

Abstract
Exciting discoveries of naturally occurring ligand-sensing and disease-linked noncoding RNAs have promoted significant interests in understanding RNA-small molecule interactions. NMR spectroscopy is a powerful tool for characterizing intermolecular interactions. In this review, we describe protocols and approaches for applying NMR spectroscopy to investigate interactions between RNA and small molecules. We review protocols of RNA sample preparations, methods for identifying RNA-binding small molecules, approaches for mapping RNA-small molecule interactions, determining complex structures, and characterizing binding kinetics. We hope this review will provide a guideline to streamline NMR applications in studying RNA-small molecule interactions, facilitating both basic mechanistic understandings of RNA functions and translational efforts in developing RNA-targeted therapeutics.
1. Introduction
The discoveries of diverse non-coding RNAs (ncRNAs) functions in the past few decades have revolutionized our understanding of the roles of RNA in biology [1–4]. These chemically simple biomolecules not only directly participate in protein synthesis [5–10], but also regulate various steps of gene expression, ranging from transcription [11–14] to translation [13–16], from chromatin remodeling [17–20] to RNA and ribonucleoprotein (RNP) trafficking [15, 21]. During these processes, it has become increasingly clear that RNA molecules, both regulatory ncRNAs [22, 23] and coding mRNAs [24], can adopt complex secondary and tertiary structures. More remarkably, these RNAs often undergo major adaptive structural changes upon recognition of specific ligands, which include proteins, DNAs, RNAs, metabolites, and even small cations and anions [25–27]. Due to their critical roles in gene regulation, dysfunctions of many RNA species have also been linked to various human diseases, including cancer, heart, and neurological diseases [28–31]. Hence, it is of significant interest and importance in delineating how RNA interacts with such a diverse set of ligands, which can not only provide mechanistic insights into their functions, but also further opens new avenues for developing therapeutics that target disease-specific RNAs [32, 33].
A broad range of biochemical and biophysical methods have been developed and applied for characterizing molecular interactions between RNA and cognate ligands. For example, binding and its associated thermodynamic properties can be characterized using methods such as electrophoretic mobility shift assay (EMSA), fluorescence-detected assays [34], isothermal titration calorimetry (ITC) [35, 36], surface plasmon resonance (SPR) [37], microscale thermophoresis [38], mass spectrometry [39–41], nuclear magnetic resonance (NMR) spectroscopy [42, 43], and others. Binding-induced structural rearrangements can be evaluated at nucleotide and molecular resolutions using methods such as, in-line probing, selective 2’-hydroxyl acylation analyzed by primer extension (SHAPE) [44, 45], pattern recognition of RNA by small molecules (PRRSM) [46], small-angle X-ray scattering (SAXS) [47], as well as single-molecule fluorescence and force microscopies [48–50]. Many of these methods can be further extended to characterize RNA-ligand interactions under cellular conditions [51–60]. Finally, molecular interactions at the atomic resolution can be obtained from high-resolution structures of RNA and their complexes determined using X-ray crystallography, NMR spectroscopy, and more recently, cryo-electron microsopy (cryo-EM) equipped with direct electron detection cameras. These methods, often complementary to each other, provide a cohort of experimental approaches that have enabled elucidations of chemical and physical basis of molecular interactions between RNA and its different types of ligands, significantly advancing our understanding of diverse RNA functions.
Among many biophysical techniques, NMR spectroscopy is a unique and powerful technique that is suitable for characterizing various aspects of biomolecular interactions [42, 43]. By applying different experimental schemes, NMR can be used to monitor weak and tight interactions, map binding sites, measure binding thermodynamics and kinetics, determine high-resolution complex structures, and characterize conformational dynamics with a wide range of timescales from picoseconds to seconds. Recently, NMR characterization of RNA-protein interactions [61] as well as RNA structural dynamics [25, 43, 62] have been extensively reviewed. Here, we review protocols and approaches for applying NMR spectroscopy to study interactions between RNA and small molecules, with a focus on developments and applications in more recent years. We discuss protocols of RNA sample preparations, methods for identifying RNA-binding small molecules, approaches for mapping RNA-small molecule interactions, determining complex structures, and characterizing binding kinetics and conformational dynamics. With the ever-growing discoveries of naturally occurring ligand-sensing RNAs [13] and disease-linked regulatory RNAs [28–31], we hope this review will provide a general guideline to streamline the application of NMR in studying RNA-small molecule interactions, facilitating both basic mechanistic understandings of RNA functions and translational efforts in developing RNA-targeted therapeutics.
2. Materials and Methods
2.1. RNA sample preparation
2.1.1. RNA sample production
A standard biomolecular NMR experiment typically requires a relatively large amount (>50 nmoles) of purified RNA. To achieve this requirement, three different approaches can be applied: solid-phase chemical synthesis, in vitro transcription, and in vivo transcription. Solid-phase chemical synthesis uses phosphoramidites as building materials for making RNA samples. For RNA oligos < 20 nucleotides, this approach is often the method of choice due to the limited abilities of the other two enzymatic approaches in directly generating short RNA oligos. RNAs from commercial resources are often generated using this approach. One major advantage of this approach is that chemical modifications can be easily incorporated in specific sequence locations as well as specific chemical positions. For example, a potential junctional RNA motif was unveiled by NMR with the introduction of a N6-methyladenosine (m6A) modification to the adenine residue next to a 5’ bulge [63]. Site-specific modifications have also enabled nitroxide incorporation in RNA for paramagnetic relaxation enhancement (PRE) studies [64, 65]. The developments of stable isotope labeled RNA phosphoramidites in recent years have further expanded the application of solid-phase synthesis in generating RNA samples needed for heteronuclear NMR experiments [66]. However, the coupling efficiency of each chemical step remains a major challenge for effective synthesis of long RNA oligos, as the yield is inverse-exponentially proportional to the length of RNA, making this method far less cost effective than enzymatic approaches for producing long RNAs. Recently, a chemo-enzymatic synthesis approach was developed that utilizes 13C/15N-labeled nucleoside 3’,5’ bisphosphates, T4 RNA ligase 1, shrimp alkaline phosphatase, and T4 RNA ligase 2 [67]. This method enables efficient site-specifically labeling in long RNAs that are otherwise difficult.
In vitro transcription with RNA polymerases is currently the most widely used method for generating large quantity of RNA samples for NMR studies [42, 61], In vitro transcription requires a short list of reagents, including RNA polymerase, DNA template, rNTPs, reaction buffers composed of magnesium, Tris, DTT, as well as inorganic pyrophosphatase (IPP) for maintaining effective magnesium concentrations. T7 RNA polymerase is the most commonly used enzyme in In vitro transcription. It can be obtained commercially or expressed and purified in-house from E. coll cells. DNA templates less than 100 nucleotides can be obtained commercially, whereas longer templates need to be generated by PCR, ligating shorter DNA pieces, or linearizing templated plasmids. For In vitro transcription using T7 polymerase, a specific promoter sequence (CTAATACGACTCACTATAG) needs to be appended to the 5’-end of the sensing strand, and the underlined G residue at the 3’-end of the promoter marks the start of transcription. In case of single-stranded DNA oligos being commercially obtained, DNA templates need to be further prepared by annealing sense and antisense strands to generate double-stranded DNAs. Alternatively, DNA templates can be prepared by annealing the short T7 promoter directly onto the full-length anti-sense strand, which reduces the cost of generating double-stranded DNA by bypassing the full-length DNA sense strand. It is worth noting that, in our experience, fully complemented double stranded DNA templates often provide better yields in transcription. Unlike solid-phase chemical synthesis, In vitro transcription uses rNTPs as building materials for RNA. Here, not only are rNTPs relatively inexpensive, there are also a broad range of isotope labeled rNTPs that are essential for advanced NMR characterization. For example, there are commercially available uniformly 15N and 13C/15N labeled rNTPs for multidimensional heteronuclear NMR measurements, position-specific 13C labeled rNTPs [68] for characterizing conformational dynamics, commercially available partially deuterated (2H) rNTPs as well as 19F labeled rATP [69] for studying large RNA molecules. In addition, by introducing a subset of isotope-labeled rNTPs into otherwise isotope-unlabeled rNTPs during in vitro transcription, nucleotide-specific labeled RNA samples can be prepared. Prior to sample preparation, it is often useful to carry out small-scale (50 μl) test reactions, where Tris and magnesium concentrations are optimized to achieve maximal yield. The optimal condition can be directly scaled up for a large-scale (10 ml) transcription, which typically generates a sufficient amount (~500 nmoles) of RNA for NMR.
Lastly, a large amount of RNA can also be generated using in vivo transcription by E. coli cells with recombinant plasmids [70, 71]. Here, the recombinant plasmid encodes a highly efficient transcription unit, which contains a strong lipoprotein gene promotor, a tRNA scaffold, and a ribosomal RNA operon transcription terminator. The RNA of interest is inserted into the anticodon stem of the tRNA scaffold, which serves to not only promote overexpression of the target RNA but also protect it from degradation by cellular RNases. Similar to protocols for preparing isotope-labeled proteins, 13C, 15N, and/or 2H enriched minimal medium can be used for E. coli growth, producing uniformly isotope-labeled RNA samples. If the tRNA scaffold does not interfere with the structure and function of the target RNA, the chimeric RNA sample can be used directly for NMR studies. Alternative, the RNA of interest can be dissected out from the tRNA scaffold by hybridizing DNA oligos with tRNA sequences, followed by RNase H cleavage. It has been shown that in vivo transcription can generate ~500 nmoles of RNA per 1 L E. coli cells, which is similar to the yield of a 10 ml in vitro transcription.
2.1.2. RNA sample purification
RNAs generated from above methods are not immediately suitable for applications in NMR studies due to contaminations, such as chemicals, DNA templates, enzymes, unincorporated rNTPs, short abortive RNA transcripts, as well as non-templated nucleotide additions to target RNA transcripts. These reactions need to be purified to ensure sample homogeneity. The most widely used purification method is denaturing polyacrylamide gel electrophoresis (PAGE), which can provide single-nucleotide resolution for RNAs that have suitable sizes for NMR studies. Here, depending on the length of the RNA, polyacrylamide gels are prepared in 8M urea with acrylamide concentration ranging between 10 to 20%. To achieve good separation, the target RNA often needs to migrate towards the bottom quarter of the gel, which can be estimated from the position of loading dye. Gel pieces containing the target RNA are cut out from the large gel, and subsequently, the target RNA is extracted from the gel using either passive “crush and soak” or active electroelution with the Elutrap system. In our hands, the later method provides superior recovery efficiency for maximal sample production. The extracted RNA is further purified with a strong anion exchange chromatography column, such as the HiTrap Q HP column, to remove residual acrylamide contaminations.
