

Abstract
Neuropathic pain (characterized by hyperalgesia and allodynia to mechanical and thermal stimuli) causes cellular changes in spinal dorsal horn neurons, some of which parallel those in synaptic plasticity associated with learning. Ubiquitin C-terminal hydrolase (UCH) appears to play a key role in long-term facilitation inAplysia. The cooperation of UCH with the proteolytic enzyme complex known as the proteasome is required for the degradation of a number of signaling molecules within the cell that may remove normal restraints on synaptic plasticity. We have used electrophysiology, in situ hybridization histochemistry, semiquantitative RT-PCR, Western blotting, and in vivobehavioral reflex analysis to investigate the ubiquitin–proteasome system in a model of neuropathic pain. In neuropathic animals, ionophoretic application of selective proteasome inhibitors attenuated dorsal horn neuron firing evoked by normally innocuous brush or cold stimuli and by noxious mustard oil stimuli. In control animals, only mustard oil-evoked responses were inhibited. Intrathecal administration of proteasome inhibitors attenuated hyperalgesia and allodynia in neuropathic rats. Expression of UCH-L1 (a rat homolog ofAplysia neuronal UCH and of the human UCH-L1, also known as PGP 9.5) and its mRNA were selectively increased within the ipsilateral dorsal horn of neuropathic rats, supporting the idea of a role for the ubiquitin–proteasome system in nociceptive processing. Proteasome inhibitors selectively attenuate allodynic and hyperalgesic responses in neuropathic pain, with some reduction in normal nociceptive, but not non-nociceptive responses, and potentially represent a novel therapeutic strategy for neuropathic pain.
Keywords: neuropathic pain, spinal cord, proteasome, ubiquitination, hyperalgesia, allodynia
After damage to the distal parts of primary afferent neurons that transmit nociceptive information, their abnormal and repetitive firing is known to induce a state of neuronal hyperexcitability within the spinal dorsal horn, referred to as “central sensitization.” A characteristic consequence of this is the development of accentuated pain-related behaviors, reflecting hyperalgesia and allodynia in response to thermal and mechanical stimulation. In animal models of neuropathic pain, these are associated with various neurochemical and neuroanatomical changes occurring within the nociceptive pathway itself (Hökfelt et al., 1994; Woolf et al., 1995; Chung et al., 1996; Cameron et al., 1997).
A number of the changes associated with central sensitization in response to sustained nociceptive activation are similar to those observed in animal models of synaptic plasticity, such as long-term potentiation (LTP) in the mammalian hippocampus. Both LTP and central sensitization trigger activity-dependent changes in neuronal excitability via mechanisms including the post-translational modification of membrane-bound proteins such as the NMDA receptor. Downstream targets common to both processes may include Ca2+/calmodulin-dependent kinases, protein kinase C (PKC), protein kinase A (PKA), and nitric oxide synthase (Bliss and Collingridge, 1993; Mayer et al., 1999). A partially analogous process of long-term facilitation (LTF) of synaptic responses occurs in Aplysia sensory pathways, where a number of the molecular changes underlying LTF are evoked in common by sensory neuron injury (Walters and Ambron, 1995). In the LTF model, sustained sensory neuron activation induces the activation of PKA with the dissociation of PKA regulatory subunits and the translocation of the constitutively active free catalytic subunits to the cell nucleus (Chain et al., 1999). The ubiquitin–proteasome system appears to play a vital role in the regulation of PKA activity by degrading the regulatory subunits of PKA, which normally maintain PKA in an inactive state. Stimulus-induced degradation of PKA regulatory subunits is thus thought to lead to persistent activation of the kinase and maintenance of the synapse in a facilitated state (Greenberg et al., 1987; Chain et al., 1999).
The ubiquitin–proteasome system is a major non-lysosomal proteolytic pathway that degrades diverse cellular proteins, including a number of proteins with important roles in the regulation of cell growth or function (Coux et al., 1996; Rolfe et al., 1997; Hershko and Ciechanover, 1998). For example, protein degradation by this pathway is important for the control of PKA activity (Hegde et al., 1993). The functional activity of the proteasome is enhanced by the UCH family of isopeptidases, which appear to play a crucial role in maintaining activity by preventing accumulation of inhibitory polyubiquitin chains (Wilkinson, 1997). The present study explores the contribution made by the ubiquitin–proteasome system to the central sensitization, hyperalgesia, and allodynia that characterize intractable neuropathic pain.
MATERIALS AND METHODS
Chronic constriction injury to the sciatic nerve
All experiments were performed in accordance with the United Kingdom Animals (Scientific Procedures) Act, 1986. Adult male Wistar rats (200–350 gm, Charles River, Kent, UK) were anesthetized with sodium pentobarbital (Sagatal 0.06 ml/100 gm, i.p.; Rhône Merieux, Essex, UK) and supplemented with halothane/O2 (Zeneca, Cheshire, UK). Under aseptic conditions, the right sciatic nerve was exposed proximal to the trifurcation, at a mid-thigh level, and four chromic cat gut ligatures (4:0; Ethicon, Edinburgh, UK) were tied to loosely constrict the nerve [chronic constriction injury (CCI)], as viewed under 40× magnification (Bennett and Xie, 1988). The overlying muscle and skin were closed with sutures (4:0; Ethicon), and the animals were allowed to recover for 72 hr before reflex testing recommenced. Sham-operated animals underwent the same surgical procedure, but no ligatures were placed around the nerve.
Behavioral tests
Behavioral testing was performed before surgery to establish a baseline for comparison with post-surgical values. Inspections were made regularly for signs of autotomy, which was rarely observed. CCI animals were always assessed for the presence of thermal hyperalgesia, cold, and mechanical allodynia before being used for further study in other tests. Only in occasional cases did reflex sensitization fail to develop, and in those examples technical factors appeared to be responsible. The following behavioral reflex tests were performed as described in detail previously (Blackburn-Munro et al., 1999; Dickinson et al., 1999). Thermal hyperalgesia was monitored using noxious radiant heat (30–55°C; Hargreaves' thermal device, Linton Instruments, Diss, UK) applied to the mid-plantar glabrous surface of the hindpaw. The withdrawal response latency was characterized as a brief paw flick recorded to the nearest 0.1 sec; a standard cutoff latency of 20 sec prevented tissue damage.
Mechanical allodynia was measured as the threshold for paw withdrawal in response to graded mechanical stimuli applied to the mid-plantar glabrous surface of the hindpaw using calibrated von Frey filaments (Stoelting, Wood Dale, IL). Threshold was defined as the pressure (force per unit area) that caused foot withdrawal five times in every 10 applications, repeated at 1–2 sec intervals. The pressure applied to the hindlimb by the von Frey filaments is calibrated as force (milliNewtons) divided by the area over which it is applied (millimeters squared).
To detect the presence of cold allodynia, rats were placed in a perspex box with an elevated aluminum floor covered with iced water, sufficient to immerse both glabrous and hairy skin of the hindpaw (3–4°C) (Bennett and Xie, 1988). Once placed in the box, rats were allowed 10 sec to acclimatize. The number of seconds the animal raised its hindpaw above the water over a 20 sec period was recorded. This was repeated four times at 10 min intervals to establish a mean suspended paw elevation time (SPET) for each rat. Animals determined as being at the peak of neuropathy, 12–14 d after CCI surgery, were then used in the experiments described below.
Intrathecal drug treatment
The effects of intrathecal administration of the selective proteasome inhibitors MG-132 (Rock et al., 1994) and epoxomicin (Sin et al., 1999) on behavioral reflex responses were examined. Before determining the effects of these inhibitors, baseline measurements were recorded in rats [normal (i.e., naive), n = 8; neuropathic, n = 26] both before and at the peak of neuropathy. A minimum of four measurements were made for each test session, for each sensory test. Rats were anesthetized briefly with halothane/O2 and injected intrathecally at the L5 level of the spinal cord with 5 nmol of MG-132 or 0.75 nmol of epoxomicin in saline with 0.5% dimethylformamide (50 μl), using a 25 gauge needle microsyringe. Injection of vehicle had no discernable effect on behavioral reflexes. The identity of the drug was concealed from the experimenter to eliminate bias, and dye injections (Pontamine Sky Blue) were performed in separate experiments to define the correct site of injection. Testing began 15 min after injection (a delay to allow for full recovery of reflex function after anesthesia) and was performed at 5 min (thermal hyperalgesia and mechanical allodynia) or 10 min intervals (cold allodynia). Testing continued until recovery to pre-injection values.
