

Abstract
Bradykinin has long been known to excite sympathetic neurons via B2 receptors, and this action is believed to be mediated by an inhibition of M-currents via phospholipase C and inositol trisphosphate-dependent increases in intracellular Ca2+. In primary cultures of rat superior cervical ganglion neurons, bradykinin caused an accumulation of inositol trisphosphate, an inhibition of M-currents, and a stimulation of action potential-mediated transmitter release. Blockade of inositol trisphosphate-dependent signaling cascades failed to affect the bradykinin-induced release of noradrenaline, but prevented the peptide-induced inhibition of M-currents. In contrast, inhibition or downregulation of protein kinase C reduced the stimulation of transmitter release, but not the inhibition of M-currents, by bradykinin. In cultures of superior cervical ganglia, classical (α, βI, βII), novel (δ, ε), and atypical (ζ) protein kinase C isozymes were detected by immunoblotting. Bradykinin induced a translocation of Ca2+-independent protein kinase C isoforms (δ and ε) from the cytosol to the membrane of the neurons, but left the cellular distribution of other isoforms unchanged. This activation of Ca2+-independent protein kinase C enzymes was prevented by a phospholipase C inhibitor. The bradykinin-dependent stimulation of noradrenaline release was reduced by inhibitors of classical and novel protein kinase C isozymes, but not by an inhibitor selective for Ca2+-dependent isoforms. These results demonstrate that bradykinin B2receptors are linked to phospholipase C to simultaneously activate two signaling pathways: one mediates an inositol trisphosphate- and Ca2+-dependent inhibition of M-currents, the other one leads to an excitation of sympathetic neurons independently of changes in M-currents through an activation of Ca2+-insensitive protein kinase C.
Keywords: rat superior cervical ganglion neurons, noradrenaline release, bradykinin, M-type K+ channels, protein kinase C, phospholipase C
Bradykinin is formed through the actions of kallikreins from plasma kininogens in response to various noxious stimuli. The nonapeptide acts as a potent mediator of inflammation and pain through its effects on peripheral sensory neurons. On one hand, the stimulation of nociceptive nerve terminals elicits the painful sensation, on the other hand, bradykinin induces the release of neuropeptides that then contribute to the inflammatory response (Bhoola et al., 1992). Nevertheless, the sympathetic nervous system may also contribute to these actions because bradykinin-induced hyperalgesia (Levine et al., 1986) and plasma extravasation (Miao et al., 1996) are abolished after sympathectomy. In addition, the sympathetic nervous system may also be involved in both the protective as well as the noxious effects that bradykinin has been suggested to exert within the cardiovascular system (Dell'Italia and Oparil, 1999).
Almost 40 years ago, bradykinin was found to cause sympathoexcitation attributable to its depolarizing action on sympathetic ganglia (Lewis and Reit, 1965; Trendelenburg, 1966). For three decades, the mechanisms underlying this effect remained unknown, but then Jones et al. (1995) demonstrated that the peptide depolarized superior cervical ganglion (SCG) neurons of the rat through an inhibition of M-type K+ (KM) channels. These ion channels are opened in the subthreshold voltage range for action potentials and become completely activated when neurons are further depolarized. Hence, activatedKM channels keep neurons polarized, and closure of these ion channels causes depolarization and increased action potential discharge (Brown, 1983; Marrion, 1997). Recently, the signaling cascade mediating the inhibition ofKM channels by bradykinin has also been elucidated: B2 bradykinin receptors are linked to G-proteins of the Gq family, most likely G11, and it is the α subunits that mediate the closure of KM channels (Jones et al., 1995; Haley et al., 1998, 2000a). Via these G-proteins, phospholipase C-β4 is stimulated (Haley et al., 2000b) to mediate the synthesis of inositol trisphosphate (IP3), which then causes liberation of Ca2+ from intracellular stores (Cruzblanca et al., 1998). Finally, cytosolic Ca2+ concentrations in the submicromolar to low micromolar range directly blockKM channels (Selyanko and Brown, 1996).
Apart from inhibiting KM channels, bradykinin was shown to either enhance (Cox et al., 2000) or reduce (Starke et al., 1977) sympathetic transmitter release via presynaptic sites of action, depending on the species and tissues investigated. In primary cultures of rat SCG neurons, activation of B2 bradykinin receptors causes action potential-dependent transmitter release (Boehm and Huck, 1997). This effect is selectively potentiated byKM channel blockers, but not by blockers of other types of K+ channels. In addition, KM channel blockers were found to stimulate transmitter release from sympathetic neurons in the absence of any other secretagogue stimulus (Kristufek et al., 1999). Therefore, it appeared obvious to assume that peptide-induced noradrenaline release was also mediated by the signaling cascade that finally leads to KM channel blockade. However, the present results demonstrate that bradykinin also uses another signaling cascade to excite sympathetic neurons, which includes Ca2+-independent protein kinase C (PKC), but bypasses KM channels.
MATERIALS AND METHODS
Primary cultures of rat superior cervical ganglion neurons. Primary cultures of dissociated SCG neurons from neonatal rats were prepared as described before (Boehm, 1999). Briefly, ganglia were dissected from 2- to 6-d-old Sprague Dawley rat pups, cut into three or four pieces and incubated in collagenase (1.5 mg/ml; catalog #9891; Sigma, Vienna, Austria) and dispase (3.0 mg/ml; catalog #165859; Boehringer Mannheim, Mannheim, Germany) for 20 min at 36°C. Subsequently, the ganglia were trypsinized (0.25% trypsin; catalog #3703; Worthington, Freehold, NJ) for 15 min at 36°C, dissociated by trituration, and resuspended in DMEM (catalog #041–01885M; Invitrogen, Gaithersburg, MD) containing 2.2 gm/l glucose, 10 mg/l insulin, 25000 IU/l penicillin, and 25 mg/l streptomycin (catalog #043–05140D; Invitrogen), 10 μg/l nerve growth factor (catalog #0436050; Invitrogen), and 5% fetal calf serum (catalog #011–0620H; Invitrogen). Thereafter, the cells were plated onto 5 mm discs for superfusion experiments and onto 35 mm culture dishes (catalog #153066; Nunc, Naperville, IL) for electrophysiological experiments, both coated with rat tail collagen (Biomedical Technologies, Stoughton, MA). For the determination of cellular inositol phosphates, cells were plated onto 24 multiwell plates (Nunc; 200,000 cells per well) coated with poly-d-lysine (Sigma). All cultures were kept in a humidified 5% CO2 atmosphere at 36°C for 4–8 d. On day 1 after plating, the medium was completely exchanged, and after 4–5 d, the medium was exchanged again, and the serum was omitted.
Measurement of inositol trisphosphate. Measurement of inositol polyphosphate formation in SCG cultures was determined as described before (Bofill-Cardona et al., 2000). Cultures were prelabeled with 7 μCi/ml of myo-[1,2-3H]inositol for 24 hr. Thirty minutes before the stimulation with bradykinin, the cells were incubated in PBS (137 mm NaCl, 2.7 mm KCl, 4.3 mmNa2HPO4, 1.47 mmKH2PO4, pH 7.4) containing 0.2% bovine serum albumin and 10 mm LiCl. Thereafter, the cells were stimulated with 1 μmbradykinin in PBS containing 10 mm LiCl for 1 min, and the incubations were terminated by replacing the buffer by 0.4 ml of 5% trichloroacetic acid. Extracts were collected, and the trichloroacetic acid was removed by washing two times with four volumes of water-saturated diethyl ether. After neutralization with 20 mm Tris base, samples were placed on Dowex AG 1×8 columns, and fractions containing inositol, inositol monophosphate, inositol diphosphates, and inositol trisphosphates, respectively, were sequentially eluted as described (Bofill-Cardona et al., 2000) and probed for their radioactive contents by liquid scintillation counting (Packard Tri-Carb 2100 TR). The radioactivity in the inositol trisphosphate fraction was expressed as percentage of the radioactivity in the inositol fraction.
Electrophysiology. Experiments were performed at room temperature (20–24°C) on the somata of single SCG neurons using the perforated-patch modification of the patch-clamp technique (Rae et al., 1991), which prevents rundown of IM(Boehm, 1998). Patch pipettes were pulled (Flaming-Brown puller; Sutter Instruments, Novato, CA) from borosilicate glass capillaries (Science Products, Frankfurt/Main, Germany) and front-filled with a solution consisting of (in mm): K2SO4 (75), KCl (55), MgCl2 (8), and HEPES (10), adjusted to pH 7.3 with KOH. Then, electrodes were back-filled with the same solution containing 200 μg/ml amphotericin B (in 0.8% DMSO), which yielded tip resistances of 1–3 MΩ. The bathing solution contained (in mm): NaCl (140), KCl (6.0), CaCl2 (2.0), MgCl2 (2.0), glucose (20), and HEPES (10), adjusted to pH 7.4 with NaOH. Tetrodotoxin (TTX; 0.5 μm) was included to suppress voltage-activated Na+ currents. Bradykinin, thapsigargin, and protein kinase C activators or inhibitors were applied via a DAD-12 drug application device (Adams & List, Westbury, NY), which permits a complete exchange of solutions surrounding the cells under investigation within <100 msec (Boehm, 1999). IM relaxations were evoked once every 20 sec by 1 sec hyperpolarizing voltage steps from −30 to −55 mV; the difference between current amplitudes 20 msec after the onset of hyperpolarizations and 20 msec before re-depolarization was taken as a measure for IM. Amplitudes obtained during the application of test drugs (b) were compared with those measured before (a) and after (c) application of these drugs by calculating 200b/(a+ c) = % of control or 100 − (200b/[a + c]) = % inhibition (Boehm, 1998).
