

Abstract
When using nuclear magnetic resonance (NMR) to assist in chemical identification in complex samples, researchers commonly rely on databases for chemical shift spectra. However, authentic standards are typically depended upon to build libraries experimentally. Considering complex biological samples, such as blood and soil, the entirety of NMR spectra required for all possible compounds would be infeasible to ascertain due to limitations of available standards and experimental processing time. As an alternative, we introduce the in silico Chemical Library Engine (ISiCLE) NMR chemical shift module to accurately and automatically calculate NMR chemical shifts of small organic molecules through use of quantum chemical calculations. ISiCLE performs density functional theory (DFT)-based calculations for predicting chemical properties—specifically NMR chemical shifts in this manuscript—via the open source, high-performance computational chemistry software, NWChem. ISiCLE calculates the NMR chemical shifts of sets of molecules using any available combination of DFT method, solvent, and NMR-active nuclei, using both user-selected reference compounds and/or linear regression methods. Calculated NMR chemical shifts are provided to the user for each molecule, along with comparisons with respect to a number of metrics commonly used in the literature. Here, we demonstrate ISiCLE using a set of 312 molecules, ranging in size up to 90 carbon atoms. For each, calculation of NMR chemical shifts have been performed with 8 different levels of DFT theory, and with solvation effects using the implicit solvent Conductor-like Screening Model. The DFT method dependence of the calculated chemical shifts have been systematically investigated through benchmarking and subsequently compared to experimental data available in the literature. Furthermore, ISiCLE has been applied to a set of 80 methylcyclohexane conformers, combined via Boltzmann weighting and compared to experimental values. We demonstrate that our protocol shows promise in the automation of chemical shift calculations and, ultimately, the expansion of chemical shift libraries.

Electronic supplementary material
The online version of this article (10.1186/s13321-018-0305-8) contains supplementary material, which is available to authorized users.
Keywords: Chemical shift, Density functional theory, DFT, Metabolomics, NMR, NWchem, Python, Quantum chemistry
Background
Metabolomics is being increasingly applied in biomedical and environmental studies, despite the technical challenges facing comprehensive and unambiguous identification of detected metabolites [1–3]. The capability to routinely measure and identify even a modicum of biologically important molecules within all of chemical space—greater than 1060 compounds [4]—remains a grand challenge in biology. The prevention and treatment of metabolic diseases, determining the interactions between plant and soil microbial communities, and uncovering the building blocks that led to abiogenesis will all strongly depend on confidently identifying small molecules, and thus understanding the mechanisms involved in the complex processes of metabolic networks [5–7]. The current gold standard for chemical identification requires matching chemical features to those measured from an authentic chemical standard. However, this is not the case with the vast majority of molecules. For example, only 17% of compounds found in the Human Metabolome Database (HMDB) and less than 1% of compounds found in exposure chemical databases like the U.S. Environmental Protection Agency (EPA) Distributed Structure-Searchable Toxicity (DSSTox) Database [8] can be purchased in pure form [9, 10]. Although analytical techniques like nuclear magnetic resonance (NMR) spectroscopy [11–13] and mass spectrometry (MS) [14–16] have been applied for the identification of metabolites and to build libraries [17–21], determining the complete composition of entire metabolomes is still non-trivial for both technical and economic reasons. In this regard, libraries constructed of experimentally obtained data are too limited, expensive, and slow to build, even for libraries with thousands of metabolites [22–25].
The most practical approach expand reference libraries for comprehensive identification of compounds detected in metabolomics studies is through in silico calculation of molecular attributes. Molecular properties that can be both accurately predicted computationally and consistently measured experimentally may be used in “standards free” metabolomics identification approaches. The metabolomics community has made many advances in calculations of measurable chemical attributes, such as chromatographic retention time [26, 27], tandem mass spectra [28–30], ion mobility collision cross section [31, 32], and NMR chemical shifts [33]. Recently, high throughput computation of chemical properties has been demonstrated using machine learning approaches [34–37]. These tools are a good resource for the metabolomics community, however, machine learning methods are limited by the size and scope of the initial training set, and thus ultimately limited by the number of authentic chemical standards available for purchase. In contrast, structure-based approaches, utilizing first principles of quantum chemical calculations, leverage our understanding of the underlying chemistry and physics to directly predict chemical properties of any chemically valid molecule. Thus, quantum chemical calculations enable us to overcome the reliance on authentic chemical standards in metabolomics. In this study, we focus on expanding the utility of density functional theory (DFT), a widely used electronic structure approach, which has been applied to predict NMR chemical shifts [38–41]. DFT enables examination of molecular conformers [42–45] and allows custom solvent conditions [46–48]. Ultimately, computational modeling can be used in the rapid identification and study of thousands of metabolites, culminating in in silico metabolome libraries of multiple chemical properties. Furthermore, the same tools that can be used to aid identification of small molecules in complex samples can also be used for structure confirmation and correction. For example, we recently used the tool described in this manuscript to help correct the misidentification of the isoflavonoid wrightiadione to the actual structure as an isobaric isostere, the alkaloid tryptanthrin [49].
Metabolomics researchers unfamiliar with DFT or similar calculations may find the application of quantum chemical calculations complicated or challenging to apply quickly, and thus avoid these techniques. To this end, and to help bring DFT calculations to large sets of small organic molecules relevant to the mainstream metabolomics community, we have developed a Python-based workflow and analysis package, the ISiCLE (in silico Chemical Library Engine) NMR chemical shift module employs DFT methods through use of NWChem [50], a high-performance quantum chemistry software package developed at Pacific Northwest National Laboratory (PNNL). The module automates calculations of NMR chemical shifts, including solvent effects, via the COnductor-like Screening Model (COSMO) [51] of user-specified NMR-active nuclei for a given set of molecules for multiple DFT methods. ISiCLE also calculates the corresponding errors if experimental values are available. In this paper, we describe ISiCLE’s NMR module, provide a working tutorial example, demonstrate its use through the calculation of chemical shifts for a large set of small molecules, and, finally, show how ISiCLE can be applied to rapidly calculate chemical shifts of arrays of Boltzmann-weighted conformers to yield high accuracy chemical shift calculations.
Methods
In silico Chemical Library Engine (ISiCLE)—NMR module
ISiCLE is a Python module that provides straightforward automation of DFT using NWChem, an open source, high-performance computational quantum chemistry package, developed at Pacific Northwest National Laboratory (PNNL), for geometry optimization and chemical shift and solvent effect calculations. Figure 1 shows a schematic representation of ISiCLE. For typical use, ISiCLE requires only a list of molecules and a list of desired levels of DFT theory from the user. For more advanced use cases, users may adjust NWChem parameters by modifying the provided .nw template file.
Fig. 1.
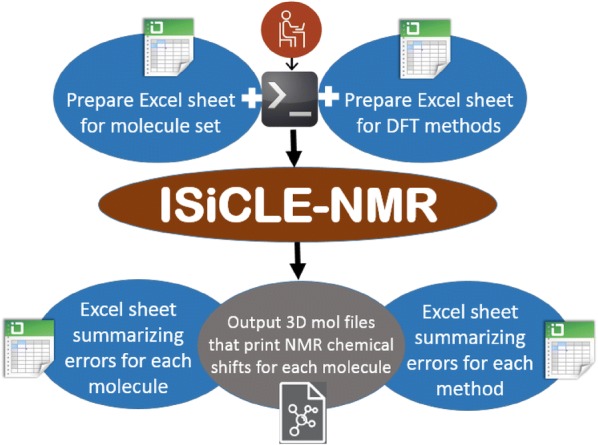
Schematic representation of inputs and outputs of the ISiCLE NMR module
Here, we describe each step of a typical ISiCLE run (see Fig. 2 for a general workflow for using the ISiCLE NMR module).
Fig. 2.
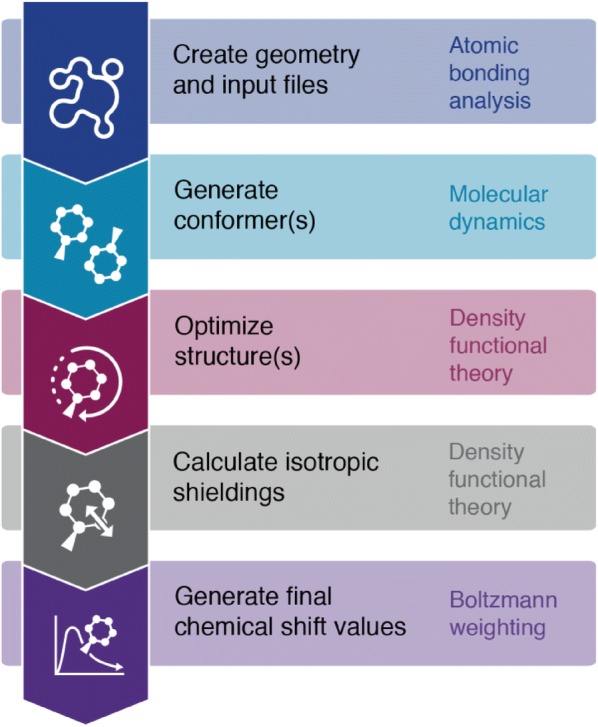
The step-by-step conceptual workflow for the ISiCLE NMR module. Conformer generation with Boltzmann weighting is optional and will be automated in subsequent versions. Please see github.com/pnnl/isicle for the latest versions
To start, users must prepare File A, containing a list of molecules, and File B, containing a list of DFT combinations, which both are required to be in Excel format (.xls or .xlsx). File A must contain all input molecules either as (i) International Union of Pure and Applied Chemistry (IUPAC) International Chemical Identifier (InChI) strings [52, 53] or (ii) XYZ files, a free-format text file having XYZ coordinates of atoms. In subsequent versions, alternative file formats will be supported, such as TSV for inputs and outputs.
Once prepared, the user runs ISiCLE. First, ISiCLE opens File A for the input molecules. OpenBabel, an open-source chemical informatics toolbox available with Python wrappers [54, 55], is called to generate geometry files. For InChI inputs, OpenBabel generates .xyz files for each molecule, unless .xyz files are provided, and converts InChI to InChIKey for naming files (otherwise, the base names of XYZ files are used for naming subsequent files). Next, OpenBabel applies the Merck molecular force field (MMFF94) [56] to generate a rough three-dimensional (3D) structure for each molecule, resulting in associated .mol files. ISiCLE then prepares NWChem input files based on the specified DFT methods, solvents, shielding parameters and regarding task directives given by the user-prepared File B. Finally, ISiCLE submits the appropriate files to, if relevant, a remote NWChem installation (typically on a non-local, networked, high-performance computer), and then retrieves the output files once the calculations are complete. Additional information and further details about ISiCLE is provided in Additional file 1 (S1). Note that future versions of ISiCLE will automatically generate conformers of a given molecule, as part of the seamless pipeline.