Single neuron electrophysiological recording experiments
After induction of anesthesia with halothane/O2, rats (normal, n = 8; sham, n = 4; neuropathic, n = 11) underwent jugular vein and tracheal cannulations. Anesthesia was then maintained using intravenous α-chloralose (60 mg/kg) and urethane (1.2 mg/kg). Supplementary doses of α-chloralose were given as required. Core temperature was maintained at 37–38°C, and O2 (0.1 l/min) was passed over the end of the cannula to enrich the inspired air. A laminectomy (segments L3–L6) was performed, as described previously (Blackburn-Munro and Fleetwood-Walker, 1997). Extracellular recordings were made from single, multireceptive neurons (laminas III–V), ipsilateral to the nerve injury in neuropathic animals and bilaterally in unoperated, naïve control rats via the central barrel (4m NaCl, pH 4.0–4.5) of a multibarreled glass microelectrode (tip size 4–5 μm). One side barrel contained 1m NaCl, pH 4.5, for automatic current balancing using a Neurophore BH2 Ionophoresis System (Medical Systems Corporation). The remaining barrels contained drugs for ionophoretic application of drug: the selective proteasome inhibitors lactacystin (Fenteany et al., 1995) and MG-132 (both in 0.5% aqueous dimethylformamide, pH 4.5, at concentrations of 500 and 200 μm, respectively; Biomol, Plymouth Meeting, PA). Drugs were ejected using positive currents of 5–60 nA, and a retention current of −12 nA was applied to each side barrel when not in use to minimize drug leakage. The resistance of the side barrels was monitored regularly, and electrodes with resistance values in excess of 45 MΩ were rejected.
The effects of the inhibitors were examined on evoked responses of neurons to sensory stimulation in both naive and neuropathic animals. Sustained activation of neurons was obtained by the following stimuli, described in detail elsewhere (Dickinson et al., 1999): (1) a rotating motorized innocuous brush for low threshold mechanical stimulation and (2) an intense cold stimulus provided by a Peltier device [5°C for 10 sec, from 32°C (ramp rate 5°C/sec), over a surface area of 1 cm2; Medical Instruments, Yale University, New Haven, CT]. The stimuli were repeated every 1–2 min to give reproducible peaks of activity. (3) Topical application of the C-fiber-selective chemical algogen, mustard oil [allyl isothiocyanate (Aldrich Chemical Company), 7.5% solution in paraffin oil, applied up to seven times at 5 min intervals].
Drugs were ejected ionophoretically in a step-wise manner (usually in increments of 10 nA, over a range of 5–60 nA) every 1–2 min, until clear effects on the neuronal firing rate were observed; if none was observed by 60 nA after 2 min duration, the drug application was terminated. For the majority of neurons, both drugs were tested on responses to all three sensory stimuli.
Action potential discrimination enabled data to be digitized and downloaded onto a computer to give a record of cell firing rate measured as action potentials per second. After subtraction of the “background” activity, the firing rate after drug application was expressed as the percentage change from control responses of the neuron before drug application.
Determination of levels of mRNA for ubiquitin C-terminal hydrolase-L1
mRNA analysis by RT-PCR. Relevant segments of spinal cord were dissected into ipsilateral and contralateral sides and immersed in liquid nitrogen. Total RNA was extracted using Trizol (Invitrogen, Paisley, UK) according to the manufacturer's guidelines. The cDNAs were obtained using SuperScript II Moloney's murine leukemia virus reverse transcriptase (Invitrogen) according to the manufacturer's protocol. Briefly, 5 μg DNase-treated total RNA was added to reverse transcription buffer (50 mm Tris-Cl, pH 8.3, 75 mmKCl, 5 mm MgCl2) containing 17 U RNAsin (Invitrogen), 10 mm dNTPs, and 0.5 μm poly(dT)12–18 primer. Semiquantitative PCR was performed for rat ubiquitin C-terminal hydrolase-L1 (UCH-L1)/PGP 9.5 homolog (Wilkinson et al., 1989; Kajimoto et al., 1992) and for the metabolic housekeeping enzyme, glyceraldehyde-3-phosphodehydrogenase (GAPDH). An excess of UCH amplification primers (forward: 5′-GCT GCT GCT GTT TCC CCT CAC; reverse: 5′-AGA CCT TGG CGG CAT CCT G) (MWG Biotech) (0.5 μm final concentration), supplemented with 5 UTaq DNA polymerase (Invitrogen), and 2 μl of the first-strand reaction mix, were used for each amplification. PCR cycling conditions were as follows: 1 cycle of 5 min at 94°C, and 22 cycles of 45 sec at 94°C, 30 sec at 56°C, 1 min at 72°C. This resulted in a linear amplification of the 324 base pair (bp) UCH-L1 DNA fragment, as quantified by direct measurements of fluorescence of the amplified fragments using a digital imaging system (UVP) and Scan-Analysis software (Biosoft). A second round of PCR was performed in the presence of 3′-NH2-linked GAPDH primers in an 8:2 molar ratio with standard GAPDH primers: forward, 5′-GGC AGC CCA GAA CAT CAT CC; reverse, 5′-CAG CCC CAG CAT CAA AGG TG. This produced levels of fluorescence comparable to the 324 bp UCH-L1 PCR fragment using PCR conditions identical to those above.
After coamplification of the target UCH-L1 and GAPDH sequences, equal amounts of reaction products were analyzed by gel electrophoresis in a 2% (w/v) agarose gel containing 0.5 μg/ml ethidium bromide. Relative levels of UCH-L1 mRNA expression were calculated by expressing relative levels of UCH-L1 mRNA fluorescence as a percentage of GAPDH mRNA fluorescence.
In situ hybridization histochemistry. Neuropathic animals and age-matched naïve controls were deeply anesthetized with halothane/O2 (4% for induction and maintenance), and a laminectomy (L3–L6) was performed for spinal cord removal. Tissue was mounted on a cryostat chuck (OCT Embedding Matrix, CellPath Ltd., Wales, UK) and snap frozen in isopentane (BDH, Poole, UK) at −40 to −45°C. Frozen transverse sections of spinal cord (10 μm) were taken at −19°C in a cryostat and thaw-mounted onto poly-lysine-coated microscope slides (BDH).
Two oligonucleotides (synthesized by Oswel DNA Service) complementary and specific to nucleotides 299–333 and 658–693 of rat UCH-L1 mRNA were used (Kajimoto et al., 1992). The oligonucleotides were 3′ end-labeled with deoxyadenosine α[35S]-triphosphate (specific activity <1250 Ci/mol; DuPont NEN) using terminal deoxynucleotidyl transferase (Promega). In situ hybridization histochemistry was performed using methodology described previously (Blackburn-Munro and Fleetwood-Walker, 1999). Controls used to demonstrate specificity of the respective oligonucleotide consisted of (1) pretreating sections with RNase A (1 mg/ml; Sigma, Poole, UK) for 1 hr before hybridization and (2) coincubation of the 35S-labeled oligonucleotide in the hybridization medium with a 100-fold excess of unlabeled oligonucleotide.
Image analysis
Cell counts. The number of cells positively hybridized for UCH-L1 within lateral and midway between lateral and medial (“mediolateral”) locations of laminas I–V was calculated at 40× magnification (total grid area 175 × 175 μm). Cells were considered to be positively labeled if the silver grains showed a dense pattern around the nucleus and were fivefold denser than a typical nonexpressing cell within the same field area or background levels. The total number of positively hybridized cells was determined for each spinal cord section (n = 10 sections), and these values were used to calculate the mean number of positively hybridized cells per unit area, in the lumbar spinal segments L3–L6 (naïve,n = 4; neuropathic, n = 4).
