

Abstract
Axonal retraction is induced by different types of physiological cues and is responsible for the elimination of mistargeted axons. There is broad agreement that alterations in the cytoskeleton underlie axonal retraction. The prevailing view is that axonal retraction involves a wholesale depolymerization of microtubules and microfilaments. However, axons retracting physiologically display a very different morphology than axons induced to retract by experimental depolymerization of microtubules. Experimental depolymerization of microfilaments actually prevents retraction rather than causing it. We have proposed an alternative hypothesis, namely that axonal retraction involves a backward retreat of cytoskeletal elements rather than their wholesale depolymerization. In the present study, we sought to test this hypothesis with regard to microtubules. When a donor of nitric oxide was applied to cultured chick sensory neurons, the majority of axons retracted dramatically within 30–60 min. Retracting axons were characterized by an enlarged distal region, a thin trailing remnant, and sinusoidal bends along the shaft. Quantitative immunofluorescence analyses showed no detectable loss of microtubule mass during retraction, even with regard to the most labile microtubules. Instead, microtubules were reconfigured into coiling and sinusoidal bundles to accommodate the shortening of the axon. Stabilization of microtubules by taxol did not prevent the retraction, even at concentrations of the drug that actually caused microtubule levels to increase. The retractions induced by nitric oxide were remarkably similar to those observed when motor proteins are manipulated, suggesting that these retractions may result from alterations in the activities of the motors that configure microtubules.
Keywords: microtubule, axon, neuron, retraction, nitric oxide, actin
The normal functioning of the nervous system depends on precise connections that wire individual neurons together into an orderly network. Establishment of the neural network involves both progressive events, which include neurogenesis and axonal outgrowth, and regressive events, which include neuronal death and axonal retraction (Cowan et al., 1984; Bernstein and Lichtman, 1999). Selective retraction of newly formed axons occurs extensively in the developing nervous system, where it functions as an alternative mechanism to neuronal death to eliminate inappropriate axonal projections. Examples of such selective axonal retraction are seen in the withdrawal of all but one axon from each muscle fiber (Gan and Lichtman, 1998) and the withdrawal of occipital neuronal axons projecting to the spinal cord (Stanfield et al., 1982). In addition, the normal growth and pathfinding of most axons involve alternate bouts of retraction and elongation (Kaethner and Stuermer, 1992; Godement et al., 1994), and a variety of external cues have been identified that can cause axons to retract (for review, see Tessier-Lavigne and Goodman, 1996). It is known that axonal growth involves the elaboration of the cytoskeletal arrays that provide architectural support for the axon, and there is broad agreement that the cytoskeleton must undergo alterations to elicit retraction. However, the nature of these alterations remains largely unknown.
The prevailing view is that cytoskeletal elements depolymerize extensively during physiological axonal retraction (Song and Poo, 1999). This view is largely based on pharmacological studies showing that wholesale microtubule depolymerization can indeed cause axons to retract (Yamada et al., 1970; Daniels, 1973). However, axons retracting because of microtubule loss display a thin beaded morphology that is very different from axons retracting physiologically, which generally thicken distoproximally and show sinusoidal bends rather than beads along their length (Bandtlow et al., 1990; Bastmeyer and Stuermer, 1993; Dent and Meiri, 1998; Nagashima et al., 1999). Furthermore, pharmacological depletion of microfilaments actually prevents axons from retracting rather than causing them to retract (Solomon and Magendantz, 1981; Joshi et al., 1985). We have proposed a different model for how the cytoskeleton is altered during physiological axonal retraction. In our model, instead of extensive depolymerization, microtubules and microfilaments are reconfigured such that they retreat backward in the retracting axon. Our model is based on recent studies that indicate that molecular motors in neurons are capable of configuring and translocating cytoskeletal polymers in much the same way that motors function in the mitotic spindle (Baas and Ahmad, 2001). In the present study, we have focused specifically on microtubules and performed a battery of tests to ascertain whether physiological axonal retraction involves wholesale depolymerization or reconfiguration of the microtubule polymers. Nitric oxide, widely recognized to induce axonal retractions during the development of the vertebrate nervous system (Cramer et al., 1998; Ernst et al., 2000), was used to induce retractions of axons in cultures of chick sensory neurons.
MATERIALS AND METHODS
Cell culture. Cultures of chick sensory neurons were prepared as described previously (Yu and Baas, 1995; Ahmad et al., 2000). Briefly, dorsal root ganglia were dissected from embryonic day 10 chicks and digested in 0.25 mg/ml trypsin and collagenase (both from Worthington Biochemical, Lakewood, NJ) at 37°C for 15 min. After neutralization of the enzymes with 10% fetal bovine serum (FBS; Hyclone, Logan, UT), the ganglia were triturated into a single-cell suspension with a Pasteur pipette, which had been fire polished to obtain a narrow opening. Cells were then diluted in a modified L-15-based medium containing Leibovitz's L-15 (Invitrogen, Grand Island, NY), 0.6% glucose (Sigma, St. Louis, MO), 2 mml-glutamine (Invitrogen), 100 U of penicillin and 100 μg of streptomycin per milliliter (Sigma), 0.6% methylcellulose (Dow Chemical, Midland, MI), 10% FBS, and 25 ng/ml nerve growth factor (Upstate Biotechnology, Lake Placid, NY). Cells were plated onto prephotoetched glass coverslips (Bellco Glass, Vineland, NJ), which had been attached to the bottom of a 35 mm plastic Petri dish into which had been drilled a 1 cm hole. The photoetched marks on the coverslip assist in relocating individual cells during the experimental procedures. Cells were kept at 37°C in normal air. By 16–20 hr after plating, neurons had generated distinguishable axons that were suitable for the experiments.
Experimental treatments. The nitric oxide donor 3-(2-hydroxy-1-methyl-2-nitrosohydrazino)-N-methyl-1-propanamine (NOC-7) was purchased from Calbiochem (La Jolla, CA). A 100 mm stock solution of NOC-7 in 100 mm NaOH was frozen in aliquots. At the time of the experiment, aliquots of the NOC-7 stock solution were thawed quickly, diluted to 1 mm in the modified L-15-based medium but without NGF, and warmed to 37°C. The medium from the cultures was removed and replaced with the medium deficient in NGF but containing NOC-7. Cultures were then incubated for various times depending on the particular experiment. Nocodazole (prepared from a 10 mm stock solution in DMSO and diluted to a final concentration of 10 μg/ml) and taxol (prepared from a 10 mm stock solution in DMSO and diluted to a final concentration of 100 nm) were prepared in complete medium and applied to cells in the same manner. When NOC-7 was added to taxol-treated cultures, the medium contained NGF during the first 30 min in taxol alone, and then the NGF was removed during subsequent treatment with taxol and NOC-7 together.