For each molecule, ISiCLE generates MDL Molfiles (.mol) [57] that contain isotropic shieldings and NMR chemical shifts. ISiCLE exports isotropic shieldings for each molecule and appends them to a MDL Molfile in the same atomic order of the original XYZ files. Then, ISiCLE converts isotropic shieldings to NMR chemical shifts by subtracting the isotropic shielding constants for the specified nuclei of the molecule of interest from those of a reference compound computed at the same level of theory (Eq. 1). For this manuscript, tetramethylsilane (TMS) is used as a reference compound. The experimental chemical shifts of TMS are assigned a value of zero, thus the calculation of NMR chemical shifts needs only isotropic shieldings of TMS [58–61]. Any molecule can be used as reference in ISiCLE as long as it has the specified nuclei and its experimental (or calculated) chemical shifts are supplied. It is explained in detail in a supplemental tutorial how a user inputs experimental data. The equation for calculating chemical shifts from isotropic shieldings is:
| 1 |
where and are the chemical shifts of atom i (of the molecule of interest) and the reference molecule, respectively. and are the isotropic shielding constants of atom i and the reference molecule, respectively.
ISiCLE also calculates errors in NMR chemical shifts if experimental data is provided in the MDL Molfiles in a required way as explained in the tutorial. The errors are quantified in terms of mean absolute error (MAE) (Eq. 2), corrected mean absolute error (CMAE) (Eq. 3), root mean square error (RMSE) (Eq. 4), and maximum absolute error (Eq. 5).
| 2 |
| 3 |
| 4 |
| 5 |
where N is the total number of chemical shifts, and and are the lists of calculated and experimental chemical shits, respectively.
Empirical scaling of isotropic shieldings or NMR chemical shifts is the most common approach to remove systematic errors. If experimental data is provided, ISiCLE uses two optional approaches for its linear regression method, where slope and intercept values are derived from (i) regression of computed NMR chemical shifts versus experimental NMR chemical shifts using (Eq. 6), and/or (ii) regression of computed isotropic shieldings versus experimental NMR chemical shifts using (Eq. 7).
| 6 |
| 7 |
where is the list of isotopic shielding constants of molecules.
Alternatively, if the user does not provide experimental NMR chemical shifts, ISiCLE can scale NMR chemical shifts using provided intercept and slope values. The scaled NMR chemical shifts are appended to MDL Molfiles.
A detailed description of InChIs and InChIKeys, and why they were chosen, can be found in the Additional file 1 (S2). Similarly, justification for the use of MDL Molfiles is explained in Additional file 1 (S2). In the next version, ISiCLE will be compatible with other file formats, such as the NMReDATA [62] format that has been recently designed for NMR data use. To help ease the use of our data, we provide NMReDATA files for the demonstration set in the Additional file 2.
Furthermore, installation details for OpenBabel and other required Python packages are provided in the tutorial (see Additional file 2). The Windows-based tutorial provides step-by-step instructions for running ISiCLE for the first time, including information for installation of packages, properly preparing input files, running a calculation, and obtaining output files. The tutorial includes example molecules with anticipated output files for use as a practice set and for benchmarking purposes. It is designed to guide users of ISiCLE and NWChem in the use of the input files and scripts, demonstrated using three small molecules: methanol, methyl-isothiocyanate, and nitromethane. Calculation time may vary (depending on network speed, local computational power, etc.), but it is expected to take less than 10 min.
Demonstration set
For an initial demonstration of ISiCLE, we have compiled a molecule set of 312 compounds from previous studies: Alver [63], Asiri et al. [64], Bally and Rablen [65], Bagno et al. [66], Borkowski et al. [67], Coruh et al. [68], Fulmer et al. [69], Hill et al. [70], Izgi et al. [71], Karabacak et al. [72], Krishnakumar et al. [73–75], Kwan and Liu [45], Li et al. [76], Lomas [77], Osmialowski et al. [78], Parlak et al. [79], Perez et al. [80], Rablen et al. [81], Sarotti and Pellegrinet [82, 83], Sebastian et al. [84], Seca et al. [52], Senyel et al. [85, 86], Sridevi et al. [87], Tormena and da Silva [88], Vijaya and Sankaran [89], Watts et al. [53], Wiitala et al. [90, 91], Willoughby et al. [92], and Yang et al. [93]. We aimed to cover a broad chemical space and distribution of sizes. Our criteria also included the existence of all 1H and/or 13C NMR experimental data in chloroform solvent, referenced to TMS at room temperature, for comparisons. Note that the NMR spectra of each molecule set were not recorded at the same magnetic field strengths. A summary of the demonstration set compounds are given in Table 1. Detailed information about the individual sets is given in Additional file 1 (S3).
Table 1.
Demonstration set sources and details
| References | # of molecules | Ave. atoms per molecule | Ave. H per molecule | Ave. C per molecule | Types of molecules |
|---|---|---|---|---|---|
| Alver [63] | 1 | 24 | 11 | 8 | Boron-based compound |
| Asiri et al. [64] | 1 | 33 | 12 | 16 | Organic photochromic compound |
| Bagno et al. [66] | 4 | 44 | 21 | 18 | Small organic molecules with constrained conformations |
| Barkowski et al. [67] | 15 | 83 | 50 | 31 | Pentacyclic terpenoids (fernenes) |
| Coruh et al. [68] | 1 | 25 | 8 | 13 | Heterocyclic aromatic compound |
| Fulmer et al. [69] | 33 | 14 | 9 | 4 | Commonly used NMR solvents |
| Hill et al. [70] | 1 | 70 | 33 | 30 | Complex drug with multiple chemical groups and one stereocenter |
| Izgi et al. [71] | 1 | 24 | 15 | 8 | Molecule with cyclohexene (C6H10) attached to ethylamine (C2H7N) |
| Karabacak et al. [72] | 1 | 17 | 8 | 7 | Planar benzene ring with attached B(OH)2 and two F groups |
| Krishnakumar et al. [74] | 2 | 16 | 4 | 6 | Agrochemical intermediate compounds with planar rings |
| Krishnakumar et al. [73] | 2 | 16 | 7 | 7 | Nitrotoluene derivatives |
| Krishnakumar et al. [75] | 2 | 17 | 6 | 7 | Phenol derivatives |
| Kwan and Liu [45] | 1 | 42 | 22 | 18 | Natural product |
| Li et al. [76] | 78 | 11 | 4 | 5 | Molecules with a large number of connected substituent groups |
| Lomas [77] | 15 | 18 | 12 | 5 | Saturated alcohols |
| Osmialowski et al. [78] | 28 | 28 | 11 | 14 | Substituted phenacylpyridines (ketimine forms) and tautomers |
| Parlak et al. [79] | 1 | 26 | 4 | 12 | Polyfluoroaromatic compound with two rings |
| Perez et al. [80] | 2 | 22 | 10 | 8 | Chloropyrimidine species |
| Rablen et al. [81] | 80 | 6 | 4 | 11 | Rigid organic compounds with constrained conformations |
| Sarotti and Pellegrinet [82, 83] | 66 | 15 | 8 | 6 | Low polarity compounds with constrained conformations |
| Sebestian et al. [84] | 1 | 26 | 11 | 14 | Phenyl cyanide compound with two planar phenyl rings |
| Seca et al. [52] | 4 | 40 | 24 | 65 | Light petroleum extracts |
| Senyel et al. [85] | 1 | 23 | 13 | 9 | A structural element of many pharmaceutical drugs |
| Senyel et al. [86] | 1 | 28 | 18 | 8 | 3-Piperidino-propylamine molecule |
| Sridevi et al. [87] | 1 | 21 | 8 | 10 | Chromene, a two ringed planar compound |
| Tormena and da Silva [88] | 3 | 17 | 8 | 8 | Para-X-sub’ed (X=H, CH3O, & NO2) aromatic carbonyl compounds |
| Vijaja and Sankaran [89] | 1 | 47 | 24 | 20 | Azine |
| Watts et al. [53] | 6 | 45 | 21 | 18 | Coniferol, a building block of lignin, stereoisomers and conformers |
| Wiitala et al. [90] | 43 | 11 | 6 | 3 | Organic compounds |
| Wiitala et al. [91] | 7 | 24 | 14 | 8 | Cis-/trans-forms of 2-, 3-, and 4-methylcyclohexanols |
| Willoughby et al. [92] | 2 | 22 | 14 | 7 | Cis-/trans-diastereomers of 3-methylcyclohexanol |
| Yang et al. [93] | 2 | 57 | 28 | 23 | Complex natural products |
| This study’s demonstration set | 312 | 20 | 10 | 8 | Small- to medium-sized organic molecules with constrained conformations |
Details (i.e. experimental conditions, total and average number of nuclei, molecule types and classes) for each molecule set are given in Additional file 1 (S3)
Computational details
As a first demonstration of ISiCLE, a benchmark study was performed with 8 different DFT methods to predict 1H and 13C NMR chemical factors for the calculations of chemical shifts in chloroform. Each compound was optimized with the Becke three-parameter Lee–Yang–Parr (B3LYP) hybrid functional [94–96] and the 6-31G(d) split-valence basis set [97]. This level of theory in geometry optimization was chosen because of its broad application in the literature for organic molecules [98, 99]. Isotropic magnetic shielding constants were calculated with the 4 different functionals, BLYP [94, 95], B3LYP [97–99], B35LYP, and BHLYP [100]. DFT methods were selected with different Hartree–Fock (HF) ratios: BLYP (0% HF), B3LYP (20% HF), B35LYP (35% HF), BHLYP (50% HF). Each method was tested with 2 different correlation-consistent Dunning basis sets (double-zeta cc-pVDZ [101] or triple-zeta cc-pVTZ [101]). All basis sets were obtained from the Environmental Molecular Sciences Laboratory (EMSL) Basis Set Exchange [102–104]. For each optimized geometry, 1H and 13C NMR chemical shifts were computed relative to TMS using the Gauge Including Atomic Orbitals (GIAO) formalism [105]. Chloroform solvation effects were simulated using COSMO.