While denaturing PAGE provides excellent separation capability that enables purifying target RNA from n-1/n+1 transcript, this approach is rather labor intensive and time consuming. Hence, various chromatographic methods, coupled with elegant construct designs, have been developed to facilitate efficient and effective RNA sample purification. To alleviate impurities arising from inhomogeneous 3’-end transcription, cis-acting ribozymes, such as hammerhead ribozyme [72], can be inserted at the 3’-end of the target RNA transcript. During transcription, the full-length transcript, despite having a heterogeneous 3’-end, undergoes self-cleavage and generates the target RNA with homogeneous length. The RNA product can then be purified from the reaction mix using anion exchange high-performance liquid chromatography (HPLC) under high temperature (85–90°C) [73], weak anion exchange fast-performance liquid chromatography (FPLC) [74], or size-exclusion gel filtration FPLC [75] under native conditions. In addition to liquid chromatography, affinity chromatography can also be applied, where affinity tags that are specific to DNA [76], proteins [77, 78], and various resins [70, 79]. Upon purifying from affinity columns, affinity tags can be further cleaved using DNAzymes, ribozymes, and RNases to generate the desired RNA with homogeneous length.
2.1.3. RNA sample condition
The final step in sample preparation is to exchange purified RNA into proper buffers, volumes, and concentrations for NMR studies. A typical NMR buffer for RNA sample contains 10 – 100 mM monovalent salt (such as sodium and potassium) and 10 mM phosphate buffer at pH 6.5. The monovalent salt is added to counterbalance negatively charged RNA backbones. Often, millimolar magnesium is added to ensure proper folding of RNA, which can be evaluated using native gels. The relatively low pHs are needed to ensure effective NMR detection of imino and amino proton signals, as these solvent-exchangeable protons have fast rates of exchange with water. For a standard 5mm NMR tube, a volume of ~ 500 μl is needed for effective NMR shimming to ensure magnetic field homogeneity across the sample. A small sample volume of ~ 300 μl can also be used in Shigemi tubes, where the reduced sample depth is supplemented with glass that matches the magnetic susceptibility of D2O. While higher sample concentrations can significantly reduce NMR experimental time, it is typically recommended to keep sample concentrations below 1.5 mM to reduce potential RNA dimerization and even oligomerization. Finally, for NMR experiments that involve characterizing proton resonances close to water signals, such as sugar protons, RNA samples in D2O can be prepared by lyophilizing the corresponding H2O sample and re-dissolving the dry pellet in the same volume of 99.996% D2O.
2.2. Identifying RNA-binding small molecules
Prior to physicochemical characterizations of intermolecular interactions, it is quintessential to first identify small molecules that specifically bind to the RNA of interest. For metabolite-sensing RNA riboswitches, cognate ligands are often identified and validated during their biochemical characterizations, and specific types of those riboswitches are subsequently annotated [13]. For other RNAs of interest, in particular disease-linked regulatory RNAs, RNA-binding small molecules are often identified from a large pool of chemical libraries via high-throughput screening (HTS). Despite having lower throughput relative to HTS, NMR spectroscopy is also a powerful tool in identifying and validating small molecules that interact with biomolecules, and has played a significant role in protein-targeted drug discovery [80]. Excellent reviews have been published in recent years, which provide thorough discussions of various NMR experiments in identifying protein-binding small molecules as well as evaluating strengths and liabilities of individual methods [80–82]. Since many of these methods are based on observing ligand NMR signals, the nature of a target, whether it is a protein or a RNA, has minor influence on experimental setups of these methods, enabling their direct applications in identifying RNA-binding small molecules. Here, we provide a brief overview of these common methods and focus on some recent developments that are specific for identifying and optimizing RNA-binding small molecules.
2.2.1. NMR-based experimental screening
Saturation transfer difference (STD) NMR spectroscopy [83] is one of the most widely used NMR methods in drug discovery, such as fragment-based drug discovery (FBDD) screening for protein targets (Fig. 1A) [80]. STD experiment builds upon magnetization transfer between biomolecules, such as proteins and RNAs, and small ligands. First, the biomolecular NMR signals that resonate at distinct frequencies from those of the ligands are selectively saturated. Via spin diffusion, these selective saturations are transferred to the remaining signals of the biomolecule. If a ligand binds the biomolecule, its NMR signals can also be saturated due to intermolecular nuclear Overhauser effect (NOE). Continuous irradiation and dynamic exchange of the ligand in its free and bound states result in reduction of the bulk magnetization of this ligand. In contrast, for any ligands that do not interact with the biomolecule, their NMR signals are minimally affected by the irradiation of biomolecular NMR signals. The ‘difference’ comes from subtracting between two NMR spectra – with and without saturation – where the resulting spectrum only displays signals from ligands that interact with the biomolecule. Hence, STD experiment can efficiently screen a pool of small molecules and identify binding-competent ligands. Despite being a powerful tool, some limits exist for STD-based screening. First, in order to effectively saturate ligand signals, the rate for ligand to exchange between its bound and free states needs to be in the intermediate to fast regime. With such a requirement of binding kinetics, identified ligands often have binding affinities in the sub-μM-to-mM range, where tight binders often evade detection. Second, an effective saturation transfer also benefits from a high proton density in target biomolecules. Relative to protein, proton density in RNA is about 2-fold lower, making STD less viable in screening RNA-binding small molecules [84]. Despite these limitations, STD has been successfully employed in characterizing RNA-binding small molecules [85–87]. In addition, since the majority of protons in RNA are solvent non-exchangeable, carrying out STD measurement of RNA in D2O instead of H2O not only has minimal perturbations on proton density, but also benefits from reduced R1 relaxation rate that enhances STD effect as well as re-gaining NMR signals closer to water resonance that are otherwise less accessible [85].
Figure 1. Methods for screening small-molecule RNA interactions via NMR.
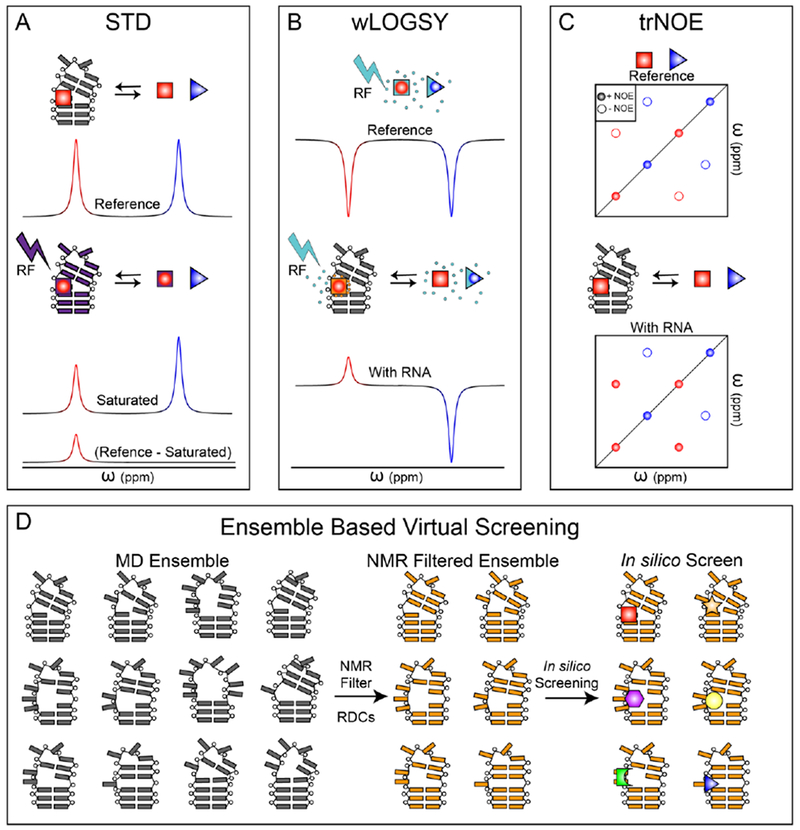
(A-C) For the three schematics, blue triangle indicates a non-binding small-molecule and red square indicates an RNA-binding small-molecule. (A) For STD, an initial reference spectrum of compound cocktail is obtained (top). The RNA is then saturated with an RF pulse and NOE transfer of saturation occurs to RNA-bound small molecules (middle). The saturated spectrum is subtracted from the reference spectrum and the result is intensity only for small-molecules that were saturated due to RNA binding (bottom). (B) A reference spectrum where RNA is absent is obtained by saturating water with an RF pulse. Rapidly tumbling small-molecules develop negative NOEs (top). The experiment is repeated in the presence of RNA. Water molecules in the binding pocket tumble slowly and the resulting NOEs are positive for RNA-binding small-molecules (bottom). (C) A reference spectrum where RNA is absent is obtained. Rapidly tumbling small-molecules develop negative intramolecular NOEs (top). The experiment is repeated in the presence of RNA. Small-molecules in the binding pocket tumble slowly and the resulting intramolecular NOEs are positive for RNA-binding small-molecules (bottom). (D) An RNA ensemble is generated via molecular dynamics simulations (left). Those conformations that best fit RDCs are then combined into an ‘NMR-filtered’ ensemble (middle). This data driven ensemble is then used for ultra-high throughput in silico screening (right).
Water-ligand observed via gradient spectroscopy (wLOGSY) is another popular NMR method used in small molecule screening (Fig. 1B) [88, 89]. Similar to STD, wLOGSY also utilizes intermolecular NOEs to identify ligands that interact with biomolecules. Here, instead of irradiating magnetization of a target RNA, bulk H2O magnetization is excited and partially transferred to ligands. In the absence of RNA, water magnetization is transferred to ligands via intermolecular water-ligand NOEs, and negative peaks are observed for these free ligands due to their rapid tumbling rates. In the presence of RNA, however, water magnetization is transferred to RNA-bound ligands via multiple mechanisms, particularly intermolecular NOEs between water and RNA-ligand complex as well as chemical exchanges between water and various labile protons in the complex. Due to a much slower tumbling rate of biomolecular complex, the sign of NOE transfer for RNA-bound ligands is opposite to their free counterparts, and these RNA-bound ligands display positive peaks. Hence, by comparing wLOGSY NMR spectra in the presence and absence of the target RNA, RNA-ligands can be easily identified as those having inverse wLOGSY signals [84]. Similar to STD, wLOGSY also has limited abilities in screening for tight binders. However, it has been shown that wLOGSY has better sensitivity than STD for screening RNA-targeted small molecules [84].