Silver grain counting. To assess any changes in the relative expression of UCH-L1 mRNA after chronic constriction injury, the mean silver grain density per positively labeled cell was measured using Image 1.44 software (Improvision) with video input from a charge-coupled device camera (Sony, Tokyo, Japan) mounted on aZeiss Axioscope microscope (40× magnification). For each section, counts were made both ipsilateral and contralateral to the nerve injury in lateral and medial zones. A minimum of five positively hybridized cells was counted in each region. A pixel count was obtained, which was converted to silver grain number via a predetermined calibration procedure. The mean number of silver grains per cell was calculated for each section (n = 10 sections). Then the mean number of silver grains per cell could be calculated. In total, 500 positively hybridized cells were counted within one side of the spinal dorsal horn for each animal.
Western blot analysis of the expression of ubiquitin C-terminal hydrolase. Hemisected L3–L6 spinal cord samples (naive, n = 4; neuropathic, n = 4; sham, n = 4) were homogenized into standard Laemmli buffer and denatured at 100°C for 5 min. Extracts were separated by electrophoresis on precast 10% polyacrylamide gels (Bio-Rad, Hemel Hempstead, UK), transferred to polyvinylidene difluoride membrane, and blocked overnight at 4°C in 4% Marvel, 0.1% Tween 20. Blots were probed with primary antibody to UCH-L1/PGP 9.5 (Affiniti; 1:5000) or GAPDH (Chemicon; 1:750) and detected by peroxidase-linked secondary antibody enhanced chemiluminescence.
Ex vivo PKA activity assays. After lumbar laminectomy under anesthesia, rats, (naïve, n = 3; neuropathic, n = 3) were treated by topical application of agents to the dorsal surface of the spinal cord. Epoxomicin (15 μm), MG-132 (100 μm),or vehicle (0.5% dimethylformamide in saline) was applied in a volume of 500 μl for 1 hr before rapid removal of segments L3–L6 to cold buffer on ice. cAMP-evoked PKA enzymatic activity and constitutive activity (thought to reflect PKA previously activatedin situ) were measured by the following procedure, a modification of those of Roskoski (1983) and Corbin (1983). Hemisected spinal cord samples were rapidly homogenized on ice in 20 mm Na HEPES, pH 7.5, with 5% glycerol, 0.25% BSA, 1 mm EGTA, 1 mmdithiothreitol, 1 mm 4-(2-aminoethyl) benzene sulfonyl fluoride (Alexis Corporation, Nottingham, UK), 2 μg/ml aprotinin, 10 μg/ml leupeptin, 2 μg/ml pepstatin, 50 μg/ml soybean trypsin inhibitor, 25 mm Na β-glycerophosphate, 1 mm Na orthovanadate, 1 mm NaF, 1 μm calyculin A, 1 μm cypermethrin (Calbiochem, Nottingham, UK), and 500 μm isobutyl methylxanthine. All reagents were from Sigma unless indicated otherwise. Aliquots of homogenate were incubated for 10 min at 30°C (linear range of assay) with 100 μm kemptide as substrate, 100 μm ATP (with [33P] α-ATP to 0.2 μCi per tube; DuPont NEN, Dreiech, Germany), 10 mmMgCl2, and 10 μm cAMP and/or 1 μm PKI6–22amide (Calbiochem) as appropriate. Assays were terminated with cold TCA (to 10%), centrifugation, and spotting of the supernatant onto P81 phosphocellulose paper. Samples were washed extensively in 75 mmH3PO4 and dried before scintillation counting. Positive controls were performed with purified bovine heart PKA catalytic subunit, and each homogenate was assayed in parallel for cAMP-evoked activity (in the absence or presence of PKI6–22 amide), constitutive activity (in the absence or presence of PKI6–22 amide), and zero time incubation blanks. Authentic (PKI6–22amide-sensitive) PKA-activity was always 86–97% of the recorded activity for constitutive and cAMP-evoked conditions, and zero time blanks were always <2% of total activity.
Subcellular distribution of specific [3H] phorbol dibutyrate binding sites. After topical application of drugs and homogenization in the same ice-cold medium as for PKA assays above, spinal cord (L3–L6) homogenates were assayed for membrane/cytosol content of specific [3H] phorbol dibutyrate binding sites as described previously (Johnson et al., 1996). Briefly, after centrifugation at 12,000 × g for 20 min, the membrane pellet was resuspended in 50 mm Tris HCl, pH 7.4, with 4 mg/ml fatty acid-free BSA (Tris BSA), and aliquots were incubated (30 min at 37°C) with 5 nm[3H] phorbol 12,13-dibutyrate (0.03 μCi per tube; DuPont NEN) with or without 10 μm unlabeled phorbol dibutyrate to define nonspecificity. Aliquots of the supernatant cytosolic fraction were added to Tris BSA containing 1 mmCaCl2, 10 mmMgCl2, and 0.5 mg/ml sonicated phosphatidylserine (Sigma) before equivalent incubation with ligand. Bovine γ-globulin (to 0.6 mg/ml) and polyethylene glycol-8000 (to 20%) in 50 mm Tris HCl were added to cytosol samples and incubated for 20 min at 4°C to precipitate proteins. After centrifugation at 12,000 × g for 20 min, all samples were aspirated, and radioactivity in the pellets was measured by liquid scintillation counting.
RESULTS
Behavioral reflex studies
Neuropathic animals showed behavioral alterations after CCI that were consistent with previous data from our laboratory (Blackburn-Munro and Fleetwood-Walker, 1999; Dickinson et al., 1999). Development of neuropathic behaviors became measurable at 8 d after surgery, with peak changes at 12–14 d. Most experiments were performed using CCI animals taken at 12–14 d after surgery, although some samples for time course studies were taken at 4 and 8 d.
Sensory responses of single dorsal horn neurons: application of inhibitors of the proteasome complex
Most of the recorded neurons were multi-receptive, responding to both innocuous and noxious stimuli. They were estimated to be located within dorsal horn laminas III–V at depths of ∼200–700 μm from the surface of the spinal cord that were measured using electrode contact at the dorsal surface and dye spot deposition in histological sections after experimentation. To facilitate neuronal classification and obtain sustained neuronal activation after mustard oil application, only neurons with receptive fields on the hairy skin of the hindlimb were investigated. Neuronal activation to application of either innocuous brush or mustard oil stimuli to the peripheral receptive field was found in the majority of recorded neurons in both normal and neuropathic animals showing peak behavioral reflex changes indicative of neuropathy. However, activation of neurons to cold stimuli (4°C) were found only in neuropathic animals, which is in agreement with the observation that it is only neuropathic animals that develop a paw withdrawal response to cold water (4°C). The spontaneous firing rates of neurons were low (1–2 Hz), and those in response to the stimuli were between 12 and 50 Hz. For each neuron, in the majority of cases, the rate of firing in response to the different stimuli, before drug application, was within 15 Hz. No significant change in activity was ever observed during control ejection of saline, pH 7.0, or the vehicle (0.5% aqueous dimethylformamide, pH 4.5) at currents up to 60 nA for 5 min. In naive animals, ionophoretic application of the proteasome inhibitors lactacystin or MG-132 caused partial inhibition of mustard oil-induced activity but no significant inhibition of brush-evoked activity (Fig.1a,b; Table1). The effects of lactacystin were similar in sham-operated animals (Table 1).
Fig. 1.