Sample preparation for fluorescence microscopy. To prevent detachment of axons from the relatively poorly adherent plain glass coverslip, cells were simultaneously fixed and extracted for fluorescence microscopy using a method described previously (Ahmad et al., 2000). Effective extraction of free tubulin was demonstrated by the marked diminution of fluorescence intensity for tubulin in nocodazole-treated cultures. Briefly, cells were fixed and extracted for 15 min in a solution containing 60 mm1,4-piperazinediethanesulfonic acid, 25 mm HEPES, 10 mm EGTA, and 2 mmMgCl2, pH 6.9, as well as 4% paraformaldehyde, 0.15% glutaraldehyde, and 0.2% Triton X-100. Cells were then rinsed extensively in PBS, pH 7.4. To stain for microtubules alone, fixed cells were exposed to a blocking solution containing 1% bovine serum albumin in PBS for 30 min and then incubated for 1 hr at room temperature with a 1:100 dilution of a monoclonal anti-β-tubulin antibody that had been conjugated directly to Cy3 (Sigma). After extensive rinsing in PBS, the cultures were mounted in a medium that reduces photobleaching (0.212% N-propylgallate in 50% glycerol and 50% PBS). To double-label microtubules and microfilaments, the same staining protocol was followed except that the incubation was with a combination of the tubulin antibody and a 1:40 dilution of Alexa Fluor 488 phalloidin (Molecular Probes, Eugene, OR). To double-label tyrosinated and total microtubules, cells were first incubated at 4°C overnight with a rat anti-tyrosinated tubulin antibody named YL 1/2 (Serotec, Raleigh, NC) at a 1:100 dilution. The next day, the cultures were exposed for 1 hr to an FITC-conjugated goat anti-rat IgG (1:200) at 37°C and then exposed, after extensive rinsing, to the Cy3-conjugated β-tubulin antibody (1:100) for 1 hr at room temperature. The FITC secondary antibody (Jackson ImmunoResearch, West Grove, PA) was found to have minimal cross-reactivity with mouse antigens in control experiments.
Light microscopy. For phase-contrast microscopy, cells were observed with an Axiovert 200M inverted microscope (Zeiss, Munchen-Hallbergmoos, Germany), and images were acquired using a charge-coupled device (Zeiss AxioCam b/w) and AxioVision 3.0 software (Zeiss). All of the fluorescence work was performed on the same microscope, except for the experiment involving tyrosinated microtubules. The FITC and Cy3 filters on our Axiovert 200M have relatively narrow excitation and emission spectra, which minimizes potential bleed-through between the channels. Nevertheless, because Cy3 and FITC have slightly overlapping spectra, we also empirically confirmed that there was no detectable bleed-through of our samples visualized at our microscope and software settings. As with the phase-contrast work, fluorescence images were captured by the ZeissAxioCam running on the AxioVision 3.0 software.
For the experiments involving double-labeling of tyrosinated and β-tubulin, our goal was to obtain a ratio image, each pixel of which displays the ratio of fluorescence intensity between the two channels. To accomplish this, we used the Zeiss Pascal laser-scanning confocal microscope. The tyrosinated tubulin (FITC) and β-tubulin (Cy3) scans were captured sequentially (rather than simultaneously), using configuration and laser attenuation settings that were kept the same for all imaged cells. The pinhole of the confocal was maximally opened, because we did not obtain optical sections for this application. Any bleed-through between the FITC and Cy3 channels was eliminated by activating only the appropriate excitation wavelength and photomultiplier (and leaving that of the other channel off). The fluorescence ratio per pixel of the FITC and Cy3 images was calculated and presented as a third image by using the ratio function of the laser-scanning microscope 5 Pascal software (version 2.8). All images were processed in the same way so that they could be compared quantitatively. The ratio images were exported into NIH Image (version 1.62; NIH freeware) and were assigned pseudocolors that represent the relative ratio values. We chose the pseudocolor scale termed “fire-1,” in which the highest intensity is shown in white, the lowest intensity is shown in black, and intermediate intensities are shown in shades of yellow and red.
Quantification of fluorescence intensity. In the single-label experiments, the total microtubule mass in the retracted axons was compared with that in the control axons. Microtubule mass was quantified by measuring the fluorescence intensity of β-tubulin staining. Fluorescence images of both the retracted axons and control axons were captured at the same camera and microscope settings. Overexposure and underexposure were avoided by using the color-coded live window in the AxioVision 3.0 software. The total fluorescence intensity of each observed axon was measured as arbitrary fluorescence units (AFU) using the NIH Image 1.62 software. Because axons of different lengths were included in our analyses, we calculated the fluorescence intensity per unit length (AFU per micrometer) of the axons, which was obtained by dividing the total AFU by the length of the same axon. The means of the fluorescence intensity per unit length of the retracted axons and control axons were compared using the two-tailed, unpaired Student's t test. A p value of <0.05 was used as the standard for statistical significance in this study. All statistical analyses and graphing were done using Prism 3.0 software (GraphPad Software, San Diego, CA). For the experiments involving taxol, images were displayed in the same quantitative pseudocolor format used to display the ratio images (as noted above). So that all of the quantitative studies could be compared relative to one another, the mean for the control microtubule levels in each experiment was adjusted to an AFU of 100, and other values were adjusted accordingly.
Final processing of all images was done using Adobe PhotoShop 6.0 (Adobe Systems, San Jose, CA).
RESULTS
Morphology of axons exposed to nitric oxide
Nitric oxide is a physiological factor that is released as a gas within the nervous system. Several lines of evidence indicate that nitric oxide is a key factor for inducing large-scale retractions of mistargeted axons during the pruning of the developing nervous system (Cramer et al., 1998; Mize et al., 1998). In a recent study, Ernst et al. (2000) reported that donors of nitric oxide introduced into the medium of cultures of chick sensory neurons release nitric oxide gas, and that this results in the retraction of a portion of the axons. These studies primarily used the nitric oxide donor known as 3-morpholino-sydnonimine (SIN-1), which was shown to induce retractions at concentrations that did not typically induce cell death or any detectable nonspecific damage to the neurons. We found that NOC-7, another potent nitric oxide donor (Ernst et al., 2000), was even more effective at inducing retractions and even less apt to cause nonspecific damage at fairly high concentrations (as assessed using the criteria of Ernst et al., 2000). In addition, we found that axonal retractions were more robust and dramatic if the neurons were cultured on plain glass as opposed to plastic or a substrate such as polylysine or laminin (Ahmad et al., 2000). All of the quantitative studies reported here were performed using NOC-7, but a small number of qualitative studies with SIN-1 produced essentially the same results. Dissociated chick sensory neurons generate multiple neurites in culture, all of which are axonal as indicated by their length, uniform diameter, and expression of axonal markers (Smith, 1998). We selected for our studies axons of medium thickness, each with a total length of ∼50–200 μm, which sometimes included one to four primary axonal branches.