Measurement of [3H]noradrenaline release. [3H]noradrenaline uptake and superfusion were performed as described (Boehm, 1999). Culture discs with dissociated neurons were incubated in 0.05 μm[3H]noradrenaline (specific activity, 52.0 Ci/mmol) in culture medium supplemented with 1 mm ascorbic acid at 36°C for 1 hr. After labeling, culture discs were transferred to small chambers and superfused with a buffer containing (in mm): NaCl (120), KCl (6.0), CaCl2 (2.0), MgCl2 (2.0), glucose (20), HEPES (10), fumaric acid (0.5), Na-pyruvate (5.0), ascorbic acid (0.57), and desipramine (0.001), adjusted to pH 7.4 with NaOH. Superfusion was performed at 25°C at a rate of ∼1.0 ml/min. Collection of 4 min superfusate fractions was started after a 60 min washout period to remove excess radioactivity.
To investigate the mechanisms of bradykinin-evoked noradrenaline release, [3H] overflow was induced by the inclusion of the peptide in the superfusion buffer for 2 min, unless indicated otherwise. Cell-permeable phorbol esters, in contrast, were included for periods of 12 min to stimulate [3H] overflow. Modulatory agents, such as U73122, U73343, thapsigargin, or diverse protein kinase C inhibitors were added to the buffer after 50 min of superfusion (i.e., 10 min before the start of sample collection). The buffer then remained unchanged until the end of experiments. To control for unspecific effects of these modulatory agents, tritium overflow was also elicited by the application of 36 monophasic rectangular electrical pulses (0.5 msec, 50 mA, 50 V/cm) at 0.3 Hz. At the end of experiments, the radioactivity remaining in the cells was extracted by immersion of the discs in 2% (v/v) perchloric acid followed by sonication. Radioactivity in extracts and collected fractions was determined by liquid scintillation counting (Packard Tri-Carb 2100 TR). Radioactivity released in response to electrical field stimulation from rat sympathetic neurons after labeling with tritiated noradrenaline under conditions similar to those of the present study had previously been shown to consist predominantly of the authentic transmitter and to contain only small amounts (≤15%) of metabolites (Schwartz and Malik, 1993). Hence, the outflow of tritium measured in this study was assumed to reflect the release of noradrenaline and not that of metabolites.
The spontaneous (unstimulated) rate of [3H] outflow was obtained by expressing the radioactivity of a collected fraction as percentage of the total radioactivity in the cultures at the beginning of the corresponding collection period. Stimulation-evoked tritium overflow was calculated as the difference between the total [3H] outflow during and after stimulation and the estimated basal outflow that was assumed to decline linearly throughout experiments. Therefore, basal outflow during periods of stimulation was assumed to equate the arithmetic mean of the samples preceding and those after stimulation, respectively. The difference between the total and the estimated basal outflow was expressed as a percentage of the total radioactivity in the cultures at the beginning of the respective stimulation (% of total radioactivity). The amount of bradykinin-evoked tritium release varies considerably between different preparations of rat SCG. Therefore, tritium overflow in the presence of release modulating agents, such as thapsigargin or PKC inhibitors, was always compared with that obtained within the same SCG preparation in the absence of these drugs. To directly compare effects of different modulatory agents on bradykinin-evoked overflow as determined in different preparations, the values obtained in the presence of these modulators were expressed as percentage of the corresponding control values within the same preparation.
Immunoblotting. After 7 d in culture, SCG cells were washed once with ice-cold PBS, and then 100 μl boiling Laemmli sample buffer were added. Before this, the cells were incubated in 1 μm 4-α-phorbol, phorbol-12-myristate-13-acetate (PMA) in culture medium for the indicated periods of time, when appropriate. As control tissue, whole brains or hearts were dissected from 3- to 6-d-old rats, cut into small pieces, frozen with liquid nitrogen, and homogenized in lysis buffer (20 mm Tris-HCl, 0.5 mmEGTA, 2 mm EDTA, 2 mmdithiotreitol, 0.5 mm p-methylsulfonyl fluoride, 10 μg/ml leupeptin, pH 7.5). These rat brain and heart preparations were diluted in Laemmli sample buffer to yield a concentration of ∼1 μg/μl protein. Approximately equal amounts (as determined by staining with Ponceau S) of SCG cell lysate and rat brain or heart lysate were separated by electrophoresis through SDS polyacrylamide minigels, transferred to nitrocellulose, and tested for different PKC isoforms with the following antibodies (Santa Cruz Biotechnology, Santa Cruz, CA): anti-PKC α (SC-8393), anti-PKC βI (SC-209), anti-PKC βII (SC210-G), anti-PKC γ (SC-211), anti-PKC ε (C-15), anti-PKC δ (SC-213), anti-PKC ζ (C-20), and anti-PKC μ (A-20). For visualization, horseradish peroxidase-linked anti-mouse (1:10,000), anti-rabbit (1:20,000; both from Amersham Biosciences, Freiburg, Germany), and anti-goat (1:30,000) IgGs (Santa Cruz Biotechnology) as well as the SuperSignal reagent (Pierce, Rockford, IL) were used.
Immunocytochemistry. Our routine methods of immunocytochemical staining of SCG neurons have been described before (Boehm, 1999). Cultures were washed with PBS, exposed to different stimuli (1 μm bradykinin for 30 sec or 0.1 μm PMA for 10 min), and immediately fixed with 4% paraformaldehyde in PBS for 20 min. After permeabilization with 0.1% Triton X-100 (in PBS for 5 min) and incubation in PBS containing 2% bovine serum albumin and 5% horse serum, cultures were stained first with PKC isoform-specific primary antibodies (as above; dilution 1:200) and then with CY3-conjugated secondary antibodies (dilution 1:1000). Images of the immunostained SCG neurons were obtained using a confocal laser-scanning microscope (LSM 410; Zeiss, Oberkochen, Germany) and stored digitally. Cells incubated only in secondary antibodies were used as controls for the specificity of immunoreactivity.
Statistics. Results are presented as arithmetic means ± SEM; n = number of cultures in inositol phosphate assays and release experiments, and of single cells in electrophysiological experiments. Significances of differences between data points were evaluated by the nonparametric Mann–WhitneyU test.
Materials.(−)-[7,8-3H]Noradrenaline was obtained from Amersham Biosciences; amphotericin B, bradykinin, thapsigargin, and desipramine from Sigma; PMA, GF 109203X, staurosporine, as well as GÖ 7874, GÖ 6983, and GÖ 6976 from Calbiochem (Bad Soden, Germany); U73122 and U73343 from Research Biochemicals (Natick, MA); bulk chemicals were from Merck (Vienna, Austria). Water-insoluble drugs were first dissolved in DMSO and then diluted into buffer to yield final DMSO concentrations of up to 0.1%, which were also included in control solutions. At these concentrations, DMSO did not affect any of the parameters investigated.
RESULTS
Bradykinin causes accumulation of inositol trisphosphate, inhibition of IM, and stimulation of noradrenaline release
Previously, three major types of excitatory cellular responses have been observed with bradykinin in rat SCG neurons: (1) accumulation of IP3 (del Rio et al., 1999) accompanied by increases in intracellular Ca2+(Cruzblanca et al., 1998), (2) inhibition ofKM channels (Jones et al., 1995), and (3) induction of transmitter release (Boehm and Huck, 1997). To corroborate these results, SCG cultures were first labeled with [3H]myoinositol and then stimulated with 1 μm bradykinin for 1 min. This led to a threefold increase in the accumulation of radioactivity within the fraction of IP3. When phospholipase C was blocked by U73122 (1 μm), this bradykinin-dependent accumulation of IP3 was reduced (Fig.1A). Second, SCG neurons were patch-clamped at a membrane potential of –30 mV to activate IM, and 1 sec hyperpolarizations to −55 mV were applied to causeIM deactivation. Outward currents as well as IM deactivation were greatly reduced in the presence of 1 μm bradykinin (Fig. 1B). Third, SCG cultures were labeled with [3H]noradrenaline, superfused with a physiological salt solution, and exposed to 1 μm bradykinin for 10 min. During these 10 min, there was a transient increase in the rate of tritium outflow (Fig.1C).
Fig. 1.

Effects of bradykinin in rat SCG neurons.A, Generation of IP3 in response to 1 μm bradykinin applied for 60 sec in either the presence or absence of 1 μm of the phospholipase C inhibitorU73122. Radioactivity in the IP3 fraction was calculated as percentage of the radioactivity in the fraction of inositol, and the values obtained in the presence of bradykinin are expressed as percentage of the values obtained in its absence. **p < 0.01 between the effects of bradykinin in the absence and presence of U73122; n = 7–11.B, IM was measured by the amphotericin B-perforated patch technique in a SCG neuron, and the current traces shown were obtained by clamping the cell at −30 mV and by applying 1 sec hyperpolarizing voltage steps to −55 mV; the recordings were performed before (control), during (bradykinin), and after (washout) the application of 1 μmbradykinin. C, Primary cultures of SCG were labeled with [3H]noradrenaline and superfused. Subsequent to a 60 min washout period, 2 min fractions of superfusate were collected, and 1 μm bradykinin was present for 10 min, as indicated by the bar. The graph shows the time course of fractional [3H] outflow calculated as percentage of the total radioactivity in the cells; n = 6.
The observation of these three effects in parallel led us to investigate whether they also are causally interrelated. Because the accumulation of IP3 was virtually abolished by the phospholipase C inhibitor U73122, we tested the effects of this drug on the inhibition of IM and on the stimulation of noradrenaline release. In neurons treated with 1 μmU73122 for 15 min, the concomitant application of 1 μm bradykinin reducedIM by 0.15 ± 1.8% (n = 4). In neurons treated with the inactive analogU73343, however, the peptide caused a 54.7 ± 10.9% inhibition ofIM (n = 4;p < 0.05). These results corroborate that the inhibition of IM by bradykinin involves phospholipase C (Cruzblanca et al., 1998; Haley et al., 2000b). However, it was not possible to determine whether phospholipase C was also involved in the stimulation of noradrenaline release, because both U73122 and U73343 caused a marked increase in the spontaneous outflow of radioactivity and thereby occluded the subsequent induction of tritium overflow by either electrical fields or bradykinin (data not shown).