Transferred NOE spectroscopy (trNOESY) [90, 91] has also been used in screening RNA-targeted small molecules [84], where NOE peaks are observed and evaluated (Fig. 1C). Unlike wLOGSY, trNOESY measures intra-molecular NOE cross peaks of ligands, and the experiment is carried out in a two-dimensional (2D) manner. In the absence of RNA, the fast tumbling rates of free ligands give rise to negative intramolecular NOE cross peaks. In the presence of RNA, the RNA-bound ligands experience much slower tumbling rates, and their intra-molecular NOE cross peaks are positive. Similar to the analysis of wLOGSY data, RNA-binding ligands can be identified as those having inverse trNOE signals. In addition, 2D trNOESY also offers the opportunity to analyze structural features of RNA-binding ligands in their bound states, as intramolecular 1H-1H distances within the ligand can be obtained from intensities of NOE cross peaks.
In STD, wLOGSY, and trNOE experiments, the concentration of RNA is typical in the range of 10 – 50 μM, and the small molecules are present in large access (i.e. 1mM). This experimental setup not only reduces the amount of RNA needed for screening, but also ensures NMR spectra being dominated by small molecule signals. While 2D 1H-1H NOESY spectra are recorded in trNOE, simple 1D 1H NMR spectra are used for STD and wLOGSY. Among these three approaches, wLOGSY has also been shown to be the preferable method for screening RNA-binding small molecules with better sensitivity and spectroscopic simplicity [84].
Besides these NOE-based approaches, other ligand-detected NMR techniques have been developed for screening protein-targeted small molecules [80]. For example, the transverse relaxation property (T2) of a ligand can be used to identify its propensity for binding biomolecules [92]. In contrast to the long T2 in its free state, a biomolecule-bound ligand experiences a dramatically reduced T2 as being part of a larger complex with a slower tumbling rate. Hence, 1H T1ρ experiments can be applied to measure transverse relaxation rates of a ligand in the presence and absence of a target biomolecule, where ligands displaying significant T2 reductions are those that can bind. Fluorinated small molecules provide another avenue for NMR-based screening. 19F is NMR active with a large gyromagnetic ratio and near 100% natural abundance [80]. Similar to the 1H T1ρ approach, libraries of fluorinated compounds can be effectively screened by measuring T2 relaxation with 19F CPMG experiment in the presence and absence of the target biomolecule. Recently, 19F-based NMR fragment screening has been applied in discovering fluorinated ligands that bind specially to telomeric RNA G-quadruplexes (TERRA) [86]. It is worth noting that these relaxation-based experiments are generally not as sensitive as the NOE-based experiments mentioned above and the magnitude of the effect also depends on the size of the target of interest.
While these conventional NMR techniques are generic and applicable to proteins, DNAs and RNAs, Asensio and co-workers have recently developed an elegant fragment-based combinatorial method for screening and optimizing polyamine scaffolds as selective DNA and RNA binders [93]. Here, regioisomer libraries are first generated by reductive amination of selected polyamines. Via microdialysis assays, the libraries are then evaluated for selectivity on the target RNA against a nontarget RNA for nonspecific interactions. The bound ligands are released by digesting the RNAs, and 13C-labeled methyl groups are introduced to the polyamine scaffolds. After derivatization, each polyamine regioisomer incorporates four -N13Me2 and a single - N13MeR groups. Remarkably, the 13C chemical shifts of methyl groups in -N13MeR upfield shift 4–5 ppm with respect to those in -N13Me2, which provide the key NMR signatures for analyzing highly similar polyamine derivatives. Indeed, the authors demonstrated that mixtures up to 21 pseudo-trisaccharide derivatives can produce HSQC spectra with tractable 13C methyl signals. With this novel labeling strategy, 13C methyl intensities from -N13MeR groups are quantified and compared between target and nontarget RNA samples for evaluating selective binders. With advanced NMR spectrometers equipped with cryogenic probes, this approach can be applied for screening with ligand concentrations as low as 2 μM. The authors have demonstrated their combinatorial method on aminoglycoside kanamycin-B, and identified several kanamycin derivatives with improved selectivity and/or affinity for ribosomal A-site RNA.
2.2.2. NMR-assisted virtual screening
Relative to experimental screening techniques, structure-based virtual screening (VS) [94] provides a powerful alternative approach that can rapidly and inexpensively expand compound libraries and generate compounds that selectively dock into pockets observed in structured RNAs. Successful VS implementation requires not only a well-developed force field that can robustly depict RNA-ligand interactions, but also an accurate high-resolution structural knowledge of the target RNA for pocket identification. However, these requirements can be challenging for virtual screening of RNA drug targets [95]. In particular, a hallmark of RNA is its conformational flexibility, and it often undergoes large conformational changes upon adaptive ligand recognitions [25–27]. Hence, static high-resolution structures from X-ray crystallography or NMR cannot faithfully represent possible conformations that are dynamically sampled by the target RNA. An alternative approach is to treat the target RNA as an ensemble of structures, and each individual structure is subject to VS [96]. However, generating robust structural ensembles from a static RNA structure using molecular dynamics (MD) simulations can also be challenging due to underdeveloped force fields for RNA and the rugged energy landscapes of RNA.
NMR spectroscopy not only is a powerful method for high-resolution structure determination, but also provides a comprehensive set of tools for characterizing conformational dynamics at the atomic resolution [25, 43, 62]. By combining NMR measurements with MD simulations, Al-Hashimi and co-workers have developed and demonstrated the utility of ensemble-based virtual screening (EBVS) for discovering RNA targeted small molecules (Fig. 1D) [97, 98]. Here, MD simulations are first carried out to generate a large pool of RNA structures. Subsequently, high-quality NMR residual dipolar couplings (RDCs), which provide long-range angular constraints and are sensitive to internal motions with timescales ranging from pico- to milli-seconds, are used to select conformations from this pool to generate an ensemble of structures that recapitulate the experimentally measured RDCs. Finally, this structure ensemble is subject to computational docking against virtual small-molecule libraries. Like any VS, the identified small-molecule binders need to be further experimentally validated in their binding properties using biochemical and/or biophysical methods. By applying this EBVS approach on human HIV-1 transactivation response element (TAR) RNA, the authors have successfully discovered selective bioactive small molecules that inhibit TAR-Tat interactions in vitro, one of which inhibits Tat-mediated activation of the HIV-1 long terminal repeat by 81% in T-cell lines [97]. More recently, Al-Hashimi and coworkers further demonstrated the importance of NMR data in generating accurate structural ensembles, which in turn significantly enrich libraries with true hits during VS [98].
2.3. Mapping RNA-small molecule interactions
While RNA-binding molecules can be identified from ligand-observed NMR techniques, these screening results provide limited information on how a ligand interacts with its target RNA. Such knowledge, which is essential for understanding the mechanism of recognition and rational design, can be readily obtained by monitoring NMR chemical shift perturbation (CSP) of the target RNA upon ligand binding. The chemical shift of an NMR signal is probably one of the most sensitive measurements for probing interactions [99]. Any perturbations of the local environment of an NMR-active nucleus, due to either direct ligand interaction or ligand-induced structural changes, will lead to chemical shift changes of its NMR signal. Unlike the ligand-observed NMR experiments, the target-observed NMR measurements require relatively large amount of RNA (>50 nmoles), where naturally abundant (1H and 31P) or isotope-enriched (13C and 15N) nuclei are monitored and compared for the target RNA in its ligand-free (apo) and ligand-bound (holo) states. Figure 2 summarizes chemical shift ranges of NMR observable nuclei in RNA that have been deposited in BMRB. The wide distribution of observable NMR resonances enables comprehensive characterization of intermolecular RNA-ligand interactions. While proton chemical shifts are mainly clustered by chemical moieties of bases and sugars, 13C/15N isotope-labeling and heteronuclear NMR experiments can greatly reduce spectral overlap for mapping binding at atomic resolution. In theory, any NMR experiments that contain chemical shift information can be used for CSP analysis. These nucleic acids NMR experiments as well as resonance assignment protocols for RNA have been comprehensively reviewed [42]. In the following, we highlight some of the most commonly used NMR experiments for mapping RNA-ligand interactions. Their applications in determining ligand binding constants are also discussed.
Figure 2. NMR chemical shifts of observable nuclei in RNA.
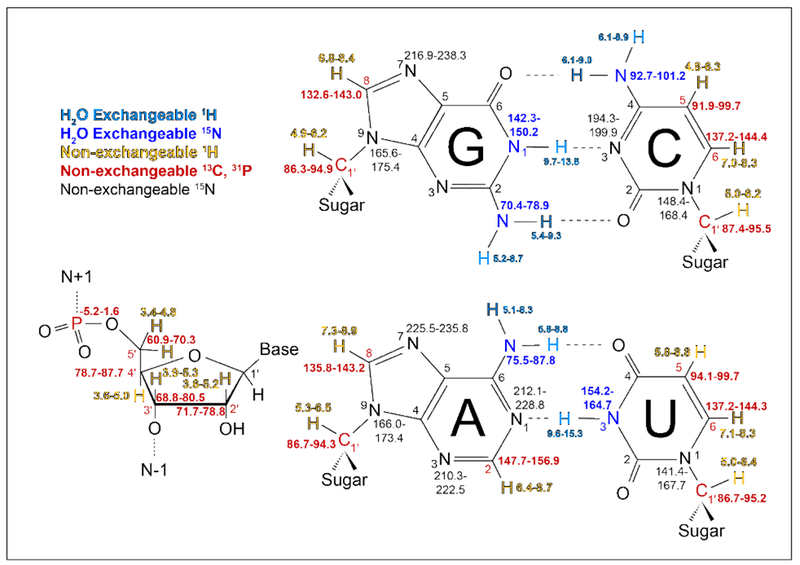
Water exchangeable observable atoms, in blue, require H2O sample conditions to observe base pairing. Non-exchangeable atoms, in red and gold, can be studied in H2O or D2O with the exception of H2’-H5”, which have spectral overlap with H2O and require D2O conditions. Chemical shifts from BMRB (http://www.bmrb.wisc.edu/) nucleic acids density histograms (density >0.05 for H, >0.02 for C/N/P).
