some combination of both (Tefferi and Vardiman, 2009). Loss-of-function mutations in the TET2 gene are seen in ~20% of MDS cases and almost 50% of chronic myelomonocytic leukemia (CMML) patients (Abdel-Wahab et al., 2009; Delhommeau et al., 2009; Kosmider et al., 2009; Song et al., 2013). While most mutations lead to loss of the entire protein, some are only missense mutations involving either the catalytic or non-catalytic domains (Abdel-Wahab et al., 2009; Sato et al., 2016). Deletion of Tet2 in mice increases HSC self-renewal and restricts differentiation, causing CMML-like disease with aggressive metastases (Li et al., 2011; Moran-Crusio et al., 2011). In addition, loss of TET2 has been linked to lymphoid disorders, as a fraction of Tet2 knockout (KO) mice developed aberrant lymphopoiesis, and somatic mutations of TET2 have been found in subtypes of lymphoid malignancies (e.g., angioimmunoblastic T lymphomas, ~33%; diffuse large B cell lymphomas, ~10%; and mantle cell lymphoma, 4%) (Asmar et al., 2013; Dominguez et al., 2018; Lemonnier et al., 2012; Mouly et al., 2018; Pan et al., 2017; Quivoron et al., 2011; Reddy et al., 2017).
The catalytic activity of Tet2 is mainly confined to its C terminus, which alone can promote DNA hydroxylation (Tahiliani et al., 2009). Tet2 has also been implicated in formation of chromatin regulatory complexes with O-linked N-acetylglucosamine transferase (OGT) and histone deacetylases (HDACs) to regulate transcription and histone modifications (Chen et al., 2013; Williams et al., 2011; Wu et al., 2011; Zhang et al., 2015). Thus far, the contributions of Tet2 to hematopoiesis have largely been studied using conventional or conditional KO mouse models that lack the entireTet2 protein. Such an approach, despite interrogating the full functions of Tet2, fails to define or distinguish between its catalytic and non-catalytic requirements in a physiologically relevant context. In this study, to comprehensively dissect endogenous and physiologically relevant Tet2 functions during normal and abnormal hematopoiesis in vivo, we generated Tet2 catalytically inactive mice by mutating the enzymatic pocket of the protein and conducted a comparative analysis of the resulting Tet2 mutant (Mut) mice with Tet2 KO mice. While both groups developed symptoms of hematological disorders early in life and had similar survival rates, differences in the disease spectrum were found between the cohorts; Tet2 KO mice developed both myeloid and lymphoid disorders with increased hematopoietic stem and progenitor cell (HSPC) fractions. In contrast, in Tet2 Mut mice, aberrant hematopoiesis was restricted largely to the myeloid lineage. Our study establishes that the role of Tet2 in hematopoietic system goes beyond its catalytic functions and identifies distinct non-catalytic requirements for Tet2 in the HSPC homeostasis, as well as in the onset of aberrant lymphopoiesis.
RESULTS
Generation of Tet2 Catalytic Mut Mice
To establish the distinct contributions of catalytic functions of Tet2 to hematopoietic homeostasis, we developed a mouse strain lacking only the catalytic activity of Tet2. We generated a Tet2 catalytic-deficient allele (Tet2m) by introducing amino acid substitutions H1367Y and D1369A in the catalytic domain of Tet2 protein using CRISPR/Cas9-based gene editing in embryonic stem cells (ESCs) (Figure 1A). A properly targeted heterozygous Tet2m/+ ESC clone was injected into blastocysts to generate chimeric mice (Figure S1A), which were bred to wildtype (WT) C57/BL6 mice to first generate Tet2m/+ and then, after intercrossing, Tet2m/m mice (here after referred to as Tet2 Mut) (Figures 1B and 1C). The Tet2 Mut mice, like a previously used strain of Tet2−/− mice (Tet2 KO) (Li et al., 2011), were born in expected Mendelian ratios and grew normally in a 129Sv-C57/BL6 mixed background (Figures S1B–S1D). The expression of Tet2 was comparable in Tet2 Mut and WT HSPCs (Lin−Sca-1+c-Kit+ cells) at the mRNA level (Figure 1D) and in total bone marrow at the protein level (Figure 1E). 5hmC levels in DNA isolated from 2-month-old Tet2 Mut bone marrow cells was reduced by ~60% and was similar to the level present in age-matched Tet2 KO cells (Figure 1F). The Tet2 KO mice lack all functions of the enzyme, while the Tet2 Mut mice only lack the enzymatic activity of the protein. Therefore, Tet2 Mut mice, in conjunction with Tet2 KO and WT mice, comprise a viable platform for comparative physiological, cellular, and molecular analyses to dissect the catalytic-dependent and independent functions of Tet2 in HSPCs and MDS etiology (Figure 1G).
Figure 1. Generation of Tet2 Catalytic Mutant (Mut) Mouse and Characterization of Its Hematological Phenotypes.

(A) Tet2 wild-type (WT) and catalytic Mut proteins (top) and alleles (bottom).
(B and C) Analysis of the genotypes of Tet2 catalytic Mut mice by RFLP (restriction fragment length polymorphism) (B) and Sanger sequencing (C).
(D) Tet2 mRNA levels in Lin−Sca-1+c-Kit+ (LSK) cells of Indicated genotypes quantified by qRT-PCR. Data are normalized to Gapdh. Note the absence of Tet2 mRNA in knockout (KO) but its normal expression in Mut samples.
(E) Tet2 protein levels in total bone marrow isolated from 4-month-old mice assessed by western blot. Quantified Tet2 signal intensity normalized to actin signal intensity is shown. Note the absence of Tet2 protein in KO but its normal and comparable expression in Mut and WT samples.
(F) Levels of 5hmC in the bone marrow of the indicated genotypes, assessed by dot blot and quantified as average of three separated experiments (relative to WT control samples) (n = 3).
(G) Overview of experimental platform using Tet2 WT, Mut, and KO mice.
(H) Blood counts of Tet2 Mut and KO mice (4 months old, n = 8).
(I) Representative smears of peripheral blood (PB) of 10-month-old Tet2 Mut (ii), Tet2 KO with MDS-like myeloid (iii), and lymphoid (iv) phenotypes and WT control mice (i). Images of representative dysplastic hematopoietic cells found in Tet2 Mut and KO mice are also shown (right; b–j, higher magnification images). Tet2 Mut and KO mice show dysplastic erythroid cells (polychromatophilic, blue arrowhead; Howell-Jolly bodies, black arrowhead; other poikilocytes [e.g., echinocytes], outlined arrowhead; immature erythroid cells, blue-outlined arrowhead). Dysplastic myeloid cells (d and f) and dysplastic platelets (giant platelet, g), as well as immature B cells (black arrows [h and i]) are shown. All images are taken at ×100 magnification. Scale bars, 10 μm.
(J) Representative flow data for myeloid and lymphoid lineages in PB from Tet2 Mut mice (middle) and Tet2 KO mice with myeloid and B cell phenotypes (right) (10 months old). Flow data for WT control are also shown (left).