Typical effects of ionophoretic application of the proteasome inhibitor lactacystin on stimulus-evoked sensory responses of single dorsal horn neurons in naive and neuropathic animals. In naive animals, lactacystin had no effect on brush-evoked activation of dorsal horn neuron cell firing rate, measured as action potentials per second (A/s), but it caused a selective attenuation of mustard oil-evoked activation of dorsal horn neuron cell firing (a, b). In contrast, in neuropathic animals, lactacystin reduced dorsal horn neuron firing evoked by application of brush, mustard oil, and cold stimuli to the neuronal receptive field area (c–e). Thebar (bottom right) indicates 1 min duration. Lines above the firing records indicate the duration of the drug application by ionophoresis, close to the recorded cell. This is measured in nanoamperes (nA) of current used to eject the drug and was increased sequentially by 10 nA every 1–2 min.
Table 1.
Effects of ionophoretically applied proteasome inhibitors on sensory responses of single dorsal horn neurons
| Drug | Condition | % of stimulus-evoked response remaining | ||
|---|---|---|---|---|
| Brush | Mustard oil | Cold | ||
| Lactacystin | 87.5 ± 5.9 | 58.6 ± 7.1* | ||
| Naive | (n = 8) | (n = 7) | ||
| (10–60 nA) | (10–60 nA) | |||
| 79.2 ± 1.6 | 50.1 ± 6.2* | |||
| Sham | (n = 3) | (n = 4) | ||
| (50–60 nA) | (10–40 nA) | |||
| 60.8 ± 4.2* | 34.1 ± 5.1 | 58.7 ± 4.7* | ||
| Neuropathic | (n = 4) | (n = 6) | (n = 3) | |
| (20–60 nA) | (20–40 nA) | (10–30 nA) | ||
| MG-132 | 76.4 ± 2.5 | 57.1 ± 8.8* | ||
| Naive | (n = 4) | (n = 4) | ||
| (10–40 nA) | (20–40 nA) | |||
| 66.4 ± 5.4* | 31.6 ± 2.1* | 48.3 ± 7.4* | ||
| Neuropathic | (n = 4) | (n = 7) | (n = 3) | |
| (30–60 nA) | (10–30 nA) | (10–30 nA) | ||
Data are expressed as mean ± SEM (n values in parentheses). Using mean firing rates over 15 sec bins, the statistical significance of differences between maximally inhibited firing rates in the presence of drug and firing rates immediately before ionophoresis were calculated by Kruskal–Wallis one-way ANOVA, followed by apost hoc Dunn's test (
p < 0.05). The ranges of ionophoretic currents required to reach a maximal depression of firing rate are indicated. Further increases in currents beyond these values caused greater inhibition.
In neuropathic animals, however, the drugs caused a greater percentage reduction in all responses, inhibitory effects on brush responses now becoming statistically significant (Fig. 1c–e; Table 1). The de novo responses to cold, which appeared only in neuropathic animals, were also significantly inhibited by both lactacystin and MG-132.
Behavioral reflex changes after intrathecal administration of the proteasome inhibitors MG-132 and epoxomicin
Intrathecal administration of the proteasome inhibitors MG-132 (5 nmol/50 μl) and epoxomicin (0.75 nmol/50 μl) in rats exhibiting peak neuropathic behavioral changes significantly reversed thermal hyperalgesia and mechanical and cold allodynia over a 90 min period (Fig. 2a–c). Control intrathecal administration of vehicle in CCI rats had no significant effect on baselines (Fig. 2a–c). The intrathecal administration of proteasome inhibitors in naive rats had no significant effect on baseline values (data not shown).
Fig. 2.

The effects of intrathecal administration of the proteasome inhibitors epoxomicin and MG-132 on reflex withdrawal responses to noxious thermal, innocuous mechanical, and innocuous cold stimuli in CCI rats. Data are presented as the mean paw withdrawal latency (in seconds) to noxious thermal stimulation (a,b), paw withdrawal threshold in milliNewtons per millimeters squared (mN/mm2) to mechanical stimulation (c, d), and suspended paw elevation time in seconds [SPET(s)] to cold water (at 4°C) (e, f), before and after intrathecal administration of epoxomicin (0.75 nmol/50 μl) (a–c) and MG-132 (5 nmol/50 μl) (d–f), respectively (atarrow). Injection of vehicle (0.5% dimethylformamide in saline) had no detectable effect on reflexes. Paw withdrawal latency from noxious heat and innocuous cold stimuli ipsilateral to CCI showed significant differences between pre-drug and post-drug administration values (†p < 0.05; one-way ANOVA, followed by a Dunnett's post hoc test). Significant differences between contralateral and ipsilateral paw are indicated (*p < 0.05; Student's paired t test). Paw withdrawal thresholds to mechanical stimulation ipsilateral to CCI showed significant differences between pre-drug and post-drug administration values (†p < 0.05; Kruskal–Wallis one-way ANOVA, followed by a post hocDunn's test). Significant differences between contralateral and ipsilateral paw withdrawal thresholds are indicated (*p< 0.05; Mann–Whitney U test).
mRNA analysis by RT-PCR
A semiquantitative RT-PCR approach was used for the screening of the UCH-L1 gene, the expression of which may be altered in a neuropathic state. Using the samples described previously, changes in the abundance of PCR product were normalized against GAPDH levels (Fig.3a). GAPDH has previously been shown to be a stable and consistent housekeeping standard in cases of spinal cord and brain insult (Medhurst et al., 2000). RT-PCR performed on cDNA extracted from the spinal cord of rats using a PCR mixture containing primers to UCH-L1 and GAPDH generated two distinct PCR products of 324 and 292 bp, respectively (Fig. 3a). Analysis was performed on points in the PCR process at which exponential amplification was observed (whereby the abundance of both UCH and GAPDH is directly proportional to the level of respective mRNA in the sample). Concurrent PCR on samples for which reverse transcriptase was omitted produced no detectable amplification products (data not shown). A statistically significant increase (mean + 47%) in the relative abundance of UCH-L1 mRNA was seen in the ipsilateral as compared with the contralateral spinal cord hemisection at 14 d (Fig. 3a). No significant changes in mRNA levels were observed between spinal cord sections of sham-operated or naive rats or ipsilateral to CCI in animals taken 4–8 d after nerve ligation.
Fig. 3.
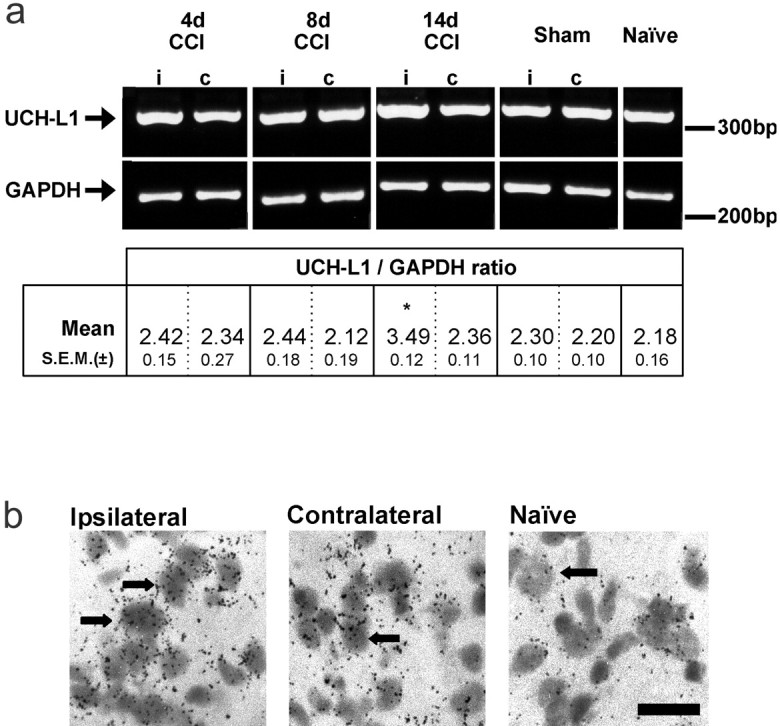
Determination of levels of mRNA for ubiquitin C-terminal hydrolase-L1 (UCH-L1). a, Relative abundance of UCH-L1 mRNA from spinal cord of CCI-treated rats as assessed by semiquantitative RT-PCR normalized to the signal obtained for the cellular housekeeping enzyme GAPDH. Densitometric analysis indicated that the relative UCH-L1 expression was similar in all conditions except in tissue ipsilateral to CCI at 14 d, where expression was significantly greater than that in contralateral samples (*p < 0.05 by paired Student's ttest; n = 3). Further analysis of the changes at 14 d after CCI was made by in situ methods. Seeb and Table 2 for quantification data. b,In situ hybridization histochemistry to show UCH-L1 mRNA expression in lamina I of the rat medial dorsal horn. High-power, light-field photomicrographs showing that expression of mRNA for UCH-L1 was higher ipsilateral to CCI after 14 d, compared with contralateral tissue and naive dorsal horn. Scale bar, 100 μm.