2.3.1. NMR measurements of chemical shift perturbation
Imino 1H NMR spectroscopy.
Solvent-exchangeable imino protons, namely H1 of guanidines and H3 of uridines, are one of the most widely used NMR probes for monitoring RNA folding and ligand binding [86, 100–102]. Despite constituting less than 5% of all protons in RNA, imino protons serve as key hydrogen bond donors that participate in diverse base pairing interactions, many of which are perturbed during ligand-binding processes. Chemical shifts of imino protons range between 9.6 to 15.3 ppm (Fig. 2), which are downfield shifted from all other protons in RNA. In addition, any imino protons that are not structurally protected from water undergo solvent exchange, a process that broadens NMR resonances and renders these signals invisible in standard 1H NMR experiments. With distinct chemical shift ranges and limited spectroscopic overlap, imino protons not only can be assigned more efficiently and unambiguously than other proton resonances, they can also be readily monitored in a simple 1D manner without applying multi-dimensional NMR experiments or incorporating 15N isotope labeling. 2D imino 1H-1H NOESY can also be carried out, which not only provides enhanced resolution but also generates distance information from NOE cross peaks for structural characterizations. However, due to unstructured imino protons being NMR “invisible”, the imino 1H experiments cannot robustly characterize RNA-ligand interactions when ligand binding sites are located in structurally flexible regions, such as apical loops and bulges.
Total Correlated Spectroscopy (TOCSY).
2D 1H-1H TOCSY is another common NMR experiment for monitoring RNA-ligand interactions without the need for isotope enrichment [103–105]. With strong spin-spin coupling, TOCSY produces through-bond correlations between H5 and H6 protons of uridines and cytosines with high sensitivity. Good TOCSY spectra can be obtained in a few hours for RNA samples with low mM concentrations. As can be seen (Fig. 2), H5-H6 cross peaks reside in a distinct chemical-shift range, and are typically well resolved even for large RNAs. In addition, since both H5 and H6 are carbon-bonded solvent-nonexchangeable protons, pyrimidines in unstructured loops and bulges can also be robustly monitored for binding in TOCSY spectra, which are complementary to those imino-based NMR characterizations. For better spectral quality and resolutions, TOCSY measurements on H5-H6 cross peaks are generally carried out using D2O samples, and a mixing time of 40 – 50 ms is typically employed.
Heteronuclear Single Quantum Correlation Spectroscopy (HSQC).
HSQC is the backbone of many modern biomolecular NMR experiments. By encoding an additional heteronuclear dimension, overlapping 1H peaks can be further resolved. For RNA, 13C-1H, 15N-1H, and 31P-1H HSQC experiments can be used to thoroughly characterize RNA-ligand interactions. Given the range of chemical shifts (Fig. 2), C8H8 of purines, C6H6 of pyrimidines, and C2H2 of adenines are often monitored in a single 13C-1 H HSQC spectrum; C5H5 of pyrimidines are monitored in a single 13C-1H HSQC spectrum; sugar C1’H’ of all residues are monitored in a single 13C-1H HSQC spectrum; and the remaining sugar CH resonances, i.e. C2’H2’, C3’H3’, C4’H4’, C5’H5/H5”, can be monitored in a single constant-time 13C-1H HSQC spectrum. Imino (N1H1 and N3H3) and amino (NH2) can also be monitored but in separate 15N-1H HSQC spectra. Ligand-interactions with RNA backbone can also be characterized using 31P-1H HSQC, which correlates ribose protons H3’, H5’ and H5” to adjacent phosphates. Due to the limited chemical shift dispersions of sugar protons and phosphates in RNA, resonances in 31P-1H HSQC spectrum are typically too overlapped to be informative. However, any presence of distinct31P-H peaks can immediately indicate unique structures and/or interactions at the corresponding backbone sites. These HSQC experiments also have different requirements for isotope enrichment. Since 31P is naturally NMR active, no special labeling is needed for 31P-1H HSQC experiment. Due to low natural abundance (0.4%) and low gyromagnetic ratio (1/10 that of proton), 15N-labeling is required for 15N-1H HSQCs on RNA. With a combination of relative higher natural abundance (1.1%) and larger gyromagnetic ratio (1/4 that of proton), 13C-1H HSQC experiments can be recorded without isotope enrichments with an acquisition time of several hours on samples with milli-molar concentrations. It is therefore preferable to prepare 13C-labeled RNA samples , which significantly reduces acquisition time and provides much better sensitivity. It should be noted that the above conventional NMR HSQC experiments are discussed to highlight basic principles for CSP using heteronuclear NMR approaches. Recently, 15N-1H BEST-TROSY (band-selective excitation short – transient – transverse relaxation-optimized spectroscopy) [106], 15N-1H SOFAST-HMQC (heteronuclear multiple quantum correlation) [107], and 13C-1H SOFAST-HMQC [108] experiments have been developed for nucleic acids. These sensitive fast-pulsing experiments can provide similar spectroscopic characterizations on chemical shift perturbation but with much higher time efficiency.
2.3.2. NMR characterization of ligand binding affinity
Beside mapping intermolecular RNA-ligand interactions, NMR chemical shift perturbations are also often used to obtain ligand-binding affinities. Here, instead of only comparing chemical shift differences between apo and holo states, RNA chemical shifts are monitored as a function of the ligand addition to the RNA sample. Since ligand binding is a dynamic and reversible process, three different CSP behaviors can occur, which correspond to fast, intermediate, and slow exchange regimes (Fig. 3). These regimes are defined by the relative values of the exchange rate of ligand binding (or kex = kon [L] + koff) and the chemical shift differences between apo and holo state (or Δω = ωapo – ωholo). When kex >> Δω, the binding process is in the fast regime of chemical exchange, and population averaged chemical shifts are observed as a function of added ligand concentration (Fig. 3). When kex << Δω, the binding process is in the slow regime of chemical exchange, and we observe disappearance of apo resonances and appearance of holo resonances as ligand being titrated (Fig. 3). When kex ~ Δω, the binding process resides the so-called intermediate regime of chemical exchange. Once the process falls into the intermediate exchange, NMR signals shift but also get broadened upon ligand titration. When the titration approaches the mid-point, RNA signals can even be broadened beyond detection. These NMR signals eventually reappear and migrate toward the holo-state chemical shifts. When applying the chemical shift titration approach to obtain binding affinity, the intermediate exchange regime should be avoided as resonances cannot be observed during the titration process. Since this spectroscopic behavior occurs at kex ~ Δω, experimental conditions can be optimized to shift the exchange to either fast or slow regimes. For example, raising or lowering temperatures can tune kex, whereas Δω can be modulated by running titration experiments on NMR spectrometers with different magnetic field strengths.
Figure 3. NMR chemical shift titration and exchange regimes.
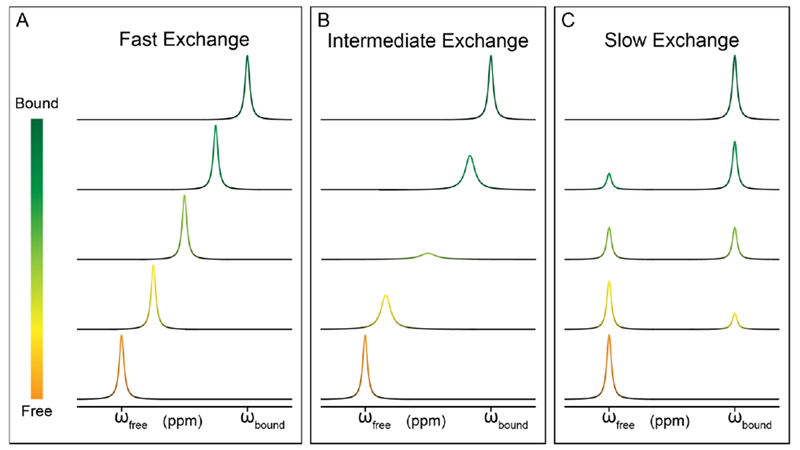
(A) Transition from free (orange) to bound (dark green) when in the fast exchange regime. (B) Transition from free to bound when in the intermediate exchange regime. (C) Transition from free to bound when in the slow exchange regime.
The observed NMR data can then be fit as a function of ligand concentration to extract an apparent ligand-binding affinity (Kd). If the ligand binding occurs in the fast exchange regime, the titration curve can be analyzed using the following equation,
| [1] |
Here, [R] is the total RNA concentration in the NMR tube, [L] is the total concentration of added ligand, Δδ[L] (=δ[L] – δapo) is the difference between the observed chemical shift at [L] and the apo-state chemical shift, and Δδmax (= δholo – δapo) is the maximal observable chemical shift change, which is the difference between the apo and holo chemical shifts. If the ligand binding occurs in the slow exchange regime, the titration curves of apo and holo resonances can be analyzed using the following two equations, respectively,
| [2] |
| [3] |
Here, [R] is the total RNA concentration in the NMR tube, [L] is the total concentration of added ligand, Iapo is the apo peak intensity in the absence of ligand, Iapo,[L] is the apo peak intensity at [L], Iholo,[L] is the holo peak intensity at [L], and Iholo is the holo peak intensity in its fully-bound state with an excess amount of ligand. The apo and holo intensities can also be fitted simultaneously to improve fitting accuracy. It is also worth noting that the extracted apparent Kds from different resonances may not match. Since chemical shifts can be perturbed via either direct ligand interaction or ligand-induced structural changes, different residues could have different dependence on ligand concentrations, resulting in different apparent binding affinities.
2.4. NMR characterization of RNA-small molecule structures
Chemical shift perturbation provides a powerful approach for characterizing RNA-ligand interactions. However, as discussed above, these changes can be induced through different mechanisms, hence, detailed chemical basis for RNA-ligand interactions can remain elusive. This knowledge can be obtained by ultimately determining a high-resolution structure of the RNA-ligand complex. NMR is a well-established biophysical tool for solving high-resolution structures of RNA and its complexes with proteins and ligands [109–121]. An excellent review has been published recently that thoroughly discusses protocols of RNA structure determination by NMR [61]. In the following, we want to highlight one NMR technique that can be used to specifically obtain structural insights of RNA-ligand interactions.