After the addition of NOC-7, four different types of axonal behaviors were observed and were defined as follows: “axonal retraction” was defined as the shortening of the axon by >5 μm, “growth-cone collapse only” was defined as the shortening of the axon by <5 μm but with the collapse of the growth cone from a broad structure to a narrow structure, “axonal elongation” was defined as the continued growth of the axon, and “no change” was defined as axons that do not show any of the above three behaviors; these axons showed no significant change in length or growth-cone morphology during the treatment. Notably, all of these four behaviors were represented in control cultures treated with NOC-7 carrier only, but the proportions of axons in each category changed on exposure of the cultures to the nitric oxide donor. In control cultures, 30.6% of 50 axons from 15 neurons retracted, 2% showed growth-cone collapse only, 65.3% elongated, and 2% showed no change. After 30 min of exposure to NOC-7, 67% of 273 axons from 100 neurons retracted, 19% showed growth-cone collapse only, 10% elongated, and 4% showed no change. These data are summarized in Figure1D. Measurement of axonal length showed that axons retracted an average of 41 ± 2.8 μm (mean ± SEM) during the first 30 min in NOC-7, whereas axons of control cells elongated an average of 2.3 ± 5.8 μm in 30 min. (Rates of axonal growth in control cultures were much faster during the next 30 min, suggesting that the exchange of medium within itself stalled the growth somewhat.) Axons continued to retract with additional time in NOC-7, but given that retraction was so dramatic in neurons exposed for only 30 min, we performed all of our cytoskeletal studies on neurons at this time point.
Fig. 1.

The morphology of axons induced to retract by NOC-7. Cultures of chick sensory neurons were treated with 1 mm NOC-7 (nitric oxide donor). The majority of the axons in the culture showed significant retraction in response to the NOC-7 treatment over the first 30 min, as shown here. Three examples are shown in A–C, wherein the first panel of each pair shows the axons immediately before the addition of NOC-7, and the second panel of each pair shows the axons 30 min after the addition of NOC-7. The morphology of the retracted axons is characterized by a retraction bulb (arrows), a trailing remnant (white arrowheads), and sinusoidal bends along the axonal shaft (black arrowheads). D, The percentages of different axonal behaviors after NOC-7 treatment, compared with the percentages for control cultures (for more details, see Results). Scale bar, 20 μm.
Our studies focused specifically on the axons that showed retraction during the first 30 min of NOC-7 treatment. Phase-contrast images of these axons show that most of them are characterized by an enlarged distal region, a thin trailing remnant, and sinusoidal bends along the shaft of the axon (Fig. 1). This retraction morphology resembles that of axons retracting in vivo, which are also characterized by a distal enlargement termed the “retraction bulb” (Riley, 1981; Gan and Lichtman, 1998). The trailing remnant was not included in the length measurements.
Changes in cytoskeletal configuration during axonal retraction
In a first set of experiments on the cytoskeleton, we wanted to obtain a global sense of cytoskeletal configuration in retracting axons. For these experiments, after axons retracted in response to NOC-7, cultures were extracted and fixed simultaneously, as outlined in Materials and Methods, and then double-labeled for microtubules and microfilaments. The axons stained for both of these cytoskeletal elements in both control and NOC-7-treated axons (Fig.2). In control axons, microtubules appeared as a continuous dense bundle along the length of the axon that oftentimes splayed apart in the region of the growth cone. Staining for microfilaments was generally dim along the length of the axon but was bright in the peripheral regions of the growth cone. In the axons induced to retract by NOC-7, the distal enlarged region was particularly brightly stained for microtubules. Microtubules showed coiling and sinusoidal bending both in the distal enlargement and along the shaft, suggesting a backward retreat and reconfiguration of microtubules to accommodate shortening of the axon. Microfilaments were redistributed into a clump in the distal region of the axonal shaft, as would be expected if the peripheral growth-cone microfilaments folded inward during growth-cone collapse. Interestingly, these microtubule and microfilament reconfigurations were also observed in axons just starting to show signs of retraction, even before a detectable reduction in the length of the axon. Figure2B shows such an axon, which displays changes in microtubule and microfilament configuration that are similar to those seen for an axon that has retracted substantially (Fig. 2C), both of which are notably different from control axons showing no retraction (Fig. 2A). Qualitatively, there appears to be no detectable reduction in the total fluorescence intensity of either microtubules or microfilaments during axonal retraction. Additional dramatic examples of the coiling and bending of microtubules during axonal retraction are shown in Figure3.
Fig. 2.

Reconfiguration of cytoskeletal components in axons exposed to NOC-7. Double fluorescence labeling of microtubules and microfilaments reveals reconfiguration of both cytoskeletal components in axons exposed to NOC-7. For each axon, the first fluorescence image shows microtubules, the second image shows microfilaments, and the third imageshows a color overlay in which microtubule staining isred and microfilament staining is green.A, In a control axon, microtubules appear as a tight straight bundle, and microfilaments are concentrated in the growth cone. B, This axon was fixed immediately after the growth cone had collapsed in response to NOC-7, before any retraction of the axonal shaft occurred. Bending of the microtubules and redistribution of the microfilaments are already observed at this stage. C, This axon was fixed after retraction was well underway. Microtubules are reconfigured into coiled and sinusoidal bundles, and microfilaments are concentrated in the most distal region of the retracted axon. B, C, Phase-contrast images before and after retraction are shown at a lower magnification. Scale bars, 20 μm.
Fig. 3.

Microtubule reconfiguration in axons induced to retract by NOC-7. Single-label studies for microtubules in retracting axons are shown here. Phase-contrast images are shown (at a lower magnification) above corresponding β-tubulin images.Numerals represent the minutes after NOC-7 addition to the culture. A, Filamentous microtubule bundles are present in the retraction bulb (arrows).B, Microtubules have accumulated in a distal–proximal manner in the retracted region (the arrow shows the brightest region at the tip of the retraction bulb). The coiling of microtubule bundles in the shaft corresponds to the contour of the axon (arrowhead). C, Sinusoidal bending of microtubule bundles accommodates the shortening of the axon (arrows). Scale bars, 20 μm.