For a second demonstration of ISiCLE, the NMR chemical shifts, along with frequency calculations (and subsequent Boltzmann weighting), two sets of axial and equatorial conformers (40 conformers each) of methylcyclohexane were processed. We performed in vacuo molecular dynamics (MD) simulations, using the sander MD software program from AmberTools (version 14) [106], to generate 80 conformers of the methylcyclohexane compound. These conformers were generated in four stages. First, the initial geometries of axial and equatorial conformers were taken from the study of Willoughby et al. [92]. Second, a short energy minimization run was performed to relax the initial structure and to remove any non-physical atom contacts. Third, a short 50 ps MD run was performed (in 0.5 fs time steps) to heat the structure from 0 to 300 K, without non-bonded cutoffs. In the fourth step, we performed 8 simulated annealing cycles, where each cycle was run for 1600 ps in 1 fs MD steps with the following temperature profile: heating from 300 to 600 K (0–300 ps), equilibration at 600 K (300–800 ps), cooling from 600 to 300 K (800–1100 ps), and equilibration at 300 K (1100–1600 ps). Ten conformers from the equilibration stage at 300 K, of each simulated annealing cycle, were randomly selected to obtain the 80 conformers. After the conformers were obtained, M06-2X was used with the basis set of 6-31 + G(d,p) for the geometry optimization and frequency calculations and B3LYP with 6-311 + G(2d,p) method for the calculations of NMR chemical shifts. Relative free energies of the conformations and Boltzmann weighted NMR chemical shifts were compared to those found in the literature [92, 107–109].
All results shown in this manuscript were generated using the Cascade high-performance computer (1440 compute nodes, 23,040 Intel Xeon E5-2670 processor cores, 195,840 Intel Xeon Phi 5110P coprocessor cores, and 128 GB memory per compute node [110]), in EMSL (a U.S. national scientific user facility) located at PNNL. Cascade is available for external users through a free, competitive proposal process. ISiCLE can utilize local clusters or high-performance computing resources available to the user. NWChem is freely available and can be downloaded from the website [111, 112].
Results and discussion
NMR chemical shift calculations have been used successfully to identify new molecules, determine metabolite identifications, and eliminate structural misassignments [59, 113]. In the last two decades, many research groups have performed benchmark DFT studies on the accuracy of optimized molecular geometry [92, 114–116], functionals [117, 118], basis sets [88, 119], and solvation models [90, 120, 121] for NMR chemical shifts [60, 122–124]. Each group uses a molecule set focusing on a unique chemical class [78, 125–129] and several groups have recommended different exchange–correlation (XC) energy functionals with a different basis set for a particular condition or suitable to specific chemical functionalities and properties [70, 77, 130–133]. The prevailing opinion is that reliable isotropic NMR chemical shifts strongly depend on accurate calculations of molecular geometries and inclusion of HF exchange in selected DFT methods, to an extent [134, 135]. On the other hand, the size of the basis set does not increase the accuracy after a point [136, 137].
The ISiCLE software can be installed locally. As seen in Fig. 1, it requires only two input files, prepared in Excel: a sequence of InChI or XYZ molecule geometry files, and a sequence of DFT methods of the user’s choice. Preparation of NWChem “run files,” 3D molecule geometry files, and/or Linux/Unix shell script “drivers” are not required. As output, ISiCLE prints isotropic shielding, calculated by NWChem, and calculated chemical shifts with respect to a reference molecule and/or application of a user-specified linear regression technique. ISiCLE is a promising tool contributing to standards-free metabolomics, which depends on the ability to calculate properties for thousands of molecules and their associated conformers.
Application 1: chemical shift calculations for a demonstration set of molecules
To test ISiCLE, we generated a set of 312 molecules. This set is large relative to other metabolomic molecule sets found in the literature, which in our literature survey averaged 34 molecules (Table 1). Our molecule set ranges from small- to large-sized molecules (number of carbon atoms ranging from 1 to 90), and experimental 13C and 1H NMR data in chloroform were available for each of them. Our set also spans a wide array of chemical classes, including acetylides, alkaloids, benzenoids, hydrocarbons, lipids, organohalogens, and organic nitrogen and oxygen compounds. ISiCLE was used to successfully perform DFT calculations for this set under chloroform solvation using eight different levels of DFT theory (4 different functionals and 2 basis sets for 13C and 1H).
A total of 2494 carbon nuclei and of 3127 hydrogen nuclei were calculated for all 312 molecules of the demonstration set and compared with experimental data. Deviation bars indicating MAE and MAXAE are plotted for each method in Fig. 3. For both 13C and 1H NMR chemical shifts, the MAE of each method with cc-pVTZ is higher than those with cc-pVDZ. For 13C, the MAE of each method with cc-pVTZ (7–10 ppm) is higher than those with cc-pVDZ (5–6 ppm). MAE of methods with a larger basis set deviate more compared to those with a smaller basis set. The smallest deviations are observed for B3LYP and B35LYP, both in MAE and MAXAE results. The same situation is observed for 1H NMR chemical shifts as well: MAE of each method with cc-pVTZ (~ 0.35 ppm) is higher than those with cc-pVDZ (~ 0.30 ppm). In contrast to 13C NMR chemical shifts, 1H NMR chemical shifts are better predicted with methods using larger basis sets (cc-pVTZ). Although the error differences among each method may be too low to confidently identify the outperforming method, B3LYP/cc-pVDZ is the most successful combination in the calculation of 13C and 1H NMR chemical shifts for our application shown here.
Fig. 3.

Mean absolute errors (MAE) and maximum absolute errors (MAXAE) of chemical shifts for the demonstration set. The grey bars represent MAE, the black bars represent MAXAE. For all methods, geometries are optimized at B3LYP/6-31G(d) in chloroform
Figure 4 shows computational costs of DFT combinations for the demonstration set. We found that the smaller basis set (cc-pVDZ) in the calculation of both 13C and 1H NMR chemical shifts was an acceptable compromise between accuracy and computational performance, compared with the larger cc-pVTZ basis. This finding is similar to a recent benchmark study [138] that showed B3LYP/cc-pVDZ is a reliable combination, balancing accuracy with computational cost in 13C chemical shifts calculation. The larger basis set (cc-pVTZ) took 2–3 times longer to complete than cc-pVDZ (in terms of total CPU time). The computational times of the isotropic shielding and chemical shift calculations for this demonstration set are given in the file of DemonstrationSet_CPUtimes.xlsx in the Additional file 2.
Fig. 4.
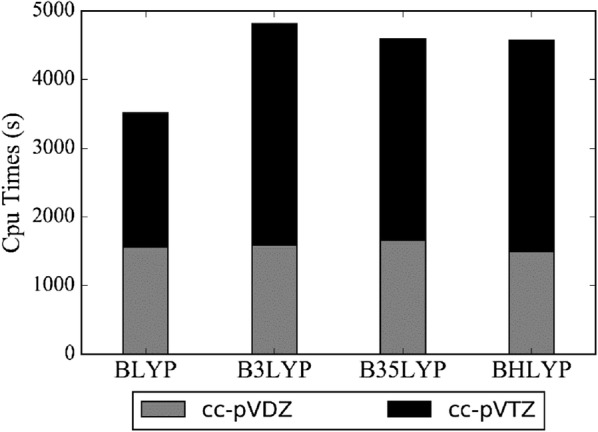
Computational costs of DFT methods performed for the demonstration set. Each bar is for two DFT methods with basis sets of cc-pVDZ and cc-pVTZ. The grey bars represent CPU times for the methods with cc-pVDZ and the black bars represent those with cc-pVDZ and the black bars represent those with cc-pVTZ
Effect of scaling by linear regression
We performed the most general approach to error reduction, empirical scaling. Our molecule set has 1554 and 1830 experimental 13C and 1H NMR chemical shifts, respectively. It provides confidence for applying linear regression effectively as it reduces the possibility of overfitting. Empirical scaling was applied to the data obtained with the best combination, B3LYP/cc-pVDZ, using two different relationships: computed shifts versus experimental chemical shifts (Eq. 6), and computed isotropic shieldings versus experimental shifts (Eq. 7). Once the empirical scaling was applied, the accuracy for 13C chemical shifts and 1H chemical shifts improved by 0.7 and 0.11 ppm, respectively. Our computed NMR chemical shifts and shieldings deviate from unity (desired slope = 1) by 0.02 for both 13C and 1H NMR chemical shifts. Linear fits with correlation coefficients of 0.99 (Fig. 5a, b) and 0.93 (Fig. 5c, d) for 13C and 1H NMR chemical shifts, respectively, were observed, which also shows that B3LYP/cc-pVDZ is able to produce data free from random error. Results of linear regression to the 13C and 1H NMR chemical shifts obtained by other DFT methods are given in the Additional file 1 (S5).
Fig. 5.

Linear correlation plots of a 13C and c 1H isotropic shielding values, and b 13C and d 1H NMR chemical shifts versus experimental NMR chemical shifts. Chemical shifts are calculated using the GIAO/B3LYP/cc-pVDZ//B3LYP/6-31G(d) level of theory for the demonstration set in CDCl3 (312 molecules (1554 carbons and 1830 hydrogens)). R2 indicates the correlation coefficient
Detailed look at 13C NMR chemical shifts
The carbon (13C) magnetic shieldings and chemical shifts derived from the various DFT methods are highly correlated, as shown by a correlation coefficient of 0.99 (Fig. 5). The inclusion of a scaling factor enhances the performance of theoretical calculations with B3LYP/cc-pVDZ//B3LYP/6-31G(d) and decreases the MAE in 13C NMR chemical shifts for this set by approximately 13%.
There has been a trend toward using multiple references, such that each molecule should be referenced to a molecule with similar properties to improve accuracy of NMR chemical shifts [123, 134, 139]. Sarotti et al. examined the influence of the reference compound used in the 13C [83] and 1H [82] NMR chemical shift calculations over a set of organic compounds, all of which were included in our calculations. They recommended the use of benzene and methanol as a reference standard in the calculations of chemical shifts of sp-sp2- and sp3- hybridized carbon atoms, respectively, instead of TMS for all type of carbon atoms [140]. Propelled by the discussion in the study of Grimblat et al. [141] about the distribution of the errors observed in sp2- and sp3- carbons, we determined the distribution of the data of chemical shifts of sp2- (933 carbons) and sp3- (745 carbons) hybridized carbons (Fig. 6, the sp2- and sp3- derived series of carbons show two separate chemical shift distributions and two separate error distributions over a much larger variety of compounds than Grimblat et al. For our demonstration set, both errors between calculated and experimental sp2- and sp3- chemical shifts more closely resemble a Student’s t-distribution [58, 142], rather than a normal distribution [138, 143]. The correlation coefficients of the errors of sp2- and sp3- carbons are 0.93 and 0.78, and 0.98 and 0.95 for Student’s t-distribution and normal distribution, respectively.
Fig. 6.