Blockade of IP3 receptors and depletion of Ca2+ stores does not affect bradykinin-induced noradrenaline release
IP3 triggers increases in intracellular Ca2+ by activation of IP3 receptors on the endoplasmic reticulum, which permit efflux of Ca2+ into the cytosol (Wilcox et al., 1998). Accordingly, IP3-dependent responses in SCG neurons can be blocked by IP3receptor antagonists, such as heparin (Cruzblanca et al., 1998) and xestospongin C (Bofill-Cardona et al., 2000). The cell-permeable IP3 antagonist xestospongin C has been reported to efficiently prevent bradykinin-evoked increases in intracellular Ca2+ in PC12 cells when these had been treated with the blocker for 30 min (Gafni et al., 1997). However, bradykinin-induced release of [3H]noradrenaline from SCG cells that had been incubated in 10 μm xestospongin C for 1 hr, followed by a 1 hr washout period, was not significantly different from the peptide-induced release of the radiotracer from cells that had been treated with vehicle (0.1% DMSO) only (Fig.2A,C). Likewise, electrically evoked tritium overflow was not different whether cells had been treated with xestospongin C or DMSO (data not shown). To find out whether this xestospongin C treatment was, in principle, capable of interfering with the bradykinin-dependent signaling cascade in SCG neurons, the cultures were incubated in this agent or in DMSO again, and subsequently the effect of bradykinin onIM was tested (Fig.2B,C). In xestospongin C-treated cells, bradykinin failed to cause a significant inhibition ofIM (1.8 ± 2.4% inhibition;n = 6), whereas in DMSO-treated cells, this inhibition amounted to 38.8 ± 10.7% (n = 7). For comparison, the muscarinic agonist oxotremorine M (10 μm) reduced IMin xestospongin C-treated cells by 60.2 ± 10.0% (n = 7) and in DMSO-treated cells by 56.2 ± 10.0% (n = 6; p > 0.7). Thus, the blockade of IP3 receptors impeded only the inhibition of IM, but not the stimulation of transmitter release, by bradykinin.
Fig. 2.
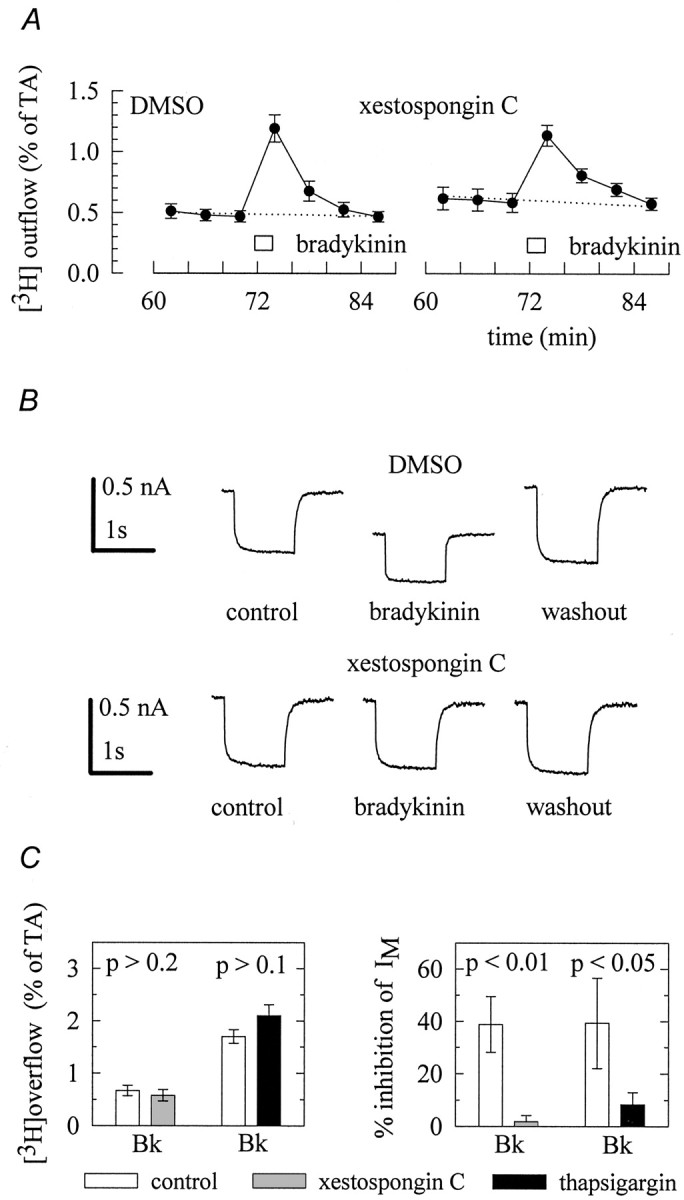
IP3 receptors and intracellular Ca2+ stores are involved in the inhibition ofIM but not in the stimulation of transmitter release. A, Cultures were labeled with [3H]noradrenaline in culture medium containing either 10 μm xestospongin C or the appropriate solvent (0.1% DMSO). Thereafter, the cells were superfused, and subsequent to a 60 min washout period 4 min fractions of superfusate were collected. A 1 μm concentration of bradykinin was present for 2 min as indicated by the bar. The graph shows the time course of fractional [3H] outflow calculated as percentage of the total radioactivity (% of TA) in the cells;n = 6. B, Cultures were treated with 10 μm xestospongin C or with the appropriate vehicle (0.1% DMSO) for 60 min, followed by a 60 min incubation in regular medium. Thereafter, IM was activated by clamping neurons at −30 mV and deactivated by applying one second hyperpolarizing voltage steps to −55 mV. The traces shown were obtained before (control), during (bradykinin), and after (washout) the application of 1 μm bradykinin. Csummarizes the effects of 10 μm xestospongin C or 0.3 μm thapsigargin on bradykinin-induced tritium overflow (left) and inhibition of IM(right). Xestospongin was applied as described above. In superfusion experiments, thapsigargin was present from min 50 onward, and in electrophysiological experiments bradykinin was first tested in the absence of thapsigargin and then reapplied to the same cell in the continuing presence of thapsigargin and subsequent to a 15 min treatment with the Ca2+-ATPase inhibitor. Results obtained in the presence of thapsigargin are compared with those obtained in its absence. p values for significances of differences between the results obtained with or without xestospongin and thapsigargin, respectively, are indicated above thebars; n = 9–12 for tritium overflow and 4–7 for the inhibition of IM, respectively.
This result was corroborated by using thapsigargin, a Ca2+-ATPase inhibitor that depletes the Ca2+ stores of SCG neurons (Foucart et al., 1995). In accordance with previous results (Cruzblanca et al., 1998), this drug significantly reduced the inhibitory action of bradykinin on IM. Nevertheless, bradykinin-induced noradrenaline release was not affected by the Ca2+-ATPase inhibitor (Fig.2C). Taken together, an IP3-dependent liberation of Ca2+ from intracellular stores is involved in the inhibition ofIM, but not in the stimulation of transmitter release.
Bradykinin-induced noradrenaline release is attenuated by PKC inhibitors
The signaling pathway of phospholipase C is bifurcated, because the enzyme catalyzes not only the synthesis of IP3, but also that of diacylglycerol, which then activates PKC (Exton, 1996). We therefore tested whether the kinase inhibitor staurosporine might interfere with the effects of bradykinin. In the presence of 0.3 μm staurosporine, bradykinin-induced noradrenaline release was reduced by >50% (Fig.3A,C). This effect of the kinase inhibitor was specific for the stimulation by the peptide, because electrically evoked tritium overflow was not different in the presence (4.3 ± 0.6% of total radioactivity;n = 12) and absence of this drug (4.5 ± 0.5% of total radioactivity; n = 12), respectively. The inhibition of IM by bradykinin was not altered by 1 μm staurosporine (Fig.3B,C), and the same was true for its inhibition by oxotremorine M (data not shown).
Fig. 3.