Quantitative analyses of microtubule levels in retracting and control axons
To quantitatively assess microtubule levels in control and retracting axons, we obtained images of axons under identical microscope, camera, and software settings and determined the fluorescence intensity per micrometer length of the axon. We also included in these studies axons that had undergone retraction in response to treatment with nocodazole, a potent microtubule depolymerizing agent. Nocodazole was applied at 10 μg/ml for 30 min before simultaneous extraction and fixation. Figure4 shows that the morphology of the axons retracting in response to nocodazole is markedly different from that of the axons retracting in response to NOC-7. Nocodazole-induced axonal retraction is characterized by the formation of multiple bead-like enlargements along the shaft and shortening and withering of the axon. Also, in some cases, nocodazole treatment induced the formation of fine lateral extensions from the axonal shaft [as has been observed previously in some studies in which axons were treated with antimicrotubule drugs (Bray et al., 1978; Joshi et al., 1986)]. Microtubule staining was very dim in these axons and showed no filamentous appearance (Fig. 4, last panel).
Fig. 4.
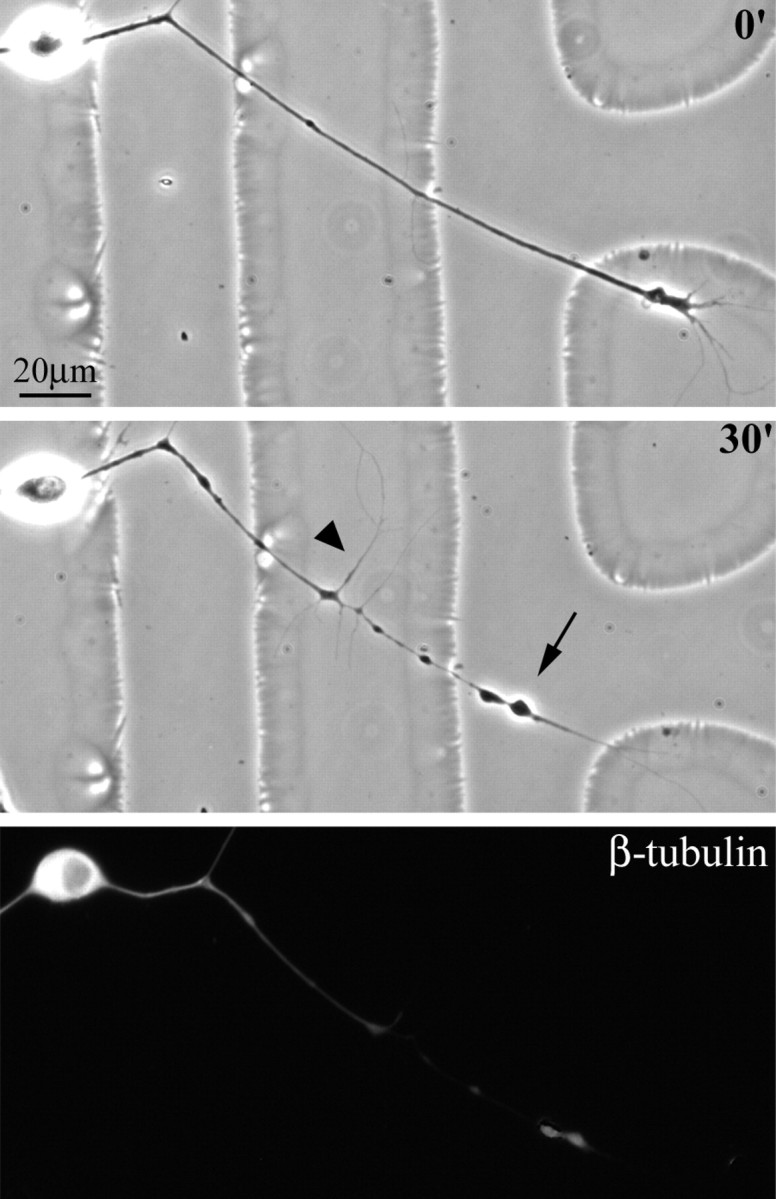
Retraction of axons in response to nocodazole. Axons treated for 30 min with nocodazole retracted with a morphology distinct from that of axons retracting in response to NOC-7. Treatment with 10 μg/ml nocodazole induced axons to retract with a morphology characterized by bead formation along the axon (arrow), outgrowth of fine lateral extensions (arrowhead), and the withering of the axonal shaft. Immunofluorescence staining of β-tubulin revealed a dramatic depletion of microtubules in axons treated with nocodazole. Scale bar, 20 μm.
Figure 5 shows the results of quantitative analyses on the levels of microtubules in control axons and axons induced to retract by either NOC-7 or nocodazole. Two values were calculated for each of the two types of retracting axons: (1) one was obtained by dividing the total fluorescence intensity by the axonal length after retraction, and (2) the other was obtained by dividing the total fluorescence intensity by the axonal length before retraction. Figure 5A (together with the figure legend) explains how these quantitative measurements were obtained. In NOC-7-treated cultures, the mean AFU per micrometer calculated for the length after retraction is significantly higher than that of control axons, whereas the mean AFU per micrometer calculated for the original length is essentially the same as that of the control axons (Fig. 5B). In contrast, nocodazole-treated cultures showed a significant reduction in microtubule levels to less than half of control levels (Fig.5C). These results show that there is significant microtubule depolymerization during axonal retraction induced by nocodazole, but that there is no detectable loss of microtubules during axonal retraction induced by NOC-7. The nocodazole experiment also demonstrates that our cofixation-extraction method indeed extracted free tubulin effectively, which is necessary for accurate assessment of polymer levels.
Fig. 5.

Quantitative data on microtubule levels in axons induced to retract by treatment with either NOC-7 or nocodazole. The method used to quantify microtubule levels is shown inA. Phase-contrast images of a representative neuron show two axons before and after 30 min of treatment with 1 mmNOC-7. Both axons retracted significantly. For each axon, the original length (Lo) and length after retraction (Lr) were measured. After simultaneous fixation/extraction to preserve microtubules while releasing free tubulin, neurons were immunostained for β-tubulin. Total fluorescence intensity within the axon (It) and background fluorescence intensity in a region alongside the axon (Ib) were measured in AFU. The difference between It and Ib represents the fluorescence intensity from the axon, adjusted for any nonspecific background fluorescence. To enable us to compare the microtubule levels in axons of different lengths, we calculated for each axon the fluorescence per micrometer of axonal length (AFU per micrometer) by dividing the (It − Ib) by either Lo and Lr for treated axons (see the equations) and by Lo only for control axons. B, Quantification of NOC-7-treated axons compared with controls. C, Quantification of nocodazole-treated axons compared with controls.B, C, The first bar in eachgraph shows control microtubule levels in parallel cultures prepared identically at the same time as the experimental cultures for the immunofluorescence analyses. The second bar in each graph shows the microtubule levels (per micrometer) in drug-treated axons, calculated for Lo. The third bar in each graph also shows microtubule levels in treated axons, but calculated for Lr. Separate control axons were analyzed for each experiment, and their mean AFU per micrometer values were normalized to 100 so that data from the different experiments could be compared. The two-tailed Student'st test (∗p < 0.05) shows a significant difference between control axons and NOC-7-treated axons when the calculations are performed using Lr. However, there is no significant difference between the control and the NOC-7-treated axons when Lo is used. In the case of the nocodazole-treated axons, there is a significant difference between controls and the treated axons calculated in both ways. Thus, there is no detectable microtubule loss in axons induced to retract by NOC-7; in contrast, there is dramatic microtubule loss in axons induced to retract by nocodazole. Scale bar, 20 μm.