Nuclear Overhauser effect spectroscopy (NOESY) is the cornerstone of NMR-based structure determination methods. 1H-1H NOESY generates through-space correlations between protons that are, generally, less than 6 Å apart. Since the intensity of NOESY cross peak depends on the distance between the paired protons, 1H-1H NOESY data are often thoroughly analyzed to obtain an extensive set of proton-proton distance constraints, which is the foundation of NMR determination of biomolecular structures (Fig. 4A). However, NOESY spectra of RNA are often difficult to analyze due to severe spectral overlap, making dissection of intermolecular RNA-ligand NOEs from crowded NOESY spectra more challenging. More than a decade ago, Feigon and coworkers developed a suite of four 2D-filtered/edited NOESY experiments for chemical shift assignments of large RNAs and RNA-protein complexes (Fig. 4B-E) [122]. This approach, which allows selective detection of NOEs between protons that are bonded to isotopically labeled carbons/nitrogens (referred to as labeled protons) and protons that are bonded to unlabeled carbons/nitrogens (referred to as unlabeled protons), can be readily applied to specifically obtain intermolecular NOEs between RNA and ligand. Since 13C/15N labeled RNA may be readily obtained at this stage of NMR study, an RNA-ligand complex sample can be prepared with 13C/15N labeled RNA and natural abundant ligand. In the F1fF2e NOESY, a filter is applied prior to f1 evolution, which ensures only unlabeled protons are present in the f1 dimension; subsequently, an edit is applied prior to f2 evolution, which ensures only labeled protons can be detected in the f2 dimension (Fig. 4B). As a result, the F1fF2e NOESY only detects intermolecular NOE cross peaks between labeled RNA and unlabeled ligand, significantly simplifying data analysis. Similarly, F2f NOESY can be applied to obtain intermolecular NOE cross peaks between labeled RNA and unlabeled ligand as well as intramolecular NOE cross peaks within unlabeled ligand (Fig. 4C). With the knowledge of chemical shift assignments, the RNA-ligand interface can be unambiguously identified. Furthermore, intermolecular RNA-ligand distances can also be obtained by analyzing NOE peak intensities, facilitating structural modeling of the binding pocket.
Figure 4. Filtered/edited NOESY for structural characterization of RNA-ligand interactions.
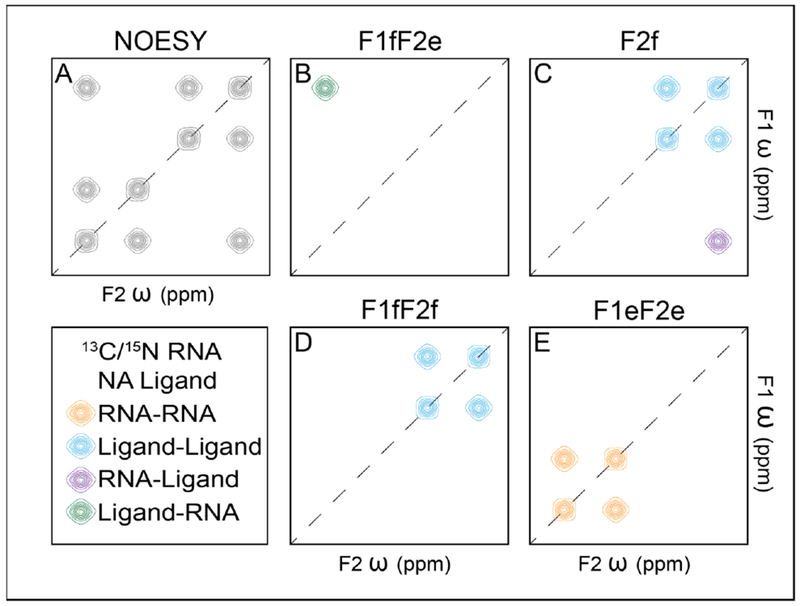
(A) The standard NOESY allows for the development of a NOE between all proximal protons. (B) The F1fF2e filters labeled signals then edits unlabeled signals giving rise to cross-peaks from unlabeled ligand to labeled RNA. (C) F2f filters labeled signals after NOEs have been developed giving rise to labeled RNA to unlabeled ligand cross-peaks and unlabeled ligand to unlabeled ligand peaks. (D) F1fF2f filters labeled signals prior to NOE development and after giving rise only to peaks from unlabeled ligand to unlabeled ligand. (E) F1eF2e edits unlabeled signals prior to NOE development and after giving rise only to peaks from labeled RNA to labeled RNA. In the figure, NA is an abbreviation for natural abundance.
2.5. NMR characterization of ligand binding kinetics
Kinetics are an important aspect of RNA-ligand interactions. Characterizing binding kinetics can facilitate understanding the biological role of a given RNA-ligand complex as well as optimizing a specific ligand binding process. While NMR has been well-established in characterizing high-resolution structures and dynamics of biomolecules, NMR is also a powerful tool for measuring kinetics of an exchange process. For example, the rates of base pair opening processes in RNA have been obtained with imino/amino proton exchange experiments [123]. Kinetic properties of non-equilibrium ligand-dependent riboswitch folding have been measured using time-resolved NMR [124, 125]. ZZ-exchange NMR spectroscopy can characterize equilibrium exchange processes that occur at subsecond-to-second timescales, providing that all exchange states are sufficiently populated for detection (>10%) [126–132]. Both thermodynamics (populations) and kinetics (rates of exchange) of the exchange process can be extracted from time-dependent ZZ-exchange profiles. Recently, via monitoring RNA signals in the apo and holo states, ZZ-exchange spectroscopy has been used to measure on and off rates of ligand-binding processes in riboswitches [130, 132].
In the past few years, exciting developments of relaxation dispersion (RD) techniques have further extended the ability of NMR in characterizing equilibrium exchange processes in RNA at microsecond-to-subsecond timescales [43, 133, 134] (Fig. 5). Built upon NMR chemical exchange properties, these techniques enable accurate characterization of highly skewed exchange processes that involve conformational states too sparsely populated (as little as ~0.5%) and transiently lived (as short as tens-of-microseconds) to be detected by conventional NMR techniques. By analyzing spin-lock-power dependent RD profiles, thermodynamics and kinetics of the exchange process can be obtained. Moreover, chemical shifts of the excited conformational states can also be extracted from RD profiles, providing structural insights that are otherwise inaccessible. These exciting NMR techniques and associated RD profiles are highlighted in Figure 5. Briefly, Carr-Purcell-Meiboom-Gill (CPMG) RD spectroscopy can be used to characterize exchange processes that occur at the rate of exchange (kex = kon + koff) between ~200 – ~2,000 s−1 [135, 136]. In order to reduce extensive carbon-carbon scalar couplings in RNA, CPMG RD is often applied to samples with site-specific isotope labeling [93, 128, 129, 137, 138]. Chemical exchange saturation transfer (CEST) spectroscopy can be used to characterize exchange processes that occur at the rate of exchange between ~20 – ~5,000 s−1 [131, 138–143]. Here, uniformly 13C/15N labeled samples can be directly used without complications, while site-specific isotope labeling schemes have been shown to be able to further improve experimental sensitivity [138]. Low spin-lock field rotating-frame R1ρ RD[131, 144–147] can be used to characterize exchange processes that occur at a much broader rate of exchange between ~60 – ~40,000 s−1. Similar to CEST experiments, uniformly 13C/15N labeled samples can be directly used for quantifying the exchange process.
Figure 5. NMR relaxation dispersion techniques for measuring ligand-binding kinetics.
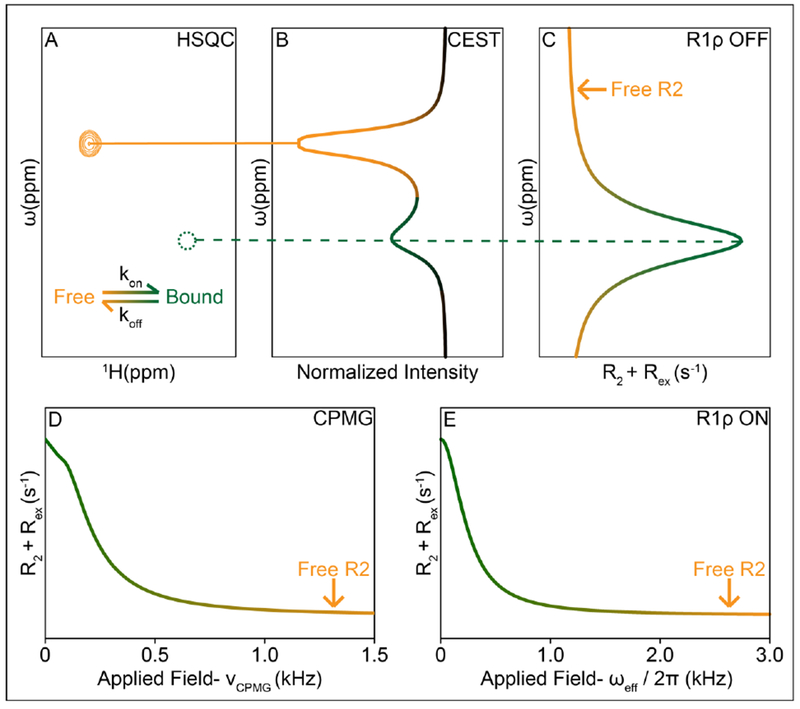
(A) HSQC of free and bound states undergoing exchange, green may be an NMR invisible state, undetectable in the HSQC. (B) Simulated CEST curve shows a major state dip at the location of the free state, and a smaller dip at the bound state (C) Simulated R1ρ off-resonance curve shows a peak indicating higher R2 values at the location of the bound state, while the free state is evident in the R2 limits of the plot. (D) CPMG and (E) R1ρ on-resonance curves show an increase in R2 due to exchange with the bound state.