(K) Percentages ± SEM of CD11b+ (left), c-Kit+ (middle), and B220lowCD19+CD43+ (in Gr-1−CD4/8− cells, right) cells in the PB in Tet2 Mut (n = 6) and KO (n = 8) mice (10 months old).
Error bars indicate SD unless otherwise specified. n.c. stands for no significant change. See also Figure S1.
Analysis of Peripheral Blood of Tet2 Mut Mice
At 4 months of age, Tet2 Mut mice exhibited normal blood counts of the peripheral blood (PB) (Figure 1H). A trend of slightly higher white blood cell (WBC) counts was found in Tet2 KO mice, but they had a normal range of red blood cell (RBC) and platelet counts (Figure 1H). Likewise, a mild increase in CD11b+ cells was observed in Tet2 KO mice, but flow cytometric analysis of PB revealed no significant dyspoiesis in either Tet2 KO or Tet2 Mut mice (Figure S1E).
At 10 months of age, Tet2 Mut mice showed no significant blood count abnormalities (except for mild reductions in Hgb and Hct). In contrast, Tet2 KO mice exhibited higher WBC counts with moderate anemia and lower platelet counts (data not shown). PB smear specimens revealed that Tet2 Mut mice had multilineage dysplastic features, much like those found in human MDS (Giagounidis and Haase, 2013; Goasguen and Bennett, 1992; Komrokji et al., 2010; Wang, 2011). Dysplasia in monocytic lineages was predominant, but defective erythroid maturation and dysplastic platelets and neutrophils were also noted (Figures 1I and S1F). Flow cytometry analysis confirmed these myeloproliferative phenotypes in PB of Tet2 Mut mice (Figures 1J, 1K, and S1G). As previously reported (Ko et al., 2011; Li et al., 2011; Moran-Crusio et al., 2011; Quivoron et al., 2011), one-third of Tet2 KO mice developed human MDS-like phenotypes (Figures 1J and 1K; KO [myeloid]). However, in a few Tet2 KO mice (<5%), a moderately increased frequency of CD3e+ cells alone was the predominant phenotype (Figure S1H; KO [T cell]). Importantly, a significant portion (more than one-fourth) of Tet2 KO mice in study succumbed to aberrant lymphopoiesis, and these B cell phenotypes were more prevalent than dysplasia in myeloid or erythroid lineages (Figures 1J and 1K). Morphological analysis found immature forms of B lymphocytes in the PB of these mice. These were of small to medium size (cell diameter is 1.5–2 times that of an erythrocyte) and displayed scanty, round, and basophilic cytoplasm and clumped nuclear chromatin with relatively round nuclear outline, which are comparable morphological features to human chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) (Figure 1I, KO [B cell] and Figure S1F, KO [lymphoid]). Flow cytometry assays showed an increase in B220mid cells, which are CD19+CD43+, implying defective differentiation in the B cell lineage (Figures 1J and 1K).
Characterization of Hematopoietic Phenotypes in Tet2 Mut and Tet2 KO Bone Marrow
At 4 months of age, Tet2 Mut mice displayed a skewed myeloid/lymphoid ratio in the bone marrow (Figures 2A, S2A, and S2B). Similarly, Tet2 KO mice with aberrant lymphopoiesis predominantly showed maturation defects in the B cell lineage, and these phenotypes became more striking with age (Figures 2B–2D, S2C, and S2D). Flow cytometric analysis of the bone marrow confirmed myeloproliferative phenotypes in 10-month-old Tet2 Mut mice (Figures 2B and S2D). Notably, Tet2 KO mice with myeloid disorders displayed similar myeloid proliferative phenotypes with analogous onset and severity (Figure S2D). In contrast, a portion of Tet2 KO mice succumbed to B-lymphoid disorders, and a clear expansion of Gr-1−CD4/8−B220midCD19+CD43+IgM+ abnormal B cells was observed in the bone marrow. These B cells reside in CD5+, phenotypic B-1a lymphocytes, which have been suggested to relate to human CLL (Figure 2B; Yu et al., 2015). These defective B-1a homeostasis are consistent with the CLL-like morphological abnormalities in PB from Tet2 KO mice (Figures 1K, 2B, and S2D).
Figure 2. Analysis of Tet2 Mut and KO Mouse Bone Marrow.

(A) Percentages of the indicated fractions in the bone marrow of Tet2 Mut and KO mice (4 months old) (n = 6).
(B) Bone marrow flow analysis for myeloid and lymphoid cells from Tet2 Mut and KO mice (10 months old).
(C and D) Representative flow data for myeloid progenitors and HSPCs in the bone marrow from Tet2 Mut and KO mice (10 months old). Percentages of the indicated fractions (percentage in Lin−Sca1−c-Kit+ cells for CMPs, GMPs, and MEPs; percentage in Lin−CD127+ cells for CLPs; percentage in Lin− cells for LSK; and percentage in CD135−LSK cells for CD150+ HSCs) (n = 6).
(E) Tet2-KO HSPCs have increased replating capacity compared to Tet2-Mut HSPCs (10 months old, up to fourth -round replating data) (n = 3).
(F) Representative images of May-Grünwald-Giemsa-stained cytospin preparations of the colonies formed by HSPCs from Tet2 WT, Mut, and KO mice (at second replating). Metamyelocytes, myelocytes, and matured granulocytes consist of WT colonies, and immature and dysplastic myeloid cells are increased in Tet2 Mut and KO colonies (also see Figure S2H). Scale bars, 10 μm.
HSPCs, hematopoietic stem and progenitor cells; LSK, Lin−c-Kit+Sca-1+; MEP, megakaryocyte-erythrocyte progenitor, Lin−Sca-1−c-Kit+CD16/32low/negCD34low/neg; GMP, granulocyte-monocyte progenitor, Lin−Sca-1−c-Kit+CD16/32highCD34+; CMP, common myeloid progenitor, Lin−Sca-1−c-Kit+CD16/32lowCD34+; CLP, common lymphoid progenitor, Lin−IL7Rα+c-KitmidSca-1+. Error bars indicate SD. n.c. stands for no significant change. See also Figure S2.
The skewed myeloid/lymphoid ratio observed in bone marrow of Tet2 Mut mice was also found at the progenitor stage; the myeloid-progenitor-enriched fractions (Lin−Sca-1−c-Kit+ cells) increased, while the lymphoid progenitors (common lymphoid progenitor [CLP]) were mildly decreased (Figures 2C, 2D, and S2C). MEP (megakaryocyte-erythroid progenitor) levels decreased and GMP (granulocyte-macrophage progenitor) levels increased in Tet2 Mut mice (Figures 2C and 2D). Tet2 KO mice with lymphoid phenotypes also displayed increased frequency of myeloid-progenitor-enriched fractions, but these increases were mainly attributed to higher numbers and frequency of MEPs. Common myeloid progenitor (CMP) levels decreased, while CLP levels were relatively maintained (Figures 2C and 2D). HSPCs were then analyzed. At 10 months, a higher number and frequency of LSK (Lin−Sca-1+c-Kit+) cells and CD135−LSK cells were found in Tet2 KO mice, but not in Tet2 Mut mice (Figures 2C, 2D, and S2D; data not shown). Furthermore, the frequency of HSC-enriched fractions (CD150+CD48−CD135−LSK cells) increased in Tet2 KO mice, regardless of having a myeloid or lymphoid phenotype (Figures 2C, 2D, and S2D).