In situ hybridization histochemistry
In accordance with the expression of UCH-L1 protein throughout eukaryotic neural and neuroendocrine cell types and in line with its role in proteasomal functioning, the mRNA for rat UCH-L1 was distributed widely in spinal cord of both naive and neuropathic animals. After CCI, there were distinct changes in the expression of mRNA for UCH-L1 in the dorsal horn ipsilateral to nerve injury, compared with contralateral and naive values. The total number of neurons positively expressing mRNA for UCH-L1 was significantly increased in the ipsilateral dorsal horn, both medially in laminas I, II, and III and laterally in laminas I and II (Fig. 3b; Table 2). Silver grain density was significantly increased mediolaterally in laminas I, II, III, and V and laterally in laminas I, II, and III (Fig. 3b; Table 2). The significant increments in numbers of hybridizing cells and silver grain density ranged from 22–28 to 17–41%, respectively. Within the ventral horn of normal animals, labeling of motoneurons was especially intense but showed no significant change after CCI treatment (data not shown).
Table 2.
In situ hybridization histochemistry for UCH-L1 mRNA expression after CCI
| Laminas | Mediolateral | Lateral | ||||
|---|---|---|---|---|---|---|
| CCI/ipsilateral | CCI/contralateral | Naive | CCI/ipsilateral | CCI/contralateral | Naive | |
| Mean number of hybridizing cells per section in spinal dorsal horn | ||||||
| LI | 35.5 ± 1.2* | 26.6 ± 0.5 | 25.9 ± 1.1 | 32.2 ± 1.4* | 24.4 ± 1.1 | 22.5 ± 2.5 |
| LII | 35.2 ± 1.8* | 25.5 ± 1.2 | 25.5 ± 1.4 | 34.2 ± 1.8* | 28.2 ± 2.1 | 30.1 ± 1.3 |
| LIII | 37.1 ± 1.9* | 30.5 ± 1.2 | 30.5 ± 1.5 | 36.5 ± 3.1 | 32.5 ± 2.5 | 35.6 ± 2.2 |
| LIV | 42.2 ± 2.2 | 38.2 ± 3.2 | 38.8 ± 3.4 | 41.1 ± 2.1 | 40.5 ± 1.4 | 40.1 ± 1.3 |
| LV | 38.9 ± 1.4 | 36.5 ± 1.8 | 37.5 ± 2.2 | 32.5 ± 1.2 | 32.1 ± 0.6 | 32.4 ± 1.0 |
| Mean number of silver grains per cell | ||||||
| LI | 14.5 ± 1.3* | 10.3 ± 0.8 | 8.8 ± 1.3 | 12.4 ± 1.3* | 9.2 ± 1.2 | 8.8 ± 0.4 |
| LII | 14.5 ± 1.2* | 11.6 ± 0.8 | 11.1 ± 1.2 | 13.6 ± 1.2* | 10.3 ± 0.9 | 10.0 ± 0.6 |
| LIII | 29.8 ± 2.3* | 21.5 ± 1.6 | 19.2 ± 2.3 | 29.5 ± 2.5* | 24.8 ± 2.1 | 24.8 ± 1.9 |
| LIV | 39.4 ± 2.2 | 35.1 ± 2.1 | 34.9 ± 2.2 | 35.8 ± 2.2 | 33.8 ± 1.8 | 32.0 ± 1.9 |
| LV | 40.0 ± 1.8* | 31.0 ± 2.1 | 32.0 ± 1.8 | 39.2 ± 3.1* | 33.4 ± 2.1 | 32.0 ± 2.0 |
Data show both the mean number of cells expressing UCH-L1 mRNA and the mean density of silver grains. Ipsilateral values were compared with both the contralateral side in CCI animals taken at peak behavioral change and with naive, unoperated values (
p < 0.05; one-way ANOVA, followed by a Dunnett's post hoc test). For all laminas analyzed, there was no significant alteration in the relative expression of UCH-L1 mRNA when comparing contralateral CCI values with naive values.
Western blot analysis to monitor changes in protein levels of UCH-L1 after CCI
To determine whether the increase in UCH-L1 mRNA after CCI leads to an overall increase in the translation of its protein product, we performed Western blot analysis using an antibody specific to UCH-L1. Densitometry of immunoreactive bands revealed an ipsilateral increase of 27–31% after CCI when compared with contralateral, sham-operated, and naive sample values. Samples were normalized in relation to levels of the established cellular housekeeper enzyme GAPDH, and values were expressed as the percentage relative GAPDH expression (Fig.4a,b).
Fig. 4.

Western blot analysis of the expression of UCH-L1 protein. Western blots of spinal cord samples from neuropathic, naive, and sham-operated rats. a shows an increase in UCH-L1 protein expression ipsilateral (Ipsi) to CCI in neuropathic animals. No change from naive samples was observed in the sham-operated or contralateral side (Contra) of CCI samples. b represents UCH-L1 expression as a percentage of GAPDH expression in terms of relative gray scale values after quantitative densitometry of ECL films. Data are presented as mean ± SEM (n = 3; *p < 0.05, from control contralateral values; Student's paired ttest).
Regulation of constitutive spinal cord PKA activity by the ubiquitin–proteasome system
To obtain a readout of the function of the ubiquitin–proteasome system in spinal cord, we monitored the enzymatic activity of PKA, the activity of which is known to be regulated through degradation of the regulatory subunits by the proteasome (Hegde et al., 1997; Chain et al., 1999). Degradation of regulatory subunits, leaving a relative excess of unrestrained catalytic subunits, is thought to lead to an elevated level of constitutive enzyme activity, which can be monitored by ex vivo enzyme assays (Roskoski, 1983). Table3 shows that the fraction of PKA activity that was constitutive was significantly elevated in spinal cord ipsilateral to CCI and that this elevation was prevented by local administration of the selective proteasome inhibitors epoxomicin and MG-132 to the spinal cord. This matches our other molecular and physiological data in suggesting that the activity of the ubiquitin–proteasome system is elevated in response to nerve injury.
Table 3.
Effects of the topical administration of the proteasome inhibitors epoxomicin and MG-132 on constitutive activation of protein kinase A induced by CCI
| Topical administration | PKA activity of spinal cord homogenates (constitutive activity as % of total) | ||
|---|---|---|---|
| Naive control | CCI | ||
| Ipsilateral | Contralateral | ||
| Nil | 9.5 ± 1.0 | 15.7 ± 1.23-150 | 8.5 ± 0.5 |
| Vehicle | 7.9 ± 0.8 | 14.4 ± 1.53-150 | 9.1 ± 0.5 |
| Epoxomicin | 8.4 ± 1.0 | 9.9 ± 0.83-151 | 8.1 ± 1.1 |
| MG-132 | — | 7.8 ± 0.23-151 | 8.6 ± 0.5 |
After topical administration under anesthesia of 500 μl of epoxomicin (15 μm), MG-132 (100 μm), or vehicle (0.5% dimethylformamide in saline), spinal segments (L3–L6) were removed and hemisected, before homogenization in cold buffer. Samples were assayed for authentic PKA [33P] phosphotransferase activity under constitutive and maximal cAMP-evoked conditions. Total authentic PKA activity was in the order of 100–200 pmol · min−1 · mg−1 tissue under these conditions and similar between ipsilateral and contralateral CCI tissue. Values are expressed as means ± SEM (n = 6–8).