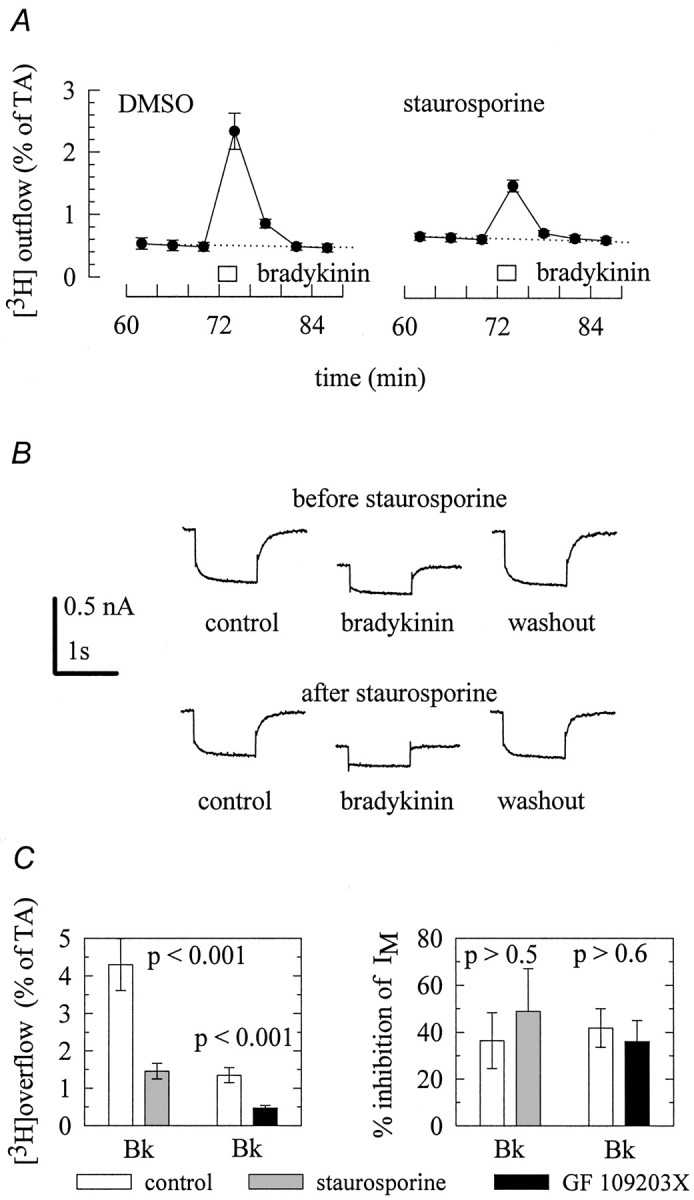
Protein kinase C is involved in the stimulation of transmitter release but not in the inhibition ofIM. A, Cultures were labeled with [3H]noradrenaline and superfused. From minute 50 on, the buffer contained either 0.3 μm staurosporine or 0.03% DMSO. Subsequent to a 60 min washout period, 4 min fractions of superfusate were collected, and 1 μm bradykinin was present for 2 min, as indicated by the bar. The graphs show the time course of fractional [3H] outflow calculated as percentage of the total radioactivity (% of TA) in the cells; n = 6. B,IM was activated by clamping a neuron at −30 mV and deactivated by applying 1 sec hyperpolarizing voltage steps to −55 mV. The top traces were obtained before (control), during (bradykinin), and after (washout) the application of 1 μm bradykinin. Then, the neuron was superfused with 1 μm staurosporine for 10 min. Thereafter, bradykinin was tested again in the continuing presence of staurosporine, and thebottom traces were obtained before (control), during (bradykinin), and after (washout) the application of the peptide.C summarizes the effects of 0.3 μmstaurosporine or 0.3 μm GF 109203X on bradykinin-induced tritium overflow (left) and inhibition ofIM (right). Both staurosporine and GF 109203X were applied as described above. Results obtained in the presence of these protein kinase C inhibitors are compared with those obtained in their absence. p values for significances of differences between the results obtained with or without staurosporine and GF 109203X, respectively, are indicated above the bars; n = 12 for tritium overflow and 4–5 for the inhibition of IM, respectively.
The inhibitory effect of staurosporine is not selective for PKC, but may also affect other enzymes, such as protein kinase A and some tyrosine kinases (Ruegg and Burgess, 1989). To corroborate that a PKC enzyme was involved in the secretagogue action of bradykinin, the above experiments were repeated with the specific PKC inhibitor GF 109203 X (0.3 μm) (Toullec et al., 1991). This drug also reduced the stimulation of tritium overflow by bradykinin, but left the inhibition of IM unaffected (Fig.3C).
Activation of PKC by phorbol esters stimulates noradrenaline release
Considering that PKC appeared to mediate the release stimulating action of bradykinin, we tested whether the application of phorbol esters was also able to induce tritium overflow. As shown in Figure4A, 0.1 μm PMA caused a marked increase in tritium outflow, and this stimulatory action of the phorbol ester was reduced in the presence of staurosporine. The secretagogue action of PMA displayed a bell-shaped concentration–response curve, with maximal effects at 0.1 μm (Fig. 4B). Because phorbol esters and diacylglycerol analogues act on a large number of targets apart from PKC, such as neuronal Ca2+ channels (Hockberger et al., 1989) and transient receptor potential channels (Hofmann et al., 1999), we did not further investigate the reasons for this type of concentration dependence and used only 0.1 μm PMA in subsequent superfusion experiments. The stimulation of noradrenaline release by this PMA concentration was significantly attenuated by the specific PKC inhibitor GF 109203X (Fig.4D).
Fig. 4.
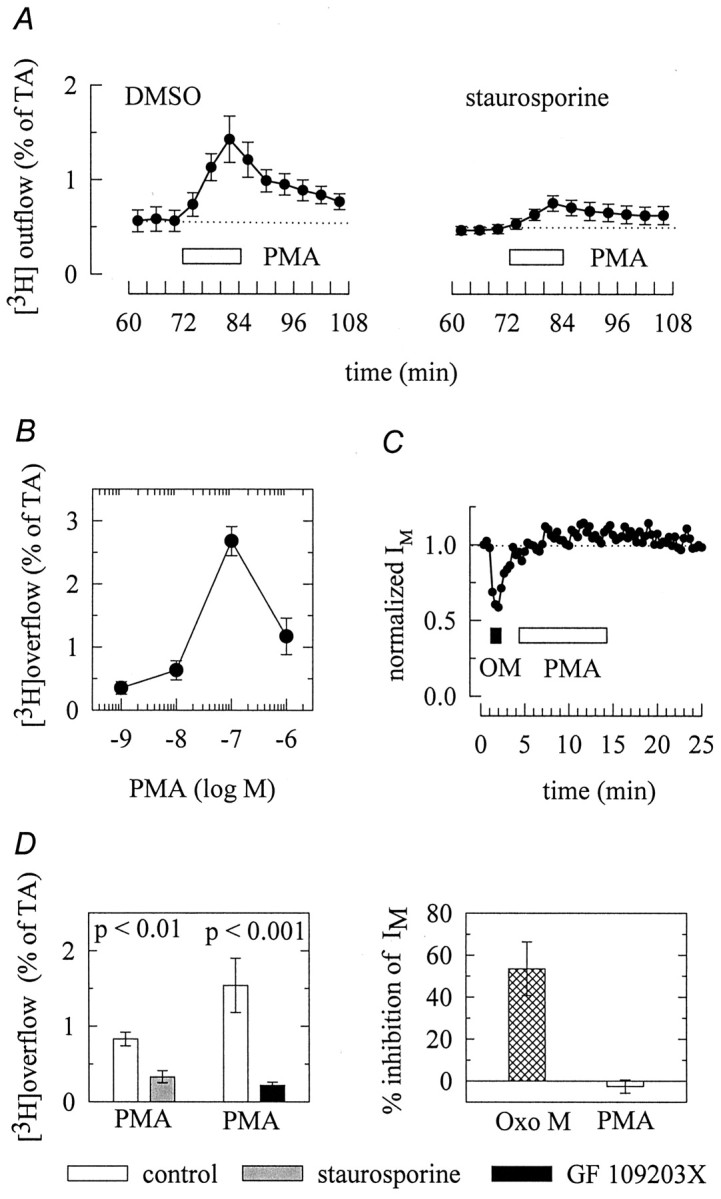
Activation of protein kinase C elicits transmitter release but does not affect IM.A, Cultures were labeled with [3H]noradrenaline and superfused. From minute 50 on, the buffer contained either 0.3 μm staurosporine or 0.03% DMSO. Subsequent to a 60 min washout period, 4 min fractions of superfusate were collected, and 0.1 μm PMA was present for 12 min, as indicated by the bar. The graphs show the time course of fractional [3H] outflow calculated as percentage of the total radioactivity (% of TA) in the cells;n = 6. B, Concentration dependence of tritium overflow elicited by PMA. Experiments were performed as shown in A; n = 9. C,IM was activated by clamping neurons at −30 mV and deactivated by applying 1 sec hyperpolarizing voltage steps to −55 mV. The graph shows the time course ofIM deactivation amplitudes normalized to the first amplitude determined. A 10 μm concentration of oxotremorine M (OM; filled bar) and 1 μmPMA (open bar) were present, as indicated by thebars; n = 3. D shows the amount of tritium overflow (left) stimulated by 0.1 μm PMA in the presence or absence of 0.3 μmstaurosporine and 0.3 μm GF 109203X, respectively, and the inhibition of IM (right) by either 10 μm oxotremorine M or 1 μm PMA. Experiments were performed as shown in A andC, respectively. p values for significances of differences between the results obtained with or without staurosporine and GF 109203X, respectively, are indicated above the bars; n = 12–13 for tritium overflow and 4 (Oxo M) to 9 (PMA) for the inhibition of IM, respectively.
As indicated above, the inhibition ofIM by bradykinin was mediated by an IP3-dependent signaling cascade. To find out whether PKC might also be involved in this effect, PMA was applied in electrophysiological experiments, but it failed to alterIM. For comparison, the muscarinic agonist oxotremorine M (10 μm) clearly reduced the IM deactivation amplitudes (Fig.4C,D).
PKC isoforms expressed in SCG neurons and their phorbol ester sensitivity
At least 11 different isoforms of PKC have been identified; these were named by Greek letters and categorized into three groups: classical (α, βI, βII, γ), novel (δ, ε, η, θ), and atypical (ζ, ι/λ) PKCs. In addition, there is the distantly related phorbol ester-sensitive PKCμ, which has been named PKD. Classical isoforms are Ca2+- and phorbol ester-sensitive, novel ones are Ca2+-insensitive but phorbol ester-sensitive, and atypical ones are Ca2+- and phorbol ester-insensitive (Way et al., 2000; Nishizuka, 2001). In sympathetic neurons of chick embryos, we detected previously phorbol ester-sensitive and -insensitive PKCs (Boehm et al., 1996). Similarly, in primary cultures of rat SCG, immunoblots with isoform-specific antibodies revealed the presence of PKC α, βI, βII, δ, ε, and ζ, but not of PKC γ or μ (Fig. 5A). Nevertheless, the antibody directed against PKC γ detected an appropriate protein in the neonatal rat brain (Fig. 5A). In contrast, the PKC μ-specific antibody failed to stain proteins of rat brain and SCG, but detected a band of ∼90 kDa when tested on blots of proteins from rat hearts (data not shown). As previously shown for sympathetic neurons of chicken (Boehm et al., 1996), antibodies against PKCs ε and ζ detected two bands of ∼90–100 kDa, whereas the other PKC isoforms appeared to migrate as single bands only (Fig.5A).