These findings prompted us to subject HSPCs from 4-month-old WT, Tet2 Mut, and KO mice to in vitro serial replating assay. We observed in the first plating that the Tet2-KO HSPCs produced fewer colonies compared to Tet2 Mut or control cells, and a slightly increased ratio of macrophage colonies was found in Tet2 Mut colonies (Figure S2E). However, after a third round of replating, Tet2-KO HSPCs exhibited increased colony-forming capacity compared to Tet2 Mut and WT controls, and this phenotype was still pronounced in the next rounds of replating (Figure S2F). By 10 months of age, these phenotypes were even more evident; when Tet2 Mut or KO colonies were replated, both Tet2 Mut and KO cells produced more colonies in the second plating, and maintained this ability through serial replatings (Figures 2E and S2G). Notably, a comparison of third and fourth round of replating revealed that Tet2-KO HSPCs can produce a higher number of colonies than Tet2-Mut HSPCs (Figure 2E). Microscopic analysis of colonies formed after the second replating confirmed an increase in immature and dysplastic myeloid cells in Tet2 Mut and KO colonies (Figures 2F and S2H). These analyses established that the non-catalytic functions of Tet2 have distinct contributions to HSPC biology.
Distinct Disease Spectrum of Tet2 Mut and KO Mice
Both Tet2 Mut and KO mice exhibited splenomegaly and swollen lymph nodes, with an ~10-fold increase in spleen weight over age-matched controls (Figures 3A and 3B; not shown). The increased spleen sizes in Tet2 Mut mice and in Tet2 KO mice with myeloid phenotypes were attributed to myeloid infiltration (Figures 3C, 3D, and S3; data not shown). In Tet2 KO mice with lymphoid phenotypes, immature B cells had increased in the spleen. Spleens were infiltrated with small to medium-sized, regular nuclear-contoured lymphoid cells in a diffuse pattern (Figures 3C, 3D, and S3). These phenotypes were even more evident at 10 months of age (Figures S3B and S3C).
Figure 3. Characterization of Hematological Malignancies in Tet2 Catalytic Mut Mice.

(A and B) Spleen weight (n = 3–6; A, left). Representative images of spleens from Tet2 Mut mice with myeloid phenotypes and Tet2 KO mice with lymphoid phenotypes (A, right) (10 months old). Representative images of lymph nodes from Tet2 Mut mice with myeloid phenotypes (B).
(C and D) Representative flow data for myeloid and lymphoid lineages in spleens from Tet2 Mut mice with myeloid phenotypes (C, middle) and Tet2 KO mice with lymphoid phenotypes (C, bottom) (10 months old). The percentages of indicated fractions in spleens from Tet2 Mut and Tet2 KO mice are shown (D, n = 6).
(E and F) Kaplan-Meier survival curves of overall survival (E) and splenomegaly-free survival (F) of Tet2 Mut and KO mice. The number of mice in each group are shown in parenthesis. Log-rank test was used to generate p values.
(G) Disease spectrum in Tet2 Mut and KO mice (n = 16, 18, respectively).
(H and I) Schematic of transplantation of diseased bone marrow (H, top). Survival curve of transplanted mice (H, bottom, n = 10 for Mut, 9 for WT and KO). All recipient mice transplanted with Tet2 Mut bone marrow developed lethal myeloid malignancies within 6 weeks after bone marrow transplantation (BMT) (H, bottom). Representative flow cytometric analysis for myeloid and lymphoid lineages in PB of recipient mice transplanted with Tet2 Mut (I. top) or KO (I. bottom) bone marrow, 3 weeks after BMT are shown.
Error bars indicate SD. n.c. stands for no significant change. See also Figure S3.
Next, we compared the onset of hematological disorders and other signs of poor health in Tet2 Mut with Tet2 KO mice. The Tet2 KO and Tet2 Mut cohorts displayed similar overall survival, with 50% of animals dying by ~450 days and majority by ~800 days, while WT mice survived beyond 2 years (Figure 3E). Likewise, the two groups had similar incidence of splenomegaly, and age-matched moribund Tet2 Mut and KO mice had no significant differences in spleen size or weight (Figures 3A and 3F). However, most strikingly, despite the similar survival and disease onset of Tet2 Mut and KO mice, the disease spectrum of Tet2 Mut was different from that of Tet2 KO mice: while Tet2 KO mice developed both myeloid and lymphoid malignancies (44.4% and 38.9% [8/18 and 7/18], respectively), 78.5% (11/14) of Tet2 Mut mice in our cohort displayed blood phenotypes restricted to the myeloid lineage by 10 months (Figure 3G).
These malignancies were readily transplantable. All recipient mice transplanted with Tet2 Mut donor marrow cells initiated the lethal forms of myeloid disorders within 6 weeks (Figure 3H), and the resulting hematological phenotypes were similar to those observed in primary Tet2 Mut mice. Monocyte-enriched CD11b+ cells were increased, accompanied by dysplasia and c-Kit+ myeloid blasts (Figure 3I; data not shown). Likewise, recipient mice transplanted with donor marrows from Tet2 KO mice with B cell phenotypes developed lethal lymphoid disorders within 6 weeks, similar to the ones observed in primary Tet2 KO mice (Figures 3H and 3I).
Distinct Gene Expression Changes in Tet2 Mut versus KO HSPCs
To gain molecular insights into the catalytic and non-catalytic functions of Tet2 in HSPCs, we analyzed the gene expression profiles of 2-month-old Tet2 Mut, Tet2 KO and WT HSPCs (LSK) and Lin− cells by RNA sequencing (RNA-seq) (Figure 4A–4F and S4A). We found 167 differentially expressed genes (DEGs) in Tet2-KO LSK (60 up, 107 down) compared to 568 DEGs in Tet2-Mut LSK cells (244 up, 344 down) (Figure 4A). Likewise, there were 197 DEGs in Tet2-KO Lin− cells (106 up, 91 down) versus 180 DEGs in Tet2-Mut Lin− cells (97 up, 83 down) (Figure 4D). Notably, in both LSK and Lin− cells, most DEGs were changed by less than two fold, suggesting that in young progenitor cells gene expression changes due to either Tet2 deficiency or loss of its catalytic activity are mostly subtle. However, comparison of the DEGs by clustering analysis (Figures 4B and 4E, left panels) or Venn diagrams (Figures 4B and 4E, right panels, and Figure S4B) revealed that the LSK and Lin− cells from Tet2 Mut and KO mice exhibited distinct gene expression profiles. Consistently, Gene Ontology enrichment analysis of DEGs in Tet2 Mut and KO samples identified both common and distinct Gene Ontology (GO) terms, including cell proliferation, differentiation, gene regulation, and apoptosis (Figures 4C and 4F). This analysis suggests that complete loss of Tet2 or loss of its catalytic activity alone leads to subtle but distinct changes in the gene expression programs of HSPCs.