While RNA-detected RD experiments have been used to characterize conformational exchange between apo and holo states to develop insights into ligand recognitions by riboswitches, carrying out RD experiments on ligands can further enable direct characterization of the ligand-binding mechanism. Recently, Kreutz, Tollinger, and co-workers have applied ligand-detected CPMG RD to study binding kinetics of preQ1 ligand to the class I preQ1 riboswitch [148]. Here, a low amount of isotope unlabeled riboswitch was added to a 15N-modified preQ1 ligand sample, creating a population-skewed exchange system, where the free ligand remains highly populated and the RNA-bound ligand is sparsely populated. By analyzing 15N CPMG RD profiles measured on free preQ1 ligand, the authors were able to directly access the off rate of preQ1 binding and also the population of the preQ1 ligand that binds to the RNA.
3. Perspective
Despite being composed of four chemically similar building blocks, RNAs can fold into sophisticated structures and recognize specific small molecules to carry out a growing plethora of functions, as evidenced with diverse naturally occurring metabolite-sensing riboswitches [13]. The growing discoveries of disease-linked ncRNAs have further promoted great interests and efforts in developing RNA-target therapeutics. Last year marks the first FDA-approved RNA-targeted drug, which is based on RNAi technology. These efforts have also led to recent successes on identifying bioactive small-molecule inhibitors that target structured FMN riboswitch [149] and self-splicing group II intron [150], demonstrating that highly structured RNAs can indeed be outstanding targets for drug discovery. Furthermore, the presence of excited conformational states in RNA, which have been unveiled in recent years by NMR RD techniques, promises novel drug targets, as these states have remained ‘hidden’ from conventional techniques. Hence, the ability to systematically characterize RNA and its interactions with small molecules is important not only for understanding basic mechanisms of ligand-dependent RNA functions but also for evaluating potential RNA-binding small molecules as lead compounds. NMR spectroscopy has been established as a powerful tool in protein-targeted drug discovery [80]. With ongoing developments of NMR techniques that are dedicated to meet unique requirements of RNA, we believe NMR spectroscopy will play similar, probably even more important, roles in facilitating discoveries and developments of novel RNA-targeted small molecule therapeutics.
Highlights.
RNA participates in diverse cellular processes where misregulation leads to disease
RNA is an important target for developing small molecule therapeutics
NMR provides a toolbox that meets distinct biophysical needs in drug discovery
NMR can screen RNA-binding small molecules and map interactions at atomic level
NMR can measure thermodynamic, kinetic, and structural properties of interactions
Acknowledgements
We thank members of the Zhang lab for critical comments. This work was supported by an NIH grant (R01 GM114432) and an NSF grant (CAREER MCB1652676).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Kruger K et al. , Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena, Cell 31 (1982) 147–157. [DOI] [PubMed] [Google Scholar]
- [2].Guerrier-Takada C et al. , The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme, Cell 35 (1983) 849–857. [DOI] [PubMed] [Google Scholar]
- [3].Mattick JS, RNA regulation: a new genetics?, Nat. Rev. Genet. 5 (2004) 316–323. [DOI] [PubMed] [Google Scholar]
- [4].Sharp PA, The centrality of RNA, Cell 136 (2009) 577–580. [DOI] [PubMed] [Google Scholar]
- [5].Ban N et al. , Placement of protein and RNA structures into a 5 A-resolution map of the 50S ribosomal subunit, Nature 400 (1999) 841–847. [DOI] [PubMed] [Google Scholar]
- [6].Clemons WM Jr. et al. , Structure of a bacterial 30S ribosomal subunit at 5.5 A resolution, Nature 400 (1999) 833–840. [DOI] [PubMed] [Google Scholar]
- [7].Tocilj A et al. , The small ribosomal subunit from Thermus thermophilus at 4.5 A resolution: pattern fittings and the identification of a functional site, Proc. Natl. Acad. Sci. U.S.A. 96 (1999) 14252–14257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cate JH et al. , X-ray crystal structures of 70S ribosome functional complexes, Science 285 (1999) 2095–2104. [DOI] [PubMed] [Google Scholar]
- [9].Ban N et al. , The complete atomic structure of the large ribosomal subunit at 2.4 A resolution, Science 289 (2000) 905–920. [DOI] [PubMed] [Google Scholar]
- [10].Nissen P et al. , The structural basis of ribosome activity in peptide bond synthesis, Science 289 (2000) 920–930. [DOI] [PubMed] [Google Scholar]
- [11].Long Y et al. , How do lncRNAs regulate transcription?, Sci. Adv 3 (2017) eaao2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ransohoff JD, Wei Y, Khavari PA, The functions and unique features of long intergenic non-coding RNA, Nat. Rev. Mol. Cell. Biol. 19 (2018) 143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Breaker RR, Riboswitches and the RNA world, Cold Spring Harb. Perspect. Biol 4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Serganov A, Nudler E, A decade of riboswitches, Cell 152 (2013) 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mortimer SA, Kidwell MA, Doudna JA, Insights into RNA structure and function from genome-wide studies, Nat. Rev. Genet. 15 (2014) 469–479. [DOI] [PubMed] [Google Scholar]
- [16].Gebert LFR, MacRae IJ, Regulation of microRNA function in animals, Nat. Rev. Mol. Cell. Biol. 20 (2019) 21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Brown CJ et al. , The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus, Cell 71 (1992) 527–542. [DOI] [PubMed] [Google Scholar]
- [18].Brockdorff N et al. , The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus, Cell 71 (1992) 515–526. [DOI] [PubMed] [Google Scholar]
- [19].Rinn JL et al. , Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs, Cell 129 (2007) 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rinn JL, lncRNAs: linking RNA to chromatin, Cold Spring Harb. Perspect. Biol 6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang X, Shan SO, Fidelity of cotranslational protein targeting by the signal recognition particle, Annu. Rev. Biophys. 43 (2014) 381–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Garst AD, Edwards AL, Batey RT, Riboswitches: structures and mechanisms, Cold Spring Harb. Perspect. Biol. 3 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Serganov A, Patel DJ, Metabolite recognition principles and molecular mechanisms underlying riboswitch function, Annu. Rev. Biophys. 41 (2012) 343–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mustoe AM et al. , Pervasive regulatory functions of mRNA structure revealed by high-resolution SHAPE probing, Cell 173 (2018) 181–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Rinnenthal J et al. , Mapping the landscape of RNA dynamics with NMR spectroscopy, Acc. Chem. Res. 44 (2011) 1292–1301. [DOI] [PubMed] [Google Scholar]
- [26].Dethoff EA et al. , Functional complexity and regulation through RNA dynamics, Nature 482 (2012) 322–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mustoe AM, Brooks CL, Al-Hashimi HM, Hierarchy of RNA functional dynamics, Annu. Rev. Biochem. 83 (2014) 441–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cooper TA, Wan L, Dreyfuss G, RNA and disease, Cell 136 (2009) 777–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ryan BM, Robles AI, Harris CC, Genetic variation in microRNA networks: the implications for cancer research, Nat. Rev. Cancer 10 (2010) 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lorenzen JM, Thum T, Long noncoding RNAs in kidney and cardiovascular diseases, Nat. Rev. Nephrology 12 (2016) 360–373. [DOI] [PubMed] [Google Scholar]
- [31].Salta E, De Strooper B, Noncoding RNAs in neurodegeneration, Nat. Rev. Neuroscience 18 (2017) 627–640. [DOI] [PubMed] [Google Scholar]
- [32].Disney MD, Dwyer BG, Childs-Disney JL, Drugging the RNA World, Cold Spring Harb. Perspect. Biol 10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Warner KD, Hajdin CE, Weeks KM, Principles for targeting RNA with drug-like small molecules, Nat. Rev. Drug Discovery 17 (2018) 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].St-Pierre P et al. , Fluorescence tools to investigate riboswitch structural dynamics, Biochim. Biophys. Acta 1839 (2014) 1005–1019. [DOI] [PubMed] [Google Scholar]
- [35].Kumar GS, Basu A, The use of calorimetry in the biophysical characterization of small molecule alkaloids binding to RNA structures, Biochim. Biophys. Acta 1860 (2016) 930–944. [DOI] [PubMed] [Google Scholar]
- [36].Gilbert SD, Mediatore SJ, Batey RT, Modified pyrimidines specifically bind the purine riboswitch, J. Am. Chem. Soc. 128 (2006) 14214–14215. [DOI] [PubMed] [Google Scholar]
- [37].Hendrix M et al. , Direct observation of aminoglycoside-RNA interactions by surface plasmon resonance, J. Am. Chem. Soc. 119 (1997) 3641–3648. [DOI] [PubMed] [Google Scholar]
- [38].Moon MH et al. , Measuring RNA-ligand interactions with microscale thermophoresis, Biochemistry 57 (2018) 4638–4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sannes-Lowery KA, Griffey RH, Hofstadler SA, Measuring dissociation constants of RNA and aminoglycoside antibiotics by electrospray ionization mass spectrometry, Anal. Biochem. 280 (2000) 264–271. [DOI] [PubMed] [Google Scholar]
- [40].Rosu F, De Pauw E, Gabelica V, Electrospray mass spectrometry to study drug-nucleic acids interactions, Biochimie 90 (2008) 1074–1087. [DOI] [PubMed] [Google Scholar]
- [41].Rizvi NF et al. , Discovery of selective RNA-binding small molecules by affinity-selection mass spectrometry, ACS Chem. Biol. 13 (2018) 820–831. [DOI] [PubMed] [Google Scholar]
- [42].Furtig B et al. , NMR spectroscopy of RNA, Chembiochem 4 (2003) 936–962. [DOI] [PubMed] [Google Scholar]