Fig. 5.
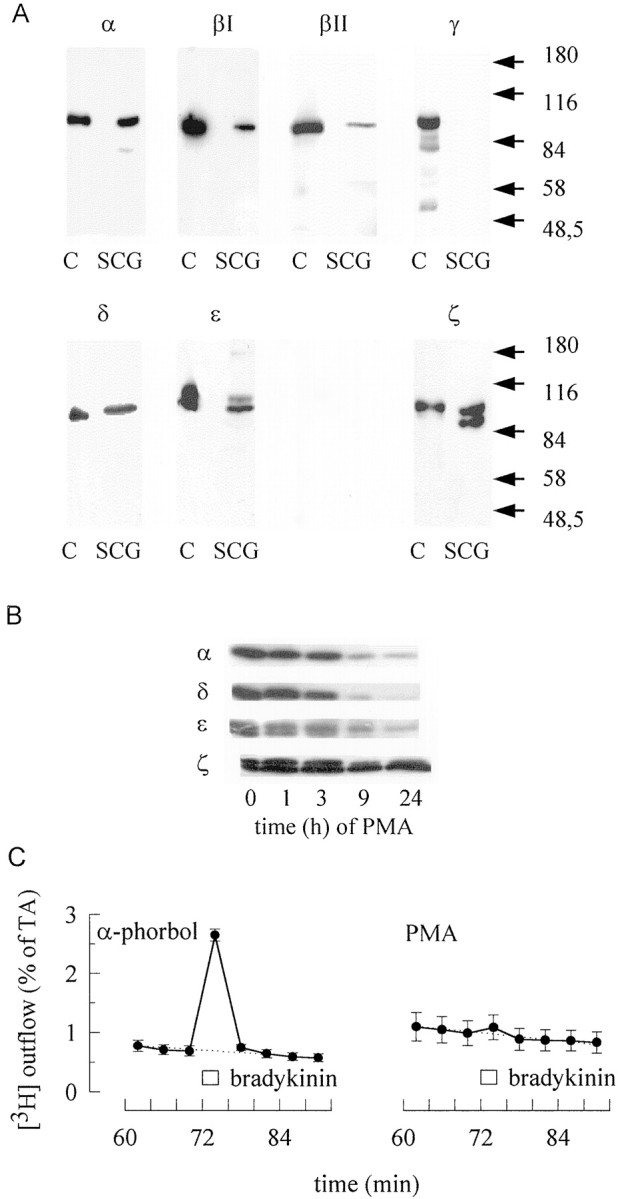
Protein kinase C isoforms in rat SCG and their role in bradykinin-evoked transmitter release. A,Lysates of rat cortex (C) or SCG cell cultures were separated by SDS-PAGE, transferred to nitrocellulose membranes, and stained with antibodies directed against the indicated subtypes of PKC. Approximate positions of molecular weight markers are indicated.B, Before SDS-PAGE, SCG cultures were treated with 1 μm PMA for the indicated periods of time. The blots were stained with antibodies directed against the indicated subtypes of PKC.C, Cultures were treated with either 1 μmPMA or 1 μm 4-α-phorbol for 24 hr, labeled with [3H]noradrenaline, and superfused. Subsequent to a 60 min washout period, 4 min fractions of superfusate were collected, and 1 μm bradykinin was present for 2 min, as indicated by the bar. The graphs show the time course of fractional [3H] outflow calculated as percentage of the total radioactivity (% of TA) in the cells; n = 6.
As stated above, PKC isoforms can be classified by their phorbol ester-sensitivity (Way et al., 2000; Nishizuka, 2001). Phorbol ester-sensitive PKCs of sympathetic neurons are not only activated by these tumor promoters, but also become downregulated during prolonged exposure (Boehm et al., 1996). To determine whether the secretagogue action of bradykinin involved phorbol ester-sensitive or -insensitive PKC isoforms, SCG cultures were treated with 1 μm PMA for up to 24 hr. This treatment reduced the levels of typical, as exemplified by PKC α, and novel PKCs, as exemplified by PKC δ and ε, but left the atypical PKC ζ almost unaltered (Fig.5B). When SCG cultures had been treated with 1 μm PMA for 24 hr, bradykinin (1 μm) triggered the release of 0.15 ± 0.02% of total radioactivity (Fig. 5C). In cultures treated with inactive 4-α phorbol, however, bradykinin-induced overflow amounted to 1.94 ± 0.12% of total radioactivity (n = 6; p < 0.01). In contrast, electrically evoked release of tritium was not different in cultures treated with either PMA or 4-α phorbol (data not shown). Hence, bradykinin apparently triggers transmitter release through an activation of phorbol ester-sensitive PKCs.
Bradykinin activates Ca2+-independent PKC isoforms
The phorbol ester-sensitive PKCs comprise Ca2+-dependent classical (α, βI, βII, γ) as well as Ca2+-independent novel (δ, ε, η, θ) isoforms (Way et al., 2000; Nishizuka, 2001). To elucidate which PKCs might become activated by bradykinin, we studied the cellular distribution of several PKCs in SCG neurons, because activation of PKCs in sympathetic (Boehm et al., 1996) and sensory (Cesare et al., 1999) neurons is accompanied by a translocation of the enzyme from the cytosol to the plasma membrane. In accordance with this, PMA (0.1 μm for 10 min) caused redistribution of all classical (α, βΙ, and βΙΙ,) and novel (δ and ε) PKCs into the membrane, as exemplified by the staining pattern of antibodies directed against the PKC isoforms α and ε (Fig.6A). The distribution of the atypical PKC ζ, however, was not affected by PMA (Fig.6A). Bradykinin (1 μm for 30 sec), in contrast, induced a translocation of PKCs δ and ε (Fig.5A,B) from the cytosol into the membrane, but left the distribution of all other PKC isoforms virtually unaltered (Fig. 5B). This indicates that bradykinin activates only Ca2+-independent PKCs.
Fig. 6.
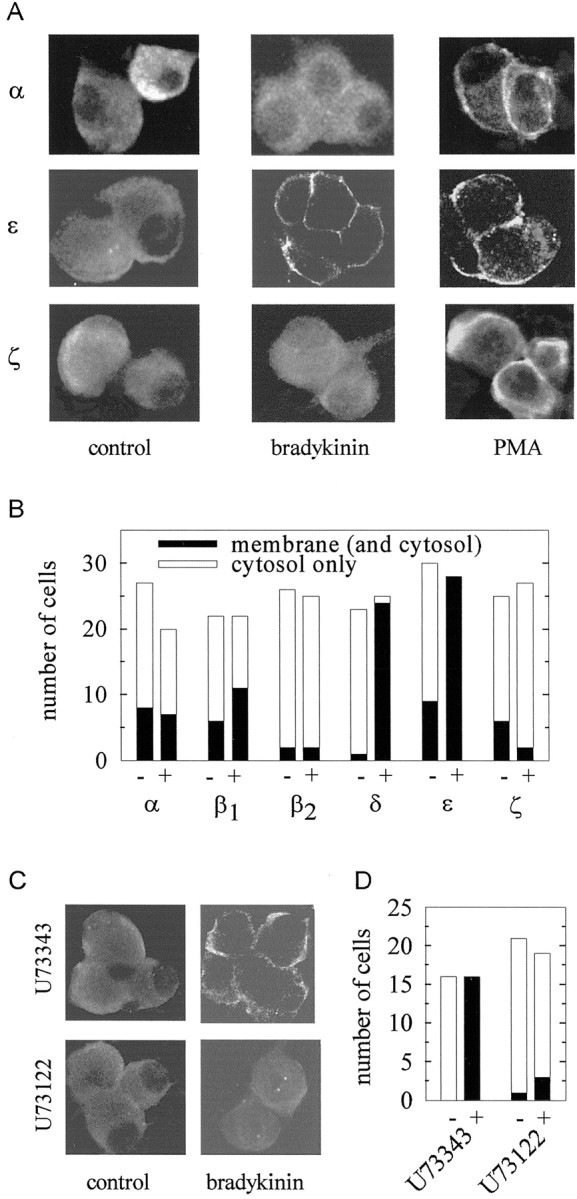
Activation of PKC isoforms in rat SCG neurons by phorbol esters and bradykinin. A, SCG cultures were either treated with bradykinin (1 μm for 30 sec) or with PMA (0.1 μm for 10 min) or remained untreated (control) and were then fixed, permeabilized, and stained with antibodies directed against the indicated subtypes of PKC. The pictures show confocal fluorescence images of two to four neurons. Note differences in the distribution of immunoreactivity between cytoplasm and membranes. B, Cultures were either treated with bradykinin (1 μm for 30 sec; +) or remained untreated (−) and were then fixed, permeabilized, and stained with antibodies directed against the indicated subtypes of PKC. Anonymous confocal fluorescence images were evaluated for predominant cytosol or membrane staining, and the graph shows the number of single cells evaluated and the number of cells that displayed a preponderance of either cytosolic or membranous immunoreactivity.C, SCG cultures were first incubated in either U73122 orU73343 (both at 10 μm for 10 min) and then stimulated with bradykinin (1 μm for 30 sec) or remained unstimulated (control). Thereafter, the cultures were fixed, permeabilized, and stained with an antibody directed against PKC ε. The pictures show confocal fluorescence images of two to four neurons. D, Cultures were treated as described in C (+, bradykinin; −, control). Anonymous confocal fluorescence images were evaluated for predominant cytosol or membrane staining, and the graph shows the number of single cells evaluated and the number of cells that displayed a preponderance of either cytosolic or membranous immunoreactivity.