Chemical shifts of sp2- and sp3- hybridized carbon atoms. a Chemical shifts, b associated errors. Chemical shifts were calculated using the B3LYP/cc-pVDZ//B3LYP/6-31G(d) level of theory in CDCl3
Furthermore, we looked for the bonded neighbors of each carbon and hydrogen extensively in Fig. 7. For carbon shifts, error was measured for carbon (n = 1709), chlorine (n = 149), fluorine (n = 8), hydrogen (n = 1161), nitrogen (n = 199), oxygen (n = 251) and sulfur (n = 20) attachments. The largest deviations occur in carbon–chlorine and carbon–sulfur attachments with MAEs of 11.2 and 5.8 ppm and MAXAE 39.7 and 16.5 ppm, respectively. The study by Li et al. [76], which used a set of chlorinated carbons, reports the same conclusion: calculation accuracy decreases as the size of the basis set used increases, but improvement was obtained after linear regression corrections for B3LYP/6-31 + G(d,p) with slope of 0.98. Other than chlorine and sulfur, carbon-hydrogen attachments also make the 13C NMR chemical shift DFT calculations deviate significantly from experimental values, with MAE of 4.7 and 3.9 ppm and with MAXAE of 51.6 and 34.9 ppm. Carbon was found in rings in 70% of the cases, and these carbons show a MAE of 4.6 ppm. Also, the MAE of 13C NMR chemical shift is 3.9 ppm for carbons bonded to a hydrogen atom but reaches 9.7 ppm in all other cases.
Fig. 7.

Chemical shift prediction errors for different functional groups. a 13C NMR chemical shifts, b 1H NMR chemical shifts. All molecules are from the demonstration set and are calculated using the GIAO/B3LYP/cc-pVDZ//B3LYP/6-31G(d) level of theory in chloroform
Oxygen and nitrogen attachments to carbon led to 13C NMR chemical shifts with MAE up to 3.1 and 4.2 ppm and MAXAE of 32.5 and 23.6 ppm, respectively. Interestingly, half the C–O attachments found in ring-form had chemical shifts with a MAE of 2.8 ppm, compared to the chemical shifts of C–O attachments not found in a ring, which had a relatively higher MAE of 3.2 ppm, leading to a percent difference of 14.1%. NMR chemical shifts of C–N attachments, present in a ring or not, show close MAE of 4.19 and 4.26 ppm, respectively, a percent difference of 1.6%. This is to be expected, since C–O attachments are expected to show some deviation in chemical shift due to the polarization of the electron distribution caused by the high electronegativity of oxygen, while nitrogen atoms have a lower electronegativity, leading to a lower deviation in C–N chemical shifts.
The correlation plot (Fig. 5a, b) shows a linear pattern with only minor deviations of the predicted 13C shieldings or 13C chemical shifts from the fitted line. It is verified by the correlation coefficient of 0.99, as observed in previous studies [130, 144], that the deviation of the slope from unity within the range of 0.95 and 1.05 is an indicator of a reliable method. However, when placed in subgroups of different attachment types, distant outliers are observed, with some more than 15 ppm away (Fig. 7a). Most outliers are observed in the C–C and C–H attachments, respectively. C–Cl and C–N chemical shifts have a high occurrence of outliers, which may be due to the chemical properties of chlorine and nitrogen such as being remarkably close to first ionization energies. We suspected that some cases of high calculation errors could be due to the consideration of only a single conformer. For the highest accuracy, proper conformational sampling must be considered, as demonstrated below in “Application 2: Boltzmann-weighted NMR chemical shifts of methylcyclohexane” section
Detailed look at 1H NMR chemical shifts
Proton (1H) chemical shifts are significantly affected by intermolecular interactions, particularly in aqueous states, especially compared to 13C chemical shifts. Agreement with experimental values improves as empirical linear scaling is performed for 1H chemical shifts. GIAO/B3LYP/cc-pVDZ//B3LYP/6-31G(d) yields scaled 1H chemical shifts in chloroform solution having a MAE of 0.30 ppm in comparison with solution experimental values. The 1H chemical shifts in the range of 10–17 ppm show the largest deviation, occurring higher than 5 ppm.
In Fig. 7b, error bars are shown for 1H chemical shifts when the hydrogen attaches to carbon (n = 1793), nitrogen (n = 17), and oxygen (n = 41). Oxygen-bound hydrogen nuclei have the largest errors (up to 10 ppm), which is to be expected due to the electronegative property of oxygen atoms, as discussed in the previous section. It is followed by less electronegative nitrogen-bound hydrogen atoms, with an MAE of 0.71 ppm and a MAXAE of 2.25 ppm. About 95% of the 1H NMR chemical shifts calculated for this set are from H–C attachments. These chemical shifts had a MAE of 0.27 ppm and a MAXAE of 4.41 ppm. The high occurrence of outliers could be evidence of how 1H NMR chemical shifts are sensitive to intermolecular interactions.
H–O attachments are highly sensitive (to concentration, solvent, temperature, etc.), and it is non-trivial to determine the NMR chemical shift value of arbitrary protons experimentally as well as predict them by using a single, “catch all” DFT method, which explains the relatively low correlation coefficient of 0.93 (Fig. 5c–d). For future studies, we may need to consider the use of different DFT methods, including the use of explicit solvation, particularly in the calculation of 1H NMR chemical shifts in the presence of H–O attachments.
Application of empirical scaling to functional groups are given in detail in the Additional file 1 (see S7 and S8).
Cross-validation
As a final assessment for the data collected with the demonstration set, we assessed the stability and accuracy of the linear regression approach using cross-validation [145]. Cross-validation is a technique mostly used in prediction problems to evaluate how much a given model generalizes to an independent set of data. Specifically, we performed Monte Carlo cross-validation [146, 147]. The procedure of application of Monte Carlo cross-validation method is explained in Additional file 1 (S9).
We observed that the estimated linear model parameters (i.e. slope and intercept) from the training set do not differ from that of the entire set. Therefore, the predictive linear model is stable to be accurately estimated and the subsets of 13C and 1H NMR chemical shifts generalize well to the groups that are not represented in the training fold.
Application 2: Boltzmann-weighted NMR chemical shifts of methylcyclohexane
Metabolites were experimentally interrogated using solution-state NMR, where the observed signal arises from the combined signals of present conformers. It is routine that NMR chemical shifts calculations are carried out on a single dominant conformer. However, it is well known that metabolites do not comprise a single conformer in solution and are instead found in a collection of various conformers [148], and the accuracy of NMR chemical shifts heavily depends on molecular geometries and conformation consideration [46]. It has been shown that for the highest accuracy NMR chemical shift calculations, consideration of conformers is critical, even for relatively small molecules [149]. As a second demonstration of ISiCLE, a conformational analysis based on DFT was performed on a set of 80 conformers of methylcyclohexane using a Boltzmann distribution technique. Boltzmann weighting determines the fractional population of each conformer based on its energy level [92]. High-throughput and straightforward DFT-based NMR chemical shift calculations of all 80 methylcyclohexane conformers was performed by ISiCLE then compared to experimental values.
It has been shown by Willoughby et al. [92] that the effects of molecular flexibility on NMR chemical shifts can be captured by Boltzmann weighting analysis, as demonstrated with methylcyclohexane (Fig. 8). Methylcyclohexane is a well-studied small molecule [150–154]. It is flexible, composed of a single methyl group attached to a six-membered ring, and known to exist as an assembly of two chair conformers. There are two distinct conformations of which chair–chair interconversion is rapid and dominated by equatorial to axial conformation. We weighted 40 axial and 40 equatorial conformers in chloroform and obtained a relative free energy of 1.99 kcal/mol with NWChem, similar to calculations using Gaussian [155] by Willoughby et al. [92], and similar to experimental findings (1.73 kcal/mol [156], 1.93 kcal/mol] [108]) and computed (2.15–2.31 kcal/mol [107] and 1.68–2.48 kcal/mol [109]) values. We compared the Boltzmann-weighted 1H and 13C chemical shifts (a ratio of 3% axial to 97% equatorial) to experimental values reported by Willoughby et al. [92]. The Boltzmann-weighted, scaled MAE was 0.017 ppm for 1H chemical shifts (), similar to the experimental value of 0.018 ppm in the study of Willoughby et al. Also, the MAE for 13C chemical shifts was 4.4 ppm and decreases to 0.8 ppm when the chemical shifts were scaled () (Fig. 9). Further details can be found in the Additional file 1 (S10).
Fig. 8.

The a equatorial and b axial structures of methylcyclohexane
Fig. 9.
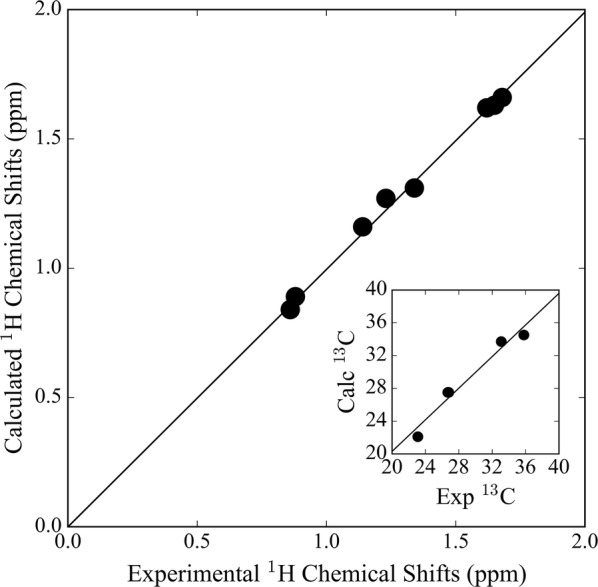
Experimental and scaled chemical shifts (ppm) of methylcyclohexane
Conclusions
We introduce the first release of ISiCLE, which predicts NMR chemical shifts of any given set of molecules relevant to metabolomics for a given set of DFT techniques. ISiCLE calculates the unscaled or scaled NMR chemical shifts (depending on the user’s choice of DFT method) and writes the data to appended MDL Molfiles. It also quantifies the error in calculated NMR chemical shifts if the user provides experimental values.
The functionality of ISiCLE is demonstrated on a molecule set consisting of 312 molecules, with experimental chemical shifts reported in chloroform solvent. 1H and 13C NMR chemical shifts were calculated using 8 different levels of DFT (BLYP, B3LYP, B35LYP, BHLYP and, cc-pVDZ, and ccpVTZ), referenced to TMS in chloroform by carrying initial geometry optimizations out at B3LYP/6-31G(d) for all molecules. The optimal combination for this set was found to be B3LYP/cc-pVDZ//B3LYP/6-31G(d) with mean absolute error of 0.33 and 3.93 ppm for proton and carbon chemical shifts, respectively. We show that DFT calculations followed by linear scaling do in fact provide an analytically useful degree of accuracy and reliability. Finally, we used ISiCLE for the calculation of NMR chemical shifts of 80 Boltzmann-weighted conformers of methylcyclohexane and compared our results with earlier studies in the literature.