Quantitative analyses on levels of microtubules enriched with tyrosinated tubulin during axonal retraction
Previous studies have established that axons contain populations of stable microtubule polymer and labile microtubule polymer (Baas and Black, 1990), and that the most distal region of the axon contains polymer that is potentially even more labile than that found elsewhere in the axon (Ahmad et al., 1993). The labile polymer undergoes more rapid bouts of subunit turnover, and this is true regardless of whether there is net assembly or disassembly of microtubules. With time after assembly, the β-tubulin that comprises the microtubules is gradually detyrosinated such that older, more stable regions of the microtubules contain less tyrosinated tubulin, whereas the more labile regions of the microtubules contain higher levels of tyrosinated tubulin (for discussion, see Baas et al., 1991). The fact that we observed no reduction of microtubule mass during axonal retraction suggests that both the stable and labile classes of polymer are not undergoing any significant loss. Nevertheless, because the labile polymer is normally undergoing more rapid dynamics, it would be more likely to be lost than the stable polymer. Indeed, previous qualitative observations suggest that there may be a partial loss of tyrosinated microtubules at the tips of axons retracting in response to other factors (but also see Fan et al., 1993; Fritsche et al., 1999). Therefore, to be particularly cautious, we focused one set of analyses specifically on the most labile polymer in the distal retracting regions of the axons. Because the distal regions of axons often stain more brightly for total tubulin, as well as tyrosinated tubulin, it is necessary to obtain a ratio image that reflects the amount of tyrosinated tubulin relative to the total tubulin in the microtubules (for details, see Yu et al., 1994). We examined ≥25 control axons, all of which consistently showed an enrichment in the ratio of tyrosinated/total tubulin in the microtubules occupying the distal region of the axon. This enrichment was also observed in 25 of 25 axons undergoing retraction in response to NOC-7, confirming that even the most labile microtubules are not undergoing any significant loss during retraction. These results are shown in quantitative pseudocolor in Figure6.
Fig. 6.

Analyses on the ratio of tyrosinated/total tubulin in microtubules during axonal retraction. A, Control axons. B, Axons retracting in response to treatment for 30 min with NOC-7. The first panel of each set shows staining for tyrosinated tubulin, whereas the second panel shows staining for total β-tubulin. Tyrosinated tubulin staining is brighter distally, but so too is total tubulin. Thethird panel of each set shows a ratio image of tyrosinated/total tubulin (see Materials and Methods and Results). The ratio image is displayed in a pseudocolor scale in whichwhite is the most intense and black is the least intense. (There is no white in these axons; hence,yellow is the brightest.) Note that the ratio is clearly highest distally in the control axons, indicating a concentration of microtubules particularly rich in tyrosinated tubulin. This same distal concentration is observed in retracting axons, indicating no loss in the distal enrichment of these tyrosinated tubulin-rich microtubules. Scale bar, 20 μm.
Effects of taxol treatment on axonal retraction induced by NOC-7
The results described thus far show that there is no detectable decrease in microtubule levels during axonal retractions induced by NOC-7, but that the microtubules change their configuration from a straight bundle into arrays of coiled and bended microtubules. The simplest interpretation of these results is that the microtubules do not undergo significant depolymerization during retraction but instead retreat backward. However, on the basis of the results presented thus far, we cannot completely dismiss the possibility that the microtubules are depolymerized in a wholesale manner and then repolymerized into coiled and bended arrays. To investigate this further, we exposed the axons for 30 min to 100 nm taxol, a drug that stabilizes microtubules against disassembly and promotes excess assembly. At this relatively low concentration, the taxol treatment in and of itself did not alter the morphology of the axons, which continued to grow normally over the 30 min of treatment (Fig.7A). Quantitative immunofluorescence analyses showed that the average microtubule levels within these axons increased by ∼30%. When taxol-treated axons were exposed to NOC-7 for 30 min (in the continuous presence of taxol), the axons showed dramatic retraction. The retractions occurred with the characteristic morphology of axons retracting to NOC-7 in the absence of taxol, with a classic retraction bulb, trailing remnant, and sinusoidal bends along the shaft (Fig. 7A) [in some other culture systems, higher doses of taxol have been reported to inhibit axonal retractions, but available evidence suggests that this may be caused by other effects of the drug rather than by the stabilization of microtubules (George et al., 1988; McNeil et al., 1999)]. Quantification of microtubule levels showed that the retracted axons have levels of microtubules that are statistically identical to those treated with taxol only (i.e., ∼30% higher than controls). Fluorescence images are shown in quantitative pseudocolor in Figure7B, and the data are displayed graphically in Figure7C. These results demonstrate that axonal retraction does not require microtubule depolymerization to occur, and that the same kind of microtubule reconfiguration occurs during retraction when microtubules are prevented from depolymerizing. Moreover, they show that these changes can occur even when the polymer levels are actually increasing above normal.
Fig. 7.