The inhibition of IM by bradykinin was found to be mediated by phospholipase C, as indicated by the action of the specific inhibitor U73122, which abolished this bradykinin effect (see above and Cruzblanca et al., 1998). To reveal whether the activation of PKCs by bradykinin was also mediated by phospholipase C, SCG cultures were incubated in either U 73122 or the inactive analogU73343 (both at 10 μm for 10 min) and then stimulated with the peptide (1 μm bradykinin for 30 sec). The subsequent investigation of the cellular distribution of PKC ε revealed that the active phospholipase C inhibitor prevented the bradykinin-induced translocation of the enzyme, whereas the inactive analog failed to do so (Fig. 6C,D). Thus, the activation of Ca2+-independent PKCs by bradykinin involves phospholipase C.
Bradykinin-induced noradrenaline release is attenuated by inhibitors of Ca2+-independent, but not of Ca2+-dependent, PKC isoforms
Several PKC inhibitors have been synthesized that can be used to differentiate between various subtypes of PKC on a functional level (Way et al., 2000). We used GÖ 6976, GÖ 6983, and GÖ7874 to find out whether it was Ca2+-independent PKC subtypes that were involved in the secretagogue actions of bradykinin. GÖ 6976 inhibits only Ca2+-dependent PKC isoforms, but not PKC δ and ε (Martiny-Baron et al., 1993). GÖ 6983 and GÖ 7874, in contrast, are potent, but unspecific inhibitors of most, if not all, PKC isoforms (Way et al., 2000). GÖ 6976 (0.3 μm) did not alter the amount of bradykinin-evoked tritium overflow from SCG neurons (Fig.7B,C), whereas GÖ 6983 (Fig. 7A,C) and GÖ7874 (Fig.7C) caused ∼50% inhibition. When transmitter release was induced by the direct activation of PKCs by phorbol esters, however, all three PKC inhibitors reduced the stimulated release of radioactivity to approximately the same extent (Fig. 7C). For comparison, electrically evoked tritium overflow was not affected by any of these PKC inhibitors (data not shown). Thus, the secretagogue action of bradykinin appears to involve only Ca2+-independent PKC isoforms, whereas that of PMA involves Ca2+-dependent as well as -independent isoforms.
Fig. 7.
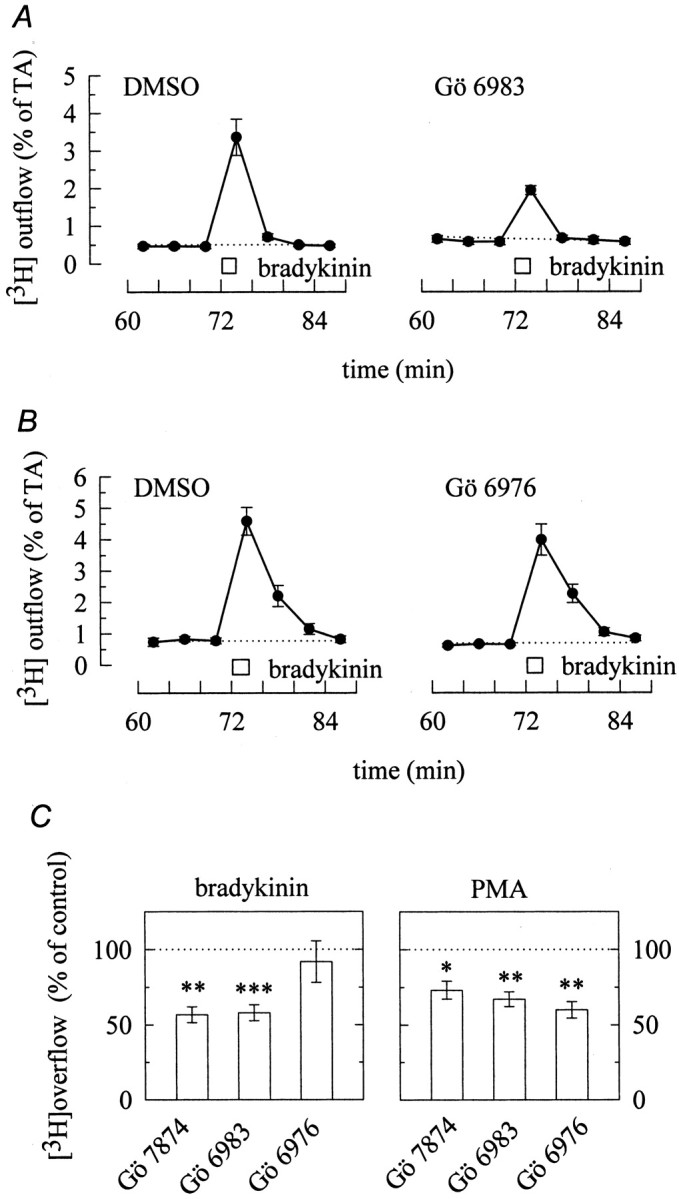
Ca2+-independent protein kinase C isoforms mediate the stimulation of transmitter release by bradykinin. A, Cultures were labeled with [3H]noradrenaline and superfused. From minute 50 on, the buffer contained either 0.3 μm GÖ 6983 or 0.03% DMSO. Subsequent to a 60 min washout period, 4 min fractions of superfusate were collected, and 1 μm bradykinin was present for 2 min as indicated by the bar. The graphs show the time course of fractional [3H] outflow calculated as percentage of the total radioactivity (% of TA) in the cells; n = 6. B, Cultures were labeled with [3H]noradrenaline and superfused. From minute 50 on, the buffer contained either 0.3 μmGÖ 6976 or 0.03% DMSO. Subsequent to a 60 min washout period, 4 min fractions of superfusate were collected, and 1 μmbradykinin was present for 2 min, as indicated by the bar. The graphs show the time course of fractional [3H] outflow calculated as percentage of the total radioactivity (% of TA) in the cells; n = 6. Csummarizes the effects of GÖ 7874, GÖ 6983, and GÖ 6976 on tritium overflow induced either by bradykinin (left; 1 μm for 2 min) or by PMA (right; 0.1 μm for 12 min). GÖ 7874, GÖ 6983, and GÖ 6976 were applied as described above. Results obtained in the presence of these protein kinase C inhibitors are expressed as percentage of those obtained in their absence. *,**p < 0.05 and p < 0.01 for the significances of differences between the results obtained in the absence or presence of GÖ 7874, GÖ 6983, and GÖ 6976, respectively; n = 11–18 for bradykinin and 6–9 for PMA.
DISCUSSION
Bradykinin is well known to potently excite sympathetic ganglia (Lewis and Reit, 1965; Trendelenburg, 1966), which finally leads to the exocytotic release of sympathetic transmitters from the neurons onto effector cells (Boehm and Huck, 1997). The excitatory action at the level of the ganglia has been suggested to be mediated by an inhibition of KM channels (Jones et al., 1995), and the same mechanism has also been implicated in the excitation of parasympathetic neurons by this peptide (Mochidome et al., 2001). Previously, bradykinin had been reported to inhibitIM in two neuronal cell lines, N1E-115 (Higashida and Brown, 1986) and PC12 (Villarroel, 1989). Thus, the inhibition of KM channels appeared to be the predominant mechanism in neuronal excitation by bradykinin. In sensory neurons, however, a PKC-mediated induction of a cation conductance (Burgess et al., 1989) has been suggested to be involved in the action of the peptide. In line with the latter results, the present experiments demonstrate that bradykinin excites sympathetic neurons independently of changes in IM, through an activation of Ca2+-independent PKC.
As a measure for the excitation of SCG neurons, we quantified the amount of [3H]noradrenaline that is released in the presence of bradykinin. This bradykinin-evoked transmitter release is abolished when action potential propagation is prevented by tetrodotoxin and is thus not mediated by presynaptic receptors, but rather by bradykinin receptors located at the somatodendritic region of the neurons (Boehm and Huck, 1997). The receptor that mediates the secretagogue action of bradykinin is a B2 receptor (Boehm and Huck, 1997) and is thus identical to the receptor that mediates the inhibition ofIM (Jones et al., 1995). Nevertheless, experimental procedures that abolished the inhibition ofIM left the stimulation of noradrenaline release by bradykinin unaffected: this was observed for the blockade of IP3 receptors by xestospongin C and for the depletion of intracellular Ca2+ stores by thapsigargin. In accordance with previous results (Cruzblanca et al., 1998), these data indicate that an IP3-dependent signaling cascade mediated the inhibition of IM. However, this signaling cascade appeared not to be involved in the stimulation of transmitter release.
Apart from inhibiting IM and from triggering noradrenaline release, bradykinin was found to stimulate phospholipase C, which led to the accumulation of IP3. During activation, phospholipase C also generates another second messenger, diacyglycerol, which, in turn, activates PKC (Exton, 1996). Bradykinin-evoked noradrenaline release was markedly reduced in the presence of PKC inhibitors, and activation of PKC by PMA triggered release. Thus, the excitatory action of bradykinin that led to the exocytotic release of noradrenaline was mediated by PKC. However, activation of PKC did not alterIM, nor did inhibition of PKC change the inhibitory effect of bradykinin onIM. Taken together, activation of bradykinin B2 receptors of SCG neurons initiates two signaling cascades, one that leads to the inhibition ofIM via IP3-dependent increases in intracellular Ca2+, and another one that finally triggers transmitter release via an activation of PKC. Previously, both bradykinin and phorbol esters were found to induce transmitter release from neuroblastoma glioma hybrid cells independently of membrane depolarization (Higashida, 1988). In SCG neurons, in contrast, the peptide as well as the PKC activator dioctanoylglycerol stimulate transmitter release in a tetrodotoxin-sensitive manner (Boehm et al., 1997; Vartian et al., 2001). Thus, PKC-mediated noradrenaline release from sympathetic neurons involves depolarization of neuronal somata and subsequent action potential propagation, but these effects do not rely on an inhibition of IM.