Figure 4. Gene Expression Profiling of Tet2 Mut and KO HSPCs.

(A) MA plot of RNA-seq analysis of 2-month-old LSK cells as log2 fold change versus mean gene expression levels. Differentially expressed genes (DEGs) are depicted in red. Mut or KO groups are compared to WT.
(B) Heatmap of differentially expressed genes identified by RNA-seq across LSK cells of indicated genotypes. Genes differentially expressed in at least one of the comparisons are displayed. Intensity values are log2 transformed (left) and colors represent relative expression across samples. Venn diagram illustrating the overlap between all DEGs in Tet2-Mut and Tet2-KO LSK cells (right).
(C) Selected terms from GO and KEGG analysis of DEGs in LSK cells. Data are plotted with colors for −log10 of p value and bubble sizes for numbers of genes.
(D) MA plot of RNA-seq analysis of 2-month-old Lin− cells as log2 fold change versus mean gene expression levels. DEGs are depicted in red (compared to WT).
(E) Heatmap (left) of DEGs identified by RNA-seq across Lin− cells of indicated genotypes. Genes differentially expressed in at least one of the comparisons are displayed. Venn diagram illustrating the overlap between all DEGs in Tet2-Mut and Tet2-KO Lin− cells (right).
(F) Selected terms from GO and KEGG analysis of DEGs in Lin− cells. Data are plotted with colors for −log10 of p value and bubble sizes for numbers of genes.
(G) Comparison of DEGs with Tet2 ChIP-seq peaks in bone marrow, revealing that nearly half of DEGs in LSK cells and approximately one-third of DEGs in Lin− cells are bound by Tet2 (i.e., direct targets of Tet2). Tet2 ChIP-seq data were from a previous study (see STAR Methods)
(H) Classification of Tet2 peaks at DEGs revealing that Tet2 binding is enriched at the promoters of Tet2-bound DEGs.
(I) Relative expression levels of Hoxa9 and Gata2 mRNA in LSK or Lin− cells from mice of indicated genotypes (compared to WT control samples), quantified by qRT-PCR (n = 3). Data normalized to Gapdh.
(J and K) Overexpression of Gata2 in the bone marrow of Tet2 Mut or KO mice with MDS- or CLL-like hematopoietic disorder, respectively, followed by competitive BMT (mixture of test cells [Ly45.2] with competitor [Ly45.1/45.2] in a 1:3 ratio). Percentage of donor-derived cells in PB of recipient mice transplanted 3 weeks after BMT is shown in (J) (n = 4), and the survival curve of transplanted mice is shown in (K) (n = 8, 12, and 10 for WT, Mu, and KO, respectively).
(L) Model summarizing the effects of loss of Tet2 versus loss of its catalytic activity alone on myeloid and lymphoid lineage homeostasis and development of malignancies.
Error bars indicate SD. n.c. stands for no significant change. See also Figure S4.
Next, we integrated the RNA-seq data with the available Tet2 chromatin immunoprecipitation sequencing (ChIP-seq) datasets from WT mouse bone marrow (Deplus et al., 2013) and compared Tet2-bound genes with the list of DEGs. Approximately 50% of DEGs in Tet2-Mut and Tet2-KO LSK cells, as well as ~30% of DEGs in Tet2-Mut and Tet2-KO Lin− cells, were bound by Tet2 (i.e., direct targets of Tet2) (Figures 4G and S4C). Classification of Tet2 peak location at these direct target genes revealed that over 80% of Tet2 binding occurred at promoters (Figure 4H). These genes with Tet2 binding at promoters included several regulators of HSCs and lineage specifiers. Among them, two transcription factors, Hoxa9 and Gata2, were significantly downregulated in Tet2-KO, but not in Tet2-Mut, LSK, or Lin− cells, respectively (Figure 4I). Other regulators of HSCs such as Gata1 and Spi1 were not affected, while Tet1 was downregulated in both Tet2-Mut and Tet2-KO LSK cells, and Tet3 expression was marginally reduced in Tet2-KO LSK cells (Figure S4D). Next, we examined whether the reexpression of selected identified downstream effectors of non-catalytic roles of Tet2, such as Gata2, can rescue disease phenotypes observed in Tet2 KO mice. Gata2 overexpression in Tet2 KO bone marrow cells ameliorated lymphoid disease phenotypes and conferred significant survival advantage (Figures 4J, 4K, S4E, and S4F). However, in Tet2 Mut bone marrow cells, it failed to rescue myeloid phenotypes (Figure 4K). Collectively, our findings suggest that the Tet2 catalytic activity primarily regulates the myeloid lineage, while its non-catalytic functions modulate the lymphoid lineage. As such, loss of Tet2 enzymatic activity alone predominantly leads to myeloid malignancies, while complete loss of Tet2 influences the hemostasis of both myeloid and lymphoid lineages, leading to aggressive malignancies of cell types of both lineages (Figure 4L).
DISCUSSION
Tet2 loss causes progressive defects in hematopoiesis, such as increased HSC self-renewal, and has been implicated in both myeloid and lymphoid disorders (Asmaret al., 2013; Dominguez et al., 2018; Lemonnieretal., 2012; Mouly et al., 2018; Pan et al., 2017; Quivoron et al., 2011; Reddy et al., 2017). The analysis of our cohorts of Tet2 Mut and KO mice have provided three lines of evidence that together establish the biological requirements of the non-catalytic functions of Tet2 to HSPC maintenance and tumor suppression: (1) loss of Tet2, compared to loss of its catalytic activity alone, leads to greater expansion of HSPCs and enhanced colony formation; (2) Tet2 Mut mice predominantly develop myeloid malignancies, while Tet2 KO mice develop both myeloid and lymphoid disorders; and (3) despite similar 5hmC levels, Tet2-KO and Tet2-Mut HSPCs have different gene expression profiles, and Tet2-KO HSPCs express reduced levels of HSC lineage specifiers Gata2 and Hoxa9. These findings provide conceptual advances to our understanding of requirements of Tet2 in hematopoiesis. The current dogma primarily attributes the contributions of Tet2 in HSPCs and hematological disorders to its enzymatic or DNA demethylase activity (Li et al., 2011; Moran-Crusio et al., 2011; Quivoron et al., 2011; Tahiliani et al., 2009). Our work shifts this “paradigm” by associating the catalytic and non-catalytic roles of Tet2 to distinct disease etiologies involving the myeloid and lymphoid lineages, respectively.
Increased incidence of aberrant lymphopoiesis is a distinct feature of Tet2 loss that is not seen in Tet2 Mut mice, suggesting that Tet2 non-catalytic roles are involved in regulation of lymphopoiesis. Interestingly, ~30% of Tet2 KO mice showed the accumulation of phenotypic B-1a lymphocytes, B220lowCD19+IgM+CD43+CD11blowCD5+ cells, and developed B cell neoplasms that are comparable to human CLL. We note that mantle cell lymphoma is also a CD5+ B cell neoplasm, and the abnormal B cells found in Tet2 KO mice are CD23mid positive (Figure S3B). However, these cells showed no Ccnd1 upregulation (data not shown), and the pathological and morphological features observed in Tet2-loss-induced neoplasms classify them as a CLL-like disorder. Intriguingly, TET2 mutations are rare (~2.3%) in human CLL, but there are also several reports of loss of TET2 function in B cell lymphomagenesis with blockade of plasma cell differentiation (Asmar et al., 2013; Dominguez et al., 2018; Lemonnier et al., 2012; Mouly et al., 2018; Pan et al., 2017; Quivoron et al., 2011; Reddy et al., 2017).