- [43].Bothe JR et al. , Characterizing RNA dynamics at atomic resolution using solution-state NMR spectroscopy, Nat. Methods 8 (2011) 919–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Merino EJ et al. , RNA structure analysis at single nucleotide resolution by selective 2’-hydroxyl acylation and primer extension (SHAPE), J. Am. Chem.Soc. 127 (2005) 4223–4231. [DOI] [PubMed] [Google Scholar]
- [45].Wang B, Wilkinson KA, Weeks KM, Complex ligand-induced conformational changes in tRN(Asp) revealed by single-nucleotide resolution SHAPE chemistry, Biochemistry 47 (2008) 3454–3461. [DOI] [PubMed] [Google Scholar]
- [46].Eubanks CS et al. , Small molecule-based pattern recognition to classify RNA structure, J. Am. Chem. Soc. 139 (2017) 409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chen Y, Pollack L, SAXS studies of RNA: structures, dynamics, and interactions with partners, Wiley interdisciplinary Rev. RNA 7 (2016) 512–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bokinsky G, Zhuang X, Single-molecule RNA folding, Acc. Chem. Res. 38 (2005) 566–573. [DOI] [PubMed] [Google Scholar]
- [49].Li PT, Vieregg J, Tinoco I Jr., How RNA unfolds and refolds, Annu. Rev. Biochem. 77 (2008) 77–100. [DOI] [PubMed] [Google Scholar]
- [50].Savinov A, Perez CF, Block SM, Single-molecule studies of riboswitch folding, Biochim. Biophys. Acta 1839 (2014) 1030–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hansel R et al. , Evaluation of parameters critical for observing nucleic acids inside living Xenopus laevis oocytes by in-cell NMR spectroscopy, J. Am. Chem. Soc. 131 (2009) 15761–15768. [DOI] [PubMed] [Google Scholar]
- [52].Bao HL, Xu Y, Investigation of higher-order RNA G-quadruplex structures in vitro and in living cells by (19)F NMR spectroscopy, Nat. Protoc 13 (2018) 652–665. [DOI] [PubMed] [Google Scholar]
- [53].Tyrrell J et al. , The cellular environment stabilizes adenine riboswitch RNA structure, Biochemistry 52 (2013) 8777–8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Spitale RC et al. , RNA SHAPE analysis in living cells, Nat. Chem. Biol. 9 (2013) 18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Watters KE, Abbott TR, Lucks JB, Simultaneous characterization of cellular RNA structure and function with in-cell SHAPE-Seq, Nucleic Acids Res. 44 (2016) e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Tian S, Das R, RNA structure through multidimensional chemical mapping, Q. Rev. Biophys. 49 (2016) e7. [DOI] [PubMed] [Google Scholar]
- [57].Rzuczek SG et al. , Precise small-molecule recognition of a toxic CUG RNA repeat expansion, Nat. Chem. Biol. 13 (2017) 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Smola MJ, Weeks KM, In-cell RNA structure probing with SHAPE-MaP, Nat. Protoc 13 (2018) 1181–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Leistra AN, Mihailovic MK, Contreras LM, Fluorescence-based methods for characterizing RNA interactions in vivo, Methods Mol. Biol 1737 (2018) 129–164. [DOI] [PubMed] [Google Scholar]
- [60].Zinshteyn B et al. , Assaying RNA structure with LASER-Seq, Nucleic Acids Res. 47 (2019) 43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Dominguez C et al. , Structure determination and dynamics of protein-RNA complexes by NMR spectroscopy, Prog. Nucl. Magn. Reson. Spectrosc. 58 (2011) 1–61. [DOI] [PubMed] [Google Scholar]
- [62].Zhao B, Zhang Q, Characterizing excited conformational states of RNA by NMR spectroscopy, Curr. Opin. Struct. Biol. 30 (2015) 134–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Liu B et al. , A potentially abundant junctional RNA motif stabilized by m(6)A and Mg(2), Nat. Comm 9 (2018) 2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Qin PZ et al. , Measuring nanometer distances in nucleic acids using a sequence-independent nitroxide probe, Nat. Protoc. 2 (2007) 2354–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Helmling C et al. , Noncovalent spin labeling of riboswitch RNAs to obtain long-range structural NMR restraints, ACS Chem. Biol. 9 (2014) 1330–1339. [DOI] [PubMed] [Google Scholar]
- [66].Wunderlich CH et al. , Stable isotope-labeled RNA phosphoramidites to facilitate dynamics by NMR, Methods Enzymol. 565 (2015) 461–494. [DOI] [PubMed] [Google Scholar]
- [67].Keyhani S et al. , Chemo-enzymatic synthesis of position-specifically modified RNA for biophysical studies including light control and NMR spectroscopy, Angew. Chem. Int. Ed. 57 (2018) 12017–12021. [DOI] [PubMed] [Google Scholar]
- [68].Longhini AP, LeBlanc RM, Dayie TK, Chemo-enzymatic labeling for rapid assignment of RNA molecules, Methods 103 (2016) 11–17. [DOI] [PubMed] [Google Scholar]
- [69].Sochor F et al. , (19)F-labeling of the adenine H2-site to study large RNAs by NMR spectroscopy, J. Biomol. NMR 64 (2016) 63–74. [DOI] [PubMed] [Google Scholar]
- [70].Ponchon L, Dardel F, Recombinant RNA technology: the tRNA scaffold, Nat. Methods 4 (2007) 571–576. [DOI] [PubMed] [Google Scholar]
- [71].Ponchon L et al. , A generic protocol for the expression and purification of recombinant RNA in Escherichia coli using a tRNA scaffold, Nat. Protoc 4 (2009) 947–959. [DOI] [PubMed] [Google Scholar]
- [72].Ferre-D’Amare AR, Scott WG, Small self-cleaving ribozymes, Cold Spring Harb. Perspect. Biol. 2 (2010) a003574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Shields TP et al. , High-performance liquid chromatography purification of homogenous-length RNA produced by trans cleavage with a hammerhead ribozyme, RNA 5 (1999) 1259–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Easton LE, Shibata Y, Lukavsky PJ, Rapid, nondenaturing RNA purification using weak anion-exchange fast performance liquid chromatography, RNA 16 (2010) 647–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].McKenna SA et al. , Purification and characterization of transcribed RNAs using gel filtration chromatography, Nat. Protoc 2 (2007) 3270–3277. [DOI] [PubMed] [Google Scholar]
- [76].Cheong HK et al. , Rapid preparation of RNA samples for NMR spectroscopy and X-ray crystallography, Nucleic Acids Res. 32 (2004) e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Kieft JS, Batey RT, A general method for rapid and nondenaturing purification of RNAs, RNA 10 (2004) 988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Batey RT, Kieft JS, Improved native affinity purification of RNA, RNA 13 (2007) 1384–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Walker SC et al. , RNA affinity tags for the rapid purification and investigation of RNAs and RNA-protein complexes, Methods Mol. Biol. 488 (2008) 23–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Gossert AD, Jahnke W, NMR in drug discovery: a practical guide to identification and validation of ligands interacting with biological macromolecules, Prog. Nucl. Magn. Reson. Spectrosc. 97 (2016) 82–125. [DOI] [PubMed] [Google Scholar]
- [81].Pellecchia M et al. , Perspectives on NMR in drug discovery: a technique comes of age, Nat. Rev. Drug Discovery 7 (2008) 738–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Sugiki T et al. , Current NMR techniques for structure-based drug discovery, Molecules 23 (2018) 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Mayer M, Meyer B, Characterization of ligand binding by saturation transfer difference NMR spectroscopy, Angew. Chem. Int. Ed. 38 (1999) 1784–1788. [DOI] [PubMed] [Google Scholar]
- [84].Johnson EC et al. , Application of NMR SHAPES screening to an RNA target, J. Am. Chem. Soc. 125 (2003) 15724–15725. [DOI] [PubMed] [Google Scholar]
- [85].Mayer M, James TL, Detecting ligand binding to a small RNA target via saturation transfer difference NMR experiments in D(2)O and H(2)O, J. Am. Chem. Soc. 124 (2002) 13376–13377. [DOI] [PubMed] [Google Scholar]
- [86].Garavis M et al. , Discovery of selective ligands for telomeric RNA G-quadruplexes (TERRA) through 19F-NMR based fragment screening, ACS Chem. Biol. 9 (2014) 1559–1566. [DOI] [PubMed] [Google Scholar]
- [87].Abulwerdi FA et al. , Selective small-molecule targeting of a triple helix encoded by the long noncoding RNA, MALAT1, ACS Chem. Biol. 14 (2019) 223–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Dalvit C et al. , Identification of compounds with binding affinity to proteins via magnetization transfer from bulk water, J. Biomol. NMR 18 (2000) 65–68. [DOI] [PubMed] [Google Scholar]
- [89].Dalvit C et al. , WaterLOGSY as a method for primary NMR screening: Practical aspects and range of applicability, J. Biomol. NMR 21 (2001) 349–359. [DOI] [PubMed] [Google Scholar]
- [90].Albrand JP et al. , Use of transferred nuclear Overhauser effects in the study of the conformations of small molecules bound to proteins, Int. J. Biol. Macromol. 1 (1979) 37–41. [Google Scholar]
- [91].Meyer B, Weimar T, Peters T, Screening mixtures for biological activity by NMR, Eu. J. Biochem. 246 (1997) 705–709. [DOI] [PubMed] [Google Scholar]
- [92].Hajduk PJ, Olejniczak ET, Fesik SW, One-dimensional relaxation- and diffusion-edited NMR methods for screening compounds that bind to macromolecules, J. Am. Chem. Soc 119 (1997) 12257–12261. [Google Scholar]
- [93].Jimenez-Moreno E et al. , Finding the right candidate for the right position: a fast NMR-assisted combinatorial method for optimizing nucleic acids binders, J. Am. Chem. Soc. 138 (2016) 6463–6474. [DOI] [PubMed] [Google Scholar]
- [94].Feixas F et al. , Exploring the role of receptor flexibility in structure-based drug discovery, Biophys. Chem. 186 (2014) 31–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Yang S, Salmon L, Al-Hashimi HM, Measuring similarity between dynamic ensembles of biomolecules, Nat. Methods 11 (2014) 552–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Amaro RE, Li WW, Emerging methods for ensemble-based virtual screening, Curr. Top. Med. Chem. 10 (2010) 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Stelzer AC et al. , Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble, Nat. Chem. Biol. 7 (2011) 553–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Ganser LR et al. , High-performance virtual screening by targeting a high-resolution RNA dynamic ensemble, Nat. Struct. Mol. Biol. 25 (2018) 425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Williamson MP, Using chemical shift perturbation to characterise ligand binding, Prog. Nucl. Magn. Reson. Spectrosc. 73 (2013) 1–16. [DOI] [PubMed] [Google Scholar]