Members of the family of PKC isozymes are characterized by their sensitivity to diacylglycerol, phorbol esters, and Ca2+: classical PKC isozymes (α, βI, βII, γ) are Ca2+-, diacylglycerol-, and phorbol ester-sensitive, novel PKC isozymes (δ, ε, η, θ) are only diacylglycerol- and phorbol ester-sensitive, and atypical PKC (ζ, ι/λ) isozymes are insensitive to these regulators, but activated, for instance, by free fatty acids (Way et al., 2000;Nishizuka, 2001). The primary cultures of SCG neurons were found to express representatives of each of these groups, namely PKC α, βI, βII, δ, ε, and ζ, but not PKC γ and μ (PKD). To find out which of these subtypes were involved in the excitatory action of bradykinin, several types of experiments were performed. (1) Long-term treatment of sympathetic neurons with phorbol esters causes downregulation of phorbol ester-sensitive PKC isozymes (Matthies et al., 1987; Boehm et al., 1996). Accordingly, SCG cultures treated with PMA for 24 hr were depleted of PKCs α, δ, and ε, but not of PKC ζ, and in these cultures bradykinin lost its secretagogue action. Hence, the effect of bradykinin was mediated by phorbol ester-sensitive PKCs. (2) Bradykinin-evoked noradrenaline release was reduced by PKC inhibitors that block all types of phorbol ester-sensitive PKCs, but not by an inhibitor that selectively blocks Ca2+-dependent PKCs. Nevertheless, all PKC inhibitors tested reduced noradrenaline release triggered by PMA and left electrically induced release unaltered. Hence, the above pattern of inhibition was specific for the excitation by bradykinin, which suggests that Ca2+-independent PKC isozymes were involved in this effect. (3) Finally, the activation of certain PKC isozymes by bradykinin was demonstrated directly: in sensory (Cesare et al., 1999) as well as sympathetic (Boehm et al., 1996) neurons, phorbol esters cause translocation of sensitive PKC isozymes from the cytosol into the plasma membrane, and this effect was also observed in the present immunocytochemical experiments. The redistribution of PKCs into certain cellular compartments is believed to parallel the activation of these enzymes (Newton and Johnson, 1998). In contrast with phorbol esters, bradykinin caused translocation of only PKC δ and ε, but not of the other isozymes. Hence, the peptide can be assumed to activate only these Ca2+-insensitive PKC isoforms. In sensory neurons, for comparison, bradykinin was reported to activate exclusively PKC ε, but not PKC δ or any other isozyme (Cesare et al., 1999).
The conclusion that bradykinin triggered transmitter release via Ca2+-independent PKC isoforms is also supported by independent results: thapsigargin, which depletes intracellular Ca2+ stores (Foucart et al., 1995), and xestospongin C, which blocks IP3receptors (Gafni et al., 1997), did not alter the amount of transmitter release induced by bradykinin, although these agents must be expected to prevent bradykinin-induced increases in intracellular Ca2+. These findings also show that the two signaling cascades initiated by bradykinin, the IP3-dependent inhibition ofIM and the PKC-mediated stimulation of transmitter release are segregated from each other: the inhibition ofIM did not require an activation of any PKC enzyme, and the activation of Ca2+-independent PKCs did not require an IP3-dependent release of Ca2+ from the endoplasmic reticulum. Nevertheless, both signaling cascades had an initial component in common, the activation of phospholipase C. This was evidenced by the fact that U73122 blocked both, the inhibition of IM (Cruzblanca et al., 1998) and the translocation of PKC ε. Previously, the enzyme mediating the inhibition of IM by bradykinin has been shown to be a phospholipase C-β4 (Haley et al., 2000b), and the link between this phospholipase and B2 bradykinin receptors are the α subunits of heterotrimeric G11-proteins (Jones et al., 1995; Haley et al., 1998, 2000a).
Apart from B2 bradykinin receptors, sympathetic neurons express other seven transmembrane receptors linked to heterotrimeric G-proteins of the Gq family, most notably angiotensin AT1 and muscarinic M1receptors (for review, see Boehm and Kubista, 2002). Activation of these latter receptors also causes depolarization (Constanti and Brown, 1981) and subsequent transmitter release (M. Mayer and S. Boehm, unpublished observation). The sympathoexcitatory actions of these three receptors are generally believed to be mediated by an inhibition of IM (Constanti and Brown, 1981; Jones et al., 1995), although at least two different signaling cascades are involved in the receptor-dependent modulation of these ion channels: bradykinin B2 receptors rely on an IP3-dependent signaling cascade to inhibitIM, whereas muscarinic M1 receptors do not (Cruzblanca et al., 1998; present results). Nevertheless, both B2 and M1 receptors are coupled to phospholipase C and trigger the synthesis of IP3 and diacylglycerol (del Rio et al., 1999; present results). Our data demonstrate that G-protein-coupled receptors linked to phospholipase C depolarize sympathetic neurons independently of changes inIM, but via the activation of Ca2+-insensitive PKC isozymes. Previously, muscarinic receptors have also been found to activate PKC in sympathetic neurons (Wakade et al., 1991; Marsh et al., 1995), although the relevant PKC isoforms have not been identified. Hence, the signaling cascade described above may also apply for the muscarinic excitation of sympathetic neurons, and muscarinic M1 receptors are known to mediate the slow component of ganglionic transmission (Brown, 1983). Therefore, Ca2+-independent PKC isozymes are not only important elements in the sympathoexcitatory action of bradykinin, as shown above, but may also be involved in the physiological regulation of neuronal signaling within the autonomic nervous system.
Footnotes
The study was supported by the “Virologiefonds” of the University of Vienna and the Austrian Science Fund (Fonds zur Fōrderung der Wissenschaftlichen Forschung; Grant P13920-MED). We are indebted to M. Freissmuth for valuable comments on this manuscript.
Correspondence should be addressed to Stefan Boehm, Department of Pharmacology, University of Vienna, Waehringerstrasse 13a, A-1090 Vienna, Austria. E-mail: stefan.boehm@univie.ac.at.
REFERENCES
- 1.Bhoola KD, Figueroa CD, Worthy K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol Rev. 1992;44:1–80. [PubMed] [Google Scholar]
- 2.Boehm S. Selective inhibition of M-type potassium channels in rat sympathetic neurons by uridine nucleotide preferring receptors. Br J Pharmacol. 1998;124:1261–1269. doi: 10.1038/sj.bjp.0701956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boehm S. ATP stimulates sympathetic transmitter release via presynaptic P2X purinoceptors. J Neurosci. 1999;19:737–746. doi: 10.1523/JNEUROSCI.19-02-00737.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boehm S, Huck S. Noradrenaline release from rat sympathetic neurons triggered by activation of B2 bradykinin receptors. Br J Pharmacol. 1997;122:455–462. doi: 10.1038/sj.bjp.0701404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boehm S, Kubista H. Fine tuning of sympathetic transmitter release via ionotropic and metabotropic presynaptic receptors. Pharmacol Rev. 2002;54:43–99. doi: 10.1124/pr.54.1.43. [DOI] [PubMed] [Google Scholar]
- 6.Boehm S, Huck S, Freissmuth M. Involvement of a phorbol ester-insensitive protein kinase C in the α2-adrenergic inhibition of voltage-gated Ca2+ current in chick sympathetic neurons. J Neurosci. 1996;16:4596–4603. doi: 10.1523/JNEUROSCI.16-15-04596.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bofill-Cardona E, Vartian N, Nanoff C, Freissmuth M, Boehm S. Two different signaling mechanisms involved in the excitation of rat sympathetic neurons by uridine nucleotides. Mol Pharmacol. 2000;57:1165–1172. [PubMed] [Google Scholar]
- 8.Brown DA. Slow cholinergic excitation—a mechanism for increasing neuronal excitability. Trends Neurosci. 1983;6:302–307. [Google Scholar]
- 9.Burgess GM, Mullaney I, McNeill M, Dunn PM, Rang HP. Second messengers involved in the mechanism of action of bradykinin in sensory neurons in culture. J Neurosci. 1989;9:3314–3325. doi: 10.1523/JNEUROSCI.09-09-03314.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cesare P, Dekker LV, Sardini A, Parker PJ, McNaughton PA. Specific involvement of PKC-ε in sensitization of the neuronal response to painful heat. Neuron. 1999;23:617–624. doi: 10.1016/s0896-6273(00)80813-2. [DOI] [PubMed] [Google Scholar]
- 11.Constanti A, Brown DA. M-Currents in voltage-clamped mammalian sympathetic neurones. Neurosci Lett. 1981;24:289–294. doi: 10.1016/0304-3940(81)90173-7. [DOI] [PubMed] [Google Scholar]