F3-150: indicates significantly greater than corresponding contralateral CCI values and naive control values (p < 0.05), by Wilcoxon test and Mann–Whitney U test, respectively.
F3-151: indicates significantly less than corresponding vehicle-treated tissue ipsilateral to CCI (p < 0.05 by Mann–Whitney U test).
Effects of proteasome inhibitor on CCI-induced translocation of [3H] phorbol dibutyrate binding sites
To investigate whether CCI-induced activation of PKC is altered by inhibition of the proteasome, we measured cytosol/membrane translocation of specific [3H] phorbol dibutyrate binding sites after topical administration of the proteasome inhibitor epoxomicin. In accordance with previous reports (Mao et al., 1993a,b), we found clear increases in specific binding of [3H] phorbol dibutyrate to the membrane rather than cytosolic fraction of tissue ipsilateral to CCI (Table4). Smaller but still statistically significant increases were seen also on the contralateral side. Intrathecal administration of epoxomicin caused no detectable alteration in the translocation of [3H] phorbol dibutyrate binding sites (either ipsilateral or contralateral to CCI) under conditions in which it was shown to reduce the ipsilateral increase in constitutive activity of PKA (Tables 3, 4).
Table 4.
Lack of effect of topical administration of the proteasome inhibitor epoxomicin on membrane translocation/activation of protein kinase C
| Topical administration | Specific [3H]PDBu binding (% associated with membrane fraction) | ||
|---|---|---|---|
| Naive control | CCI | ||
| Ipsilateral | Contralateral | ||
| Vehicle | 24.0 ± 3.7 | 61.4 ± 6.54-150 | 46.0 ± 4.94-150 |
| Epoxomicin | 26.2 ± 2.9 | 61.0 ± 4.84-150 | 48.5 ± 6.14-150 |
After topical administration of epoxomicin or vehicle under anesthesia (as in Table 3), spinal cord segments L3–L6 were removed and hemisected before homogenization in ice-cold buffer. Samples were assayed for membrane-bound and cytosolic specific [3H]PDBu binding sites. Total recovered specific binding was in the order of 6000 dpm per sample aliquot and was similar in all conditions tested. Values are means ± SEM (n = 5).
F4-150: indicates significantly greater than naive control values (p < 0.05 by Mann–Whitney U test). PDBu, Phorbal dibutyrate.
DISCUSSION
Injury in afferent nerves can elicit their sustained firing and result in phenotypic and functional changes both within dorsal root ganglia and in the dorsal horn of the spinal cord. The resulting state of central sensitization plays a key role in bringing about the hyperalgesia and allodynia, as well as sensitivity to cold, that characterize neuropathic pain states. We now provide evidence to suggest that the ubiquitin–proteasome system is important in the cellular mechanisms underlying neuropathic sensitization after CCI. The proteasome also appears to play a role in the normal spinal processing of noxious, but not innocuous, sensory stimuli. After CCI, previously innocuous stimuli such as brush and cold are perceived as noxious and correspondingly show sensitivity to blockade by proteasome inhibitors. Both the behavioral reflex studies and especially the electrophysiological experiments in animals with established CCI sensitization showed a rapid onset of activation of the proteasome inhibitors. Several structurally distinct proteasome inhibitors exhibited similar effects, suggesting that they are effective because of their actions on the proteasome. The rapidity of action suggests that ongoing degradation of a key signaling regulator (such as PKA regulatory subunits) is actively essential for the maintenance of established neuropathic sensitization. The contribution of the ubiquitin–proteasome system to sensitization was closely paralleled by increased expression (ipsilateral to nerve injury) of UCH-L1 at the level of RT-PCR, in situ hybridization (ISHH), and immunoblot. The steady-state elevation of UCH-L1 expression observed here at 14 d after CCI is less than the acute rise seen within 4 hr after LTF in Aplysia (Hegde et al., 1997). This may relate in part to the higher basal levels of UCH-L1 in mammalian neurons (Wilkinson et al., 1989). Furthermore, the CCI sensitization model differs from LTF in that changes in spinal neuronal function develop slowly and are then maintained over many days. If optimal UCH-L1 activity is required for operation of the ubiquitin–proteasome system and this pathway plays a greater functional role after CCI (as demonstrated) then even small changes in UCH-L1 such as the increment of ∼30% seen in RT-PCR, ISHH, and immunoblot methods may be functionally significant. We do not know whether the level of UCH-L1 expression is rate limiting for proteasome function here, but if that is the case (as appears to be so in Aplysia LTF), then the requirement for ongoing proteasome activity shown by the inhibitor results suggests that small increases might well result in functional change. The time course of the increase in UCH-L1 mRNA ipsilateral to CCI matched closely the development of behavioral reflex sensitization that we observed, being clearly established by 14 d after nerve ligation. No significant increases were detected at 4 or 8 d after surgery, times at which thermal hyperalgesia, mechanical allodynia, and cold allodynia were undetectable and showed <25% development of maximal effect, respectively, in these experiments. UCH-L1 plays an essential role in the recycling of ubiquitin, which is necessary to maintain adequate rates of proteasome-mediated degradation of proteins. The regulatory subunits of PKA are an example of such a known proteasome target, and their degradation is thought to lead to the formation of constitutively active PKA. In accordance with our other data, elevated levels of constitutively active PKA were found in spinal cord ipsilateral to CCI, and this increase was reversed by acute local administration of a selective proteasome inhibitor. No changes were observed in the CCI-induced cytosol to membrane translocation of PKC, an alternative signal transduction mediator, which is not considered to be directly influenced by proteasome function.
Activity-dependent synaptic changes, associated with altered states of responsiveness and “synaptic memory,” have been investigated in LTF in Aplysia (Bailey and Kandel, 1993) and in LTP in central mammalian neurons (Bliss and Collingridge, 1993). Although the precise nature and origin of these alterations differ, they may share a number of common features with the central sensitization that occurs within the spinal cord in response to chronic activation of nociceptive inputs.
Neuropathic sensitization is similar to other forms of repetitive input-induced sensitization in spinal cord (such as wind-up and inflammatory sensitization) in its dependence on NMDA receptor function (Mao et al., 1993a,b; Tal and Bennett, 1993). Although distinct neurochemical changes in these different pain states suggest that the mechanisms leading to NMDA receptor activation may differ, the NMDA receptor appears to be crucial to the sensitization phenomenon. It is not yet clear whether all forms of spinal sensitization rely on proteasome function as neuropathic sensitization does. However, the attenuation by proteasome inhibitors of both sub-acute mustard oil-evoked responses and neuropathic sensitization (which have in common a dependence on NMDA receptor-mediated events) (Heppenstall and Fleetwood-Walker, 1997) suggests that the role of the proteasome may relate closely to NMDA receptor activation. Whether the proteasome plays a role in processes leading to NMDA receptor activation or in events downstream of either the Ca2+entry-mediated or adapter protein-mediated signaling of the receptor is not yet clear. Nevertheless, the proteasome clearly represents an interesting new target in neuropathic and other forms of pain.