Establishing the mechanisms by which Tet2 deficiency leads to diverse hematological abnormalities is critical for understanding how Tet2 regulates normal hematopoiesis and how its down-regulation or mutations as seen in various cancers contribute to tumorigenesis. This is of paramount significance in therapeutic strategies for hematological disorders involving loss of TET2. Analysis of our Tet2 Mut mouse model conclusively establishes that the catalytic activity of Tet2 is the prime regulator of myeloid lineage and MDS pathogenesis. This suggests that Tet2 targets affected in MDS are regulated by the enzymatic activity of Tet2 through DNA demethylation. Consistently, DNA hypermethylation is a hallmark of hematologic disorders, and DNA demethylating agents such as 5-Aza-2′-deoxycytidine (decitabine) are used to treat higher-risk MDS and acute myeloid leukemia (Fabiani et al., 2010; Garcia-Manero and Fenaux, 2011; Gardin and Dombret, 2017; Issa et al., 2004). Likewise, vitamin C (which boosts TET enzymatic activity) has been shown to be of therapeutic benefit in MDS and CMML disease in mice haploinsufficient for Tet2 (Agathocleous et al., 2017; Cimmino et al., 2017). Since our model attributes regulation of lymphopoiesis and suppression of lymphomagenesis to the non-catalytic roles of Tet2, it is likely that such treatments might not be as effective in TET2-loss-driven lymphoid disorders. Here, we identify key transcription factors and lineage specifiers Hoxa9 and Gata2 as direct targets of Tet2 that are deregulated in Tet2 KO, but not Tet2 Mut, HSPCs. We have shown that Gata2 can rescue lymphoid-associated phenotypes observed in Tet2 KO mice. Further work will determine how these genes are regulated in a Tet2 catalytic-independent manner, possibly involving OGT and or HDAC2 (Chen et al., 2013; Zhang et al., 2015). Identification of additional catalytic and non-catalytic targets and mechanisms of Tet2 that are distinctly associated with myeloid versus lymphoid diseases will further enhance our understanding of how TET2 contributes to HSPC maintenance and suppresses hematological disorders and malignancies. This will substantially impact strategies for how TET2 should be targeted for therapeutic purposes.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
This study did not generate new unique reagents. Further information and requests for resources and reagents (including mouse strains, cell lines and plasmids generated in this study) should be directed to and will be fulfilled by the Lead Contact, Ke.I. (keisuke.ito@einstein.yu.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
A gene block carrying nucleotide substitutions in Tet2 exon 9 to introduce point mutations H1367Y and D1369A into the enzymatic pocket of Tet2 was synthesized (Table S1) and cloned into Topo PCR2.1 vector. This donor plasmid along with a pX330 plasmid expressing Cas9 and a gRNA against Tet2 exon 9 (Table S1) were used for gene editing in V6.5 mESCs (background: mixed 129/B6, sex: male) following our previously published protocols (Wang et al., 2013). These mutations only changed the conserved HRD amino acids in the iron-binding site of the catalytic domain to YRA. They are shown to render the enzyme catalytically inactive without compromising the stability or localization of Tet2, and in vitro biochemical and in vivo overexpression systems have confirmed their lack of activity on methylated DNA (Ito et al., 2010; Tahiliani et al., 2009). Targeted clones were screened by RFLP (restriction fragment length polymorphism) using a unique HaeIII restriction site introduced with silent mutations in the Tet2 mutant allele. Properly edited clones were verified by Sanger sequencing. A heterozygote Tet2+/m ESC clone was injected into mouse blastocysts at the Einstein Transgenic Core facility. Chimeric animals were bred to wild-type mice to obtain Tet2m/m (Mut) mice. Tet2−/− (KO) mice, published previously (Li et al., 2011), were obtained from Dr. Mingjing Xu (University of Maimi). Tet2 KO and Tet2 Mut mice were intercrossed to avoid any variations due to background differences between the different groups. Cohorts of Tet2+/+ (wild-type), Tet2 KO and Tet2 Mut mice (consisting of equal number of male and female animals in each group) were established and housed in an SPF (Specific-pathogen-free) barrier facility. All animals in this study included both sexes and were in a mixed 129/B6 background. Animals were examined weekly for distended belly, enlarged lymph nodes and spleen, overall mobility, and health. The peripheral blood of moribund animals was collected for analysis. Moribund animals were scarified and hematopoietic tissues including spleen, lymph nodes and bone marrow were subjected to further histological and flow cytometric evaluation to characterize the disease. All studies were performed in accordance with our IACUC approved protocols overseen by the Institute for Animal Studies at Albert Einstein College of Medicine.
METHOD DETAILS
5hmC quantification
DNA was isolated from total bone marrow of 3-month-old mice and analyzed by dot blot using anti 5hmC antibody (Active Motif) following manufacturer protocol. Breifly, DNA was spotted in replicates on Hybond N+ membranes, dried at room temperature, blocked in 5% milk and then probed with anti 5hmC antibody (Active Motif 1:10,000) for 1hr followed by three 10min washes in PBST. Membrane was probed with secondary HRP-conjugated anti rabbit antibody (1:500) for 1hr and washed three times with PBST before developing using ECL western blot detection reagent and autoradiogrpahy. 5hmC signal intensity was quantified by ImageJ software (Schneider et al., 2012) and plotted as the average of three independent experiments.
Western blot
Bone marrow from 4-month-old mice was isolated, lysed with laemmmli sample buffer, and boiled. Lysates were resolved on 6%–10% SDS-PAGE (Mini-PROTEAN electrophoresis chamber, Bio-Rad), and transferred on PVDF membranes (Mini Trans-Blot apparatus, Bio-Rad) following manufacturer’s protocols. Membranes were blocked in 5% milk in PBS with 0.1% tween (PBST) and incubated overnight at 4°C, or for 1hr at room temperature with primary antibodies (Tet2 ab124297, abcam 1:1000, and Actin AC-15, abcam 1:40000). Secondary antibody incubations (HRP-anti mouse, 401253, oranti rabbit, 401393, 1:5000, CalBiochem) were carried out for 1hr at room temperature. Protien signal was detected using ECL western blot detection reagent and autoradiogrpahy. Protein signal intensity was quantified by ImageJ. Tet2 signal was normalized to actin signal.