ISiCLE is a promising automated framework for accurate NMR chemical shift calculations of small organic molecules. Through this tool, we hope to expand chemical shift libraries, without the need for chemical standards run in the laboratory, which could lead to significantly more identifiable metabolites. Future work includes wrapping individual steps of the ISiCLE NMR module into a formal workflow management system such as Snakemake, to include better fault tolerance, modularization, and improved data provenance. Furthermore, additional chemical properties will be included, such as ion mobility collision cross section and infrared spectra. Finally, ISiCLE will be adapted to run seamlessly on cloud computer resources such as Amazon AWS, Microsoft Azure, and Google Cloud. ISiCLE is a promising tool contributing to standards-free metabolomics, which depends on the ability to calculate properties for thousands of molecules and their associated conformers.
Additional files
Additional file 1. Supporting information document.
Additional file 2. Supporting information files. Including tutorial, code, and all other files.
Authors’ contributions
YY designed the tool, gathered the dataset, performed the calculations and provided the analysis. NG provided technical support. RR conceived and supervised the project. All authors participated in the manuscript preparation. All authors read and approved the final manuscript.
Acknowledgements
This research was supported by PNNL Laboratory Directed Research and Development program, the Microbiomes in Transition (MinT) Initiative. This work was performed in the W. R. Wiley Environmental Molecular Sciences Laboratory (EMSL), a DOE national scientific user facility at the PNNL. The NWChem calculations were performed using the Cascade supercomputer at the EMSL. PNNL is operated by Battelle for the DOE under contract DE-AC05-76RL0 1830.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Project name: ISiCLE. Project home page: github.com/pnnl/isicle. Programming language: Python. Operating system(s): Platform independent. License: GNU GPL license. Free academic and non-profit research use only. All data files, source code, and tutorial are provided in the additional files.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Fiehn O. Metabolomics—the link between genotypes and phenotypes. Plant Mol Biol. 2002;48(1–2):155–171. doi: 10.1023/A:1013713905833. [DOI] [PubMed] [Google Scholar]
- 2.Sumner LW, Mendes P, Dixon RA. Plant metabolomics: large-scale phytochemistry in the functional genomics era. Phytochemistry. 2003;62(6):817–836. doi: 10.1016/S0031-9422(02)00708-2. [DOI] [PubMed] [Google Scholar]
- 3.Bino RJ, et al. Potential of metabolomics as a functional genomics tool. Trends Plant Sci. 2004;9(9):418–425. doi: 10.1016/j.tplants.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 4.Dobson CM. Chemical space and biology. Nature. 2004;432(7019):824–828. doi: 10.1038/nature03192. [DOI] [PubMed] [Google Scholar]
- 5.Nicholson JK, et al. Metabonomics: a platform for studying drug toxicity and gene function. Nat Rev Drug Discov. 2002;1(2):153–161. doi: 10.1038/nrd728. [DOI] [PubMed] [Google Scholar]
- 6.Berendsen RL, Pieterse CM, Bakker PA. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012;17(8):478–486. doi: 10.1016/j.tplants.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 7.Griffin JL, Bollard ME. Metabonomics: its potential as a tool in toxicology for safety assessment and data integration. Curr Drug Metab. 2004;5(5):389–398. doi: 10.2174/1389200043335432. [DOI] [PubMed] [Google Scholar]
- 8.Richard AM, Gold LS, Nicklaus MC. Chemical structure indexing of toxicity data on the internet: moving toward a flat world. Curr Opin Drug Discov Dev. 2006;9(3):314–325. [PubMed] [Google Scholar]
- 9.Daviss B. Growing pains for metabolomics. Scientist. 2005;19(8):25–28. [Google Scholar]
- 10.Nicholson JK, Wilson ID. Understanding ‘global’ systems biology: metabonomics and the continuum of metabolism. Nat Rev Drug Discov. 2003;2(8):668–676. doi: 10.1038/nrd1157. [DOI] [PubMed] [Google Scholar]
- 11.Beckonert O, et al. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat Protoc. 2007;2(11):2692–2703. doi: 10.1038/nprot.2007.376. [DOI] [PubMed] [Google Scholar]
- 12.Nicholson JK, Lindon JC, Holmes E. ‘Metabonomics’: understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica. 1999;29(11):1181–1189. doi: 10.1080/004982599238047. [DOI] [PubMed] [Google Scholar]
- 13.Nicholson JK, et al. 750 MHz 1H and 1H-13C NMR spectroscopy of human blood plasma. Anal Chem. 1995;67(5):793–811. doi: 10.1021/ac00101a004. [DOI] [PubMed] [Google Scholar]
- 14.Soga T, et al. Quantitative metabolome analysis using capillary electrophoresis mass spectrometry. J Proteome Res. 2003;2(5):488–494. doi: 10.1021/pr034020m. [DOI] [PubMed] [Google Scholar]
- 15.Smith CA, et al. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal Chem. 2006;78(3):779–787. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 16.Dettmer K, Aronov PA, Hammock BD. Mass spectrometry-based metabolomics. Mass Spectrom Rev. 2007;26(1):51–78. doi: 10.1002/mas.20108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith CA, et al. METLIN: a metabolite mass spectral database. Ther Drug Monit. 2005;27(6):747–751. doi: 10.1097/01.ftd.0000179845.53213.39. [DOI] [PubMed] [Google Scholar]
- 18.Wishart DS, et al. HMDB 3.0—the human metabolome database in 2013. Nucleic Acids Res. 2013;41(Database issue):D801-7. doi: 10.1093/nar/gks1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ulrich EL, et al. BioMagResBank. Nucleic Acids Res. 2008;36(Database issue):D402-8. doi: 10.1093/nar/gkm957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pence HE, Williams A. ChemSpider: an online chemical information resource. J Chem Educ. 2010;87(11):1123–1124. doi: 10.1021/ed100697w. [DOI] [Google Scholar]
- 21.Tautenhahn R, et al. XCMS Online: a web-based platform to process untargeted metabolomic data. Anal Chem. 2012;84(11):5035–5039. doi: 10.1021/ac300698c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Little JL, et al. Identification of “known unknowns” utilizing accurate mass data and ChemSpider. J Am Soc Mass Spectrom. 2012;23(1):179–185. doi: 10.1007/s13361-011-0265-y. [DOI] [PubMed] [Google Scholar]
- 23.Little JL, Cleven CD, Brown SD. Identification of “known unknowns” utilizing accurate mass data and chemical abstracts service databases. J Am Soc Mass Spectrom. 2011;22(2):348–359. doi: 10.1007/s13361-010-0034-3. [DOI] [PubMed] [Google Scholar]
- 24.Patti GJ, et al. A view from above: cloud plots to visualize global metabolomic data. Anal Chem. 2013;85(2):798–804. doi: 10.1021/ac3029745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wishart DS, et al. HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res. 2009;37(Database issue):D603-10. doi: 10.1093/nar/gkn810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Randazzo GM, et al. Enhanced metabolite annotation via dynamic retention time prediction: steroidogenesis alterations as a case study. J Chromatogr, B: Anal Technol Biomed Life Sci. 2017;1071:11–18. doi: 10.1016/j.jchromb.2017.04.032. [DOI] [PubMed] [Google Scholar]
- 27.Vinaixa M, et al. Mass spectral databases for LC/MS- and GC/MS-based metabolomics: state of the field and future prospects. TrAC Trends Anal Chem. 2016;78:23–35. doi: 10.1016/j.trac.2015.09.005. [DOI] [Google Scholar]
- 28.Bocker S. Searching molecular structure databases using tandem MS data: are we there yet? Curr Opin Chem Biol. 2017;36:1–6. doi: 10.1016/j.cbpa.2016.12.010. [DOI] [PubMed] [Google Scholar]
- 29.Allen F, Greiner R, Wishart D. Competitive fragmentation modeling of ESI-MS/MS spectra for putative metabolite identification. Metabolomics. 2015;11(1):98–110. doi: 10.1007/s11306-014-0676-4. [DOI] [Google Scholar]
- 30.Wolf S, et al. In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC Bioinform. 2010;11:148. doi: 10.1186/1471-2105-11-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng XY, et al. Structural elucidation of cis/trans dicaffeoylquinic acid photoisomerization using ion mobility spectrometry–mass spectrometry. J Phys Chem Lett. 2017;8(7):1381–1388. doi: 10.1021/acs.jpclett.6b03015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Metz TO, et al. Integrating ion mobility spectrometry into mass spectrometry-based exposome measurements: what can it add and how far can it go? Bioanalysis. 2017;9(1):81–98. doi: 10.4155/bio-2016-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Graham TR, et al. Precursor ion–ion aggregation in the Brust–Schiffrin synthesis of alkanethiol nanoparticles. J Phys Chem C. 2016;120(35):19837–19847. doi: 10.1021/acs.jpcc.6b06155. [DOI] [Google Scholar]
- 34.Zhou ZW, et al. Large-scale prediction of collision cross-section values for metabolites in ion mobility-mass spectrometry. Anal Chem. 2016;88(22):11084–11091. doi: 10.1021/acs.analchem.6b03091. [DOI] [PubMed] [Google Scholar]
- 35.Zhou ZW, et al. LipidCCS: prediction of collision cross-section values for lipids with high precision to support ion mobility-mass spectrometry-based lipidomics. Anal Chem. 2017;89(17):9559–9566. doi: 10.1021/acs.analchem.7b02625. [DOI] [PubMed] [Google Scholar]