PKA is implicated in spinal sensitization and pain (Cerne et al., 1993;Sluka, 1997). PKA may be activated downstream of receptors that lead to cAMP production (or downstream of Ca2+-mobilizing receptors, if Ca2+-activated isoforms of adenylate cyclase are present) and among many other targets is known to phosphorylate and activate both NMDA- and AMPA-type glutamate receptors (Greengard et al., 1991; Roche et al., 1996; Westphal et al., 1999). Although mutant mice with a targeted deletion of one of the PKA regulatory subunits did not show any attenuation of neuropathic sensitization (Malmberg et al., 1997), the fact that PKA catalytic activity is normally restrained by the presence of regulatory subunits makes this difficult to interpret. In the LTF model inAplysia, PKA appears to play a key role in increased responsiveness, and substantial evidence indicates that an imbalance of PKA regulatory and catalytic subunits, mediated by proteasome degradation of the former, is a crucial factor (Hegde et al., 1993;Chain et al., 1999). This matches closely the proteasome-dependent generation of constitutively active PKA that we observed ipsilateral to CCI (Table 3). In the Aplysia model, expression of the homolog of UCH-L1 is rapidly increased in the facilitation paradigm (Hegde et al., 1997), again closely paralleling the observations here with neuropathic sensitization, except that the full development of the sensitized state here occurs over a more prolonged time period. An alternative pathway that is known to be subject to proteasome regulation is that of the transcription factor, nuclear factor κB (NF-κB). NF-κB exists in a latent form in unstimulated cells, complexed to the inhibitory protein, I-κB. Inflammation-associated molecules, such as cytokines, can induce I-κB phosphorylation, leading to its ubiquitination and degradation by the proteasome (Karin and Delhase, 2000). Dissociated NF-κB can then regulate the expression of various target genes, including the inducible enzymes cyclo-oxygenase-2 and nitric oxide synthase, adhesion molecules, cytokines, and neuropeptides (O'Neill and Kaltschmidt, 1997). Although NF-κB expression decreases acutely after nerve injury (Doyle and Hunt, 1997), NF-κB immunoreactivity has been reported to increase within ipsilateral DRG neurons 2 weeks after nerve injury (Ma and Bisby, 1998), perhaps in response to cytokines and trophic factors secreted by non-neuronal cells during Wallerian degeneration. In cerebellar granule neurons, NF-κB expression appears to be regulated by glutamate acting via NMDA receptors (Guerrini et al., 1995). In pilot experiments with intrathecal administration of the selective NF-κB inhibitor parthenolide (Alexis) to CCI rats (1.5 nmol/50 μl 0.3% dimethylformamide in saline), we found no detectable change in behavioral responses to heat, cold, or mechanical stimuli (data not shown). This suggests that although certain proteasome targets like PKA may play an important role in neuropathic sensitization, others like the NF-κB/I-κB complex may not be important. Nevertheless, more extensive studies on these and other pathways will be necessary before it is clear how the proteasome plays out its key role in enabling neuropathic sensitization.
In conclusion, we have unexpectedly shown that proteasome inhibitors can selectively inhibit neuropathic sensitization, which is likely to underlie the development of chronic intractable pain after nerve injury. Importantly, proteasome inhibitors attenuate neuropathic allodynia without impairing normal sensory responses to low-intensity peripheral stimuli, a particularly advantageous therapeutic profile. Expression of a key enzyme in ubiquitin–proteasome function is increased in spinal dorsal horn ipsilateral to injury, as is the activity of a known target of the complex (PKA), which is expected to display deregulated constitutive activity after proteasome action. Proteasome-dependent activation of PKA in neuropathic sensitization closely parallels that in LTF in Aplysia. Proteasome inhibitors are under phase I trials as anti-cancer drugs (Lee and Goldberg, 1998). In many cases, advanced development of tumors leads to local inflammation and pressure trauma to afferent nerves, resulting in a neuropathic component to cancer pain. The present study predicts that in addition to any direct effect on cancer cells, proteasome inhibitors would exert a useful additional role in attenuating the central sensitization that leads to chronic hyperalgesia and allodynia.
Footnotes
This work was supported by The Wellcome Trust (S.F.-W.) and by The Medical Research Council and the University of Edinburgh for the award of Studentships (A.M. and E.G., respectively). We thank staff at Wellcome Animal Research Unit and Medical Faculty Animal Area facilities for animal husbandry.
Correspondence should be addressed to Dr. S. M. Fleetwood-Walker, Department of Preclinical Veterinary Sciences, University of Edinburgh, Edinburgh EH9 1QH, UK. E-mail: sfw@vet.ed.ac.uk.
G. Blackburn-Munro's present address: NeuroSearch A/S, 93 Pederstrupvej, #DK-2750, Ballerup, Denmark.
T. Dickinson's present address: Department of Pharmacology, Quintiles Scotland Ltd., Heriot-Watt University Research Park, Riccarton, Edinburgh EH14 4AP, UK.
REFERENCES
- 1.Bailey CH, Kandel ER. Structural changes accompanying memory storage. Annu Rev Physiol. 1993;55:397–426. doi: 10.1146/annurev.ph.55.030193.002145. [DOI] [PubMed] [Google Scholar]
- 2.Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 3.Blackburn-Munro G, Fleetwood-Walker SM. The effects of Na+ channel blockers on somatosensory processing by rat dorsal horn neurons. NeuroReport. 1997;8:1549–1554. doi: 10.1097/00001756-199705060-00001. [DOI] [PubMed] [Google Scholar]
- 4.Blackburn-Munro G, Fleetwood-Walker SM. The sodium channel auxiliary subunits beta1 and beta2 are differentially expressed in the spinal cord of neuropathic rats. Neuroscience. 1999;90:153–164. doi: 10.1016/s0306-4522(98)00415-1. [DOI] [PubMed] [Google Scholar]
- 5.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 6.Cameron AA, Cliffer KD, Dougherty PM, Garrison CJ, Willis WD, Carlton SM. Time course of degenerative and regenerative changes in the dorsal horn in a rat model of peripheral neuropathy. J Comp Neurol. 1997;379:428–442. doi: 10.1002/(sici)1096-9861(19970317)379:3<428::aid-cne8>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 7.Cerne R, Rusin KI, Randic M. Enhancement of the N-methyl-d-aspartate response in spinal dorsal horn neurons by cAMP-dependent protein kinase. Neurosci Lett. 1993;161:124–128. doi: 10.1016/0304-3940(93)90275-p. [DOI] [PubMed] [Google Scholar]
- 8.Chain DG, Casadio A, Schacher S, Hegde AN, Valbrun M, Yamamoto N, Goldberg AL, Bartsch D, Kandel ER, Schwartz JH. Mechanisms for generating the autonomous cAMP-dependent protein kinase required for long-term facilitation in Aplysia. Neuron. 1999;22:147–156. doi: 10.1016/s0896-6273(00)80686-8. [DOI] [PubMed] [Google Scholar]
- 9.Chung K, Lee BH, Yoon YW, Chung JM. Sympathetic sprouting in the dorsal root ganglia of the injured peripheral nerve in a rat neuropathic pain model. J Comp Neurol. 1996;376:241–252. doi: 10.1002/(SICI)1096-9861(19961209)376:2<241::AID-CNE6>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 10.Corbin JD. Determination of the cAMP-dependent protein kinase activity ratio in intact tissues. Methods Enzymol. 1983;99:227–232. doi: 10.1016/0076-6879(83)99057-2. [DOI] [PubMed] [Google Scholar]
- 11.Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem. 1996;65:801–847. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- 12.Dickinson T, Mitchell R, Robberecht P, Fleetwood-Walker SM. The role of VIP/PACAP receptor subtypes in spinal somatosensory processing in rats with an experimental peripheral mononeuropathy. Neuropharmacology. 1999;38:167–180. doi: 10.1016/s0028-3908(98)00171-3. [DOI] [PubMed] [Google Scholar]