Flow cytometry
For flow cytometric analyses, we used monoclonal antibodies specific for the following: CD135 (A2F10), CD34 (RAM34), Sca-1 (D7), CD3e (145–2C11), CD8 (53–6.72), IgM (II/41), F4/80 (BM8), CD71 (R17217), CD127 (A7R34), CD23 (B3B4), CD24 (M1/69), CD45.2 or Ly5.2 (104), and CD45.1 or Ly5.1 (A20); all were from eBioscience. CD4 (GK1.5), B220 (RA3–6B2), TER-119 (TER-119), CD11b (M1/70), CD19 (1D3), CD43 (S7) and NK-1.1 (PK136) were from BD Bioscience. CD150 (TC15–12F12.2), CD48 (HM48–1), c-Kit (2B8) and Gr-1 (RB6–8C5) were from BioLegend. A mixture of monoclonal antibodies against CD4, CD8, CD3e, B220, TER-119, CD11b, Gr-1, IgM, CD19, CD127, and NK-1.1 was used as a lineage marker (Lineage). When CLL was assessed, the antibody against CD127 was not included in the lineage mixture. In some experiments, Streptavidin-APC-eFluor 780 (47–4317) or -PerCP-Cy5.5 (45–4317) was used as a secondary antibody.
Colony formation assay
The colony formation assay was performed using MethoCult GF M3434 (Stem Cell Technologies, Vancouver, BC, Canada) according to the manufacturer’s instructions. Briefly, the flow sorted Lin−Sca-1+c-Kit+ cells (500 cells/plate, triplicate culture) were seeded into methylcellulose-based medium, and colonies were counted under microscope after 7 days incubation at 37C°, 5% CO2. GEMM, Colony-forming unit-granulocyte, erythroid, macrophage, and megakaryocyte; GM, Colony-forming unit-granulocyte and macrophage; M, Colony-forming unit-macrophage; E, Burst-forming unit-erythroid. Long-term cultures and in vitro replating assay was performed as previously reported (500 Lin−Sca-1+c-Kit+ cells/replicate, triplicate culture) (Ito et al., 2012; Ito et al., 2016; Song et al., 2013). Frequencies of cells with colony forming capacity after long-term in vitro culture were calculated using a limiting-dilution method (maximum likelihood from the proportion of negative wells measured for each input dilution of cells tested) (Purton and Scadden, 2007). In each experiment, at least 12 replicates of each dilution were performed, and a split-plot analysis of variance was used to investigate the difference between the two conditions.
Analysis of hematopoiesis
The collected peripheral blood was smeared for May-Grünwald-Giemsa staining, and/or subjected to an automated blood count. In some cases, hematological organs, including bone marrows and spleens, were sampled and subjected to evaluate the infiltration of the hematological disorders. To determine the in vivo transplantability of the disease, 1.0 × 105 bone marrow mononuclear cells (BMMNCs) from primary deceased Tet2 Mut, Tet KO mice or littermate controls (Ly5.2) were transplanted into irradiated recipient mice (Ly5.1) with 4.0 × 105 competitor BMMNCs (Ly5.1/2). Donor (Ly5.2) cells were reconstituted. Repopulation of donor myeloid and lymphoid cells were monitored, and in some experiments, donor contribution to the immature hematopoietic lineages also evaluated.
Gene expression profiling by RNA-seq analysis
LSK and Lin− cells were sorted from pooled bone marrow of two 10-week-old mice and RNA was extracted using QIAGEN RNAeasy Kit. RNA was amplified and barcoded and used to prepare libraries of two technical replicates, with ERCC spike in controls. The libraries were subjected to 150bp paired-end sequencing (Illumina HiSeq 2500 platform) at the Einstein Epigenomics core. This generated 15~40 million read pairs per sample. The reads were trimmed using the TrimGalore (v 0.4.1, https://github.com/FelixKrueger/TrimGalore) to remove adaptors and then mapped to the mouse genome (mm10) by TopHat2 software (v 2.0.13) with default parameters (Kim et al., 2013). The read pair numbers mapped to individual genes in the Refseq gene annotation (downloaded from the UCSC genome browser in 03/2017) were calculated with the software HTseq (v 0.6.1) (Anders et al., 2015) using “–s reverse” parameter. The Fragments Per Kilobase of transcript per Million (FPKMs) were calculated using the Cufflinks package (v 2.2.1) (Trapnell et al., 2010). Read counts at the genes with FPKM > 1 were determined by the DESeq2 software (Love et al., 2014) for differential expression. False Discovery Rate (FDR) < 0.05 was used for selecting differentially expressed genes (DEGs) between Tet2 Mut and wild-type or Tet2 KO and wild-type samples. Gene Ontology (GO) analysis was performed by DAVID (Jiao et al., 2012) (https://david.ncifcrf.gov/tools.jsp). Bubble plots were used to show enriched GO terms and KEGG pathways. In the bubble plots, enrichment factors were calculated as the ratio of gene counts that mapped to a certain pathway versus the total gene number of that pathway. The expression patterns of the combined DEGS were identified by hierarchical clustering and displayed as heatmap. Tet2 ChIP-seq peaks were obtained from a previous study of bone morrow (Deplus et al., 2013). They were first converted to the mm10 version using the LiftOver tool from the UCSC genome browser, and then associated to genes and separated into promoter peaks (< +/− 2kb of transcription start sites; TSSs), gene body peaks, or distal regulatory peaks (< +/−50 kb of start and end of genes). Genes associated with Tet2 peaks were considered as Tet2 bound genes and compared to the list of DEGs. The RNA-seq gene expression datasets have been deposited in the Gene Expression Omnibus database.
RT-qPCR
RNA from LSK or Lin− cells sorted from two-month-old mouse bone marrow was isolated using Qaigen RNAeasy Kit and subjected to cDNA synthesis using Superscript III First-Strand synthesis system, Invitrogen. Real time quantitative PCR was performed using SYBR green master mix (Applied Biosystems) in QunatStudio 6 Flex Real-Time PCR system following standard protocols. Relative gene expression level was analyzed by comparative Ct method and was normalized to Gapdh. Primer sequences for all genes are shown in supplemental information (Table S1).
Gata2 lentiviral preparation and transduction of bone marrow
HA-tagged mouse Gata2 was cloned into FUW-ires-Puro vector. Lentivirus was prepared by transfecting FUW-HA-mGata2 or empty vector into HEK293T cells along with viral packing plasmids (pPAX2 and pMDG) using X-tremeGENE DNA transfection reagent (Roche) following manufacturer’s protocol. Media was refreshed after 16hrs. Viral soup was harvested 24hr and 48hr later. Bone marrow isolated from moribund mice was cultured, transduced for 24hrs and then selected for 24hrs in puromycin (1ug/mL). Selected cells were subjected to hematopoietic analyses by FACS and transplantation studies as explained above.
QUANTIFICATION AND STATISTICAL ANALYSES
Data were processed and analyzed in Graphpad PRISM software. Log rank, one-way ANOVA and Student t test were utilized to determine statistical significance unless otherwise specified. P-values lower than 0.05 were considered statistically significant. Statistical methods for analysis of genome wide datasets involving RNA-seq are explained in detail under the respective sections as part of the detailed methods.