Axonal retraction induced by NOC-7 in the presence of taxol. A, Phase-contrast images of an axon before any treatment, after 30 min in taxol alone, and after 30 min of treatment with NOC-7 in the continued presence of taxol. Note that the taxol treatment did not prohibit axonal growth when applied without the NOC-7, nor did it prohibit axonal retraction after the NOC-7 was added.B, Microtubule immunofluorescence studies of three different axons: (1) a control axon, (2) an axon treated for 30 min with taxol alone, and (3) an axon treated with taxol for 30 min and then treated with both taxol and NOC-7 for an additional 30 min. Axons are shown in a quantitative pseudocolor scale in whichwhite is the most intense and black is the least intense. Note that the taxol-treated axon is brighter than the control axon. The taxol/NOC-7-treated axon is even brighter yet (per micrometer). C, Quantitative studies on fluorescence intensity, in arbitrary fluorescence units. Taxol treatment alone increased microtubule levels by ∼30% above control levels. Axons retracting in response to NOC-7 (in the continued presence of taxol) displayed a much higher yet fluorescence intensity per micrometer when calculated for length after retraction (Lr). However, when calculated for original length (Lo), the intensity per micrometer was indistinguishable from that of the axons treated with taxol alone. Thus, taxol treatment elevated total microtubule levels above controls, but this stimulation of microtubule assembly did not prohibit axonal retraction in response to NOC-7. The two-tailed Student'st test (∗p < 0.05) shows a significant difference between control axons and axons treated with taxol and a significant difference between control axons and axons treated with taxol followed by NOC-7. There was also a significant difference between axons exposed only to taxol and axons exposed to taxol followed by NOC-7, if calculations were performed using Lr. However, there was no significant difference between axons exposed only to taxol and axons exposed to taxol followed by NOC-7 if calculations were performed using Lo. Scale bars, 20 μm.
DISCUSSION
There has been great interest over the past few decades in how the cytoskeletal arrays of the axon are elaborated during its growth. This topic has been of particular interest both because of its importance and because of a great deal of heated controversy (for review and discussion, see Baas, 2002). Early models maintained that the cytoskeletal elements are transported down the axon in the form of assembled polymers and that this transport is fueled by molecules that we would today refer to as molecular motor proteins. This view was challenged in the mid-1980s by workers who felt that it was more reasonable that the polymers were stationary and that their subunit components were somehow transported down the axon. In this alternate model, the elaboration of the cytoskeletal arrays resulted from the polymerization of new cytoskeletal elements along the length of the axon during its growth. In recent years, the transport of cytoskeletal polymers has been directly observed (Dent et al., 1999; Roy et al., 2000; Wang et al., 2000) but so too has local polymer assembly (Dent et al., 1999; Kabir et al., 2001). It appears that the most satisfactory model includes both polymer transport and local assembly dynamics as key contributors to the elaboration of the axonal cytoskeletal arrays. In recent years, neuroscientists have begun to focus on the retraction of axons as an event that is equally noteworthy during the development of the nervous system. However, to date, most consideration of the cytoskeletal mechanisms involved in axonal retraction has been limited to the general concept that the polymers most probably depolymerize to accommodate the shortening of the axon during retraction.
As outlined in the introductory remarks, there are reasons to question this conclusion. Although it is true that pharmacologically depleting an axon of its microtubules can cause it to retract, the morphological features of the retraction are not at all similar to those of axons retracting in the developing nervous system or to axons retracting in the culture dish in response to physiological cues. Although some physiological cues appear to cause local actin depolymerization within growth cones (Fan et al., 1993) (for information regarding what underlies their collapse, see Fritsche et al., 1999), it is unlikely that there would be wholesale actin depolymerization within the axonal shaft. It has been shown repeatedly that axonal retraction is an actomyosin-based process and that depleting microfilaments from axons actually prohibits their retraction rather than causing it (Solomon and Magendantz, 1981; Joshi et al., 1985; Ahmad et al., 2000). In addition, recent studies have even called into question whether microfilaments are always depolymerized in growth cones during their collapse; these studies suggest that the microfilaments can also be debundled and reconfigured, with no significant net diminution in polymer levels (Fournier et al., 2000; Zhou and Cohan, 2001). Our qualitative observations suggest that microfilaments are folded inward during growth-cone collapse in response to nitric oxide, whereas other studies using nitric oxide indicate that there is some local depolymerization of actin as well (Ernst et al., 2000). It is our impression, however, that phalloidin staining favors actin bundles over debundled filaments, and, hence, we feel that additional studies are merited to fully resolve this issue. Our focus was on microtubules.
Our analyses on microtubules resulted in four major findings. First, as in previous studies using physiological inducers of axonal retraction, the morphology of the retracting axons was characterized by an enlarged distal region, a thin trailing remnant, and sinusoidal bends along the axonal shaft. There were no signs of the kind of beading and withering observed in axons depleted of microtubules with nocodazole. Second, fluorescence imaging of microtubules revealed that in the retracting axons, microtubules are still abundant but reconfigured into coiling and sinusoidal arrays to accommodate the shortening of the axon. Quantification of microtubule levels showed no detectable diminution in microtubule levels during retraction. Third, there was no reduction in the retracting axons of the distal concentration of the more labile microtubule polymer enriched in tyrosinated tubulin, indicating that even the polymer turning over most rapidly is not lost during retraction. Finally, pretreatment of the axons with 100 nmtaxol, a drug that stabilizes microtubules against disassembly and actually caused a 30% increase in microtubule levels in our studies, did not abolish axonal retraction induced by the nitric oxide donor. Thus, not only is there no loss in microtubule levels during retraction, but we can also conclude that the microtubules do not undergo any wholesale depolymerization followed by repolymerization to produce the newly configured coiled and bent microtubule arrays, because the same response is obtained under conditions in which polymer levels are actually rising.
Together, all of these observations demonstrate that there is no wholesale depolymerization of microtubules during the axonal retractions that we have observed, but instead that the pre-existing microtubules retreat backward. We cannot dismiss the possibility that there might be some depolymerization of microtubules during retraction, especially in the latter stages of axonal retraction, as the microtubules are ultimately chased back into the cell body. Another factor that should be considered is the ongoing addition of microtubules to the axon by anterograde transport from the cell body; this transport may or may not persist during axonal retraction but could potentially offset a small amount of polymer that does in fact depolymerize during retraction. Despite these caveats, our data clearly show that the early stages of axonal retraction cannot be explained in terms of polymer loss but instead appear to be the result of forces that impinge on the microtubule array to reconfigure it. Consistent with this conclusion, we reported previously that axons retract with similar characteristics when at least one microtubule-associated motor protein is experimentally manipulated (Ahmad et al., 2000). Specifically, we showed that inhibition of cytoplasmic dynein causes axons to retract with no observable loss of microtubules but with bending and coiling of the axon and the microtubules that were similar (but not identical) to the bending and coiling observed in the present study in response to nitric oxide. On this basis, and based on the fact that retraction itself is dependent on myosin-driven forces, we have proposed a new model for axonal retraction based on reconfiguration of cytoskeletal polymers by motor-driven forces (Baas and Ahmad, 2001).