- 13.Doyle CA, Hunt SP. Reduced nuclear factor kappa B (p65) expression in rat primary sensory neurons after peripheral nerve injury. NeuroReport. 1997;8:2937–2942. doi: 10.1097/00001756-199709080-00026. [DOI] [PubMed] [Google Scholar]
- 14.Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactaystin. Science. 1995;268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- 15.Greenberg SM, Castellucci VF, Bayley H, Schwartz JH. A molecular mechanism for long-term sensitization in Aplysia. Nature. 1987;329:62–65. doi: 10.1038/329062a0. [DOI] [PubMed] [Google Scholar]
- 16.Greengard P, Jen J, Nairn AC, Stevens CF. Enhancement of the glutamate response by cAMP-dependent protein kinase in hippocampal neurons. Science. 1991;253:1135–1138. doi: 10.1126/science.1716001. [DOI] [PubMed] [Google Scholar]
- 17.Guerrini L, Blasi F, Denis-Donini S. Synaptic activation of NF-kappa B by glutamate in cerebellar granule neurons in vitro. Proc Natl Acad Sci USA. 1995;92:9077–9081. doi: 10.1073/pnas.92.20.9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hegde AN, Goldberg AL, Schwartz JH. Regulatory subunits of cAMP-dependent protein kinases are degraded after conjugation to ubiquitin: a molecular mechanism underlying long-term synaptic plasticity. Proc Natl Acad Sci USA. 1993;90:7436–7440. doi: 10.1073/pnas.90.16.7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hegde AN, Inokuchi K, Pei W, Casadio A, Ghirardi M, Chain DG, Martin KC, Kandel ER, Schwartz JH. Ubiquitin C-terminal hydrolase is an immediate-early gene essential for long-term facilitation in Aplysia. Cell. 1997;89:115–126. doi: 10.1016/s0092-8674(00)80188-9. [DOI] [PubMed] [Google Scholar]
- 20.Heppenstall PA, Fleetwood-Walker SM. The glycine site of the NMDA receptor contributes to neurokinin 1 receptor agonist facilitation of NMDA receptor agonist-evoked activity in rat dorsal horn neurons. Brain Res. 1997;744:235–245. doi: 10.1016/S0006-8993(96)01065-7. [DOI] [PubMed] [Google Scholar]
- 21.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 22.Hökfelt T, Zhang X, Wiesenfeld-Hallin Z. Messenger plasticity in primary sensory neurons following axotomy and its functional implications. Trends Neurosci. 1994;17:22–30. doi: 10.1016/0166-2236(94)90031-0. [DOI] [PubMed] [Google Scholar]
- 23.Johnson MS, Simpson J, Mitchell R. Effect of phorbol 12,13-dibutyrate on ligand binding, enzyme activity and translocation of protein kinase C isoforms in the alpha T3–1 gonadotrope-derived cell line. Mol Cell Biochem. 1996;165:65–75. doi: 10.1007/BF00229746. [DOI] [PubMed] [Google Scholar]
- 24.Kajimoto Y, Hashimoto T, Shirai Y, Nishino N, Kuno T, Tanaka C. cDNA cloning and tissue distribution of a rat ubiquitin carboxyl-terminal hydrolase PGP 9.5. J Biochem (Tokyo) 1992;112:28–32. doi: 10.1093/oxfordjournals.jbchem.a123860. [DOI] [PubMed] [Google Scholar]
- 25.Karin M, Delhase M. The I kappa B kinase (IKK), NF-kappa B: key elements of proinflammatory signaling. Semin Immunol. 2000;12:85–98. doi: 10.1006/smim.2000.0210. [DOI] [PubMed] [Google Scholar]
- 26.Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 1998;8:397–403. doi: 10.1016/s0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- 27.Ma W, Bisby MA. Increased activation of nuclear factor kappa B in rat lumbar dorsal root ganglion neurons following partial sciatic nerve injuries. Brain Res. 1998;797:243–254. doi: 10.1016/s0006-8993(98)00380-1. [DOI] [PubMed] [Google Scholar]
- 28.Malmberg AB, Brandon EP, Idzerda RL, Liu H, McKnight GS, Basbaum AI. Diminished inflammation and nociceptive pain with preservation of neuropathic pain in mice with a targeted mutation of the type I regulatory subunit of cAMP-dependent protein kinase. J Neurosci. 1997;17:7462–7470. doi: 10.1523/JNEUROSCI.17-19-07462.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mao J, Price DD, Hayes RL, Lu J, Mayer DJ, Frenk H. Intrathecal treatment with dextrorphan or ketamine potently reduces pain-related behaviors in a rat model of peripheral mononeuropathy. Brain Res. 1993a;605:164–168. doi: 10.1016/0006-8993(93)91368-3. [DOI] [PubMed] [Google Scholar]
- 30.Mao J, Mayer DJ, Hayes RL, Price DD. Spatial patterns of increased spinal cord membrane-bound protein kinase C and their relation to increases in 14C-2-deoxyglucose metabolic activity in rats with painful peripheral mononeuropathy. J Neurophysiol. 1993b;70:470–481. doi: 10.1152/jn.1993.70.2.470. [DOI] [PubMed] [Google Scholar]
- 31.Mayer DJ, Mao J, Holt J, Price DD. Cellular mechanisms of neuropathic pain, morphine tolerance, and their interactions. Proc Natl Acad Sci USA. 1999;96:7731–7736. doi: 10.1073/pnas.96.14.7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Medhurst AD, Harrison DC, Read SJ, Campbell CA, Robbins MJ, Pangalos MN. The use of TaqMan RT-PCR assays for semiquantitative analysis of gene expression in CNS tissues and disease models. J Neurosci Methods. 2000;98:9–20. doi: 10.1016/s0165-0270(00)00178-3. [DOI] [PubMed] [Google Scholar]
- 33.O'Neill LA, Kaltschmidt C. NF-kappa B: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997;20:252–258. doi: 10.1016/s0166-2236(96)01035-1. [DOI] [PubMed] [Google Scholar]
- 34.Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 35.Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg AL. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78:761–771. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 36.Rolfe M, Chiu MI, Pagano M. The ubiquitin-mediated proteolytic pathway as a therapeutic area. J Mol Med. 1997;75:5–17. doi: 10.1007/s001090050081. [DOI] [PubMed] [Google Scholar]
- 37.Roskoski R. Assays of protein kinase. Methods Enzymol. 1983;99:3–6. doi: 10.1016/0076-6879(83)99034-1. [DOI] [PubMed] [Google Scholar]
- 38.Sin N, Kim KB, Elofsson M, Meng L, Auth H, Kwok BH, Crews CM. Total synthesis of the potent proteasome inhibitor epoxomicin: a useful tool for understanding proteasome biology. Bioorg Med Chem Lett. 1999;9:2283–2288. doi: 10.1016/s0960-894x(99)00376-5. [DOI] [PubMed] [Google Scholar]
- 39.Sluka KA. Activation of the cAMP transduction cascade contributes to the mechanical hyperalgesia and allodynia induced by intradermal injection of capsaicin. Br J Pharmacol. 1997;122:1165–1173. doi: 10.1038/sj.bjp.0701486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tal M, Bennett GJ. Dextrorphan relieves neuropathic heat-evoked hyperalgesia in the rat. Neurosci Lett. 1993;151:107–110. doi: 10.1016/0304-3940(93)90058-s. [DOI] [PubMed] [Google Scholar]
- 41.Walters ET, Ambron RT. Long-term alterations induced by injury and by 5-HT in Aplysia sensory neurons: convergent pathways and common signals? Trends Neurosci. 1995;18:137–142. doi: 10.1016/0166-2236(95)93891-z. [DOI] [PubMed] [Google Scholar]
- 42.Westphal RS, Tavalin SJ, Lin JW, Alto NM, Fraser ID, Langeberg LK, Sheng M, Scott JD. Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science. 1999;285:93–96. doi: 10.1126/science.285.5424.93. [DOI] [PubMed] [Google Scholar]
- 43.Wilkinson KD. Regulation of ubiquitin-dependent processes by deubiquitinating enzymes. FASEB J. 1997;11:1245–1256. doi: 10.1096/fasebj.11.14.9409543. [DOI] [PubMed] [Google Scholar]
- 44.Wilkinson KD, Lee KM, Deshpande S, Duerksen-Hughes P, Boss JM, Pohl J. The neuron-specific protein PGP 9.5 is a ubiquitin carboxyl-terminal hydrolase. Science. 1989;246:670–673. doi: 10.1126/science.2530630. [DOI] [PubMed] [Google Scholar]
- 45.Woolf CJ, Shortland P, Reynolds M, Ridings J, Doubell T, Coggeshall RE. Reorganization of central terminals of myelinated primary afferents in the rat dorsal horn following peripheral axotomy. J Comp Neurol. 1995;360:121–134. doi: 10.1002/cne.903600109. [DOI] [PubMed] [Google Scholar]