DATA AND CODE AVAILABILITY
The RNA-seq datasets have been deposited in the Gene Expression Omnibus (GEO) database (Accession number GSE132090). Mouse strains, cell lines and plasmids generated in this study will be made available upon request.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Tet2 | Abcam | Cat# ab124297; RRID: AB_2722695 |
| Anti-Actin | Abcam | Cat# AC-15; RRID: AB_1658432 |
| Anti-5hmC | Active Motif | Cat# 39770; RRID: AB_10013602 |
| Goat Anti-Rabbit IgG-HRP | CalBiochem | Cat# 401393–2ML; RRID: AB_437797 |
| Goat Anti-Mouse IgG-HRP | CalBiochem | Cat# 401253; RRID: AB_437779 |
| CD4 (GK1.5) | BD Bioscience | Cat# 553728; RRID: AB_395012 |
| B220 (RA3–6B2) | BD Bioscience | Cat# 553086; RRID: AB_394616 |
| TER-119 (TER-119) | BD Bioscience | Cat# 553672; RRID: AB_394985 |
| CD11b (M1/70) | BD Bioscience | Cat# 553309; RRID: AB_394773 |
| CD19 (1D3) | BD Bioscience | Cat# 553784; RRID: AB_395048 |
| CD43 (S7) | BD Bioscience | Cat# 560663; RRID: AB_1727479 |
| NK-1.1 (PK136) | BD Bioscience | Cat# 553163; RRID: AB_394675 |
| c-Kit (2B8) | BioLegend | Cat# 105826; RRID: AB_1626278 |
| Gr-1 (RB6–8C5) | BioLegend | Cat# 108404; RRID: AB_313369 |
| CD135 (A2F10) | eBiosciences | Cat# 12–1351–82; RRID: AB_465859 |
| CD34 (RAM34) | eBiosciences | Cat# 11–0341–85; RRID: AB_465022 |
| Sca-1 (D7) | eBiosciences | Cat# 25–5981–81; RRID: AB_469668 |
| CD3e (145–2C11) | eBiosciences | Cat# 13–0031–85; RRID: AB_466320 |
| CD8 (53–6.72) | eBiosciences | Cat# 13–0081–85; RRID: AB_466347 |
| IgM (II/41) | eBiosciences | Cat# 13–5790–85; RRID: AB_466676 |
| CD71 (R17217) | eBiosciences | Cat# 11–0711–82; RRID: AB_465124 |
| CD127 (A7R34) | eBiosciences | Cat# 12–1271–82; RRID: AB_465844 |
| CD45.2 or Ly5.2 (104) | eBiosciences | Cat# 11–0454–82; RRID: AB_465061 |
| CD45.1 or Ly5.1 (A20) | eBiosciences | Cat# 25–0453–81; RRID: AB_469628 |
| Streptavidin-APC-eFluor780 | eBiosciences | Cat# 47–4317–82; RRID: AB_10366688 |
| Streptavidin-PerCP-Cy5.5 | eBiosciences | Cat# 45–4317–82; RRID: AB_10311495 |
| Critical Commercial Assays | ||
| RNAeasy Mini kit | QIAGEN | 74104 |
| Superscript III first strand | Invitrogen | 18080–400 |
| XtremeGene 9 DNA transfection reagent | Roche | 06365787001 |
| Deposited Data | ||
| RNA-seq data | This paper | GEO: GSE132090 |
| Experimental Models: Organisms/Strains | ||
| Tet2−/− (Tet2 KO) mouse | Li et al., 2011 | N.A. |
| Tet2m/m (Tet2 Mut) mouse | This paper | N.A. |
| B6.SJL-Ptprca Pepcb/BoyJ | Jackson lab | 002014 |
| Oligonucleotides | ||
| RT-qPCR primers, see Table S1 | This paper | n/a |
| Genotyping Primers, see Table S1 | This paper | n/a |
| gRNA oligos for gene targeting, see Table S1 | This paper | n/a |
| Recombinant DNA | ||
| Topo pcr2.1 donor vector | This paper | n/a |
| pX330-Tet2-gRNA | This paper | n/a |
| FUW-ires-puro | This paper | n/a |
| FUW-HA-mGata2-ires-puro | This paper | n/a |
| Software and Algorithms | ||
| Trim galore v0.4.1 | Github | https://github.com/FelixKrueger/TrimGalore |
| Tophat v2.0.13 | John Hopkins University | https://ccb.jhu.edu/software/tophat/index.shtml |
| HTSeq v0.6.1 | Anders et al., 2015 | https://github.com/simon-anders/htseq |
| Cufflinks v2.2 | Trapnell et al., 2010 | http://cole-trapnell-lab.github.io/cufflinks/ |
| DESeq2 v1.20.0 | Love et al., 2014 | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| DAVID 6.8 | Jiao et al., 2012 | https://david.ncifcrf.gov/ |
| GraphPad Prism 7 | GraphPad | https://www.graphpad.com/ |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
Highlights.
Loss of Tet2 enhances colony replating more than loss of its catalytic activity
Tet2 catalytic mutant mice predominantly develop myeloid malignancies
Tet2 knockout mice develop both myeloid and lymphoid disorders
Tet2 catalytic mutant versus knockout HSPCs exhibit distinct gene expression profiles
ACKNOWLEDGMENTS
We thank the Einstein epigenomics, flow cytometry (NIH grant P30 CA013330), histopathology, and transgenic cores for help with RNA-seq, cell sorting, sectioning tissues, and blastocyst injections, respectively. We are grateful to M. Xu for the Tet2 knockout mouse strain and M. Yoshimoto for the critical discussions of B lymphopoiesis. We thank Q. Tang and other members of the Ito and Dawlaty labs, as well as P.S. Frenette, U. Stiedl, A. Verma, and other members of Einstein Stem Cell Institute, for helpful suggestions and discussions. M.M.D. is supported by the Sidney Kimmel Foundation, the Leukemia Research Foundation, NYSDOH/NYSTEM, and the NIH NIGMS (grant R01GM122839). Keisuke Ito is supported by the NIH NIDDK (grants R01DK98263, R01DK115577, and R01DK100689) and NYSDOH as core director of Einstein single-cell genomics/epigenomics (C029154). M.M.D. and Keisuke Ito are also supported by funds from the Ruth L. and David S. Gottesman Institute for Stem Cell and Regenerative Medicine Research at Einstein, as well as by the NIH NHLBI (grant R01HL148852 to M.M.D. and Keisuke Ito as multi-PIs). S.C. is supported by the Einstein Training Program in Stem Cell Research from the Empire State Stem Cell Fund (C30292GG). Y.Z. and D.Z. are in part supported by the NIH NEI (grants R01 EY014237 and EY012200). Keisuke Ito is a Scholar of The Leukemia and Lymphoma Society (#1360–19).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.094.