- [100].Ilgu M et al. , An adaptable pentaloop defines a robust neomycin-B RNA aptamer with conditional ligand-bound structures, RNA 20 (2014) 815–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Liu J et al. , Identification of spermidine binding site in T-box riboswitch antiterminator RNA, Chem. Biol. & Drug Design 87 (2016) 182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Keller H et al. , Adenine protonation enables cyclic-di-GMP binding to cyclic-GAMP sensing riboswitches, RNA 24 (2018) 1390–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Murchie AIH et al. , Structure-based drug design targeting an inactive RNA conformation: Exploiting the flexibility of HIV-1 TAR RNA, J. Mol. Biol. 336 (2004) 625–638. [DOI] [PubMed] [Google Scholar]
- [104].Abulwerdi FA et al. , Development of small molecules with a noncanonical binding mode to HIV-1 trans activation response (TAR) RNA, J. Med. Chem. 59 (2016) 11148–11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Prado S et al. , A small-molecule inhibitor of HIV-1 Rev function detected by a diversity screen based on RRE-Rev interference, Biochem. Pharm. 156 (2018) 68–77. [DOI] [PubMed] [Google Scholar]
- [106].Schnieders R et al. , Evaluation of (15)N-detected H-N correlation experiments on increasingly large RNAs, J. Biomol. NMR 69 (2017) 31–44. [DOI] [PubMed] [Google Scholar]
- [107].Farjon J et al. , Longitudinal-relaxation-enhanced NMR experiments for the study of nucleic acids in solution, J. Am. Chem. Soc. 131 (2009) 8571–8577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Sathyamoorthy B et al. , Development and application of aromatic [(13)C, (1)H] SOFAST-HMQC NMR experiment for nucleic acids, J. Biomol. NMR 60 (2014) 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Puglisi JD et al. , Conformation of the TAR RNA-arginine complex by NMR spectroscopy, Science 257 (1992) 76–80. [DOI] [PubMed] [Google Scholar]
- [110].Davis B et al. , Rational design of inhibitors of HIV-1 TAR RNA through the stabilisation of electrostatic “hot spots”, J. Mol. Biol. 336 (2004) 343–356. [DOI] [PubMed] [Google Scholar]
- [111].Kang M, Peterson R, Feigon J, Structural Insights into riboswitch control of the biosynthesis of queuosine, a modified nucleotide found in the anticodon of tRNA, Mol. Cell 33 (2009) 784–790. [DOI] [PubMed] [Google Scholar]
- [112].Ferner J et al. , Structures of HIV TAR RNA-ligand complexes reveal higher binding stoichiometries, Chembiochem 10 (2009) 1490–1494. [DOI] [PubMed] [Google Scholar]
- [113].Kang M, Eichhorn CD, Feigon J, Structural determinants for ligand capture by a class II preQ1 riboswitch, Proc. Natl. Acad. Sci. U.S.A. 111 (2014) E663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Miller SB et al. , A structure-based mechanism fortRNAand retroviral RNA remodelling during primer annealing, Nature 515 (2014) 591–595. [DOI] [PubMed] [Google Scholar]
- [115].Keane SC et al. , RNA structure. Structure of the HIV-1 RNA packaging signal, Science 348 (2015) 917–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Cornilescu G et al. , Structural analysis of multi-helical RNAs by NMR-SAXS/WAXS: application to the U4/U6 di-snRNA, J. Mol. Biol. 428 (2016) 777–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Shortridge MD et al. , A macrocyclic peptide ligand binds the oncogenic microRNA-21 precursor and suppresses Dicer processing, ACS Chem. Biol. 12 (2017) 1611–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Tolbert M et al. , HnRNP A1 Alters the structure of a conserved enterovirus IRES domain to stimulate viral translation, J. Mol. Biol. 429 (2017) 2841–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Pham VV et al. , HIV-1 Tat interactions with cellular 7SK and viral TAR RNAs identifies dual structural mimicry, Nat. Comm 9 (2018) 4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Shortridge MD et al. , An ultra-high affinity ligand of HIV-1 TAR reveals the RNA structure recognized by P-TEFb, Nucleic Acids Res. 47 (2019) 1523–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Weickhmann AK et al. , The structure of the SAM/SAH-binding riboswitch, Nucleic Acids Res. 47 (2019) 2654–2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Peterson RD et al. , New applications of 2D filtered/edited NOESY for assignment and structure elucidation of RNA and RNA-protein complexes, J. Biomol. NMR 28 (2004) 59–67. [DOI] [PubMed] [Google Scholar]
- [123].Russu IM, Probing site-specific energetics in proteins and nucleic acids by hydrogen exchange and nuclear magnetic resonance spectroscopy, Methods Enzymol. 379 (2004) 152–175. [DOI] [PubMed] [Google Scholar]
- [124].Furtig B et al. , Time-resolved NMR studies of RNA folding, Biopolymers 86 (2007) 360–383. [DOI] [PubMed] [Google Scholar]
- [125].Lee MK et al. , Real-time multidimensional NMR follows RNA folding with second resolution, Proc. Natl. Acad. Sci. U.S.A. 107 (2010) 9192–9197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Farrow NA et al. , A heteronuclear correlation experiment for simultaneous determination of 15N longitudinal decay and chemical exchange rates of systems in slow equilibrium, J. Biomol. NMR 4 (1994) 727–734. [DOI] [PubMed] [Google Scholar]
- [127].Wenter P et al. , Kinetics of RNA refolding in dynamic equilibrium by 1H-detected 15N exchange NMR spectroscopy, J. Am. Chem. Soc. 128 (2006) 7579–7587. [DOI] [PubMed] [Google Scholar]
- [128].Kloiber K et al. , Probing RNA dynamics via longitudinal exchange and CPMG relaxation dispersion NMR spectroscopy using a sensitive 13C-methyl label, Nucleic Acids Res. 39 (2011) 4340–4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Wunderlich CH et al. , Synthesis of (6-(13)C)pyrimidine nucleotides as spin-labels for RNA dynamics, J. Am. Chem. Soc. 134 (2012) 7558–7569. [DOI] [PubMed] [Google Scholar]
- [130].Reining A et al. , Three-state mechanism couples ligand and temperature sensing in riboswitches, Nature 499 (2013) 355–359. [DOI] [PubMed] [Google Scholar]
- [131].Zhao B, Hansen AL, Zhang Q, Characterizing slow chemical exchange in nucleic acids by carbon CEST and low spin-lock field R1rho NMR spectroscopy,J. Am. Chem. Soc. 136 (2014) 20–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Zhao B et al. , An excited state underlies gene regulation of a transcriptional riboswitch, Nat. Chem. Biol. 13 (2017) 968–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Sekhar A, Kay LE, NMR paves the way for atomic level descriptions of sparsely populated, transiently formed biomolecular conformers, Proc. Natl. Acad. Sci. U.S.A. 110 (2013) 12867–12874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Palmer AG 3rd, Chemical exchange in biomacromolecules: past, present, and future, J. Magn. Reson. 241 (2014) 3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Palmer AG 3rd, Kroenke CD, Loria JP, Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules, Methods Enzymol. 339 (2001) 204–238. [DOI] [PubMed] [Google Scholar]
- [136].Korzhnev DM, Kay LE, Probing invisible, low-populated States of protein molecules by relaxation dispersion NMR spectroscopy: an application to protein folding, Acc. Chem. Res. 41 (2008) 442–451. [DOI] [PubMed] [Google Scholar]
- [137].Johnson JE Jr., Hoogstraten CG, Extensive backbone dynamics in the GCAA RNA tetraloop analyzed using 13C NMR spin relaxation and specific isotope labeling, J. Am. Chem. Soc. 130 (2008) 16757–16769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Longhini AP et al. , Chemo-enzymatic synthesis of site-specific isotopically labeled nucleotides for use in NMR resonance assignment, dynamics and structural characterizations, Nucleic Acids Res. 44 (2016) e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Forsen S, Hoffman RA, Study of moderately rapid chemical exchange reactions by means of nuclear magnetic double resonance, J. Chem. Phys. 39 (1963) 2892. [Google Scholar]
- [140].Fawzi NL et al. , Atomic-resolution dynamics on the surface of amyloid-beta protofibrils probed by solution NMR, Nature 480 (2011) 268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Vallurupalli P, Bouvignies G, Kay LE, Studying “invisible” excited protein states in slow exchange with a major state conformation, J. Am. Chem. Soc. 134 (2012) 8148–8161. [DOI] [PubMed] [Google Scholar]
- [142].Zhao B, Zhang Q, Measuring residual dipolar couplings in excited conformational states of nucleic acids by CEST NMR spectroscopy, J. Am.Chem. Soc. 137 (2015) 13480–13483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Chen B, LeBlanc R, Dayie TK, SAM-II riboswitch samples at least two conformations in solution in the absence of ligand: Implications for recognition, Angew. Chem. Int. Ed. 55 (2016) 2724–2727. [DOI] [PubMed] [Google Scholar]
- [144].Palmer AG 3rd, Massi F, Characterization of the dynamics of biomacromolecules using rotating-frame spin relaxation NMR spectroscopy, Chem. Rev. 106 (2006) 1700–1719. [DOI] [PubMed] [Google Scholar]
- [145].Massi F et al. , NMR R1 rho rotating-frame relaxation with weak radio frequency fields, J. Am. Chem. Soc. 126 (2004) 2247–2256. [DOI] [PubMed] [Google Scholar]
- [146].Korzhnev DM, Orekhov VY, Kay LE, Off-resonance R(1rho) NMR studies of exchange dynamics in proteins with low spin-lock fields: an application to a Fyn SH3 domain, J. Am. Chem. Soc. 127 (2005) 713–721. [DOI] [PubMed] [Google Scholar]
- [147].Hansen AL et al. , Extending the range of microsecond-to-millisecond chemical exchange detected in labeled and unlabeled nucleic acids by selective carbon R(1rho) NMR spectroscopy, J. Am. Chem. Soc. 131 (2009) 3818–3819. [DOI] [PubMed] [Google Scholar]
- [148].Moschen T et al. , Ligand-detected relaxation dispersion NMR spectroscopy: dynamics of preQ1-RNA binding, Angew. Chem. Int. Ed. 54 (2015) 560–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Howe JA et al. , Selective small-molecule inhibition of an RNA structural element, Nature 526 (2015) 672–677. [DOI] [PubMed] [Google Scholar]
- [150].Fedorova O et al. , Small molecules that target group II introns are potent antifungal agents, Nat. Chem. Biol. 14 (2018) 1073–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]