- 12.Cox SL, Schelb V, Trendelenburg AU, Starke K. Enhancement of noradrenaline release by angiotensin II and bradykinin in mouse atria: evidence for cross-talk between G(q/11) protein- and G(i/o) protein-coupled receptors. Br J Pharmacol. 2000;129:1095–1102. doi: 10.1038/sj.bjp.0703167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cruzblanca H, Koh DS, Hille B. Bradykinin inhibits M current via phospholipase C and Ca2+ release from IP3 -sensitive Ca2+ stores in rat sympathetic neurons. Proc Natl Acad Sci USA. 1998;95:7151–7156. doi: 10.1073/pnas.95.12.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.del Rio E, Bevilacqua JA, Marsh SJ, Halley P, Caulfield MP. Muscarinic M1 receptors activate phosphoinositide turnover and Ca2+ mobilization in rat sympathetic neurons, but this signalling pathway does not mediate M-current inhibition. J Physiol (Lond) 1999;520:101–111. doi: 10.1111/j.1469-7793.1999.00101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dell'Italia LJ, Oparil S. Bradykinin in the heart: friend or foe? Circulation. 1999;100:2305–2307. doi: 10.1161/01.cir.100.23.2305. [DOI] [PubMed] [Google Scholar]
- 16.Exton H. Regulation of phosphoinositide phospholipases by hormones, neurotransmitters, and other agonists linked to G proteins. Annu Rev Pharmacol Toxicol. 1996;36:481–509. doi: 10.1146/annurev.pa.36.040196.002405. [DOI] [PubMed] [Google Scholar]
- 17.Foucart S, Gibbons SJ, Bronson JR, Miller RJ. Increase in [Ca2+]i by CCh in adult rat sympathetic neurons are not dependent on intracellular Ca2+ pools. Am J Physiol. 1995;268:C829–C837. doi: 10.1152/ajpcell.1995.268.4.C829. [DOI] [PubMed] [Google Scholar]
- 18.Gafni J, Munsch JA, Lam TH, Catlin MC, Costa LG, Molinski TF, Pessah IN. Xestospongins: potent membrane permeable blockers of the inositol 1, 4, 5-triphosphate receptor. Neuron. 1997;19:723–733. doi: 10.1016/s0896-6273(00)80384-0. [DOI] [PubMed] [Google Scholar]
- 19.Haley JE, Abogadie FC, Delmas P, Dayrell M, Vallis Y, Milligan G, Caulfield MP, Brown DA, Buckley NJ. The α subunit of Gq contributes to muscarinic inhibition of the M-type potassium current in sympathetic neurons. J Neurosci. 1998;18:4521–4531. doi: 10.1523/JNEUROSCI.18-12-04521.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haley JE, Delmas P, Offermanns S, Abogadie FC, Simon MI, Buckley NJ, Brown DA. Muscarinic inhibition of calcium current and M current in Gαq-deficient mice. J Neurosci. 2000a;20:3973–3979. doi: 10.1523/JNEUROSCI.20-11-03973.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haley JE, Abogadie FC, Fernandez-Fernandez JM, Dayrell M, Vallis Y, Buckley NJ, Brown DA. Bradykinin, but not muscarinic, inhibition of M-current in rat sympathetic ganglion neurons involves phospholipase C-β4. J Neurosci 20 2000b. RC105(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Higashida H. Acetylcholine release by bradykinin, inositol 1, 4, 5-trisphosphate and phorbol dibutyrate in rodent neuroblastoma cells. J Physiol (Lond) 1988;397:209–222. doi: 10.1113/jphysiol.1988.sp016996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Higashida H, Brown DA. Two polyphosphatidylinositide metabolites control two K+ currents in a neuronal cell. Nature. 1986;323:333–335. doi: 10.1038/323333a0. [DOI] [PubMed] [Google Scholar]
- 24.Hockberger P, Toselli M, Swandulla D, Lux HD. A diacylglycerol analogue reduces neuronal calcium currents independently of protein kinase C activation. Nature. 1989;338:340–342. doi: 10.1038/338340a0. [DOI] [PubMed] [Google Scholar]
- 25.Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- 26.Jones S, Brown DA, Milligan G, Willer E, Buckley NJ, Caulfield MP. Bradykinin excites rat sympathetic neurons by inhibition of M current through a mechanism involving B2 receptors and Gαq/11. Neuron. 1995;14:400–405. doi: 10.1016/0896-6273(95)90295-3. [DOI] [PubMed] [Google Scholar]
- 27.Kristufek D, Koth G, Motejlek A, Schwarz K, Huck S, Boehm S. Modulation of spontaneous and stimulation-evoked transmitter release from rat sympathetic neurons by the cognition enhancer linopirdine: insights into its mechanisms of action. J Neurochem. 1999;72:2083–2091. doi: 10.1046/j.1471-4159.1999.0722083.x. [DOI] [PubMed] [Google Scholar]
- 28.Levine JD, Taiwo YO, Collin SD, Tam JK. Noradrenaline hyperalgesia is mediated through interaction with sympathetic postganglionic neurone terminals rather than activation of primary afferent nociceptors. Nature. 1986;323:158–160. doi: 10.1038/323158a0. [DOI] [PubMed] [Google Scholar]
- 29.Lewis GP, Reit E. The action of angiotensin and bradykinin on the superior cervical ganglion of the cat. J Physiol (Lond) 1965;179:538–553. doi: 10.1113/jphysiol.1965.sp007679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marrion NV. Control of M-current. Annu Rev Physiol. 1997;59:483–504. doi: 10.1146/annurev.physiol.59.1.483. [DOI] [PubMed] [Google Scholar]
- 31.Marsh SJ, Trouslard J, Leaney JL, Brown DA. Synergistic regulation of a neuronal chloride current by intracellular calcium and muscarinic receptor activation: a role for protein kinase C. Neuron. 1995;15:729–737. doi: 10.1016/0896-6273(95)90160-4. [DOI] [PubMed] [Google Scholar]
- 32.Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D, Schachtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Go6976. J Biol Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- 33.Matthies HJG, Palfrey HC, Hirning LD, Miller RJ. Downregulation of protein kinase C in neuronal cells: effects on neurotransmitter release. J Neurosci. 1987;7:1198–1206. doi: 10.1523/JNEUROSCI.07-04-01198.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miao FJ-P, Jänig W, Levine JD. Role of sympathetic postganglionic neurons in synovial plasma extravasation induced by bradykinin. J Neurophysiol. 1996;75:715–724. doi: 10.1152/jn.1996.75.2.715. [DOI] [PubMed] [Google Scholar]
- 35.Mochidome T, Ishibashi H, Takahama K. Bradykinin activates airway parasympathetic ganglion neurons by inhibiting M-currents. Neuroscience. 2001;105:785–791. doi: 10.1016/s0306-4522(01)00211-1. [DOI] [PubMed] [Google Scholar]
- 36.Newton AC, Johnson JE. Protein kinase C: a paradigm for regulation of protein function by two membrane-targeting modules. Biochim Biophys Acta. 1998;1376:155–172. doi: 10.1016/s0304-4157(98)00003-3. [DOI] [PubMed] [Google Scholar]
- 37.Nishizuka Y. The protein kinase C family and lipid mediators for transmembrane signalling and cell regulation. Alcohol Clin Exp Res. 2001;25:3S–7S. doi: 10.1097/00000374-200105051-00003. [DOI] [PubMed] [Google Scholar]
- 38.Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- 39.Ruegg UT, Burgess GM. Staurosporine, K-252 and UCN-01: potent but nonspecific inhibitors of protein kinases. Trends Pharmacol Sci. 1989;10:218–220. doi: 10.1016/0165-6147(89)90263-0. [DOI] [PubMed] [Google Scholar]
- 40.Schwartz DD, Malik KU. Cyclic AMP modulates but does not mediate the inhibition of [3H]norepinephrine release by activation of alpha-2 adrenergic receptors in cultured rat ganglion cells. Neuroscience. 1993;52:107–113. doi: 10.1016/0306-4522(93)90186-j. [DOI] [PubMed] [Google Scholar]
- 41.Selyanko AA, Brown DA. Intracellular calcium directly inhibits potassium M channels in excised membrane patches from rat sympathetic neurons. Neuron. 1996;16:151–162. doi: 10.1016/s0896-6273(00)80032-x. [DOI] [PubMed] [Google Scholar]
- 42.Starke K, Peskar BA, Schumacher KA, Taube HD. Bradykinin and postganglionic sympathetic transmission. Naunyn Schmiedebergs Arch Pharmacol. 1977;299:23–32. doi: 10.1007/BF00508633. [DOI] [PubMed] [Google Scholar]
- 43.Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin E, Boursier E, Loriolle F. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- 44.Trendelenburg U. Observations on the ganglion-stimulating action of angiotensin and bradykinin. J Pharmacol Exp Ther. 1966;154:418–425. [PubMed] [Google Scholar]
- 45.Vartian N, Moskvina E, Scholze T, Unterberger U, Allgaier C, Boehm S. UTP evokes noradrenaline release from rat sympathetic neurons by activation of protein kinase C. J Neurochem. 2001;77:876–885. doi: 10.1046/j.1471-4159.2001.00290.x. [DOI] [PubMed] [Google Scholar]
- 46.Villarroel A. M-current suppression in PC12 cells by bradykinin is mediated by a pertussis toxin-insensitive G-protein and modulated by intracellular calcium. Brain Res. 1989;740:227–233. doi: 10.1016/s0006-8993(96)00870-0. [DOI] [PubMed] [Google Scholar]
- 47.Wakade TD, Bhave SV, Bhave AS, Malhotra RK, Wakade AR. Depolarizing stimuli and neurotransmitters utilize separate pathways to activate protein kinase C in sympathetic neurons. J Biol Chem. 1991;266:6424–6428. [PubMed] [Google Scholar]
- 48.Way KJ, Chou E, King G. Identification of PKC-isoform-specific biological actions using pharmacological approaches. Trends Pharmacol Sci. 2000;21:181–187. doi: 10.1016/s0165-6147(00)01468-1. [DOI] [PubMed] [Google Scholar]
- 49.Wilcox RA, Primrose WU, Nahorski SR, Challiss RAJ. New developments in the molecular pharmacology of the myo-inositol 1, 4, 5-trisphosphate receptor. Trends Pharmacol Sci. 1998;19:467–475. doi: 10.1016/s0165-6147(98)01260-7. [DOI] [PubMed] [Google Scholar]