REFERENCES
- Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, Malinge S, Yao J, Kilpivaara O, Bhat R, et al. (2009). Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 114, 144–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agathocleous M, Meacham CE, Burgess RJ, Piskounova E, Zhao Z, Crane GM, Cowin BL, Bruner E, Murphy MM, Chen W, et al. (2017). Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 549, 476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asmar F, Punj V, Christensen J, Pedersen MT, Pedersen A, Nielsen AB, Hother C, Ralfkiaer U, Brown P, Ralfkiaer E, et al. (2013). Genome-wide profiling identifies a DNA methylation signature that associates with TET2 mutations in diffuse large B-cell lymphoma. Haematologica 98, 1912–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Chen Y, Bian C, Fujiki R, and Yu X (2013). TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 493, 561–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimmino L, Abdel-Wahab O, Levine RL, and Aifantis I (2011). TET family proteins and their role in stem cell differentiation and transformation. Cell Stem Cell 9, 193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, Ng V, Xia B, Witkowski MT, Mitchell-Flack M, et al. (2017). Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell 170, 1079–1095.e1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Massé A, Kosmider O, Le Couedic JP, Robert F, Alberdi A, et al. (2009). Mutation in TET2 in myeloid cancers. N. Engl. J. Med 360, 2289–2301. [DOI] [PubMed] [Google Scholar]
- Deplus R, Delatte B, Schwinn MK, Defrance M, Méndez J, Murphy N, Dawson MA, Volkmar M, Putmans P, Calonne E, et al. (2013). TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 32, 645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez PM, Ghamlouch H, Rosikiewicz W, Kumar P, Béguelin W, Fontán L, Rivas MA, Pawlikowska P, Armand M, Mouly E, et al. (2018). TET2 deficiency causes germinal center hyperplasia, impairs plasma cell differentiation, and promotes B-cell lymphomagenesis. Cancer Discov. 8, 1632–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiani E, Leone G, Giachelia M, D’alo’ F, Greco M, Criscuolo M, Guidi F, Rutella S, Hohaus S, and Voso MT (2010). Analysis of genome-wide methylation and gene expression induced by 5-aza-2′-deoxycytidine identifies BCL2L10 as a frequent methylation target in acute myeloid leukemia. Leuk. Lymphoma 51, 2275–2284. [DOI] [PubMed] [Google Scholar]
- Garcia-Manero G, and Fenaux P (2011). Hypomethylating agents and other novel strategies in myelodysplastic syndromes. J. Clin. Oncol 29, 516–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardin C, and Dombret H (2017). Hypomethylating agents as a therapy for AML. Curr. Hematol. Malig. Rep 12, 1–10. [DOI] [PubMed] [Google Scholar]
- Giagounidis A, and Haase D (2013). Morphology, cytogenetics and classification of MDS. Best Pract. Res. Clin. Haematol 26, 337–353. [DOI] [PubMed] [Google Scholar]
- Goasguen JE, and Bennett JM (1992). Classification and morphologic features of the myelodysplastic syndromes. Semin. Oncol. 19, 4–13. [PubMed] [Google Scholar]
- Issa JP, Garcia-Manero G, Giles FJ, Mannari R, Thomas D, Faderl S, Bayar E, Lyons J, Rosenfeld CS, Cortes J, and Kantarjian HM (2004). Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in hematopoietic malignancies. Blood 103, 1635–1640. [DOI] [PubMed] [Google Scholar]
- Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, and Zhang Y (2010). Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466, 1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, and Zhang Y (2011). Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, Schafer ZT, Evans RM, Suda T, Lee CH, and Pandolfi PP (2012). A PML-PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med 18, 1350–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Turcotte R, Cui J, Zimmerman SE, Pinho S, Mizoguchi T, Arai F, Runnels JM, Alt C, Teruya-Feldstein J, et al. (2016). Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science 354, 1156–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao X, Sherman BT, Huang da W, Stephens R, Baseler MW, Lane HC, and Lempicki RA (2012). DAVID-WS: a stateful web service to facilitate gene/protein list analysis. Bioinformatics 28, 1805–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, Tsangaratou A, Rajewsky K, Koralov SB, and Rao A (2011). Ten-Eleven- Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc. Natl. Acad. Sci. USA 108, 14566–14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komrokji RS, Zhang L, and Bennett JM (2010). Myelodysplastic syndromes classification and risk stratification. Hematol. Oncol. Clin. North Am 24, 443–457. [DOI] [PubMed] [Google Scholar]
- Kosmider O, Gelsi-Boyer V, Ciudad M, Racoeur C, Jooste V, Vey N, Quesnel B, Fenaux P, Bastie JN, Beyne-Rauzy O, et al. ; Groupe Francophone des Myélodysplasies (2009). TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica 94, 1676–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemonnier F, Couronné L, Parrens M, Jaïs JP, Travert M, Lamant L, Tournillac O, Rousset T, Fabiani B, Cairns RA, et al. (2012). Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood 120, 1466–1469. [DOI] [PubMed] [Google Scholar]
- Li Z, Cai X, Cai CL, Wang J, Zhang W, Petersen BE, Yang FC, and Xu M (2011). Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 118, 4509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, et al. (2011). Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20, 11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouly E, Ghamlouch H, Della-Valle V, Scourzic L, Quivoron C, Roos-Weil D, Pawlikowska P, Saada V, Diop MK, Lopez CK, et al. (2018). B-cell tumor development in Tet2-deficient mice. Blood Adv. 2, 703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan F, Wingo TS, Zhao Z, Gao R, Makishima H, Qu G, Lin L, Yu M, Ortega JR, Wang J, et al. (2017). Tet2 loss leads to hypermutagenicity in haematopoietic stem/progenitor cells. Nat. Commun 8, 15102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purton LE, and Scadden DT (2007). Limiting factors in murine hematopoietic stem cell assays. Cell Stem Cell 1, 263–270. [DOI] [PubMed] [Google Scholar]
- Quivoron C, Couronné L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, Do Cruzeiro M, Delhommeau F, Arnulf B, Stern MH, et al. (2011). TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 20, 25–38. [DOI] [PubMed] [Google Scholar]
- Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, Leppa S, Pasanen A, Meriranta L, Karjalainen-Lindsberg ML, et al. (2017). Genetic and functional drivers of diffuse large B cell lymphoma. Cell 171, 481–494.e415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Wheat JC, Steidl U, and Ito K (2016). DNMT3A and TET2 in the Pre-Leukemic Phase of Hematopoietic Disorders. Front. Oncol 6, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song SJ, Ito K, Ala U, Kats L, Webster K, Sun SM, Jongen-Lavrencic M, Manova-Todorova K, Teruya-Feldstein J, Avigan DE, et al. (2013). The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell 13, 87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, and Rao A (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tefferi A, and Vardiman JW (2009). Myelodysplastic syndromes. N. Engl. J. Med 361, 1872–1885. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SA (2011). Diagnosis of myelodysplastic syndromes in cytopenic patients. Hematol. Oncol. Clin. North Am 25, 1085–1110, vii. [DOI] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, and Jaenisch R (2013). One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PA, Rappsilber J, and Helin K (2011). TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature 473, 343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, D’Alessio AC, Ito S, Xia K, Wang Z, Cui K, Zhao K, Sun YE, and Zhang Y (2011). Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature 473, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu EM, Kittai A, and Tabbara IA (2015). Chronic lymphocytic leukemia: current concepts. Anticancer Res. 35, 5149–5165. [PubMed] [Google Scholar]
- Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, Zhao D, Liu Y, Wang C, Zhang X, et al. (2015). Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 525, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq datasets have been deposited in the Gene Expression Omnibus (GEO) database (Accession number GSE132090). Mouse strains, cell lines and plasmids generated in this study will be made available upon request.