- 36.Brockherde F, et al. Bypassing the Kohn–Sham equations with machine learning. Nat Commun. 2017;8:872. doi: 10.1038/s41467-017-00839-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sarotti AM. Successful combination of computationally inexpensive GIAO C-13 NMR calculations and artificial neural network pattern recognition: a new strategy for simple and rapid detection of structural misassignments. Org Biomol Chem. 2013;11(29):4847–4859. doi: 10.1039/c3ob40843d. [DOI] [PubMed] [Google Scholar]
- 38.Forsyth DA, Sebag AB. Computed C-13 NMR chemical shifts via empirically scaled GIAO shieldings and molecular mechanics geometries. Conformation and configuration from C-13 shifts. J Am Chem Soc. 1997;119(40):9483–9494. doi: 10.1021/ja970112z. [DOI] [Google Scholar]
- 39.Casabianca LB, De Dios AC. Ab initio calculations of NMR chemical shifts. J Chem Phys. 2008;128(5):052201. doi: 10.1063/1.2816784. [DOI] [PubMed] [Google Scholar]
- 40.Auer AA, Gauss J, Stanton JF. Quantitative prediction of gas-phase C-13 nuclear magnetic shielding constants. J Chem Phys. 2003;118(23):10407–10417. doi: 10.1063/1.1574314. [DOI] [Google Scholar]
- 41.Mothana B, Ban FQ, Boyd RJ. Validation of a computational scheme to study N-15 and C-13 nuclear shielding constants. Chem Phys Lett. 2005;401(1–3):7–12. doi: 10.1016/j.cplett.2004.10.145. [DOI] [Google Scholar]
- 42.Saito H. Conformation-dependent C-13 chemical-shifts—a new means of conformational characterization as obtained by high-resolution solid-state C-13 NMR. Magn Reson Chem. 1986;24(10):835–852. doi: 10.1002/mrc.1260241002. [DOI] [Google Scholar]
- 43.Jaime C, et al. C-13 NMR chemical-shifts—a single rule to determine the conformation of calix[4]arenes. J Org Chem. 1991;56(10):3372–3376. doi: 10.1021/jo00010a036. [DOI] [Google Scholar]
- 44.Yannoni CS, et al. C-13 NMR-study of the C60 cluster in the solid-state—molecular-motion and carbon chemical-shift anisotropy. J Phys Chem. 1991;95(1):9–10. doi: 10.1021/j100154a005. [DOI] [Google Scholar]
- 45.Kwan EE, Liu RY. Enhancing NMR prediction for organic compounds using molecular dynamics. J Chem Theory Comput. 2015;11(11):5083–5089. doi: 10.1021/acs.jctc.5b00856. [DOI] [PubMed] [Google Scholar]
- 46.Malkin VG, et al. Solvent effect on the NMR chemical shieldings in water calculated by a combination of molecular dynamics and density functional theory. Chem Eur J. 1996;2(4):452–457. doi: 10.1002/chem.19960020415. [DOI] [Google Scholar]
- 47.Casanovas J, et al. Calculated and experimental NMR chemical shifts of p-menthane-3,9-diols. A combination of molecular dynamics and quantum mechanics to determine the structure and the solvent effects. J Org Chem. 2001;66(11):3775–3782. doi: 10.1021/jo0016982. [DOI] [PubMed] [Google Scholar]
- 48.Benzi C, et al. Reliable NMR chemical shifts for molecules in solution by methods rooted in density functional theory. Magn Reson Chem. 2004;42:S57–S67. doi: 10.1002/mrc.1447. [DOI] [PubMed] [Google Scholar]
- 49.Garcellano RC, et al. Isolation of tryptanthrin and reassessment of evidence for its isobaric isostere wrightiadione in plants of the Wrightia Genus. J Nat Prod. 2018 doi: 10.1021/acs.jnatprod.8b00567. [DOI] [PubMed] [Google Scholar]
- 50.Valiev M, et al. NWChem: a comprehensive and scalable open-source solution for large scale molecular simulations. Comput Phys Commun. 2010;181(9):1477–1489. doi: 10.1016/j.cpc.2010.04.018. [DOI] [Google Scholar]
- 51.Klamt A, Schuurmann G. Cosmo—a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J Chem Soc Perkin Trans. 1993;2(5):799–805. doi: 10.1039/P29930000799. [DOI] [Google Scholar]
- 52.Seca AML, et al. Chemical composition of the light petroleum extract of Hibiscus cannabinus bark and core. Phytochem Anal. 2000;11(6):345–350. doi: 10.1002/1099-1565(200011/12)11:6<345::AID-PCA540>3.0.CO;2-T. [DOI] [Google Scholar]
- 53.Watts HD, Mohamed MNA, Kubicki JD. Comparison of multistandard and TMS-standard calculated NMR shifts for coniferyl alcohol and application of the multistandard method to lignin dimers. J Phys Chem B. 2011;115(9):1958–1970. doi: 10.1021/jp110330q. [DOI] [PubMed] [Google Scholar]
- 54.The Open Babel Package version 2.3.1. http://openbabel.org. Accessed 16 Oct 2018
- 55.O’Boyle NM, et al. Open Babel: an open chemical toolbox. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Halgren TA. Merck molecular force field. 1. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem. 1996;17(5–6):490–519. doi: 10.1002/(SICI)1096-987X(199604)17:5/6<490::AID-JCC1>3.0.CO;2-P. [DOI] [Google Scholar]
- 57.Dalby A, et al. Description of several chemical-structure file formats used by computer-programs developed at Molecular Design Limited. J Chem Inf Comput Sci. 1992;32(3):244–255. doi: 10.1021/ci00007a012. [DOI] [Google Scholar]
- 58.Smith SG, Goodman JM. Assigning stereochemistry to single diastereoisomers by GIAO NMR calculation: the DP4 probability. J Am Chem Soc. 2010;132(37):12946–12959. doi: 10.1021/ja105035r. [DOI] [PubMed] [Google Scholar]
- 59.Brown SG, Jansma MJ, Hoye TR. Case study of empirical and computational chemical shift analyses: reassignment of the relative configuration of phomopsichalasin to that of diaporthichalasin. J Nat Prod. 2012;75(7):1326–1331. doi: 10.1021/np300248w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aliev AE, Courtier-Murias D, Zhou S. Scaling factors for carbon NMR chemical shifts obtained from DFF B3LYP calculations. J Mol Struct Theochem. 2009;893(1–3):1–5. doi: 10.1016/j.theochem.2008.09.021. [DOI] [Google Scholar]
- 61.Baldridge KK, Siegel JS. Correlation of empirical delta(TMS) and absolute NMR chemical shifts predicted by ab initio computations. J Phys Chem A. 1999;103(20):4038–4042. doi: 10.1021/jp990523x. [DOI] [Google Scholar]
- 62.Pupier M, et al. NMReDATA, a standard to report the NMR assignment and parameters of organic compounds. Magn Reson Chem. 2018;56(8):703–715. doi: 10.1002/mrc.4737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alver O. DFT, FT-Raman, FT-IR, solution and solid state NMR studies of 2,4-dimethoxyphenylboronic acid. C R Chim. 2011;14(5):446–455. doi: 10.1016/j.crci.2010.11.005. [DOI] [Google Scholar]
- 64.Asiri AM, et al. Synthesis, molecular conformation, vibrational and electronic transition, isometric chemical shift, polarizability and hyperpolarizability analysis of 3-(4-Methoxy-phenyl)-2-(4-nitro-phenyl)-acrylonitrile: a combined experimental and theoretical analysis. Spectrochim Acta Part A Mol Biomol Spectrosc. 2011;82(1):444–455. doi: 10.1016/j.saa.2011.07.076. [DOI] [PubMed] [Google Scholar]
- 65.Bally T, Rablen PR. Quantum-chemical simulation of H-1 NMR spectra. 2. Comparison of DFT-based procedures for computing proton–proton coupling constants in organic molecules. J Org Chem. 2011;76(12):4818–4830. doi: 10.1021/jo200513q. [DOI] [PubMed] [Google Scholar]
- 66.Bagno A, Rastrelli F, Saielli G. Predicting the NMR spectra of nucleotides by DFT calculations: cyclic uridine monophosphate. Magn Reson Chem. 2008;46(6):518–524. doi: 10.1002/mrc.2204. [DOI] [PubMed] [Google Scholar]
- 67.Borkowski EJ, Suvire FD, Enriz RD. Advances in correlation between experimental and DFT/GIAO computed C-13 NMR chemical shifts: a theoretical study on pentacyclic terpenoids (fernenes) J Mol Struct Theochem. 2010;953(1–3):83–90. doi: 10.1016/j.theochem.2010.05.007. [DOI] [Google Scholar]
- 68.Coruh A, et al. Synthesis, molecular conformation, vibrational, electronic transition, and chemical shift assignments of 4-(thiophene-3-ylmethoxy)phthalonitrile: a combined experimental and theoretical analysis. Struct Chem. 2011;22(1):45–56. doi: 10.1007/s11224-010-9694-7. [DOI] [Google Scholar]
- 69.Fulmer GR, et al. NMR chemical shifts of trace impurities: common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist. Organometallics. 2010;29(9):2176–2179. doi: 10.1021/om100106e. [DOI] [Google Scholar]
- 70.Hill DE, Vasdev N, Holland JP. Evaluating the accuracy of density functional theory for calculating H-1 and C-13 NMR chemical shifts in drug molecules. Comput Theor Chem. 2015;1051:161–172. doi: 10.1016/j.comptc.2014.11.007. [DOI] [Google Scholar]
- 71.Izgi T, et al. FT-IR and NMR investigation of 2-(1-cyclohexenyl)ethylamine: a combined experimental and theoretical study. Spectrochim Acta Part A Mol Biomol Spectrosc. 2007;68(1):55–62. doi: 10.1016/j.saa.2006.10.050. [DOI] [PubMed] [Google Scholar]
- 72.Karabacak M, et al. Experimental (UV, NMR, IR and Raman) and theoretical spectroscopic properties of 2-chloro-6-methylaniline. Mol Phys. 2009;107(3):253–264. doi: 10.1080/00268970902821579. [DOI] [Google Scholar]
- 73.Krishnakumar V, et al. Molecular structure, vibrational spectra, HOMO, LUMO and NMR studies of 2-chloro-4-nitrotoluene and 4-chloro-2-nitrotoluene. Spectrochim Acta Part A Mol Biomol Spectrosc. 2012;91:1–10. doi: 10.1016/j.saa.2012.01.038. [DOI] [PubMed] [Google Scholar]
- 74.Krishnakumar V, Barathi D, Mathammal R. Molecular structure, vibrational spectra, HOMO, LUMO and NMR studies of 1,2-dichloro-4-nitrobenzene and 2,3,5,6-tetrachloro-1-nitrobenzene based on density functional calculations. Spectrochim Acta Part A Mol Biomol Spectrosc. 2012;86:196–204. doi: 10.1016/j.saa.2011.10.024. [DOI] [PubMed] [Google Scholar]
- 75.Krishnakumar V, et al. Molecular structure, spectroscopic studies (FTIR, FT-Raman and NMR) and HOMO–LUMO analysis of 6-chloro-o-cresol and 4-chloro-3-methyl phenol by density functional theoretical study. Spectrochim Acta Part A Mol Biomol Spectrosc. 2012;97:144–154. doi: 10.1016/j.saa.2012.05.070. [DOI] [PubMed] [Google Scholar]