In our previous study, we suggested that dynein-driven forces between the microtubule and microfilament arrays may attenuate the myosin-based contractility of the axon. If this is correct, physiological signals that induce retraction might function by uncoupling cytoplasmic dynein from either the microtubules or the actin. Alternatively, the signals may simply increase the contractility of the myosin so that it overwhelms the dynein forces (e.g., by enhancing phosphorylation of myosin regulatory light chain) (Billuart et al., 2001). Another possibility is that there are additional motors, specialized kinesins, that contribute directly to the reconfiguration of the microtubules. Some support for this possibility comes from our observations that the microtubules begin to show coiling and bending even before there is any detectable actomyosin-based shortening of the axon. Additional support comes from the fact that the retractions observed with nitric oxide were not completely identical to those observed with dynein inhibition; the coiling of microtubules was more dramatic with the nitric oxide, suggesting that additional motor forces may be involved.
Of course, it is possible that different physiological signals elicit their effects by different pathways; some may affect myosins, and others may affect cytoplasmic dynein or particular kinesins. We also cannot eliminate the possibility that some inducers of retraction might cause large-scale depolymerization of microtubules; our studies have focused exclusively on nitric oxide, and there is certainly a wide variety of physiological factors that can induce axons to retract. To the best of our knowledge, however, there are no observations of axons retracting physiologically that display characteristics of widespread or large-scale microtubule depolymerization, and the results of previous analyses on other types of retracting axons, although not quantitative, are entirely consistent with the conclusions of our study (Miller et al., 1994; McNeil et al., 1999). We believe that the next important step in understanding cytoskeletal reconfiguration during axonal retraction is to elucidate how the biochemical pathways affected by physiological cues alter the forces generated by molecular motor proteins.
Footnotes
This work was supported by two grants to P.W.B. from the National Institutes of Health. We thank Gianluca Gallo and Paul Letourneau of the University of Minnesota for helpful advice. We also thank Dan Buster and Doug Baird of our institution for assistance, advice, and discussions.
Correspondence should be addressed to Peter W. Baas, Department of Neurobiology and Anatomy, Drexel University College of Medicine, 2900 Queen Lane, Philadelphia, PA 19129. E-mail peter.w.baas@drexel.edu.
REFERENCES
- 1.Ahmad FJ, Pienkowski TP, Baas PW. Regional differences in microtubule dynamics in the axon. J Neurosci. 1993;13:856–866. doi: 10.1523/JNEUROSCI.13-02-00856.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmad FJ, Hughey J, Wittmann T, Hyman A, Greaser M, Baas PW. Motor proteins regulate force interactions between microtubules and microfilaments in the axon. Nat Cell Biol. 2000;2:276–280. doi: 10.1038/35010544. [DOI] [PubMed] [Google Scholar]
- 3.Baas PW. Microtubule transport in the axon. Int Rev Cytol. 2002;212:41–62. doi: 10.1016/s0074-7696(01)12003-6. [DOI] [PubMed] [Google Scholar]
- 4.Baas PW, Ahmad FJ. Force generation by cytoskeletal motor proteins as a regulator of axonal elongation and retraction. Trends Cell Biol. 2001;11:244–249. doi: 10.1016/s0962-8924(01)02005-0. [DOI] [PubMed] [Google Scholar]
- 5.Baas PW, Black MM. Individual microtubules in the axon consist of domains that differ in both composition and stability. J Cell Biol. 1990;111:495–509. doi: 10.1083/jcb.111.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baas PW, Slaughter T, Brown A, Black MM. Microtubule dynamics in axons and dendrites. J Neurosci Res. 1991;30:134–153. doi: 10.1002/jnr.490300115. [DOI] [PubMed] [Google Scholar]
- 7.Bandtlow C, Zachleder T, Schwab ME. Oligodendrocytes arrest neurite growth by contact inhibition. J Neurosci. 1990;10:3837–3848. doi: 10.1523/JNEUROSCI.10-12-03837.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bastmeyer M, Stuermer CA. Behavior of fish retinal growth cones encountering chick caudal tectal membranes: a time-lapse study on growth cone collapse. J Neurobiol. 1993;24:37–50. doi: 10.1002/neu.480240104. [DOI] [PubMed] [Google Scholar]
- 9.Bernstein M, Lichtman JW. Axonal atrophy: the retraction reaction. Curr Opin Neurobiol. 1999;9:364–370. doi: 10.1016/s0959-4388(99)80053-1. [DOI] [PubMed] [Google Scholar]
- 10.Billuart P, Winter CG, Maresh A, Zhao X, Luo L. Regulating axon branch stability: the role of p190 RhoGAP in repressing a retraction signaling pathway. Cell. 2001;107:195–207. doi: 10.1016/s0092-8674(01)00522-0. [DOI] [PubMed] [Google Scholar]
- 11.Bray D, Thomas C, Shaw G. Growth cone formation in cultures of sensory neurons. Proc Natl Acad Sci USA. 1978;75:5226–5229. doi: 10.1073/pnas.75.10.5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cowan WM, Fawcett JW, O'Leary DDM, Stanfield BB. Regressive events in neurogenesis. Science. 1984;225:1258–1265. doi: 10.1126/science.6474175. [DOI] [PubMed] [Google Scholar]
- 13.Cramer KS, Leamey CA, Sur M. Nitric oxide as a signaling molecule in visual system development. Prog Brain Res. 1998;118:101–114. doi: 10.1016/s0079-6123(08)63203-3. [DOI] [PubMed] [Google Scholar]