- 76.Li YJ, et al. Screening and characterization of natural antioxidants in four Glycyrrhiza species by liquid chromatography coupled with electrospray ionization quadrupole time-of-flight tandem mass spectrometry. J Chromatogr A. 2011;1218(45):8181–8191. doi: 10.1016/j.chroma.2011.09.030. [DOI] [PubMed] [Google Scholar]
- 77.Lomas JS. H-1 NMR spectra of alcohols in hydrogen bonding solvents: DFT/GIAO calculations of chemical shifts. Magn Reson Chem. 2016;54(1):28–38. doi: 10.1002/mrc.4312. [DOI] [PubMed] [Google Scholar]
- 78.Osmialowski B, Kolehmainen E, Gawinecki R. GIAO/DFT calculated chemical shifts of tautomeric species. 2-Phenacylpyridines and (Z)-2-(2-hydroxy-2-phenylvinyl)pyridines. Magn Reson Chem. 2001;39(6):334–340. doi: 10.1002/mrc.856. [DOI] [Google Scholar]
- 79.Parlak C, et al. Molecular structure, NMR analyses, density functional theory and ab initio Hartree–Fock calculations of 4,4′-diaminooctafluorobiphenyl. J Mol Struct. 2008;891(1–3):151–156. doi: 10.1016/j.molstruc.2008.03.031. [DOI] [Google Scholar]
- 80.Perez M, et al. Accuracy versus time dilemma on the prediction of NMR chemical shifts: a case study (chloropyrimidines) J Org Chem. 2006;71(8):3103–3110. doi: 10.1021/jo0600149. [DOI] [PubMed] [Google Scholar]
- 81.Rablen PR, Pearlman SA, Finkbiner J. A comparison of density functional methods for the estimation of proton chemical shifts with chemical accuracy. J Phys Chem A. 1999;103(36):7357–7363. doi: 10.1021/jp9916889. [DOI] [Google Scholar]
- 82.Sarotti AM, Pellegrinet SC. Application of the multi-standard methodology for calculating H-1 NMR chemical shifts. J Org Chem. 2012;77(14):6059–6065. doi: 10.1021/jo3008447. [DOI] [PubMed] [Google Scholar]
- 83.Sarotti AM, Pellegrinet SC. A multi-standard approach for GIAO C-13 NMR calculations. J Org Chem. 2009;74(19):7254–7260. doi: 10.1021/jo901234h. [DOI] [PubMed] [Google Scholar]
- 84.Sebastian S, et al. Quantum mechanical study of the structure and spectroscopic (FT-IR, FT-Raman, C-13, H-1 and UV), first order hyperpolarizabilities, NBO and TD-DFT analysis of the 4-methyl-2-cyanobiphenyl. Spectrochim Acta Part A Mol Biomol Spectrosc. 2011;78(2):590–600. doi: 10.1016/j.saa.2010.11.028. [DOI] [PubMed] [Google Scholar]
- 85.Senyel M, Unal A, Alver O. Molecular structure, NMR analyses, density functional theory and ab initio Hartree–Fock calculations of 3-phenylpropylamine. C R Chim. 2009;12(6–7):808–815. doi: 10.1016/j.crci.2008.11.002. [DOI] [Google Scholar]
- 86.Senyel M, Alver O, Parlak C. H-1, C-13, N-15 NMR and (n)J(C, H) coupling constants investigation of 3-piperidino-propylamine: a combined experimental and theoretical study. Spectrochim Acta Part A Mol Biomol Spectrosc. 2008;71(3):830–834. doi: 10.1016/j.saa.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 87.Sridevi C, Shanthi G, Velraj G. Structural, vibrational, electronic, NMR and reactivity analyses of 2-amino-4H-chromene-3-carbonitrile (ACC) by ab initio HF and DFT calculations. Spectrochim Acta Part A Mol Biomol Spectrosc. 2012;89:46–54. doi: 10.1016/j.saa.2011.12.050. [DOI] [PubMed] [Google Scholar]
- 88.Tormena CF, da Silva GVJ. Chemical shifts calculations on aromatic systems: a comparison of models and basis sets. Chem Phys Lett. 2004;398(4–6):466–470. doi: 10.1016/j.cplett.2004.09.103. [DOI] [Google Scholar]
- 89.Vijaya P, Sankaran KR. A combined experimental and DFT study of a novel unsymmetrical azine 2-(4-methoxybenzylidene)-1-(1-(4-isobutylphenyl) ethylidene)hydrazine. Spectrochim Acta Part A Mol Biomol Spectrosc. 2015;138:460–473. doi: 10.1016/j.saa.2014.11.047. [DOI] [PubMed] [Google Scholar]
- 90.Wiitala KW, Hoye TR, Cramer CJ. Hybrid density functional methods empirically optimized for the computation of C-13 and H-1 chemical shifts in chloroform solution. J Chem Theory Comput. 2006;2(4):1085–1092. doi: 10.1021/ct6001016. [DOI] [PubMed] [Google Scholar]
- 91.Wiitala KW, et al. Evaluation of various DFT protocols for computing H-1 and C-13 chemical shifts to distinguish stereoisomers: diastereomeric 2-, 3-, and 4-methylcyclohexanols as a test set. J Phys Org Chem. 2007;20(5):345–354. doi: 10.1002/poc.1151. [DOI] [Google Scholar]
- 92.Willoughby PH, Jansma MJ, Hoye TR. A guide to small-molecule structure assignment through computation of (H-1 and C-13) NMR chemical shifts. Nat Protoc. 2014;9(3):643–660. doi: 10.1038/nprot.2014.042. [DOI] [PubMed] [Google Scholar]
- 93.Yang J, Huang SX, Zhao QS. Structure revision of hassananes with use of quantum mechanical (13)C NMR chemical shifts and UV–Vis absorption spectra. J Phys Chem A. 2008;112(47):12132–12139. doi: 10.1021/jp8072415. [DOI] [PubMed] [Google Scholar]
- 94.Becke AD. Density-functional exchange-energy approximation with correct asymptotic-behavior. Phys Rev A. 1988;38(6):3098–3100. doi: 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- 95.Lee CT, Yang WT, Parr RG. Development of the Colle–Salvetti correlation-energy formula into a functional of the electron-density. Phys Rev B. 1988;37(2):785–789. doi: 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- 96.Becke AD. Density-functional thermochemistry. 3. The role of exact exchange. J Chem Phys. 1993;98(7):5648–5652. doi: 10.1063/1.464913. [DOI] [Google Scholar]
- 97.Hehre WJ. Ab initio molecular orbital theory. New York: Wiley-Interscience; 1986. [Google Scholar]
- 98.Ruiz E, Nunzi F, Alvarez S. Magnetic communication through functionalized nanotubes: a theoretical study. Nano Lett. 2006;6(3):380–384. doi: 10.1021/nl052135e. [DOI] [PubMed] [Google Scholar]
- 99.Johnson JRT, Panas I. Water adsorption and hydrolysis on molecular Al oxides and hydroxides—solvation versus cluster formation. Phys Chem Chem Phys. 2001;3(24):5482–5488. doi: 10.1039/b106318a. [DOI] [Google Scholar]
- 100.Becke AD. A new mixing of Hartree–Fock and local density-functional theories. J Chem Phys. 1993;98(2):1372–1377. doi: 10.1063/1.464304. [DOI] [Google Scholar]
- 101.Dunning TH. Gaussian-basis sets for use in correlated molecular calculations. 1. The atoms boron through neon and hydrogen. J Chem Phys. 1989;90(2):1007–1023. doi: 10.1063/1.456153. [DOI] [Google Scholar]
- 102.Feller D. The role of databases in support of computational chemistry calculations. J Comput Chem. 1996;17(13):1571–1586. doi: 10.1002/(SICI)1096-987X(199610)17:13<1571::AID-JCC9>3.0.CO;2-P. [DOI] [Google Scholar]
- 103.Schuchardt KL, et al. Basis set exchange: a community database for computational sciences. J Chem Inf Model. 2007;47(3):1045–1052. doi: 10.1021/ci600510j. [DOI] [PubMed] [Google Scholar]
- 104.Basis Set Exchange. https://bse.pnl.gov/bse/portal. Accessed 16 Oct 2018
- 105.Dupuis M. New integral transforms for molecular properties and application to a massively parallel GIAO-SCF implementation. Comput Phys Commun. 2001;134(2):150–166. doi: 10.1016/S0010-4655(00)00195-8. [DOI] [Google Scholar]
- 106.Case DA, Babin V, Berryman JT, Betz RM, Cai Q, Cerutti DS, Cheatham TE, III, Darden TA, Duke RE, Gohlke H, Goetz AW, Gusarov S, Homeyer N, Janowski P, Kaus J, Kolossváry I, Kovalenko A, Lee TS, LeGrand S, Luchko T, Luo R, Madej B, Merz KM, Paesani F, Roe DR, Roitberg A, Sagui C, Salomon-Ferrer R, Seabra G, Simmerling CL, Smith W, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Kollman PA (2014) AMBER 14, University of California, San Francisco
- 107.Ribeiro DS, Rittner R. The role of hyperconjugation in the conformational analysis of methylcyclohexane and methylheterocyclohexanes. J Org Chem. 2003;68(17):6780–6787. doi: 10.1021/jo034421l. [DOI] [PubMed] [Google Scholar]
- 108.Abraham RJ, Ribeiro DS. Conformational analysis. Part 36. A variable temperature C-13 NMR study of conformational equilibria in methyl substituted cycloalkanes. J Chem Soc Perkin Trans. 2001;2(3):302–307. doi: 10.1039/b008712m. [DOI] [Google Scholar]
- 109.Freeman F, Kasner ML, Hehre WJ. An ab initio molecular orbital theory study of the conformational free energies of 2-methyl-, 3-methyl-, and 4-methyltetrahydro-2H-pyran. J Mol Struct Theochem. 2001;574:19–26. doi: 10.1016/S0166-1280(00)00757-0. [DOI] [Google Scholar]
- 110.Cascade Supercomputer. https://www.emsl.pnnl.gov/emslweb/instruments/computing-cascade-atipa-1440-intel-xeon-phi-node-fdr-infiniband-linux-cluster. Accessed 16 Oct 2018
- 111.NWChem: Open source high-performance computational chemistry. www.nwchem-sw.org. Accessed 16 Oct 2018
- 112.NWChem: Open source high-performance computational chemistry—Github. https://github.com/nwchemgit. Accessed 16 Oct 2018
- 113.Tuan NQ, et al. A grayanotox-9(11)-ene derivative from Rhododendron brachycarpum and its structural assignment via a protocol combining NMR and DP4 plus application. Phytochemistry. 2017;133:45–50. doi: 10.1016/j.phytochem.2016.10.010. [DOI] [PubMed] [Google Scholar]
- 114.Barone G, et al. Determination of the relative stereochemistry of flexible organic compounds by ab initio methods: conformational analysis and Boltzmann-averaged GIAO C-13 NMR chemical shifts. Chem Eur J. 2002;8(14):3240–3245. doi: 10.1002/1521-3765(20020715)8:14<3240::AID-CHEM3240>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 115.Barone G, et al. Structure validation of natural products by quantum-mechanical GIAO calculations of C-13 NMR chemical shifts. Chem Eur J. 2002;8(14):3233–3239. doi: 10.1002/1521-3765(20020715)8:14<3233::AID-CHEM3233>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 116.Remya K, Suresh CH. Which density functional is close to CCSD accuracy to describe geometry and interaction energy of small non-covalent dimers? A benchmark study using gaussian09. J Comput Chem. 2013;34(15):1341–1353. doi: 10.1002/jcc.23263. [DOI] [PubMed] [Google Scholar]