- 14.Daniels MP. Fine structural changes in neurons and nerve fibers associated with colchicine inhibition of nerve fiber formation in vitro. J Cell Biol. 1973;58:463–470. doi: 10.1083/jcb.58.2.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dent EW, Meiri KF. Distribution of phosphorylated GAP-43 (neuromodulin) in growth cones directly reflects growth cone behavior. J Neurobiol. 1998;35:287–299. doi: 10.1002/(sici)1097-4695(19980605)35:3<287::aid-neu6>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 16.Dent EW, Callaway JL, Szebenyi G, Baas PW, Kalil K. Reorganization and movement of microtubules in axonal growth cones and developing interstitial branches. J Neurosci. 1999;19:8894–8908. doi: 10.1523/JNEUROSCI.19-20-08894.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ernst AF, Gallo G, Letourneau PC, McLoon SC. Stabilization of growing retinal axons by the combined signaling of nitric oxide and brain-derived neurotrophic factor. J Neurosci. 2000;20:1458–1469. doi: 10.1523/JNEUROSCI.20-04-01458.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan J, Mansfield SG, Redmond T, Gordon-Weeks PR, Raper JA. The organization of F-actin and microtubules in growth cones exposed to a brain-derived collapsing factor. J Cell Biol. 1993;121:867–878. doi: 10.1083/jcb.121.4.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fournier AE, Nakamura F, Kawamoto S, Goshima Y, Kalb RG, Strittmatter SM. Semaphorin3A enhances endocytosis at sites of receptor-F-actin colocalization during growth cone collapse. J Cell Biol. 2000;149:411–421. doi: 10.1083/jcb.149.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fritsche J, Reber BF-X, Schindelholz B, Bandtlow CE. Differential cytoskeletal changes during growth cone collapse in response to hSema III and thrombin. Mol Cell Neurosci. 1999;14:398–418. doi: 10.1006/mcne.1999.0777. [DOI] [PubMed] [Google Scholar]
- 21.Gan WB, Lichtman JW. Synaptic segregation at the developing neuromuscular junction. Science. 1998;282:1508–1511. doi: 10.1126/science.282.5393.1508. [DOI] [PubMed] [Google Scholar]
- 22.George EB, Schneider BF, Lasek RJ, Katz MJ. Axonal shortening and the mechanisms of axonal motility. Cell Motil Cytoskeleton. 1988;9:48–59. doi: 10.1002/cm.970090106. [DOI] [PubMed] [Google Scholar]
- 23.Godement P, Wang LC, Mason CA. Retinal axon divergence in the optic chiasm: dynamics of growth cone behavior at the midline. J Neurosci. 1994;14:7024–7039. doi: 10.1523/JNEUROSCI.14-11-07024.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joshi HC, Chu D, Buxbaum RE, Heidemann SR. Tension and compression in the cytoskeleton of PC12 neurites. J Cell Biol. 1985;101:697–705. doi: 10.1083/jcb.101.3.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joshi HC, Baas P, Chu DT, Heidemann SR. The cytoskeleton of neurites after microtubule depolymerization. Exp Cell Res. 1986;163:233–245. doi: 10.1016/0014-4827(86)90576-8. [DOI] [PubMed] [Google Scholar]
- 26.Kabir N, Schaefer AW, Nakhost A, Sossin WS, Forscher P. Protein kinase C activation promotes microtubule advance in neuronal growth cones by increasing average microtubule growth lifetimes. J Cell Biol. 2001;152:1033–1044. doi: 10.1083/jcb.152.5.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaethner RJ, Stuermer CA. Dynamics of terminal arbor formation and target approach of retinotectal axons in living zebrafish embryos: a time-lapse study of single axons. J Neurosci. 1992;12:3257–3271. doi: 10.1523/JNEUROSCI.12-08-03257.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McNeil RS, Swann JW, Brinkley BR, Clark GD. Neuronal cytoskeletal alterations evoked by a platelet-activating factor (PAF) analogue. Cell Motil Cytoskeleton. 1999;43:99–113. doi: 10.1002/(SICI)1097-0169(1999)43:2<99::AID-CM2>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 29.Miller JD, Hadley RD, Hammond CE. Growth cone collapse and neurite retraction from cultured Helisoma neurons in response to antibody Fab fragments against an extracellular matrix protein. Brain Res Dev Brain Res. 1994;79:203–218. doi: 10.1016/0165-3806(94)90125-2. [DOI] [PubMed] [Google Scholar]
- 30.Mize RR, Wu HH, Cork RJ, Scheiner CA. The role of nitric oxide in development of the patch-cluster system and retinocollicular pathways in the rodent superior colliculus. Prog Brain Res. 1998;118:133–152. doi: 10.1016/s0079-6123(08)63205-7. [DOI] [PubMed] [Google Scholar]
- 31.Nagashima M, Dent EW, Shi XZ, Kalil K. Cortical neurite outgrowth and growth cone behaviors reveal developmentally regulated cues in spinal cord membranes. J Neurobiol. 1999;39:393–406. doi: 10.1002/(sici)1097-4695(19990605)39:3<393::aid-neu6>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 32.Riley DA. Ultrastructural evidence for axon retraction during the spontaneous elimination of polyneuronal innervation of the rat soleus muscle. J Neurocytol. 1981;10:425–440. doi: 10.1007/BF01262414. [DOI] [PubMed] [Google Scholar]
- 33.Roy S, Coffee P, Smith G, Liem RKH, Brady ST, Black MM. Neurofilaments are transported rapidly but intermittently in axons: implications for slow axonal transport. J Neurosci. 2000;20:6849–6861. doi: 10.1523/JNEUROSCI.20-18-06849.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith CL. Cultures from chick peripheral ganglia. In: Banker G, Goslin K, editors. Culturing nerve cells, Ed 2. MIT; Cambridge, MA: 1998. pp. 261–287. [Google Scholar]
- 35.Solomon F, Magendantz M. Cytochalasin separates microtubule disassembly from loss of asymmetric morphology. J Cell Biol. 1981;89:157–161. doi: 10.1083/jcb.89.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song HJ, Poo MM. Signal transduction underlying growth cone guidance by diffusible factors. Curr Opin Neurobiol. 1999;9:355–363. doi: 10.1016/s0959-4388(99)80052-x. [DOI] [PubMed] [Google Scholar]
- 37.Stanfield BB, O'Leary DDM, Fricks C. Selective collateral elimination in early postnatal development restricts cortical distribution of rat pyramidal tract neurones. Nature. 1982;298:371–373. doi: 10.1038/298371a0. [DOI] [PubMed] [Google Scholar]
- 38.Tessier-Lavigne M, Goodman CS. The molecular biology of axon guidance. Science. 1996;274:1123–1133. doi: 10.1126/science.274.5290.1123. [DOI] [PubMed] [Google Scholar]
- 39.Wang L, Ho CL, Sun D, Liem RKH, Brown A. Rapid movement of axonal neurofilaments interrupted by prolonged pauses. Nat Cell Biol. 2000;2:137–141. doi: 10.1038/35004008. [DOI] [PubMed] [Google Scholar]
- 40.Yamada KM, Spooner BS, Wessells NK. Axon growth: roles of microfilaments and microtubules. Proc Natl Acad Sci USA. 1970;66:1206–1212. doi: 10.1073/pnas.66.4.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu W, Baas PW. The growth of the axon is not dependent upon net microtubule assembly at its distal tip. J Neurosci. 1995;15:6827–6833. doi: 10.1523/JNEUROSCI.15-10-06827.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu W, Ahmad FJ, Baas PW. Microtubule fragmentation and partitioning in the axon during collateral branch formation. J Neurosci. 1994;14:5872–5884. doi: 10.1523/JNEUROSCI.14-10-05872.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou FQ, Cohan CS. Growth cone collapse through coincident loss of actin bundles and leading edge actin without actin depolymerization. J Cell Biol. 2001;153:1071–1083. doi: 10.1083/jcb.153.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]