- 117.Zhao Y, Truhlar DG. Improved description of nuclear magnetic resonance chemical shielding constants using the M06-L meta-generalized-gradient-approximation density functional. J Phys Chem A. 2008;112(30):6794–6799. doi: 10.1021/jp804583d. [DOI] [PubMed] [Google Scholar]
- 118.Magyarfalvi G, Pulay P. Assessment of density functional methods for nuclear magnetic resonance shielding calculations. J Chem Phys. 2003;119(3):1350–1357. doi: 10.1063/1.1581252. [DOI] [Google Scholar]
- 119.Cimino P, et al. Comparison of different theory models and basis sets in the calculation of C-13 NMR chemical shifts of natural products. Magn Reson Chem. 2004;42:S26–S33. doi: 10.1002/mrc.1410. [DOI] [PubMed] [Google Scholar]
- 120.Cramer CJ, Truhlar DG. Implicit solvation models: equilibria, structure, spectra, and dynamics. Chem Rev. 1999;99(8):2161–2200. doi: 10.1021/cr960149m. [DOI] [PubMed] [Google Scholar]
- 121.Reddy G, Yethiraj A. Implicit and explicit solvent models for the simulation of dilute polymer solutions. Macromolecules. 2006;39(24):8536–8542. doi: 10.1021/ma061176+. [DOI] [Google Scholar]
- 122.Bagno A, Rastrelli F, Saielli G. Predicting C-13 NMR spectra by DFT calculations. J Phys Chem A. 2003;107(46):9964–9973. doi: 10.1021/jp0353284. [DOI] [PubMed] [Google Scholar]
- 123.Wang B, et al. Accurate prediction of proton chemical shifts. I. Substituted aromatic hydrocarbons. J Comput Chem. 2001;22(16):1887–1895. doi: 10.1002/jcc.1139. [DOI] [Google Scholar]
- 124.Wang B, Hinton JF, Pulay P. Accurate prediction of proton chemical shifts. II. Peptide analogues. J Comput Chem. 2002;23(4):492–497. doi: 10.1002/jcc.10044. [DOI] [PubMed] [Google Scholar]
- 125.Smirnov SN, et al. Hydrogen deuterium isotope effects on the NMR chemical shifts and geometries of intermolecular low-barrier hydrogen-bonded complexes. J Am Chem Soc. 1996;118(17):4094–4101. doi: 10.1021/ja953445+. [DOI] [Google Scholar]
- 126.Benedict H, et al. Hydrogen/deuterium isotope effects on the N-15 NMR chemical shifts and geometries of low-barrier hydrogen bonds in the solid state. J Mol Struct. 1996;378(1):11–16. doi: 10.1016/0022-2860(95)09143-2. [DOI] [Google Scholar]
- 127.Gidley MJ, Bociek SM. C-13 Cp/MAS NMR-studies of amylose inclusion complexes, cyclodextrins, and the amorphous phase of starch granules—relationships between glycosidic linkage conformation and solid-state C-13 chemical-shifts. J Am Chem Soc. 1988;110(12):3820–3829. doi: 10.1021/ja00220a016. [DOI] [Google Scholar]
- 128.Buckingham AD. Chemical shifts in the nuclear magnetic resonance spectra of molecules containing polar groups. Can J Chem Rev Can Chim. 1960;38(2):300–307. doi: 10.1139/v60-040. [DOI] [Google Scholar]
- 129.Gauss J. Effects of electron correlation in the calculation of nuclear-magnetic-resonance chemical-shifts. J Chem Phys. 1993;99(5):3629–3643. doi: 10.1063/1.466161. [DOI] [Google Scholar]
- 130.Lodewyk MW, Siebert MR, Tantillo DJ. Computational prediction of 1H and 13C chemical shifts: a useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem Rev. 2012;112(3):1839–1862. doi: 10.1021/cr200106v. [DOI] [PubMed] [Google Scholar]
- 131.Flaig D, et al. Benchmarking hydrogen and carbon NMR chemical shifts at HF, DFT, and MP2 levels. J Chem Theory Comput. 2014;10(2):572–578. doi: 10.1021/ct400780f. [DOI] [PubMed] [Google Scholar]
- 132.Gregor T, Mauri F, Car R. A comparison of methods for the calculation of NMR chemical shifts. J Chem Phys. 1999;111(5):1815–1822. doi: 10.1063/1.479451. [DOI] [Google Scholar]
- 133.Teale AM, et al. Benchmarking density-functional theory calculations of NMR shielding constants and spin–rotation constants using accurate coupled-cluster calculations. J Chem Phys. 2013;138(2):024111. doi: 10.1063/1.4773016. [DOI] [PubMed] [Google Scholar]
- 134.Wu A, et al. Systematic studies on the computation of nuclear magnetic resonance shielding constants and chemical shifts: the density functional models. J Comput Chem. 2007;28(15):2431–2442. doi: 10.1002/jcc.20641. [DOI] [PubMed] [Google Scholar]
- 135.Giesen DJ, Zumbulyadis N. A hybrid quantum mechanical and empirical model for the prediction of isotropic C-13 shielding constants of organic molecules. Phys Chem Chem Phys. 2002;4(22):5498–5507. doi: 10.1039/B206245C. [DOI] [Google Scholar]
- 136.Jain R, Bally T, Rablen PR. Calculating accurate proton chemical shifts of organic molecules with density functional methods and modest basis sets. J Org Chem. 2009;74(11):4017–4023. doi: 10.1021/jo900482q. [DOI] [PubMed] [Google Scholar]
- 137.Gao HW, et al. Comparison of different theory models and basis sets in the calculations of structures and C-13 NMR spectra of [Pt(en)(CBDCA-O, O′)], an analogue of the antitumor drug carboplatin. J Phys Chem B. 2010;114(11):4056–4062. doi: 10.1021/jp912005a. [DOI] [PubMed] [Google Scholar]
- 138.Xin D, et al. Development of a (13)C NMR chemical shift prediction procedure using B3LYP/cc-pVDZ and empirically derived systematic error correction terms: a computational small molecule structure elucidation method. J Org Chem. 2017;82(10):5135–5145. doi: 10.1021/acs.joc.7b00321. [DOI] [PubMed] [Google Scholar]
- 139.Zhang Y, et al. OPBE: a promising density functional for the calculation of nuclear shielding constants. Chem Phys Lett. 2006;421(4–6):383–388. doi: 10.1016/j.cplett.2006.01.095. [DOI] [Google Scholar]
- 140.d’Antuono P, et al. A joined theoretical–experimental investigation on the 1H and 13C NMR signatures of defects in poly(vinyl chloride) J Phys Chem B. 2008;112(47):14804–14818. doi: 10.1021/jp805676q. [DOI] [PubMed] [Google Scholar]
- 141.Grimblat N, Zanardi MM, Sarotti AM. Beyond DP4: an improved probability for the stereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts. J Org Chem. 2015;80(24):12526–12534. doi: 10.1021/acs.joc.5b02396. [DOI] [PubMed] [Google Scholar]
- 142.Navarro-Vazquez A. State of the art and perspectives in the application of quantum chemical prediction of H-1 and C-13 chemical shifts and scalar couplings for structural elucidation of organic compounds. Magn Reson Chem. 2017;55(1):29–32. doi: 10.1002/mrc.4502. [DOI] [PubMed] [Google Scholar]
- 143.Ermanis K, et al. Doubling the power of DP4 for computational structure elucidation. Org Biomol Chem. 2017;15(42):8998–9007. doi: 10.1039/C7OB01379E. [DOI] [PubMed] [Google Scholar]
- 144.Pierens GK. H-1 and C-13 NMR scaling factors for the calculation of chemical shifts in commonly used solvents using density functional theory. J Comput Chem. 2014;35(18):1388–1394. doi: 10.1002/jcc.23638. [DOI] [PubMed] [Google Scholar]
- 145.Kohavi R (1995) A study of cross-validation and bootstrap for accuracy estimation and model selection. In: International joint conference on articial intelligence
- 146.Dubitzky W, Granzow M, Berrar DP. Fundamentals of data mining in genomics and proteomics. Berlin: Springer; 2007. [Google Scholar]
- 147.Xu Q-S, Liang Y-Z. Monte Carlo cross validation. Chemom Intell Lab Syst. 2001;56(1):1–11. doi: 10.1016/S0169-7439(00)00122-2. [DOI] [Google Scholar]
- 148.Li DW, Bruschweiler R. Certification of molecular dynamics trajectories with NMR chemical shifts. J Phys Chem Lett. 2010;1(1):246–248. doi: 10.1021/jz9001345. [DOI] [Google Scholar]
- 149.Di Micco S, et al. Plakilactones G and H from a marine sponge. Stereochemical determination of highly flexible systems by quantitative NMR-derived interproton distances combined with quantum mechanical calculations of C-13 chemical shifts. Beilstein J Org Chem. 2013;9:2940–2949. doi: 10.3762/bjoc.9.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hirsch JA. Table of conformational energies. Top Stereochem. 1967;1:199. [Google Scholar]
- 151.Beckett CW, Pitzer KS, Spitzer R. The thermodynamic properties and molecular structure of cyclohexane, methylcyclohexane, ethylcyclonexane and the seven dimethylcyclohexanes. J Am Chem Soc. 1947;69(10):2488–2495. doi: 10.1021/ja01202a070. [DOI] [PubMed] [Google Scholar]
- 152.Streitwieser A, Heathcock CH, Kosower E. Introduction to organic chemistry. New York: Macmillan; 1992. [Google Scholar]
- 153.Smith MB, March J. March’s advanced organic chemistry. New York: Wiley; 1992. [Google Scholar]
- 154.Solomons TWG. Organic chemistry. New York: Wiley; 2000. [Google Scholar]
- 155.Frisch MJ, et al. Gaussian 16 Rev. B.01. Wallingford: Gaussian, Inc; 2016. [Google Scholar]
- 156.Booth H, Everett JR. Experimental-determination of the conformational free-energy—enthalpy, and entropy differences for alkyl-groups in alkylcyclohexanes by low-temperature C-13 magnetic-resonance spectroscopy. J Chem Soc Perkin Trans. 1980;2(2):255–259. doi: 10.1039/p29800000255. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Supporting information document.
Additional file 2. Supporting information files. Including tutorial, code, and all other files.
Data Availability Statement
Project name: ISiCLE. Project home page: github.com/pnnl/isicle. Programming language: Python. Operating system(s): Platform independent. License: GNU GPL license. Free academic and non-profit research use only. All data files, source code, and tutorial are provided in the additional files.